

Abstract
The importance of antioxidants in maintaining homeostasis has long been accepted and includes antioxidant proteins such as, peroxiredoxin (Prx), superoxide dismutase and glutathione S transferases. Sulfiredoxin (Srx) is a recently identified antioxidant protein with a role in signaling through catalytic reduction of oxidative modifications. It was first characterized for its regulation of Prx(s) through reduction of the conserved cysteine from sulfinic to sulfenic acid, thereby impacting the role of Prx in regulation of downstream transcription factors and kinase signaling pathways. Furthermore, the reduction of sulfinic to sulfenic acid prevents further oxidation of the conserved cysteine residue to sulfonic acid, the end result of which is degradation. Srx also has a role in the reduction of glutathionylation a post-translational, oxidative modification that occurs on numerous proteins and has been implicated in a wide variety of pathologies, including Parkinson’s disease. The regulation of glutathionylation/deglutathionylation (or thiol switch) has been likened to phosphorylation/dephosphorylation, another post-translational modification involved in the regulation of signaling pathways. Unlike, the reduction of Prx over-oxidation, Srx-dependent deglutathionylation appears to be non-specific. Deglutathionylation of multiple proteins has been observed both in vitro and in vivo in response to oxidative and/or nitrosative stress. This review discusses Srx as a novel antioxidant, and focuses on its potential role in the regulation of glutathionylation/deglutathionylation pathways, that have been implicated in a growing number of disease states.
Keywords: Glutathione, Glutathionylation, Sulfiredoxin, Reactive oxygen/nitrogen species, Oxidative stress, GST
1. General introduction
Cells have evolved an intricate network of antioxidant molecules and proteins that are required to maintain homeostasis. Reactive oxygen species (ROS) are generated exogenously by environmental agents or endogenously as a consequence of normal cellular metabolic processes including, respiration. When ROS levels exceed the antioxidant capacity of cells, oxidative stress (OS) results. The toxicity of ROS is due to their ability to damage a large number of cellular constituents, of which unsaturated lipids, proteins and DNA appear most sensitive. Failure to respond to OS can result in severe damage and ultimately cell death. It is not surprising that many aerobically growing organisms possess strategies for protection against OS, termed the oxidative stress response (OSR), which involves the synthesis and activation of protective enzymes or molecules. Aerobic organisms possess both enzymatic and non-enzymatic antioxidant defense systems to protect cellular constituents and maintain a balanced cellular redox state.
As mentioned above problems arise when ROS exceed the antioxidant capacity of the cell. Homeostasis is maintained by antioxidant molecules and proteins that function to remove the stress from the cell and hence restore the redox balance. The major antioxidant enzymes in the cell include (a) super-oxide dismutase (SOD) [24], which catalyzes the conversion of the superoxide radical to H2O2 (b) catalase [32] which catalyzes the formation of O2 and H2O from hydrogen peroxide (H2O2) (c) glutathione peroxidase [7] which catalyzes the reduction of hydroperoxides using glutathione (GSH) as a reductant and (d) peroxiredoxin (Prx) [6] which reduces both H2O2 and alkyl hydroperoxide in conjunction with thioredoxin reductase (TR), thioredoxin (Trx) and NADPH.
O2•− + H2O → 2H2O (superoxide dismutase)
2H2O2 → 2H2O + O2 (catalase)
H2O2 + 2GSH → GSSG + 2H2O (glutathione peroxidase)
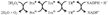
The functional diversity of small redox proteins within a cell is becoming more apparent, with roles both as antioxidant enzymes and regulators of signal transduction, allowing the cell to respond to the stressful environment. As antioxidants are the first line of defense to exogenous stress, small redox proteins that possess this dual function have been described as intracellular stress sensors. In addition to those described above, members of the glutathione S-transferase (GST) super family are emerging as critical small redox proteins that impact kinase signaling. For example, GSTπ is a negative regulator of c-jun N-terminal kinase (JNK) [1], whereas GSTμ influences the differential control of ASK1, a mitogen-activated protein kinase kinase kinase (MAPKKK). Under basal conditions the activity of ASK1 is low because of its sequestration via protein–protein interactions with no less than 3 small redox proteins, Trx, glutaredoxin (Grx) and GSTμ (Fig. 1) [27,9,29]. ROS mediates the liberation of both JNK andASK1 from its negative regulator, GST, thereby activating kinase signaling and impacting both apoptotic and growth responses.
Fig. 1.

Differential regulation of ASK1 by Trx, Grx and GSTμ. ASK1 is in complex with reduced Trx, Grx and GSTμ and is inactive under basal conditions. ROS/RNS, generated by a plethora of different stresses, leads to the activation of ASK1 through the dissociation of one or another of its small redox molecule inhibitors. The dissociation occurs concurrently with the formation of a disulfide bond with Trx, Grx or GSTμ. The activation of ASK1 results in the activation of a cascade, the end point of which is the initiation of apoptosis or stress response.
This review focuses on a novel antioxidant protein, named sulfiredoxin (Srx), that has only recently been identified and found to respond to oxidative and/or nitrosative stress.
2. Srx, a novel antioxidant protein
OSR is a term that is used to describe the signaling events that occur when the intracellular OS is higher than the anti-oxidant capacity of the cell. Srx was first identified in Saccharomyces cerevisiae and was found to be involved in the OSR [4]. Srx is not only induced in response to treatment with H2O2 but deletion of the gene leads to a decreased resistance to H2O2. Insight into its mechanism of action was clarified when Srx was identified as a binding partner to the yeast peroxiredoxin Tsa1, an antioxidant involved in the reduction of H2O2. The mechanism of many antioxidant proteins is dependent upon reactive cysteine residues that redox cycle (see above (d)). For example, the conserved cysteine residue in Tsa1 (involved in its antioxidant cycling function) is oxidized to sulfenic acid (–SOH). This oxidized cysteine residue is easily recycled, or reduced, by Trx in the thioredoxin pathway (see above). However, the further oxidation of the cysteine residue to sulfinic acid (–SO2H) until recently was thought to be an irreversible step. The identification that the sulfinic acid oxidation step could be reduced, albeit through the action of an as yet unidentified protein, added another layer of complexity to these ‘simple’ antioxidant cycling pathways. The identification of Srx as a binding partner of Tsa1, led to the discovery of Srx as the ‘unknown’ protein involved in the reduction of the over-oxidized Tsa1 from the ‘irreversible’ −SO2H to −SOH (Fig. 2).
Fig. 2.

Scheme of cysteine oxidation. The oxidation of a sulfur residue within the amino acid cysteine can result in the formation of a cysteine radical or a sulfenic, sulfinic or sulfonic acid derivative (the latter of which is irreversible). Reduction of the sulfinic to the sulfenic acid derivative in Prx is known to occur through the action of Srx. The oxidation of the sulfur residue in GSH results in the formation of glutathione sulfenate. This reactive molecule can react with a reduced protein cysteine residue (Cys–SH), either directly or indirectly through the formation of glutathione disulfide S-oxide, to form a protein mixed disulfide. Studies have also implicated Srx in the reduction of glutathionylated proteins (Cys–S–SG).
Srx is a member of a conserved family of antioxidants present in eukaryotes. Srx contains a C-terminal cysteine residue that is conserved in all family members. Studies with the yeast and human homologue show this residue is critical for its antioxidant function [4,5]. Interestingly, Srx is not apparent in prokaryotes; it is thought that this is due to the role of Srx in the restoration of over-oxidized Prx, whose counterparts in prokaryotes are not sensitive to oxidative inactivation. Hence, it is suggested that Prx inactivation and restoration by Srx is co-evolutionarily selected [4]. Prxs are both antioxidants and regulators of H2O2-mediated signaling. The family consists of six members, Prx I–IV (typical 2-Cys), Prx V (atypical 2-Cys) and Prx VI (1-Cys Prx). The cysteine residue within the active site of Srx is conserved in all family members and for this reason the role of Srx in the restoration of over-oxidized Prx is also suggested to be conserved. Indeed, Chang et al. showed that this function is conserved in the human homologue of Srx [8]. This group also showed that the restorative role of Srx was specific for typical 2-Cys Prx’s [33]. The Srx-dependent regulation of Prx implicates Srx in the regulation of H2O2-mediated signaling. Indeed, the Morgan group demonstrated a role for Srx in H2O2-mediated signaling in the fission yeast Schizosaccharomyces pombe [5]. Under conditions of low peroxide stress, the conserved cysteine residue in Tpx1 becomes oxidized which in turn causes the oxidation of Pap1 and hence its translocation to the nucleus and activation of gene expression. Under conditions of high peroxide stress Tpx1 is reduced by Srx, therefore Pap1 remains reduced and consequently remains in the cytoplasm. Hence, Srx is involved in the differential regulation of signaling in response to increasing levels of peroxide stress.
3. Glutathionylation as a post-translational modification
GSH is a critical antioxidant in the cell and its’ conjugation to electrophilic compounds is essential to the detoxification pathway. We are now beginning to realize the potentially major role that GSH plays in post-translational modifications that alter cellular response to oxidative and nitrosative stress. It is not surprising that GSH is present in millimolar concentrations in the cell. In fact, the ratio of the reduced GSH pool to glutathione disulfide (GSSG) is a redox sensor and used to quantitate the redox state. In models of OS, transient shifts in the GSH/GSSG ratio from 100 to 10 or even one have been described and found to correlate with the amount of protein mixed disulfide formation [17].
Glutathionylation is the formation of a mixed disulfide between cysteine residues in proteins and GSH (Fig. 2). This reversible post-translational modification can occur on multiple proteins when the cellular environment is subject to oxidative and/or nitrosative stress. Glutathionylation is mediated by a number of different radical species. Fig. 2 illustrates a subset of radicals, generated in the cell, that are purported to glutathionylate intracellular proteins.
Glutathionylation has been proposed to act as a protective cellular mechanism preventing proteins from terminal modifications as a consequence of exposure to ROS and reactive nitrogen (RNS) species. The fundamental relevance of glutathionylation in response to both oxidative and/or nitrosative stress in vivo has been a subject of some debate. More recent studies have shown that glutathionylation has a role in cell signaling. There is a growing list of proteins that can be glutathionylated and the functional relevance of the process is not yet fully realized [23]. The list includes protein tyrosine phosphatase 1B (PTP1B) [3], c-jun [22], GST [13], Grx, Trx [23], H-Ras [20], MEKK1 [12] and most recently PTEN [34]. It is apparent that the entire scope of glutathionylation and its implications for cellular signaling has not yet been fully explored. Identifying the proteins involved in the reversal of this modification, deglutathionylation, has provided further complexity to the roles that antioxidant proteins play.
4. Deglutathionylation as a mechanism of cellular stress regulation
Reversibility of post-translational modifications is a requisite for cellular signaling. Deglutathionylation is the removal of the GSH moiety from proteins and can occur when the environment becomes more reducing in an enzyme-dependent or -independent manner. To date, a limited number of proteins have been identified that are involved in deglutathionylation. Grx is a well-characterized protein known to be involved in both the oxidative glutathionylation and the reductive deglutathionylation steps [19]. Grx contains a conserved motif (CXXC) containing 2 cysteine residues, only one of which are required for its oxidative function with respect to glutathionylation via a monothiol mechanism of disulfide exchange. Interestingly, studies showed that the presence of both cysteine residues within the conserved CXXC motif are required for its reductive function of deglutathionylation via a dithiol mechanism of disulfide exchange.
Protein disulfide isomerase (PDI) is another protein implicated in the glutathionylation process. PDI is a large protein of five domains that is localized to the endoplasmic reticulum (ER). PDI is involved in the folding of extracellular proteins through disulfide bond formation and isomerization. It is the redox potential of the active sites within PDI that make it a good oxidant and as such is currently implicated in the oxidative glutathionylation step (for a review see [31]). Of particular interest, we and others have shown that both PDI and Grx are themselves regulated by glutathionylation [23,16] (Townsend et al., AACR 2005).
We have demonstrated a role for human Srx (Srx1) in the regulation of glutathionylation in response to oxidative and/or nitrosative stress (Findlay et al., AACR, 2005). Srx1 appears to target multiple intracellular proteins and is distinct in that it is not itself glutathionylated during the removal of GSH moieties. Srx1 is the first protein identified that has been implicated specifically in the reductive deglutathionylation step. The specificity of Srx1 may be due to the presence of only one cysteine residue within the entire human sequence. This differs from Grx, which contains two cysteine residues in a CXXC and is involved in both the oxidative glutathionylation and reductive deglutathionylation steps. In addition, the conserved cysteine residue within Grx is an acceptor for the GSH moiety during the disulfide exchange reaction, i.e. Grx becomes glutathionylated itself during the reaction. This is converse to Srx, which does not become glutathionylated during the deglutathionylation reaction.
5. Oxidative/nitrosative stress and disease
Formation of ROS and RNS is not only the result of an exogenous insult, but a consequence of normal cellular metabolism. It is the tight regulation of these processes within the cell that allows maintenance of homeostasis and consequently a limit to the amount of damage caused by these ROS/RNS. Dysregulation leads to an accumulation of ROS/RNS resulting in cellular damage. Unfortunately however, many diseases are associated with an increase in oxidative/nitrosative damage and aging itself is correlated with increased cellular damage due to the activity of intracellular ROS/RNS over time (for a review see [15]). Diseases associated with an increase in oxidative and/or nitrosative damage include Alzheimer’s, Amyotrophic Lateral Sclerosis (ALS), Atherosclerosis, Diabetes, HIV, cancer and aging. These diseases and the oxidative/nitrosative modifications associated with them are reviewed in more detail [14].
Many of these same diseases have also been linked with glutathionylation. Although the post-translational modification of gluathionylation has been known for some time, the tools to detect this modification have only recently become more readily available and user friendly. To date, although glutathionylation of multiple proteins have been detected, the effects of these modifications, if any, on disease state or progression is less known. The diseases that are correlated with increases in glutathionylation include Parkinson disease (PD), Friedreich’s ataxia (FRDA), diabetes’s (type I and II), human immunodeficiency virus (HIV), hyperlipidemia and renal cell carcinoma (for a review see [18]).
5.1. PD
PD is a progressive neurodegenerative disorder that primarily affects the dopamine neurons in the substantia nigra pars compacta (SNpc), leading to the major clinical and pharmacological abnormalities that characterize the disease. Of all the neurodegenerative diseases, evidence for a dysfunction of glutathione metabolism is strongest for PD. Insights into the pathogenesis of PD have been achieved experimentally using the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Exposure to MPTP induces PD-like symptoms in humans and causes degeneration of dopaminergic neurons in animal species [30]. This merepidine analog is metabolized to 1-methyl-4-phenylpyridinium (MPP+) by the enzyme monoamine oxidase B. MPP+ is subsequently selectively taken up by dopaminergic terminals and concentrated in neuronal mitochondria in the substantia nigra. MPP+ binds to and inhibits complex I of the mitochondrial transport chain, thereby producing the same biochemical defect as detected in SNpc of PD patients [30]. In PD monoamine oxidase-derived H2O2 was shown to inhibit mitochondrial respiration by glutathionylation of respiratory chain enzymes [10].
NADPH is an essential cofactor for the regeneration of GSH. Mitochondrial NADP+-dependent isocitrate dehydrogenase (IDPm) is a key enzyme involved in the cellular defense against oxidative damage by supplying NADPH in the mitochondria, needed for the regeneration of mitochondrial GSH. Elevation of mitochondrial NADPH and GSH by IDPm suppresses OS and ROS-mediated damage. Studies show that IDPm can be inhibited by reversible glutathionylation. More importantly, glutathionylated IDPm was isolated from brain samples of MPTP-treated mice, the PD mouse model [21]. An important role for GSH has been proposed for the pathogenesis of PD, where a decrease in GSH concentrations in the substantia nigra was observed in preclinical stages of the disease [28]. Furthermore, mitochondrial dysfunction appears to play a major role in the neurodegeneration associated with the pathology of PD.
5.2. FRDA
One of the best studied examples of disease associated glutathionylation is FRDA, an autosomal recessive and most common of the hereditary ataxia’s, caused by decreased expression of frataxin, a protein implicated in iron metabolism. Patients with FRDA have iron deposits in the heart, increased mitochondrial iron in fibroblasts, and greater sensitivity to OS by pro-oxidants such as FeCl3 and H2O2 [25]. In addition, an impairment of the antioxidant enzymes SOD and glutathione peroxidase were found in vivo together with a decreased level of free GSH in the blood of patients [26]. More recent studies have shown an increase in actin glutathionylation in fibroblasts of patients with FRDA [26]. This study shows that upon treatment of FRDA fibroblasts with reduced glutathione, a complete rescue of cytoskeletal abnormalities and cell viability is observed, suggesting that OS-induced glutathionylation of actin leads to an impairment of cytoskeletal function in FRDA fibroblasts [26]. The intracellular iron imbalance due to the frataxin deficiency leads to OS and has been proposed as the underlying pathogenesis of FRDA. OS induces actin glutathionylation and hence impairment of cytoskeletal functions. This link between iron overload and actin glutathionylation has been suggested as the underlying pathogenic mechanism that contributes to the progression of neurodegeneration in the disease.
A role of oxidized actin has also been reported in some other neurodegeneraive diseases, such as in a mouse model of ALS [11]. In addition, oxidation of actin was found to be significantly higher in brain extracts of patients with Alzheimer’s disease, suggesting that OS-induced injury may lead to the degeneration of neurons in theAlzheimer’s disease brain [2].
6. Concluding remarks
Glutathionylation is emerging as an important post-translational modification that influences a large number of proteins. The growing number of disease states that are correlated with increased glutathionylation of specific proteins highlights the need to understand the underlying mechanisms involved in the regulation of the glutathionylation and deglutathionylation pathways. The identification of Srx and its role as a novel antioxidant protein specifically involved in the deglutathionylation of proteins is an important discovery and there exists a potential role for Srx1 as a deglutathionylating enzyme in the treatment of diseases associated with an increase in protein glutathionylation.
Abbreviations:
- ALS
amylotrophic lateral sclerosis
- ER
endoplasmic reticulum
- FRDA
Friedrich’s ataxia
- Grx
glutaredoxin
- GSH
glutathione
- GSSG
glutathione disulfide
- GST
glutathione S-transferase
- H2O2
hydrogen peroxide
- IDPm
NADP+-dependent isocitrate dehydrogenase
- JNK
c-jun N-terminal kinase
- MPP+
1-methyl-4-phenylpyridinium
- OS
oxidative stress
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- OSR
oxidative stress response
- PD
Parkinson’s disease
- PDI
protein disulfide isomerase
- PTP1B
protein tyrosine phosphatase 1B
- Prx
peroxiredoxin
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SNpc
substantia nigra pars compacta
- SOD
superoxide dismutase
- Srx
sulfiredoxin
- TR
thioredoxin reductase
- Trx
thioredoxin
References
- [1].Adler V, Yin Z, et al. Regulation of JNK signaling by GSTp. EMBO J 1999;18(5):1321–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Aksenov MY, Markesbery WR. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci Lett 2001;302(2–3): 141–5. [DOI] [PubMed] [Google Scholar]
- [3].Barrett WC, DeGnore JP, et al. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 1999;38(20): 6699–705. [DOI] [PubMed] [Google Scholar]
- [4].Biteau B, Labarre J, et al. ATP-dependent reduction of cysteine–sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003; 425(6961):980–4. [DOI] [PubMed] [Google Scholar]
- [5].Bozonet SM, Findlay VJ, et al. Oxidation of a eukaryotic 2-cys peroxiredoxin is a molecular switch controlling the transcriptional response to increasing levels of hydrogen peroxide. J Biol Chem 2005;280(24):23319–27. [DOI] [PubMed] [Google Scholar]
- [6].Chae HZ, Chung SJ, et al. Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem 1994;269(44):27670–8. [PubMed] [Google Scholar]
- [7].Chance B, Sies H, et al. Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979;59(3):527–605. [DOI] [PubMed] [Google Scholar]
- [8].Chang TS, Jeong W, et al. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem 2004;279(49):50994–1001. [DOI] [PubMed] [Google Scholar]
- [9].Cho SG, Lee YH, et al. Glutathione S-transferase mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J Biol Chem 2001;276(16):12749–55. [DOI] [PubMed] [Google Scholar]
- [10].Cohen G, Farooqui R, et al. Parkinson disease: a new link between monoamine oxidase and mitochondrial electron flow. Proc Natl Acad Sci USA 1997;94(10):4890–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Collard JF, Cote F, et al. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 1995; 375(6526):61–4. [DOI] [PubMed] [Google Scholar]
- [12].Cross JV, Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J 2004;381(Pt 3):675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dafre AL, Sies H, et al. Protein S-thiolation and regulation of microsomal glutathione transferase activity by the glutathione redox couple. Arch Biochem Biophys 1996;332(2):288–94. [DOI] [PubMed] [Google Scholar]
- [14].Dalle-Donne I, Scaloni A, et al. Proteins as biomarkers of oxidative/nitrosative stress in diseases: the contribution of redox proteomics. Mass Spectrom Rev 2005;24(1):55–99. [DOI] [PubMed] [Google Scholar]
- [15].Droge W The plasma redox state and ageing. Ageing Res Rev 2002; 1(2):257–78. [DOI] [PubMed] [Google Scholar]
- [16].Fratelli M, Demol H, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci USA 2002;99(6):3505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gilbert HF. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol 1995;251:8–28. [DOI] [PubMed] [Google Scholar]
- [18].Giustarini D, Rossi R, et al. S-glutathionylation: from redox regulation of protein functions to human diseases. J Cell Mol Med 2004; 8(2):201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gravina SA, Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry 1993;32(13):3368–76. [DOI] [PubMed] [Google Scholar]
- [20].Ji Y, Akerboom TP, et al. S-nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Arch Biochem Biophys 1999;362(1):67–78. [DOI] [PubMed] [Google Scholar]
- [21].Kil IS, Park JW. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem 2005;280(11):10846–54. [DOI] [PubMed] [Google Scholar]
- [22].Klatt P, Molina EP, et al. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J 1999;13(12):1481–90. [DOI] [PubMed] [Google Scholar]
- [23].Klatt P, Pineda Molina E, et al. Novel application of S-nitrosoglutathione-sepharose to identify proteins that are potential targets for S-nitrosoglutathione-induced mixed-disulphide formation. Biochem J 2000;349(Pt 2):567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969;244(22):6049–55. [PubMed] [Google Scholar]
- [25].Pandolfo M Molecular pathogenesis of Friedreich ataxia. Arch Neurol 1999;56(10):1201–8. [DOI] [PubMed] [Google Scholar]
- [26].Piemonte F, Pastore A, et al. Glutathione in blood of patients with Friedreich’s ataxia. Eur J Clin Invest 2001;31(11):1007–11. [DOI] [PubMed] [Google Scholar]
- [27].Saitoh M, Nishitoh H, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 1998;17(9):2596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sofic E, Lange KW, et al. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci Lett 1992;142(2):128–30. [DOI] [PubMed] [Google Scholar]
- [29].Song JJ, Rhee JG, et al. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem 2002;277(48):46566–75. [DOI] [PubMed] [Google Scholar]
- [30].Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem 1993;61(4):1191–206. [DOI] [PubMed] [Google Scholar]
- [31].Wilkinson B, Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta 2004;1699(1–2):35–44. [DOI] [PubMed] [Google Scholar]
- [32].Winkler H, Adam G, et al. Co-ordinate control of synthesis of mitochondrial and non-mitochondrial hemoproteins: a binding site for the HAP1 (CYP1) protein in the UAS region of the yeast catalase T gene (CTT1). EMBO J 1988;7(6):1799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Woo HA, Jeong W, et al. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem 2005; 280(5):3125–8. [DOI] [PubMed] [Google Scholar]
- [34].Yu CX, Li S, et al. “Redox regulation of PTEN by S-nitrosothiols.”. Mol. Pharmacol; 2005. [DOI] [PubMed] [Google Scholar]