

Abstract
Immunoglobulin A nephropathy is the most common primary glomerulonephritis worldwide, and it can be associated with liver disease. However, cases of Immunoglobulin A nephropathy secondary to Wilson’s disease are very rare. A 20-year-old Japanese man presented with microscopic hematuria, proteinuria, and renal dysfunction. A renal biopsy showed mesangial cell proliferation, immunoglobulin A deposition, and electron-dense deposit in the mesangial areas, all of which are consistent with Immunoglobulin A nephropathy. Computed tomography of the abdomen showed liver atrophy and splenomegaly, and the diagnosis of Wilson’s disease was confirmed with decreased serum ceruloplasmin levels, increased urinary copper excretion, Kayser–Fleischer rings and copper deposition in the liver biopsy. The patient was treated successfully with trientine hydrochloride and zinc acetate and showed improvement in renal manifestations. Wilson’s disease is a rare cause of secondary Immunoglobulin A nephropathy. We recommend that Wilson’s disease should be considered the cause of secondary Immunoglobulin A nephropathy in juvenile patients with hematuria, proteinuria, and splenomegaly and suggest measuring the serum ceruloplasmin concentrations, urinary copper excretion, and evaluating Kayser–Fleischer rings in these patients.
Keywords: Wilson’s disease, IgA nephropathy, Trientine hydrochloride, Zinc acetate
Introduction
Immunoglobulin A nephropathy (IgAN) is the most common primary glomerulonephritis worldwide. It usually occurs between the age of 20 and 30 years, and approximately 30–40% of patients requires renal replacement therapy after 20 years of diagnosis [1, 2]. IgAN also has been reported in association with liver disease, and it accounts for 9% of all biopsy-proven cases of IgAN [3]. However, cases of IgAN associated with Wilson’s disease (WD) are scarce. Here, we report a rare case of a 20-year-old Japanese man with IgAN secondary to WD who demonstrated proteinuria, microscopic hematuria, renal dysfunction, and splenomegaly. In addition, we reviewed the literature of such cases.
Case report
A 20-year-old Japanese man was referred to our medical center with microscopic hematuria, proteinuria and worsening of the serum creatinine level after an episode of upper respiratory infection. He had a baseline creatinine level of 0.7 mg/dL. There was no complaint of arthralgia, myalgia, or abdominal pain. The patient had no history of any drug abuse or known allergies, and he never smoked or drank alcohol. Additionally, he had no family history of kidney or liver disease. On examination, his respiratory rate was 12 breaths per minute, his heart rate was 66 beats per minute, his blood pressure was 118/64 mmHg, he had a saturation of 100% in room air, and his body temperature was 36.5 °C. Notable examination findings included splenomegaly. Neurologic examination was unremarkable. There were no icteric sclerae, oropharyngeal exudate, ascites, or pretibial edema, and no stigmata of chronic liver disease. Laboratory findings (Table 1) included mild thrombocytopenia (platelet count 11.8 × 109/L) and a normal white blood cell level (7810 × 106/L). A moderately decreased kidney function (blood urea nitrogen: 12.0 mg/dL; serum creatinine: 1.51 mg/dL; estimated glomerular filtration rate: 62 ml/min/1.73 m2) was noted. Additionally, the prothrombin time was 13.2 s (control: 10–13 s), the bilirubin level was 0.8 mg/dL (normal: 0.3–1.2 mg/dL), the alanine transaminase 15 U/L (normal: 5–45 U/L), the aspartate aminotransferase level was 23 U/L (normal: 5–45 U/L), the alkaline phosphatase 406 U/L (normal: 104–398 U/L), the lactate dehydrogenase level was 109 U/L (normal: 120–245 U/L), the total protein was 7.1 g/dL, and the albumin level was 4.2 g/dL. The direct Coombs test was negative. The immunoglobulin levels were normal (IgG, 963 mg/dL; IgM, 80 mg/dL), except for mild elevation of IgA (442 mg/dl). Antinuclear antibodies and antineutrophil cytoplasm antibodies were negative, and complement levels were also normal (C3, 111 mg/dL; C4, 43 mg/dL; CH50, 48 IU/mL). Urinalysis showed mild proteinuria (0.3 g/day) with microhematuria (> 100 erythrocytes per high-power field) and a few red blood cell casts. A rapid strep test on a throat culture was negative. Viral screenings for hepatitis B and C were negative. Plain computed tomography of the abdomen showed mild liver atrophy and splenomegaly; the size of the kidneys was normal. Renal biopsy showed 25 glomeruli, there was proliferation of mesangial cells (Fig. 1), none of them (0%) had global sclerosis and 1 (4%) had cellular crescent formation. There was no interstitial change or arterial lesions (Oxford classification: M0 E0 S0 T0). Immunofluorescence microscopy showed deposition of IgA (Fig. 2) and C3 (Fig. 3). Additionally, electron microscopy showed electron-dense deposit in the mesangial areas (Fig. 4). Wilson’s disease, alpha 1 anti-trypsin deficiency, and hemochromatosis were considered because the patient presented with liver atrophy and splenomegaly without a clear etiology. Laboratory findings were notable for the low level of serum copper concentration (21 µg/day), low level of serum ceruloplasmin (4.5 mg/dL) and elevated urine copper excretion (254 µg/day); Kayser–Fleischer rings were also found in the periphery of the cornea, all of which fulfilled the definite diagnosis of Wilson’s disease based on European Association for the Study of the Liver guidelines [4]. The alpha 1 anti-trypsin level (209 mg/dL), serum ferritin levels (209 ng/dL), and transferrin saturation (26%) were normal. Wilson’s disease was confirmed by liver biopsy, which showed copper deposition (Fig. 5), and a homozygous mutation in the gene ATP7B (R778L), the most common type of mutation in Japanese patients, was confirmed on genetic testing. However, light microscopy of the kidney did not show copper deposition in the renal tubules (Fig. 6). The final diagnosis of IgA nephropathy secondary to Wilson’s disease was made. Figure 7 shows clinical course of this patient. Six months after initiating 1.5 g of trientine hydrochloride and 150 mg of zinc acetate, liver enzymes remained normal, and the serum copper concentration decreased from 21 to 15 µg/day without any adverse effect. Microhematuria disappeared. Additionally, proteinuria declined from 0.31 to 0.09 g/day, and his serum creatinine improved (1.51–1.20 mg/dl). The serum IgA level decreased from 442 to 309 mg/dl. The patient visited our outpatient clinic every 3 months and was assessed with excellent medication adherence to date. We also recommended participating in patients’ associations to support the patient and the family.
Table 1.
Laboratory data
| Hemogram and coagulation | Blood chemistry | Urinalysis |
|---|---|---|
| WBC 5500/µl (3,040 − 8,540) | TP 7.1 g/dl (6.6–8.0) | Protein 0.3 g/day |
| RBC 492 × 104/µl (378–499 × 104) | Alb 4.2 g/dl (4.1-5.0) BUN 12.0 mg/dl (8–20) |
RBC > 100 /HPF WBC 1–4 /HPF |
| Hb 13.8 g/dl (10.8–14.9) | Cre 1.30 mg/dl (0.5–0.8) | RBC casts (+) |
| Hct 36.0% (35.6–45.4) | UA 1.9 mg/dl (2.9–5.2) | |
| Plt 11.8 × 104/µl (15.0-36.1 × 104) | Na 139 mEq/l (138–146) | Other tests |
| K 4.0 mEq/l (3.6–4.9) | Serum ceruloplasmin 4.5 mg/dl (21–37) | |
| PT 13.2 s (10–13 s) | Cl 103 mEq/l (99–109) | |
| APTT 34.9% (control 28.3%) | Ca 8.9 mg/dl (8.7–10.0) | α1 anti-trypsin 209 mg/dl (150–270) |
| Direct coombs test (−) | iP 1.8 mg/dl (2.5–4.6) | |
| Infection | AST 23 IU/l (13–33) | Serum copper 21 µg/dl (66–130) |
| HBs-Ag (−) | ALT 15 IU/l (6–27) | Urine copper 254 µg/day (< 40) |
| HBs-Ab (−) | LDH 109 IU/l (119–229) | |
| HBe-Ag (−) | ALP 406 IU/l (115–379) | |
| HBc-Ab (−) | Total bilirubin 0.8 mg/dl (0.3–1.2) | |
| HCV-Ab (−) | C3 111 mg/dl (65–135) | |
| Immunological examinations | C4 43 mg/dl (13–35) | |
| CRP 0.2 mg/dl (< 0.3) | CH50 48 U/ml (28–53) | |
| IgG 963 mg/dl (870–1700) | ANA (−) | |
| IgA 442 mg/dl (90–400) | MPO-ANCA (−) | |
| IgM 80 mg/dl (35–220) | PR3-ANCA (−) | |
| C1q-immune complex (−) | ||
| Cryoglobulin (−) | ||
| Fe 59 µg/dl | ||
| TIBC 281 µg/dl | ||
| Ferritin 209 µg/dl |
Fig. 1.
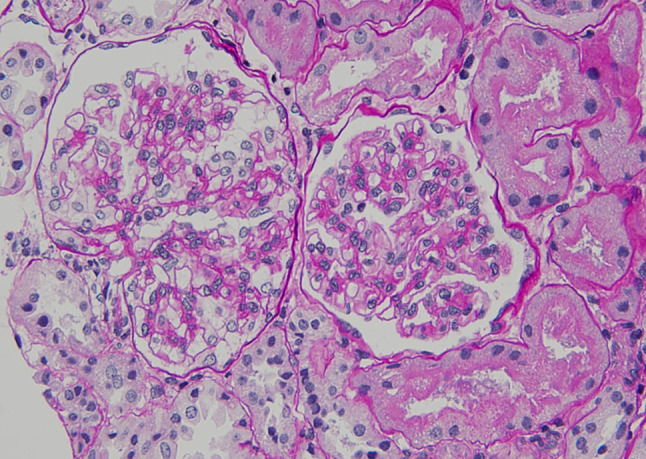
Light microscopy of the kidney showed proliferation in the mesangial matrix of the glomeruli (Hematoxylin-eosin, × 400)
Fig. 2.
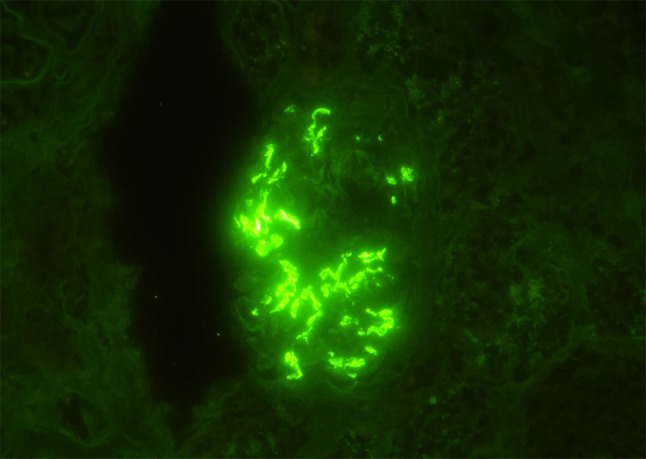
Immunofluorescence staining of the kidney showed bright granular staining in the glomeruli (IgA immunofluorescence, × 400)
Fig. 3.
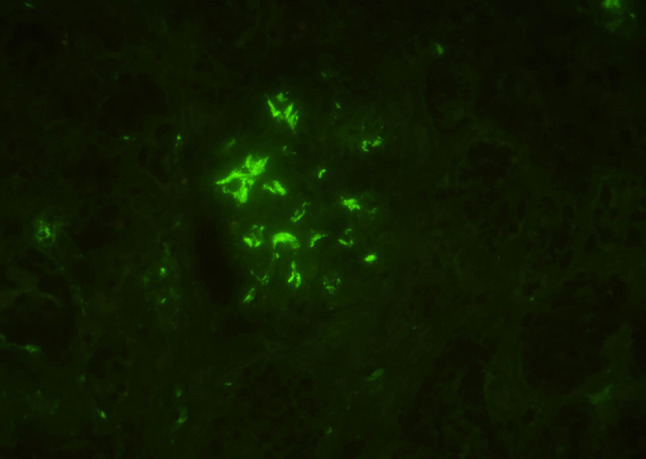
Immunofluorescence staining of the kidney showed granular staining in the glomeruli (C3 immunofluorescence, × 400)
Fig. 4.
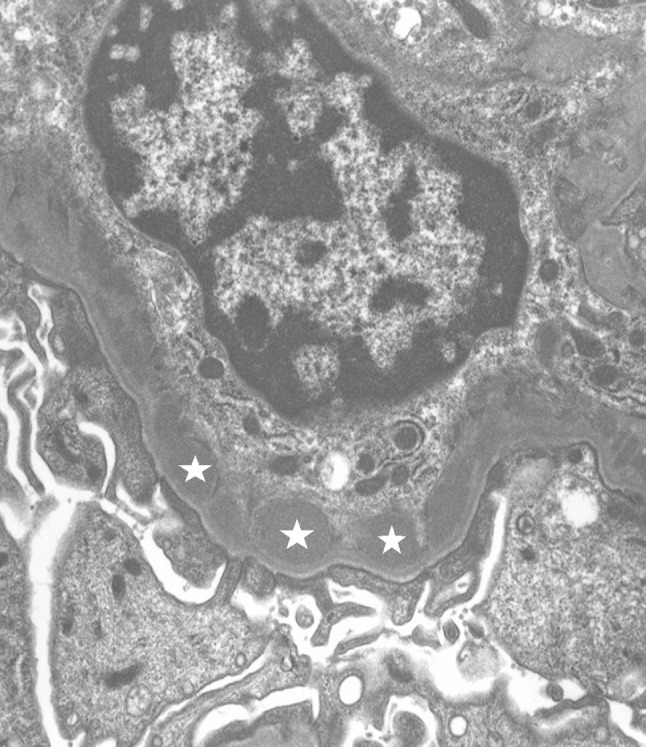
Electron microscopy showed electron-dense deposit (star) in the mesangial area (× 7500)
Fig. 5.
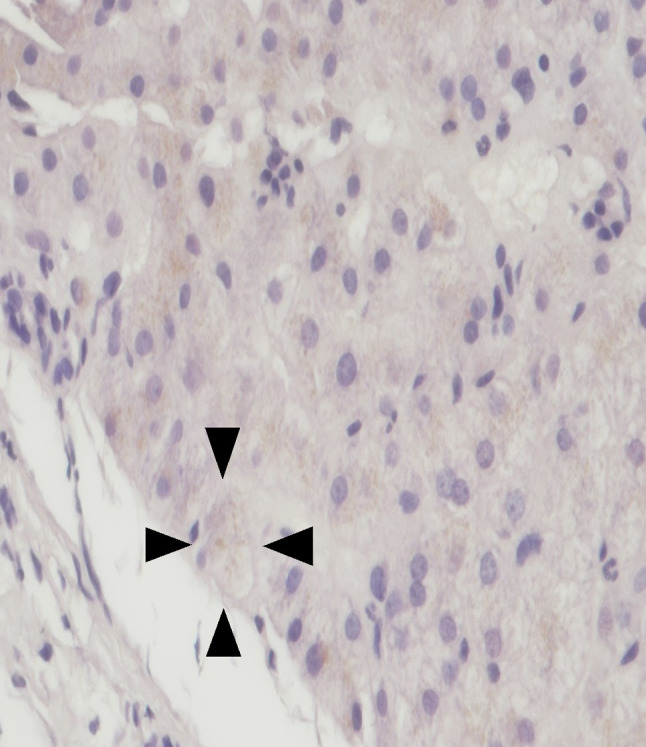
Light microscopy of the liver showed brownish copper deposition (arrowhead) (Copper, × 400)
Fig. 6.
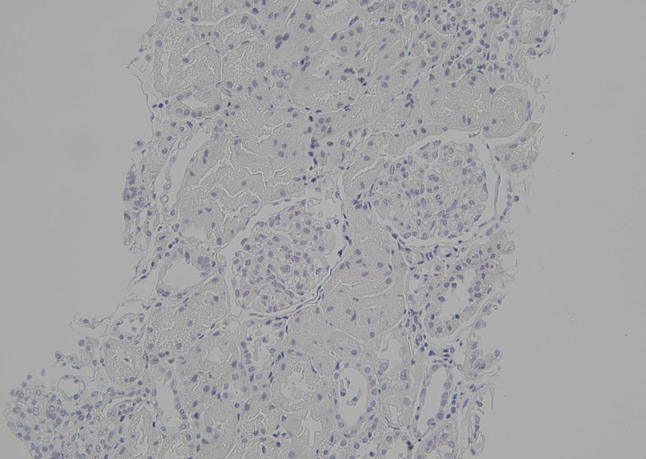
Light microscopy of the kidney showed no copper deposition (Copper, × 20)
Fig. 7.

Clinical course
Discussion
In the present case, we made two important clinical observations. First, we have reported the first adult case of IgAN secondary to WD. WD is a genetic autosomal recessive disease characterized by copper accumulation in the liver, eyes, and nerves [5]. Although several cases presenting with hypercalciuria and nephrocalcinosis were reported [6–8], renal abnormality is relatively rare. Another study reported that only 5 (6%) had both hematuria and proteinuria among 85 children with WD [9]. However, case reports of IgAN associated with WD are scarce, and only two cases in children were reported. These reports are summarized in Table 2, one concerned a 9 year-old-girl, and another concerned a child younger than 14 years old (gender unknown); both were treated with zinc sulfate [9, 10]. To our knowledge, no previous adult case of IgAN secondary to WD has been reported.
Table 2.
Case summary of IgA nephropathy secondary to Wilson’s disease
| Case | Age/Gender | SCr (mg/dl) | UP (g/day) | Medical management | Outcome |
|---|---|---|---|---|---|
| Zhuang U [9] | < 14/Unknown | Unknown | Unknown | Zinc sulfate | Unknown |
| Tu J [10] | 9/Female | Unknown | 0.09 | Zinc sulfate | After 2 mons UP (−) URBC 3/4 /HPF |
| Our case | 20/Male | 1.51 | 0.3 | Trientine HCl Zinc acetate |
UP 0.09 g/day SCr 1.20 mg/dl URBC 1–4 /HPF Urine Cu 154 µg/day Plasma Cu 15 µg/day |
SCr serum creatinine, UP urinary protein, URBC urinary red blood cells, HCl hydrochloride, mons months, HPF high-power field, Cu copper
Our second observation is that this case showed clinical improvement using chelating agents alone. A previous pediatric case reported on the short-term clinical course using zinc treatment [10]. However, to the best of our knowledge, this is the first case of IgA secondary to WD reporting successful treatment with chelating agents in an adult patient. The serum creatinine level, proteinuria, and hematuria were gradually relieved after initiating the chelating treatments, supporting the diagnosis of IgAN secondary to WD. There is a possibility that the treatment of WD improved renal hemodynamics, leading to the improvement of renal function and urine abnormalities. In terms of the pathogenesis of hepatic IgAN, several hypotheses have been suggested: high levels of circulating IgA immune complexes, a reduction in the fractional catabolism of IgA and its complexes, and dysregulation of the IgA immune system; however, the precise pathogenesis remains uncertain [3]. We may speculate on the pathogenesis of this case based on a model suggested by Novak et al. [11]. First, the space of Disse was obstructed due to WD. Second, B-cells and plasma cells produced large amounts of polymeric IgA1 and some galactose-deficient IgA1s, and the galactose-deficient IgA1s were recognized by anti-glycan IgG or IgA antibodies. Both polymeric IgA1s and galactose-deficient IgA1–IgG immune complexes were too bulky to reach the asialoglycoprotein receptors on hepatocytes, but they could pass through the larger fenestrae in glomerular capillaries overlying the mesangium, inducing glomerular injury. This speculation is supported by several reasons. First, the serum IgA levels declining after initiating chelating agents. Second, the immunofluorescence staining of IgA1 was brighter than that of IgA2 (Fig. 8). Third, galactose-deficient IgA1 (Gd-IgA1) was deposited using the Gd-IgA1-specific monoclonal antibody KM55, as shown in Fig. 9 [12].
Fig. 8.
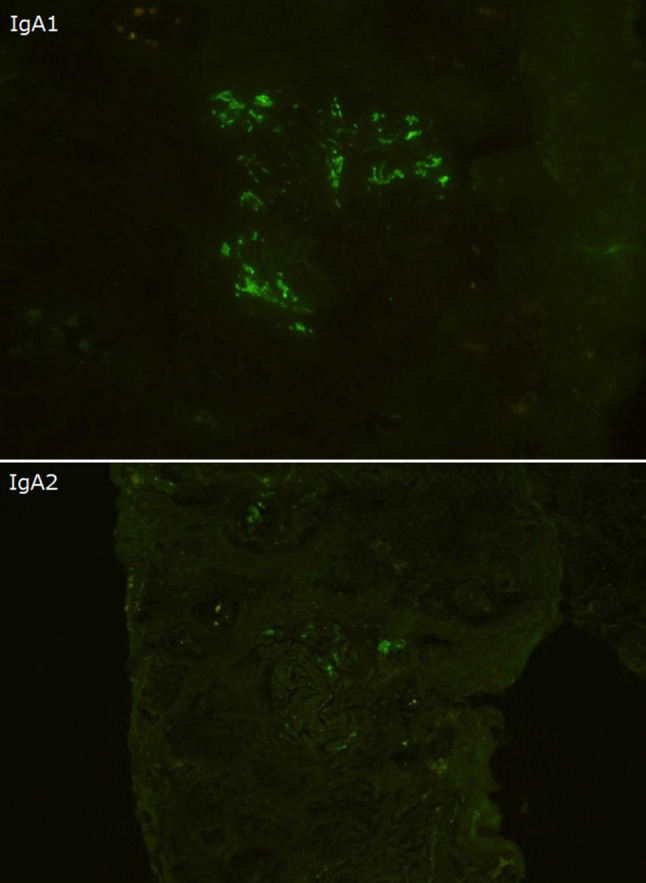
Immunofluorescence staining of IgA1 was brighter than that of IgA2 (IgA1 and IgA2 immunofluorescence, × 400)
Fig. 9.
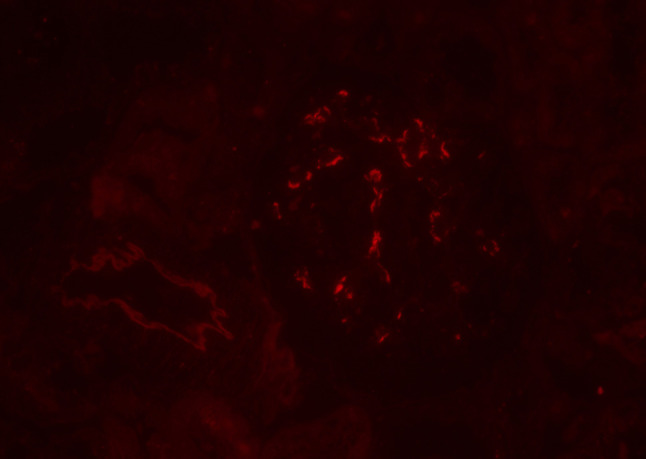
Immunofluorescence staining of Gd-IgA1-specific monoclonal antibody KM55 (immunofluorescence, × 400)
According to the Kidney Disease: Improving Global Outcomes guidelines [13], angiotensin-converting enzyme inhibitor/angiotensin II-receptor blocker and glucocorticoid therapy are recommended for patients with IgAN to control proteinuria. However, we reserved the treatments because the patient had a normal blood pressure and the urinary protein excretion was less than 1 g per day. Regarding liver transplantation in patients with WD, the revised Wilson’s disease prognostic index (RWPI) greater than 11 had a very high risk of death if liver transplantation is not performed [14, 15]. Liver transplantation was reserved for this patient because he had an RWPI of 1, and the patient was expected to improve with chelating agents alone.
This case report has several limitations. First, IgAN can be associated with WD incidentally, but the improvement in renal manifestations after giving chelating agents, the brighter immunofluorescence staining of IgA1 than that of IgA2, and the deposition of KM55 confirm the diagnosis of IgAN secondary to WD rather than mere association. Second, family screening of first-degree relatives was not performed. Molecular analysis for the same mutations in siblings is important because the probability of finding a homozygote is 25% [5]. Third, strategic treatments for IgAN secondary to WD remain unknown because of limited data, and a systematic prospective study is required in the future.
Conclusion
In summary, we report an adult case of IgAN secondary to WD, presenting with hematuria, proteinuria and renal dysfunction, who responded to treatment with trientine hydrochloride and zinc acetate. IgA nephropathy secondary to Wilson’s disease is rare, but it is an important cause of secondary IgA nephropathy in juvenile patients with splenomegaly. The diagnosis of Wilson’s disease is easy to establish by measuring a low serum ceruloplasmin concentration, increased urinary copper excretion, and Kayser–Fleischer rings. Although molecular analysis of ATP7B mutations can also be diagnostic, it does not necessarily detect all disease-producing mutations. Finally, patients with Wilson’s disease should be well-informed of a lifelong need for follow-up to monitor clinical progress and should be encouraged to comply with medications.
Conflict of interest
The authors have declared that no conflict of interest exists.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee at which the studies were conducted (IRB approval number: 2017−126) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
References
- 1.Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347(10):738–748. doi: 10.1056/NEJMra020109. [DOI] [PubMed] [Google Scholar]
- 2.Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368(25):2402–2414. doi: 10.1056/NEJMra1206793. [DOI] [PubMed] [Google Scholar]
- 3.Pouria S, Barratt J. Secondary IgA nephropathy. Semin Nephrol. 2008;28(1):27–37. doi: 10.1016/j.semnephrol.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 4.European Association for Study of liver EASL clinical practice guidelines: Wilson’s disease. J Hepatol. 2012;56(3):671–685. doi: 10.1016/j.jhep.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 6.Azizi E, Eshel G, Aladjem M. Hypercalciuria and nephrolithiasis as a presenting sign in Wilson disease. Eur J Pediatr. 1989;148:548–549. doi: 10.1007/BF00441555. [DOI] [PubMed] [Google Scholar]
- 7.Nakada SY, Brown MR, Rabinowitz R. Wilson’s disease presenting as symptomatic urolithiasis: a case report and review of the literature. J Urol. 1994;152:978–979. doi: 10.1016/S0022-5347(17)32635-6. [DOI] [PubMed] [Google Scholar]
- 8.Di Stefano V, Lionetti E, Rotolo N, La Rosa M, Leonardi S. Hypercalciuria and nephrocalcinosis as early feature of wilson disease onset: description of a pediatric case and literature review. Hepat Mon. 2012;12(8):e6233. doi: 10.5812/hepatmon.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhuang XH, MO Y, Jiang XY, Chen SM. Analysis of renal impairment in children with Wilson’s disease. World J Pediatr. 2008;4:102–105. doi: 10.1007/s12519-008-0019-5. [DOI] [PubMed] [Google Scholar]
- 10.Tu J, Chen C, Li H, Chu M, Geng H. A special case of recurrent gross hematuria: answers. Pediatr Nephrol. 2017;32:273–275. doi: 10.1007/s00467-015-3265-5. [DOI] [PubMed] [Google Scholar]
- 11.Novak J, Julian BA, Tomana M, Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. 2008;28(1):78–87. doi: 10.1016/j.semnephrol.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasutake J, Suzuki Y, Suzuki H, Hiura N, Yanagawa H, Makita Y, et al. Novel lectin-independent approach to detect galactose-deficient IgA1 in IgA nephropathy. Nephrol Dial Transpl. 2015;30:1315–1321. doi: 10.1093/ndt/gfv221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.KDIGO clinical practice guidelines for glomerulonephritis—Chap. 10: immunoglobulin A nephropathy. Kidney Int. 2012;2:209–17.
- 14.Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli-Vergani G. Wilson’s disease in children: 37-year experience and revised King’s score for liver transplantation. Liver Transpl. 2005;11(4):441–448. doi: 10.1002/lt.20352. [DOI] [PubMed] [Google Scholar]
- 15.Petrasek J, Jirsa M, Sperl J, Kozak L, Taimr P, Spicak J, Filip K, et al. Revised King’s College score for liver transplantation in adult patients with Wilson’s disease. Liver Transpl. 2007;13(1):55–61. doi: 10.1002/lt.20920. [DOI] [PubMed] [Google Scholar]