

Abstract
Abdominal Aortic Aneurysms (AAA) involve slow dilation and weakening of the aortic wall due to breakdown of structural matrix components, such as elastic fibers by chronically overexpressed matrix metalloproteinases (MMPs), primarily, MMPs-2 and −9. Auto-regenerative repair of disrupted elastic fibers by smooth muscle cells (SMCs) at the AAA site is intrinsically poor and together with chronic proteolysis prevents restoration of elastin homeostasis, necessary to enable AAA growth arrest or regression to a healthy state. Oral doxycycline (DOX) therapy can inhibit MMPs to slow AAA growth, but has systemwide side-effects and inhibits new elastin deposition within AAA tissue, diminishing prospects for restoring elastin homeostasis preventing the arrest/regression of AAA growth. We have thus developed cationic amphiphile (DMAB)-modified submicron particles (SMPs) that uniquely exhibit pro-elastogenic and anti-proteolytic properties, separate from similar effects of the encapsulated drug. These SMPs can enable sustained, low dose DOX delivery within AAA tissue to augment elastin regenerative repair. To provide greater specificity of SMP targeting, we have conjugated the DOX-SMP surface with an antibody against cathepsin K, a lysosomal protease that is highly overexpressed within AAA tissue. We have determined conditions for efficient cathepsin K Ab conjugation onto the SMPs, improved SMP binding to aneurysmal SMCs in culture and to injured vessel walls ex vivo, conjugation did not affect DOX release from the SMPs, and improved pro-elastogenic and anti-proteolytic effects due to the SMPs likely due to their increased proximity to cells via binding. Our study results suggest that cathepsin K Ab conjugation is a useful targeting modality for our pro-regenerative SMPs. Future studies will investigate SMP retention and biodistribution following targeting to induced AAAs in rat models through intravenous or catheter-based aortal infusion and thereafter their efficacy for regenerative elastic matrix repair in the AAA wall.
Keywords: Submicron particles, elastic matrix, regenerative matrix repair, matrix metalloproteinases (MMPs), cathepsin K, drug delivery
Graphical Abstract
1. Introduction
Abdominal Aortic Aneurysms (AAAs), a disease that primarily afflicts the elderly, involve slow dilation of the abdominal aorta to rupture [1]. AAAs result from slow breakdown of extracellular matrix (ECM) structures of the aortic wall (collagen, elastic fibers) by chronically overexpressed matrix metalloproteinases (MMPs) [2]. Although imaging-based screening of high-risk patient populations now enable early detection of these asymptomatic conditions, their management is currently limited to passive, imaging-based growth monitoring up to a critical, rupture-imminent stage (diameter > 5.5 cm). At this time, surgery is performed, although it is unsuitable for many patients due to high risk [3,4]. Recent preclinical and clinical studies suggest that systemic (oral) dosing with doxycycline (DOX), a broad-spectrum MMP inhibitor drug, can attenuate proteolytic activity in the AAA wall to slow AAA growth [5,6]. However, since this mode of DOX delivery can result in body-wide inhibition of MMPs, which are required for normal matrix turnover in healthy tissues, and system wide side effects, modalities for targeted and localized DOX delivery to the AAA wall are mandated [7,8]. In addition, studies have shown that high MMP inhibitory doses of orally-delivered DOX further inhibit [9,10] the inherently poor regeneration of elastic matrix by vascular smooth muscle cells (SMCs) in the AAA wall [8], deterring any prospects of restoring local elastic matrix homeostasis and hence being able to arrest or regress AAA growth.
In a prior work, we developed novel, cationic amphiphile-surface functionalized poly-lactic co-glycolic acid (PLGA) sub-micron particles (SMPs) for predictable and sustained, low dose DOX release within AAA tissues. We showed that DOX, released at ~1/100th of the systemic dose, to have significant anti-proteolytic and pro-elastogenic effects on cultured aneurysmal SMCs. These outcomes were augmented by similar effects provided by the surface-functionalized drug-free polymer carriers [11,12]. In the current study, we have sought to modify this SMP design to impart specificity of their targeting to the AAA tissue. In this context, cathepsin K, a lysosomal cysteine protease primarily involved in bone remodeling [13,14], is greatly overexpressed by activated smooth muscle cells (SMCs) within the inflamed AAA wall tissue relative to healthy aortae [15] and could represent a useful target for SMP binding within the AAA wall. Accordingly, in this study we surface-modified our SMPs to incorporate cathepsin K antibodies. Covalent reaction and adsorption-based conjugation protocols were investigated to determine the most effective in conjugating a cathepsin K polyclonal antibody (pAb). The benefits of cathepsin K pAb-modification to targeted uptake of the SMPs by elastase-injured arteries and aneurysmal rat aortic SMC (EaRASMCs) cultures stimulated with tumor necrosis factor–alpha (TNF-α; an inflammatory cytokine that upregulates cathepsin K [16]), and the impact of the likely more intimate and prolonged binding/association of these modified DOX SMPs with the cells on their elicited pro-regenerative and anti-proteolytic outcomes were also investigated.
2. Materials and Methods
2.1. Isolation and culture of SMCs from elastase perfusion-induced rat AAAs
All procedures requiring the use of animals were conducted with approval from the Institutional Animal Care and Use Committee (IACUC) at the Cleveland Clinic (ARC # 2010–0299). The Clinic’s animal facility is AAALAC-approved and has animal assurance (#A3145–01). EaRASMCs were harvested from multiple (n = 3) adult male Sprague-Dawley rats at 14 days following elastase infusion, as previously published by our lab and others [17,18]. The AAA segment was isolated from the rats following laparotomy, and the intimal layer was scraped off. The medial layer was then separated from the collagenous adventitia, dissected into ~0.5 mm long slices, and rinsed in sterile phosphate-buffered saline (PBS). Subsequently, the tissue was digested in DMEM-F12 medium (Invitrogen, Carlsbad, CA) containing 125 U/mg collagenase (Worthington Biochemicals, Lakewood, NJ) and 3 U/mg elastase (Worthington Biochemicals) for 30 min at 37 oC, centrifuged (400g, 5 min) and cultured in T-75 flasks in DMEM-F12 medium containing 10% v/v of fetal bovine serum (FBS; PAA Laboratories, Etobicoke, Ontario) and 1 % v/v of penicillin-streptomycin (PenStrep; Thermo Fisher, South Logan, UT). At 70% confluence, the primary EaRASMCs from individual rats (n = 3) were pooled and passaged. Pooled EaRASMCs of passage 2–6 were used in our experiments. In prior published studies, we have extensively characterized the EaRASMCs and demonstrated their retention of a diseased phenotype in culture for several passages [17].
Primary rat aortic SMCs (RASMCs; healthy cell controls) were isolated from aortae of multiple (n = 3) healthy Sprague-Dawley rats in a similar manner as previously described for EaRASMCs. These pooled primary cells were passaged and used at passage 2–6 in experiments.
2.2. Formulation of fluorescein-loaded, Alexa Fluor 633-loaded, DOX-loaded, and blank PLGA SMPs
Poly(DL-lactic-co-glycolic acid) (PLGA; 50:50 lactide:glycolide; inherent viscosity 0.95 – 1.2 dl/g in hexafluoroisopropanol; Durect Corporation, Birmingham, AL) SMPs loaded with fluorescein (Chemicon, Temecula, CA) or Alexa Fluor 633 (AF633) (Invitrogen) were prepared through a double emulsion solvent evaporation technique [19–21]. PLGA (50 mg) was dissolved in 2 ml of chloroform (Fisher Scientific, Fair Lawn, NJ) and 0.1 ml (1 mg/ml) of fluorescein or AF633 was added. This solution was then emulsified using a probe sonicator (Q500; QSonica LLC, Newtown, CT) for 30 s on ice at an amplitude of 20% to form a water-oil emulsion. For preparation of DOX-loaded SMPs, an aqueous DOX solution containing a 2% w/w ratio of DOX: PLGA was instead emulsified into the PLGA solution using a probe sonicator. Next, 6 ml of an aqueous phase consisting of nanopure water and the surfactant, diodecyldimethylammonium bromide (DMAB; Sigma-Aldrich, St. Louis, MO), was added and sonicated again for 30 s on ice at 20% amplitude to form the water-in oil-in water double emulsion. This double emulsion was stirred for 16 h at room temperature and then desiccated for 1 h under vacuum. The samples were separated by ultracentrifugation (35,000 rpm, 30 min; Beckman L-80, Beckman Instruments, Inc., Palo Alto, CA), washed twice with nanopure water to remove residual DMAB and unencapsulated fluorescein/AF633/DOX, sonicated, and repeat ultracentrifuged (30,000 rpm, 30 min). The samples were lyophilized for 48 h to obtain a dry powder. Samples were kept covered throughout this procedure to ensure the fluorescent dye did not undergo bleaching. Following the same methods as previously described, blank PLGA SMPs were also formulated that did not fluoresce. DMAB was again used as the surfactant and the samples were lyophilized to produce a dry power. The SMPs were finally passed through a 0.4 μm filter to obtain a sterile sample while concurrently removing any larger aggregates.
The efficiency of DOX encapsulation within the SMPs was determined by pooling the supernatants from the washing and ultracentrifugation steps for individual SMP formulations. Unencapsulated DOX in the supernatant fraction was assayed by UV spectrophotometry (SpectraMax M2, Molecular Devices, Inc., Sunnyvale, CA) using the absorbance peak of DOX at 270 nm. This peak was calibrated to a standard curve generated using serial dilutions of a 1mg/mL DOX solution to obtain a standard curve, to obtain the amount of unencapsulated DOX. The total amount of encapsulated DOX and the encapsulation efficiency were determined by subtracting the total amount of unencapsulated DOX from the known amount of DOX added during SMP formulation.
2.3. Determination of SMP size and zeta potential
Mean hydrodynamic diameters of the SMPs were determined using a dynamic light scattering technique, and their mean zeta potentials (surface charge) were determined via a phase analysis light scattering technique using a commercial particle-sizing system (PSS/NICOMP 380/ZLS, Particle Sizing Systems, Santa Barbara, CA), as previously described [11].
2.4. Preparation of cathepsin K pAb-modified SMPs
2.4.1. Adsorption
The fluorescein-SMPs were dispersed in PBS (pH 7.4, 0.5 mg/ml) and 490 μl aliquot of this was mixed with 10 μl of cathepsin K pAb (200 μg/ml; rabbit anti-rat; Santa Cruz Biotechnology, Inc., Dallas, TX). A mixture containing 490 μl of the SMP dispersion and 10 μl PBS was used as a control. These mixtures were gently stirred (4 oC, 2 h, 5 h, or 24 h), and then centrifuged (12,000g, 10 min) to separate the free antibody from SMPs. The resulting pellet containing cathepsin K pAb-adsorbed SMPs was washed twice with PBS and re-dispersed in 500 μl of PBS(pH 7.4) [22].
2.4.2. Covalent binding
A cross-linking molecule 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide HCl (EDC; Thermo Fisher, Rockford, IL.) was used to form a covalent bond between free carboxylic acid residues on the PLGA SMP surface and primary amine group on the cathepsin K pAbs [22,23]. The SMPs were dispersed in 2-(N-Morpholino)ethanesulfonic acid, pH 5.5, (MES; Sigma-Aldrich) at a concentration of 0.5 mg/ml and 490 μl of this dispersion was mixed with 10 μl of cathepsin K pAb. Subsequently, 100 ng of EDC was added to this mixture, resulting in an approximate EDC to Ab molar ratio of 10 [24]. Two controls were run concurrently: one lacking EDC and the other lacking both EDC and the antibody. These reaction mixtures were gently stirred at 24 οC for either 2 h or 5 h. The mixtures were centrifuged for 5 min at 13,200 rpm, the supernatant discarded and the SMP pellet washed two times with PBS (pH 7.4), and finally re-dispersed into PBS.
2.5. Assessment of cathepsin K pAb incorporation on SMP surface
2.5.1. Fluorescence spectroscopy to assess cathepsin K pAb binding to SMPs
A fluorescein-tagged goat anti-rabbit secondary antibody (Chemicon, Temecula, CA), was used to qualitatively determine the relative binding of the cathepsin K pAb on the surface of the SMPs. Cathepsin K pAb-conjugated SMPs were treated with the secondary antibody (4 oC, 1 h) at a 1:500 dilution. The samples were then centrifuged (12,000g, 10 min) and the pellets washed twice with PBS, to remove any unbound secondary antibody, and then re-dispersed in 500 μl of PBS. Three 150 μl aliquots per sample were placed in the wells of a microplate. The fluorescence intensity due to fluorescein was measured (λex = 493 nm and λem = 525 nm) using a SpectraMax M2e microplate reader (Molecular Devices, Sunnyvale, CA).
2.5.2. Measurement of cathepsin K pAb conjugation efficiency via fluorescence spectroscopy and confocal microscopy
The cathepsin K pAb was covalently conjugated to fluorescein-loaded SMPs over 2 h or 5 h as described previously, with proper controls. Following conjugation and washing, an Alexa Fluor (AF) 546-conjugated donkey anti-rabbit secondary Ab (1:1000 dilution; ThermoFisher Scientific), was used to fluorescently tag the SMP-bound cathepsin K pAb (25 οC, 1 h). Samples were centrifuged (12,000g, 10 min), washed twice with PBS, and re-suspended in PBS at a concentration of 0.5 mg of SMPs/ml. Again, three 150 μl aliquots per sample were added to a microplate and the fluorescence of both the fluorescein (λex = 493 nm and λem = 525 nm) and Alexa Fluor 546 (λex = 556 nm and λem = 573 nm) were measured using a microplate reader. The SMPs were aliquoted onto glass cover slips, mounted, and visualized on a confocal microscope (Leica TCS SP5 II, Leica Microsystems, Inc., Buffalo Grove, IL) to detect cathepsin K pAb-conjugated SMPs exhibiting both green (fluorescein) and deep red (AF 546) fluorescence.
2.5.3. Assessing retention of conjugated and absorbed cathepsin K pAb on SMP surface
To determine whether conjugation or adsorption resulted in sustained cathepsin K attachment to the SMP surface, Alexa Fluor 633 (Invitrogen) was loaded into the SMPs. Cathepsin K pAb modification of the SMPs was performed as described above, for both methods. An Alexa Fluor 488 goat anti-rabbit secondary antibody (1:1000 dilution; Invitrogen) was used to fluorescently tag the cathepsin K pAb. The SMPs were centrifuged (12,000g, 10 min), washed twice with PBS, re-suspended in PBS (0.5 mg SMPs/ml) and a fluorescent microscope was used to visualize the SMPs.
To determine fluorescence intensity due to SMP surface-incorporated cathepsin K Abs, a corrected total fluorescence (CTF) protocol was used. Briefly, individual SMPs were selected using the drawing tool in ImageJ to obtain an integrated density. The integrated density was used to calculate the corrected total fluorescence for FITC and AF633 at Day 1 and Day 14. The CTF is calculated by: (Integrated Density – (Mean Fluorescence of Background * Area of Selected SMP)). To determine the ratio of FITC bound to the SMP surface, the CTF of FITC (adsorption n = 132, n = 130 and conjugation n = 154, n = 207) was divided by the average CTF of AF633 for each test case.
2.6. Verification of cathepsin K overexpression and targeting by SMPs
2.6.1. Assessing cathepsin K expression by EaRASMCs
EaRASMCs (passage 3) and RASMCs (healthy cell controls) were seeded at 7.5 × 104 cells/well, and cultured for 2 weeks in six-well plates in DMEM-F12 medium containing 10% v/v FBS and 1% v/v PenStrep. Thereafter, the spent medium was replaced with DMEM-F12 medium containing 2% v/v FBS and 1% v/v PenStrep. Half of the EaRASMC cultures received medium supplemented with 100 ng/ml of TNF-α in order to simulate the inflammatory aneurysmal tissue milieu. The RASMCs did not receive TNF-α because this is not physiologically relevant.
For immunofluorescence visualization of cathepsin K expression, after a further 24 hours of culture, all cell layers were fixed in 4% v/v paraformaldehyde, permeabilized with Triton-100 (VWR International, UK) and labeled with the primary cathepsin K Ab (1:100 dilution) and a secondary AF 546-tagged secondary antibody (1:1000 dilution). DAPI (Vector Labs, Burlingame, CA) and AF488-Phalloidin (Molecular Probes, Temecula, CA) were used to stain the nuclei and actin cytoskeleton, respectively. The cell layers were imaged on a confocal microscope.
For semi-quantitative comparison of cathepsin K expression in the cell culture groups, following the 24 hour incubation period, the cell layers were harvested in RIPA buffer (Thermo Scientific) containing Halt™ protease inhibitor (Thermo Scientific) and stored in −80oC. The samples were assayed for total protein content using a bicinchonic acid (BCA) assay kit (Thermo Scientific) [25]. A 20 μl aliquot of each sample was loaded under reduced conditions into each lane of a 12% sodium dodecylsulfate polyacrylamide gel electrophoresis gel (SDS-PAGE), along with a BenchMark™ pre-stained molecular weight ladder (Invitrogen) and a C-32 whole cell lysate (Santa Cruz) as a positive control. The gels were transferred wet onto nitrocellulose membranes (Invitrogen). Subsequently, the membranes were blocked for 1 h with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE), after which they were immunolabeled (1 h, 25 οC) with the rabbit cathepsin K pAb (1:200 dilution) and a mouse monoclonal antibody against β-actin (1:1000; Sigma-Aldrich) as the loading control. Secondary antibody labeling occurred at room temperature for 1 h using IRDye® 680LT goat-anti-rabbit (1:15,000 dilution; LI-COR Biosciences) and IRDye® 800CW (1:20,000; LI-COR Biosciences). A LI-COR Odyssey scanning system was used to detect the protein bands via fluoroluminescence. The intensities of the cathepsin K bands were quantified using Image Studio® and normalized to their respective β-actin bands to allow reliable comparison between different samples in the same blot.
2.6.2. Assessing cathepsin K expression in matrix-injured arteries
Cathepsin K expression in elastase injured arteries was compared to expression of the housekeeping protein, β-actin, using western blot. Porcine carotid arteries (n = 3) were infused with either elastase (20 units/ml, Sigma) or 0.9% v/v sterile saline. The end of each artery was clamped shut and a catheter was used to infuse 1 ml of either solution. Following infusion, the arteries were incubated at 37 °C for 20 minutes. After incubation, the samples were washed with PBS to remove any residual elastase or saline. The arteries were then cut into segments of similar length (~15 mm) and flash frozen in liquid nitrogen. After storage overnight (−80 °C), the tissues were lyophilized to produce a dry sample. The dry weight of each segment was obtained. The harvesting for western blot followed the same procedure as described previously.
2.6.3. SMP targeting of cathepsin K in in vitro EaRASMC cultures
Using the covalent conjugation method, PLGA SMPs encapsulated with AF546 were conjugated to cathepsin K pAb. EaRASMCs (passage 3) were seeded at 7.5 × 104 cells per well and cultured for 13 days in six-well plates (A = 10 cm2; BD-Biosciences, Franklin Lakes, NJ). The cells were cultured in DMEM-F12 supplemented with 10% v/v FBS and 1% v/v PenStrep. On the 13th day, the cultures received low-serum medium: DMEM-F12 supplemented with 2% v/v FBS and 1% v/v PenStrep. Additionally, half of the EaRASMC culture wells received low-serum medium supplemented with 100 ng/ml of TNF-α in order to stimulate an aneurysmal or activated milieu within the culture. Following 24 h of incubation, EaRASMCs, both with and without TNF-α, received the cathepsin K pAb-conjugated AF546-encapsulated SMPs. The final concentration of SMPs was 0.5 mg/ml. After a further 24 h, the cell layers were fixed, stained with AF488-Phalloidin (green for cytoskeleton; Molecular Probes) and mounted on slides with VectaShield containing DAPI (blue for nuclei; Vector Labs, Burlingame, CA).
RASMCs (passage 3) were seeded and cultured similarly to the EaRASMCs. After 13 days, the cultures received low-serum medium, but no TNF-α. After 24 h of incubation, half of the cultures received the conjugated SMPs and the other half received the control SMPs. SMP concentration was maintained at 0.5 mg/ml. After another 24 h of incubation, the cell layers were fixed, stained, and mounted on slides, as done with the EaRASMCs.
2.6.4. Targeting cathepsin K modified SMPs to the matrix-injured artery wall
To determine the effectiveness of cathepsin K targeting in aorta, thawed porcine arteries (n=3) were infused with elastase (20 units/ml, 37°C, 20 minutes, Sigma) and rinsed with PBS. Following elastase infusion, Alexa Fluor 633-loaded SMPs, Alexa Fluor 633-loaded SMPs with cathepsin K pAb conjugation, Alexa Fluor 633-loaded SMPs with IgG Ab conjugation, or 0.9% saline were infused in the artery (1 ml, 2 mg/ml SMP). After incubation (37°C, 20 minutes), the arteries were flushed with PBS to remove any unbound SMPs. The arteries were imaged (IVIS Spectrum CT In Vivo Imager, PerkinElmer) and analyzed using spectral unmixing to measure SMP binding using Living Image® software. Briefly, spectral unmixing was used to subtract tissue autofluorescence from each sample to allow for pure AF633 SMP fluorescence. Once the tissue autofluoresence was removed, ROIs were drawn over each artery and the total radiant efficiency ([p/s]/[μW/cm2]) was measured and the fold-increase of SMPs bound was calculated.
2.7. Characterizing DOX release from cathepsin K Ab-conjugated and unconjugated PLGA SMPs in vitro
DOX release from both cathepsin K pAb-conjugated and -unconjugated SMPs was measured in PBS at 37oC. Aliquots (1 ml) containing conjugated or unconjugated DOX-SMPs (0.5 mg/ml), were collected at various time points over 45 days, centrifuged (13,000 rpm, 30 min, 4oC) and the amount of DOX in the supernatant quantified by UV spectrophotometry. The absorbance at λ = 270 nm was calibrated to a standard curve generated using serial dilutions of DOX in PBS. Following the absorbance measurements, the volume in each sample was replenished with fresh PBS. The total amount of DOX loaded in the SMPs was calculated using the same procedure as previously mentioned and the released amount was used to determine the percentage of DOX released.
2.8. Experimental design for cell culture
In order to study the effect of cathepsin K pAb-conjugated DOX-SMPs and unconjugated SMPs on cellular elastic matrix synthesis, EaRASMCs were seeded at 3 × 104 cells per well in wells of a 6-well plate, and cultured for 21 days in DMEM-F12 medium supplemented with 2% v/v FBS, 1% v/v PenStrep, and 50 ng/ml of TNF-α. The media from each well was removed and centrifuged (5 min, 12,000 RPM) to pellet the SMPs. The SMPs were re-suspended in fresh medium before being added back into each well. Culture groups included standalone EaRASMC cultures (treatment control), EaRASMCs cultured with unconjugated SMPs loaded with 2% w/w of DOX, and cathepsin K pAb-conjugated SMPs loaded with 2% v/v of DOX. In the latter cases, the SMPs were added on Day 1 following an overnight incubation at a concentration of 0.2 mg/ml.
2.9. DNA assay for cell proliferation
The DNA content of the cell layers was measured via a fluorometric assay of Labarca and Paigen [26] to determine the combined effects of the unconjugated or conjugated SMPs and released DOX on EaRASMC proliferation. The cell layers were harvested at 1 and 21 days of culture in Pi buffer, sonicated on ice, and assayed for DNA content. Cell density was calculated assuming 6 pg of DNA per cell.
2.10. Fastin assay for elastin
A Fastin assay (Accurate Scientific and Chemical, Westbury, NY) was used to quantify the amounts of elastic matrix (alkali-soluble and insoluble fractions) deposited by EaRASMCs. For each of 3 replicate samples, cell layers from 3 separate wells were harvested in Pi buffer, at 21 days of culture, and pooled. The cell layers were homogenized by sonication over ice. The cell suspension thus obtained was digested with 0.1 N NaOH (1 h, 98 ºC) and then centrifuged to yield a pellet containing mature, highly cross-linked alkali-insoluble elastin, and a supernatant fraction containing less cross-linked alkali-soluble elastin. The alkali-insoluble elastin was then converted into a soluble form prior to quantification, as the Fastin assay can only quantify soluble α-elastin. To do this, the pellet obtained after the NaOH digestion step was dried and solubilized with 0.25 M oxalic acid (1 h, 95 ºC), then pooled and centrifuge-filtered (3000 rpm, 10 min) in microcentrifuge tubes (Amicon® Ultra, 10 kDa molecular weight cut-off; Millipore, Inc., Billerica, MA). The alkali-soluble and insoluble matrix elastin fractions, as well as the tropoelastin precursors released into the cell culture medium were then measured using the Fastin assay. The amounts of elastin measured were also normalized to the corresponding DNA amounts, so as to provide an accurate comparison between the different treatments.
2.11. Western blots for MMP-2 and −9 expression
MMP-2 and −9 expression by EaRASMCs co-cultured with the cathepsin K pAb-conjugated or unconjugated DOX-SMPs were compared using western blots. At 21 days of culture, the cell layers were harvested in RIPA buffer with protease inhibitor and 3 wells were pooled per replicate (n = 3 replicates/treatment). The samples were assayed for total protein content using a bicinchonic acid (BCA) assay kit [25]. Maximum volumes of sample protein (15.6 μL) were then loaded under reduced conditions into each lane of a 10% Bis-Tris electrophoresis gel (Invitrogen), along with a SeeBlue™ pre-stained molecular weight ladder (Invitrogen) and MMP-2 and −9 standards. The gels were run in MOPS buffer (Invitrogen) for 50 minutes at 200 V, and subsequently dry transferred onto nitrocellulose membranes (iBlot® Western Blotting System, Invitrogen). As before, the membranes were blocked with Odyssey Blocking Buffer for 1 h and then immunolabeled (4 oC, 16 h) with a rabbit polyclonal antibody against MMP-2 (1:500 dilution; Abcam, Cambridge, MA) or rabbit monoclonal antibody against MMP-9 (1:500 dilution; Millipore, Billerica, MA) with a mouse monoclonal antibody against β-actin (1:1000 dilution; Sigma-Aldrich) as a loading control. Secondary labeling occurred for 1 h at room temperature using IRDye® 680LT goat-anti-rabbit (1:15,000 dilution; LI-COR Biosciences) and IRDye® 800CW goat-anti-mouse (1:20,0000 dilution; LI-COR Biosciences). A LI-COR Odyssey laser scanner was used to quantify the fluorescence of the secondary antibodies. The intensities of the active MMP-2 and MMP-9 bands on all gels were quantified using ImageJ software, expressed in terms of relative density units (RDU) and normalized to the intensity of their respective β-actin bands to enable comparison between the different test cases within the same blot. The ratios obtained for the SMP-supplemented cell layers were further normalized to that for standalone EaRASMC cultures (treatment controls).
2.12. Gel zymography for MMP-2 and −9 activity
The differential effects of cathepsin K pAb-conjugated and unconjugated DOX-SMPs on enzyme activity of MMP-2 and −9 in EaRASMC cultures was assessed using gel zymography, as previously described [17,25]. Cell layers harvested in RIPA with a protease inhibitor were loaded into each lane of a 10% zymogram gel (Invitrogen) in a volume containing 5 μg of protein, along with a SeeBlue™ pre-stained molecular weight ladder, and MMP-2 and −9 protein standards. Gels were run for 2 h at 125 V. The gels were then washed in a buffer containing 2.5% v/v Triton-X-100 for 30 min to remove sodium dodecyl sulfate (SDS) detergent, and then incubated overnight in a substrate/development buffer to activate the MMPs. The gels were stained with Coomassie Brilliant Blue solution for 45 min, and destained for 90 min, until clear bands appeared visible against the blue background of the gel. Band intensities (RDU) of the bands obtained for SMP-supplemented cultures were measured using ImageJ software, and normalized to those obtained for the SMP-untreated control cultures to determine fold changes in MMP activity. Data was acquired from 3 independent replicate gels.
2.13. Visualizing deposited elastic matrix via transmission electron microscopy (TEM)
To image the deposited elastic matrix, EaRASMCs were seeded into permanox chamber slides (5 × 104 cells per well) stimulated with 50 ng/ml TNF-α. After 21 days of culture, the test and control EaRASMC cultures (Section 2.8) were rinsed with PBS (37 °C) and fixed (5 min, 37°C, 4% w/v paraformaldehyde/ 2.5% w/v glutaraldehyde prepare in 0.1 M sodium cacodylate buffer). Following this, the cell layers were incubated in the fixative overnight at 4 °C. The samples were post-fixed in 1% w/v osmium tetroxide (1 hour), dehydrated in a graded ethanol series (50–100% v/v), embedded in Epon 812 resin, sectioned, placed on copper grids, stained with uranyl acetate and lead citrate, and imaged (using FEI Tecnai G2 Spirit) under multiple magnifications.
2.14. Statistical analysis
All experimental data presented (n = 3/condition, unless stated otherwise) are mean values with standard deviation (SD). Statistical significance of differences between mean values for different samples and conditions was evaluated using a Student’s t-test, with p ≤ 0.05 considered as statistically significant.
3. Results
3.1. Formulation and physical characterization of PLGA SMPs
The physical characteristics of PLGA SMPs, such as size and zeta potential, frequently depend on the formulation method(s) employed [27, 28]. DOX-free SMPs formulated via a single emulsion solvent evaporation method using 0.25% w/v DMAB as the stabilizer, exhibited mean hydrodynamic diameters of 295.1 ± 8.1 nm and a ζ-potential of +35.5 ± 0.7 mV (see Table 1). Encapsulating 2% w/w fluorescein or 2% w/w DOX via a double emulsion solvent evaporation technique did not significantly alter the size or surface charge of the PLGA SMPs, as also shown in Table 1. The SMPs were found to exhibit a relatively uniform size distribution and surface charge. The overall encapsulation efficiency of DOX in the PLGA SMPs was around 42%.
Table 1.
Mean hydrodynamic size and surface charge of PLGA NPs (n = 6 replicates, mean ± SD).
| Size (nm) | ζ-potential (mV) | |
|---|---|---|
| No encapsulation | 295.1 ± 8.1 | +35.5 ± 0.7 |
| 2% w/w fluorescein | 338.0 ± 9.1 | +32.9 ± 0.4 |
| 2% w/w DOX | 310.5 ± 8.4 | +36.2 ± 0.6 |
3.2. Assessment of cathepsin K antibody conjugation to SMPs
Fluorescence spectroscopy was used to compare the extent of cathepsin K antibody binding to SMPs. Increasing the time of antibody-SMP contact resulted in higher levels of antibody conjugation on the SMPs, for both the adsorption and covalent conjugation methods (Figure 1A). Extending the incubation period to 24 h did not increase RFU values beyond that measured at 5 hours, suggesting saturation of the SMP surface with bound antibodies; SMPs not conjugated with the cathepsin K antibody, but incubated with the fluorescein-tagged secondary antibody showed minimal fluorescence, indicating lack of non-specific SMP binding of the secondary probe. As seen in Figure 1B, for a 5 hour incubation period, no significant differences were noted in SMP normalized amounts of conjugated cathepsin K antibody, between the adsorption and covalent conjugation methods. The FITC SMPs incubated without AF546 showed an RFU ratio that was significant but still far lower than the ratio obtained for the test cases. These results suggest cathepsin K pAb was efficiently incorporated on the SMP surface. Confocal micrographs showed visibly greater co-localization of cathepsin K antibodies (red) with the fluorescein-encapsulated SMPs (green) conjugated via the covalent method (Figure 1E). Control SMPs not conjugated with the cathepsin K antibody but treated with the AF546-tagged secondary probe showed no auto-fluorescence in the red spectrum indicating lack of non-specific binding of the secondary probe.
Figure 1.
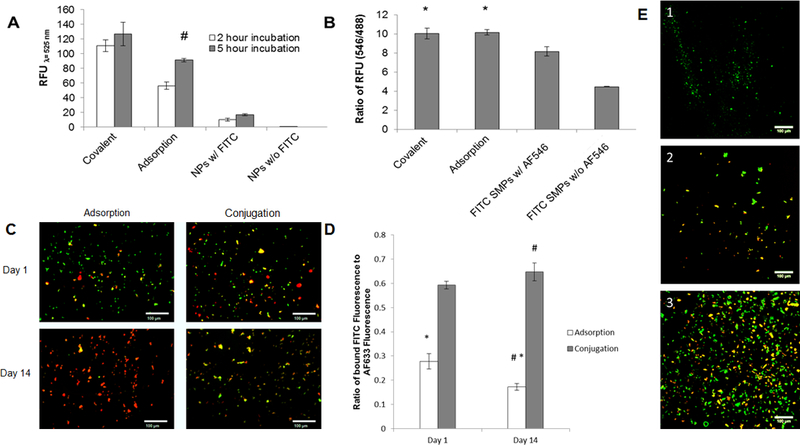
UV spectroscopy analysis of PLGA SMPs conjugated with cathepsin K antibody. Panel 1A shows effect of incubation time on conjugation of the antibody to SMPs. The cathepsin K antibody was detected with a fluorescein-tagged secondary Ab. A higher fluorescence intensity (RFU) indicates more effective antibody conjugation. Panel 1B compares relative abundance of conjugated antibodies on the SMPs. Cathepsin K antibodies were conjugated onto fluorescein-loaded SMPs over 5 hours, and were detected with secondary antibodies tagged with AF546. Values shown indicate mean ± SD of RFUs (Panel A) or of ratios of RFUs due to the fluorescein and AF546; n = 3 per case; # denotes significance of differences versus 2 h of incubation, deemed for p < 0.05; * denotes significance of differences versus control FITC SMPs treated with the AF-546-tagged secondary antibody, deemed for p< 0.05. Panels 1C, 1D show results of fluorescence microscopy analysis of cathepsin K surface modification to AF633-loaded SMPs (red). A fluorescein antibody (green) was added to visualize the cathepsin K modification. Panel C shows representative images for the adsorption and conjugation methods at day 1 and day 14. The green fluorescence demonstrates successful cathepsin K conjugation to the SMP surface. At day 14, green fluorescence associated with SMPs modified using Ab-adsorption was much lower compared to SMPs chemically conjugated with the Abs. Panel 1D shows the ratio of FITC intensity to AF633 intensity (mean ± SE; adsorption n=132, n=130 and conjugation n=154, n=207). The conjugation method bound more cathepsin K to the SMP surface for a longer period of time. # denotes significance of differences between adsorption and conjugation on day 14 deemed for p<0.05. * denotes significance of differences between day 1 and day 14 for the adsorption method deemed for p<0.05. In panel 1E, confocal micrographs compare cathepsin K antibody bound to SMPs via adsorption and covalent conjugation methods (see quantitative data in panel 1B). Conjugation was performed over 5 hours. Fluorescein (green) was encapsulated within the SMPs and the cathepsin K antibody was detected with an AF546-tagged secondary antibody (red). Panel 1E1 shows lack of red auto-fluorescence from cathepsin K antibody-conjugated SMPs not treated with the AF546-tagged secondary antibody. Panels E2 and E3 show that cathepsin K antibody was successfully conjugated to the SMPs using the adsorption- and covalent binding methods respectively. Scale bar: 100 μm (panel C), 100 μm (panel E).
Fluorescence microscopy was used to compare robustness of cathepsin K Ab attachment to the SMP surface for both the adsorption and conjugation methods (Figures 1C and 1D). Images were taken under similar conditions at day 1 and day 14. The intensity of the AF633-loaded SMPs (red) was measured as well as the intensity of the FITC-labeled cathepsin K antibody (green). The fluorescence of the SMPs can be seen in Figure 1C with a noticeable change in FITC fluorescence at day 14 for the adsorption method. To quantify the changes in fluorescence intensity, the corrected total fluorescence was calculated and a ratio of FITC to AF633 was determined. Figure 1D shows the conjugation method provides a significantly (p < 0.05) more robust attachment of the cathepsin K Ab to the SMP surface. After 14 days, there was a significant decrease in cathepsin K Ab presence on the SMP surface for the adsorption method. However, there was no significant loss of cathepsin K Ab from the SMP surface at 14 days, when covalently conjugated.
3.3. Verifying cathepsin K overexpression
3.3.1. Verifying cathepsin K overexpression in TNF-α stimulated EaRASMCs
Increased expression of cathepsin K by diseased EaRASMCs and effects of TNF-α in augmenting such expression were investigated towards simulating the aneurysmal tissue milieu in cell culture. Western blot analysis (Figure 2D) showed cathepsin K protein synthesis to be significantly higher in EaRASMC cultures stimulated with TNF-α relative to RASMCs and unstimulated EaRASMCs; only the zymogen form of cathepsin K was detected. IF labeling for cathepsin K (Figure 2A and 2B; red fluorescence), confirmed these findings. Cathepsin K was localized both on the cell membrane in the extracellular space and intracellularly as seen in Figures 2B and C. In Figure 2C, cathepsin K expression (red fluorescence) at the bottom of a cell appears minimal, but is seen to increase as we move up through the cell to the cell surface (Panel 2C3).
Figure 2.
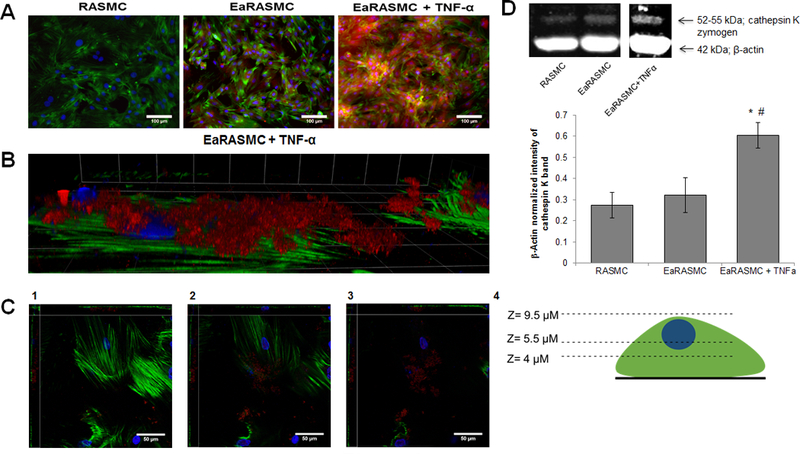
(A). IF images showing relative expression of cathepsin K by healthy, and aneurysmal SMCs, without and with TNF-α stimulation. Cathepsin K, visualized with AF-546-tagged secondary antibody, appears red while the cytoskeletal actin filaments stained with AF488 phalloidin appear green, and DAPI-stained nuclei appear blue. Scale bar: 100 μm. (B) High magnification view of EaRASMCs stimulated with TNF-α and cathepsin K visualized with AF-546-tagged secondary antibody and cytoskeletal actin stained with AF488 phalliodin. Grid: 23 μm x 23 μm. (C). Images of the EaRASMCs at different z-axis heights. (1) The bottom of the cell layer which shows minimal cathepsin K. (2) The middle of the cell layer in which cathepsin K begins to appear. (3) The top of the cells where the most cathepsin K is found. Scale bar for panels 2C1–3: 50 μm. (4) Schematic of the z-axis heights for images 2C1–3. (D). Western blot analysis for relative expression of cathepsin K by healthy, and aneurysmal SMCs, without and with TNF-α stimulation. The figure shows representative blot, indicating bands for the cathepsin K zymogen and β-actin (loading control). The plot shows β-actin normalized cathepsin K band intensity (mean ± SD; n = 3 per case; # denotes p < 0.05 compared to control RASMCs); * denotes p < 0.05 compared to TNF-α-unstimulated EaRASMCs. # indicates significance of differences versus RASMCs, deemed for p < 0.05.
3.3.2. Verifying cathepsin K overexpression in elastase-injured arteries
Differences in cathepsin K expression in healthy and matrix-injured arteries was investigated to rationalize SMP targeting using a cathepsin K Ab in abdominal aortic aneurysms, wherein such elastase-induced matrix disruption occurs. Western blot analysis (Figure 3) showed significant increase in active cathepsin K (38 kDa) expression after exposure to elastase compared to the saline control (7.85 ± 1.93 vs. 0.14 ± 0.02, p < 0.05). These findings are similar to our results showing an increased cathepsin K expression in TNF-α-activated EaRASMCs cultures evocative of an injured vascular tissue milieu and rationalize study of elastase injured arteries in the context of investigating targeted binding of cathepsin K Ab-conjugated SMPs (Figure 2D).
Figure 3.
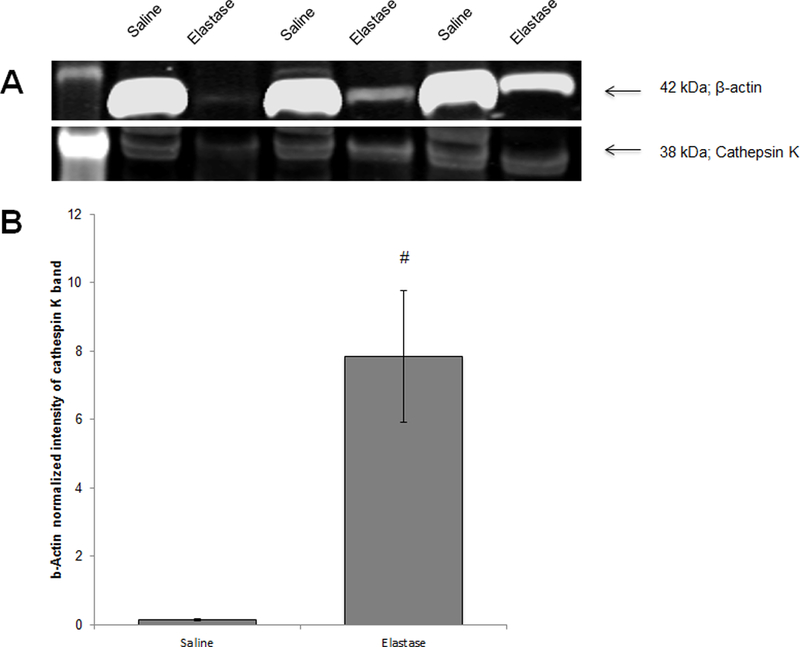
Western blot analysis for cathepsin K expression in saline and elastase treated porcine carotid arteries. Panel A shows a representative blot, showing the active cathepsin K form (38 kDa) and β-actin (loading control). Panel B shows β-actin normalized cathepsin K band intensity (mean ± SEM; n = 9 per case; # denotes significance of difference, deemed for p < 0.05.
3.4. Targeted binding of conjugated SMPs to cathepsin K expressing cells
Figure 4A shows confocal images of SMCs exposed to cathepsin K Ab-conjugated PLGA SMPs (red fluorescence) and unconjugated control SMPs. EaRASMCs stimulated with TNF-α exhibited a high amount of bound, conjugated SMPs relative to the RASMCs and EaRASMCs cultured without TNF-α activation. This indicates that the cathepsin K Ab-conjugated SMPs have a much higher binding ability correlated to the overexpression of cathepsin K in the stimulated cultures. The control cases showed a similar pattern of amount of SMPs bound to cell layers being directly related to the amount of cathepsin K expression. As previously mentioned, the EaRASMCs without TNF-α exhibit only slightly higher cathepsin K expression compared to the RASMCs (Figure 2A). This is reflected in Figure 4A by only a mildly higher binding of Ab-conjugated SMPs in the EaRASMC cultures versus the RASMC cultures. Figure 4B shows a higher magnification image of SMP-bound, TNF-α-stimulated EaRASMCs. Figure 4C provides images of this cell layer at varying z-axis heights. Figure 4C1 shows the bottom of the cell layer exhibiting minimal fluorescence due to cathepsin K-Ab modified SMPs. Moving up through the cell layer (4C2–4C3), the SMPs begin to appear within the cells. In 4C4, the SMPs are clearly visible on the cell surface.
Figure 4.
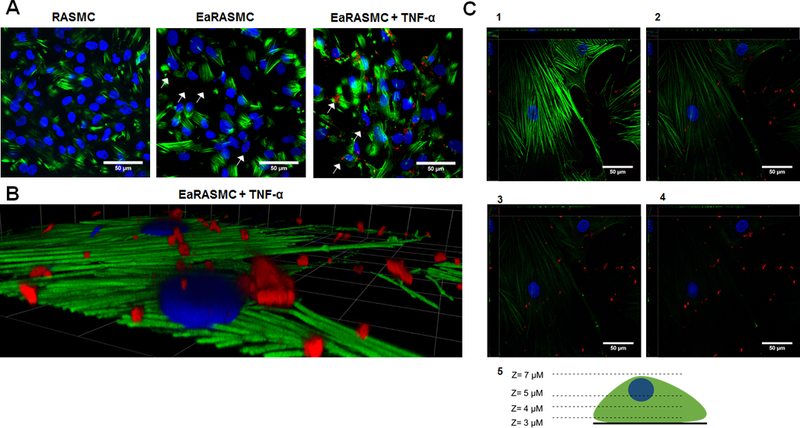
(A) Confocal micrographs showing binding of cathepsin K antibody-conjugated SMPs to healthy, and aneurysmal SMCs, without and with TNF-α stimulation. All SMPs were encapsulated with AF546, causing them to fluoresce red. A significantly higher number of SMPs bound to EaRASMCs than to the RASMCs and more still in EaRASMC cultures stimulated with TNF-α (white arrows). Cytoskeletal actin filaments, stained with AF488 phalloidin fluoresce green and DAPI-stained nuclei appear blue. (B). High magnification image of SMP localization to TNF-α stimulated EaRASMCs. (C). Images of EaRASMCs with SMPs at various z-axis heights. (1) Bottom of cell layer with minimal SMP bound. (2) and (3) Middle layers of the cell where SMPs begin to appear around the cell. (4) Top of the cell layer where SMPs are bound to the cell surface. (5) Schematic of the various z-axis heights for images 1–4. Scale bars represent 50 μm (A), 23 μm x 23 μm (B), and 50 μm (C).
Figure 5 shows fluorescence images (panel A) and a plot indicating the fold-differences (panel B) in binding of Ab-unconjugated SMPs, IgG Ab-conjugated SMPs, and cathepsin K Ab-conjugated SMPs to cathepsin K expressing wall of porcine arteries. Similar to observations in cell cultures, intensity of fluorescence due to uptake of cathepsin K Ab-conjugated SMPs in the artery wall was ~2-fold higher relative to that observed for Ab-unconjugated SMPs. Further, binding of nonspecific Ab–modified SMPs to the artery wall was not statistically different from that of the unmodified SMPs.
Figure 5.
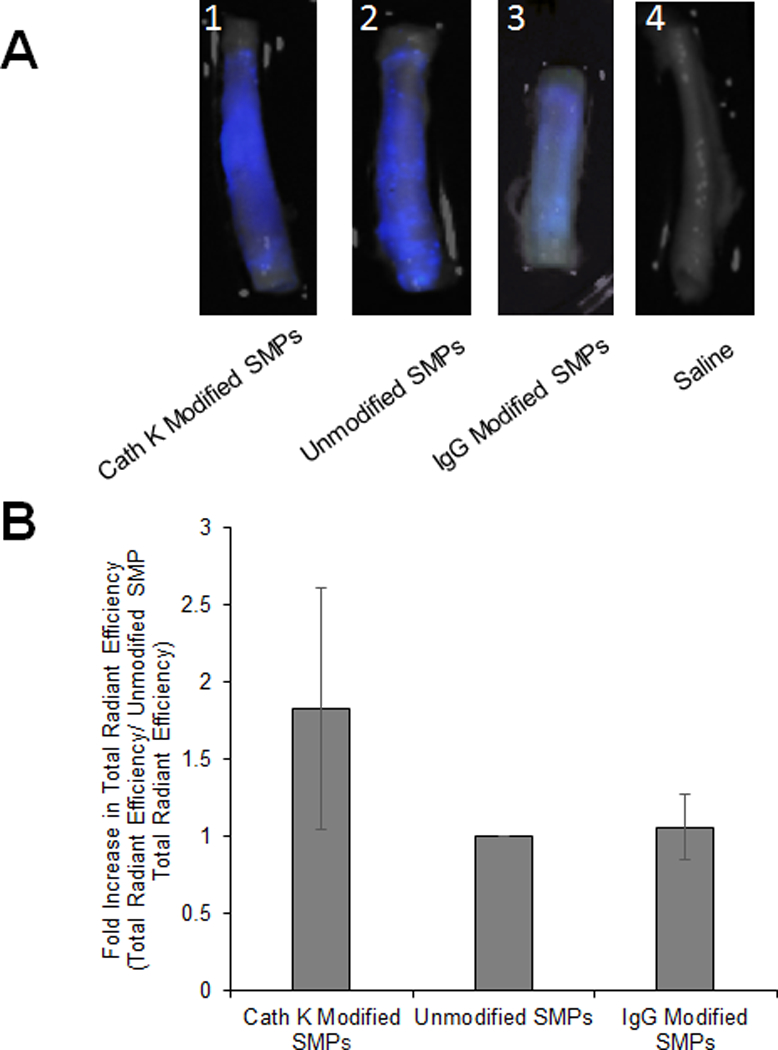
Targeting of cathepsin K Ab-conjugated SMPs to wall of elastase injured porcine carotid arteries. (A) Pseudocolor of SMP localization in elastase treated arteries with cathepsin K-Ab conjugated AF633 SMPs (1), unconjugated AF633 SMPs (2), IgG-Ab conjugated AF633 SMPs (3), and saline (4). (B) Fold difference in binding of cathepsin K Ab-conjugated SMPs, and non-specific IgG conjugated SMPs to elastase treated arteries compared to binding of unmodified SMPs (mean ± SEM; n = 6 and n = 3 for IgG-Ab conjugation). * indicates significance of differences between the cases deemed for p < 0.05.
3.5. In vitro DOX release from conjugated and unconjugated SMPs
Figure 6 shows the in vitro release of DOX from unconjugated and cathepsin K Ab-conjugated PLGA SMPs. Only a single SMP formulation incorporating 2% w/w DOX and provided at a concentration of 0.5 mg of SMPs/ml was studied. Similar to previous reports, DOX release from both unconjugated and the Ab-conjugated SMPS exhibited a biphasic release profile with an initial burst release over the first day [11, 29] followed by a slower exponential release [30, 31].
Figure 6.
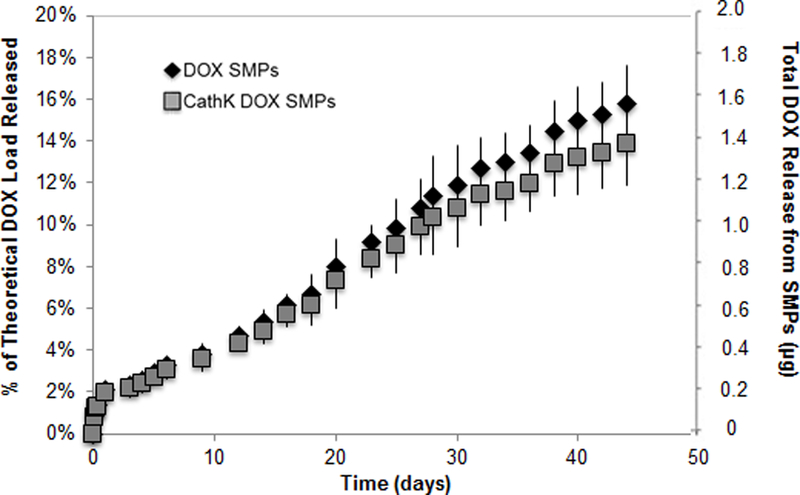
In vitro DOX release profiles from cathepsin K antibody-conjugated and unconjugated PLGA SMPs (0.5 mg/ml) loaded with 2% w/w DOX (mean ± SD; n = 3 per group).
Over the next 45 days over which DOX release was monitored, the cumulative DOX release from either case was less than 20% of the total amount of drug encapsulated. There was no significant difference in the release profiles between the Ab-conjugated and unconjugated SMPs.
3.6. Effect of conjugated, DOX-loaded SMPs on SMC proliferation and elastic matrix synthesis
Previous unpublished lab data that indicated a 5 μg/ml exogenous dose of DOX was cytotoxic to EaRASMC cultures. We chose a 2% w/w DOX encapsulation in PLGA SMPs for further in vitro study as the total DOX released over a 21 day culturing period is less than 5.0 μg/ml of DOX based on the release profiles in Figure 6. As Figure 7A indicates, DOX release from the PLGA SMPs did not hinder the proliferation of the EaRASMC cultures. While the cultures exposed to unconjugated DOX SMPs showed significantly higher proliferation (p < 0.5) than the cultures with conjugated DOX SMPs, cell proliferation in both sets of SMP-treated cultures were not statistically different from the SMP-untreated cultures.
Figure 7.
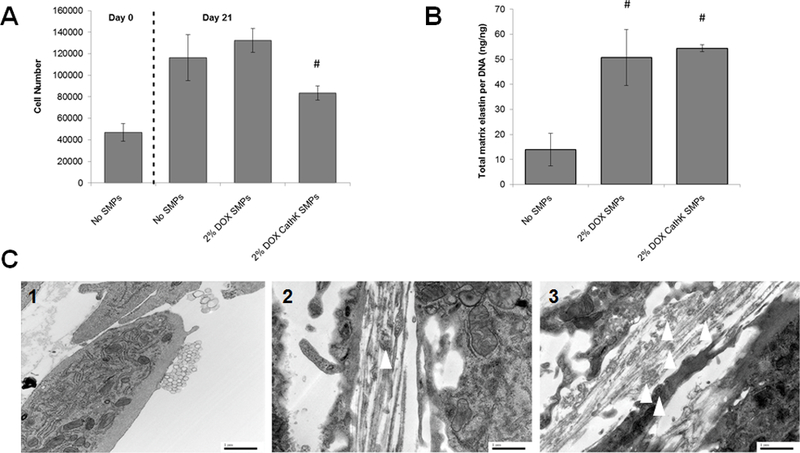
(7A) Proliferation of EaRASMCs is not impacted by coculture with DOX-SMPs (cathepsin K Ab-conjugated or unmodified), although cell proliferation in the cultures treated with the Ab-conjugated SMPs was significantly lower than that cultured with the unmodified SMPs. DOX loading within the SMPs was 2% w/w. SMP concentration in the cultures was 0.2 mg/ml. Cells were harvested at 1 and 21 days after initial seeding (mean ± SD; n = 3 cultures per group; # denotes significance of differences versus unconjugated DOX SMPs, deemed for p< 0.05). (7B) Cathepsin K antibody conjugation of DOX-SMPs does not alter their pro-elastogenic effects on cultured EaRASMCs. SMP-untreated EaRASMC cultures were investigated as treatment controls. All cell layers were cultured for 21 days. Amounts of deposited elastic matrix, comprised of both the alkali-soluble and alkali-insoluble elastin fractions were normalized to DNA content of the respective cell layers (mean ± SD; n =3 cultures per group; # denotes significance of differences relative to treatment controls deemed for p < 0.05). (7C) Transmission electron micrographs showing effects of cathepsin K-Ab-conjugated and unconjugated DOX-SMPs on elastic matrix deposition in TNF-α activated SMC cultures. Elastic matrix deposition was sparse in the SMP-untreated cultures and few amorphous elastin deposits and no mature fibers were seen (1). Numerous forming elastic fibers were seen in the SMP-treated cultures (2), with a greater number of amorphous elastin deposits (white arrows) associated with the microfibrillar components in the cultures that received the cathepsin-K Ab-modified DOX-SMPs (3). Scale bars: 1 μm.
Results of the Fastin assay presented in Figure 7B, showed that on a DNA content (or cell number) normalized basis, elastic matrix deposition in cultures receiving unmodified DOX-releasing SMPs was significantly higher (p < 0.05) than in SMP-untreated cultures; matrix deposition was also unaffected by cathepsin K Ab-conjugation to the SMPs. The TEM results presented in Figure 7C closely mirror the outcomes of the Fastin assay in demonstrating that cathepsin K Ab-conjugated and unconjugated DOX SMPs increase generation of elastic matrix compared to NP-untreated EaRASMC cultures. While only sparse elastin fibrils and microfibrils were seen in the control cultures (Figure 7C1), significantly greater presence of forming elastic fibers associated with amorphous elastin deposits were seen in the SMP-treated cultures. No significant differences in elastic matrix deposition were noted between the cultures receiving Ab-conjugated and unconjugated SMPs.
3.7. Effect of DOX-loaded SMP conjugation on MMP-2 & −9 expression and activity
A representative Western blot image for MMP-2 expression in EaRASMC cultures supplemented with the different SMP formulations is shown in Figure 8A. The active MMP-2 bands across all conditions were normalized to their respective β-actin loading control band. The data from cultures receiving unconjugated or cathepsin K Ab-conjugated DOX SMPs is shown normalized to the control EaRASMC cultures receiving no DOX SMPs in Figure 8B. For both cases, the DOX released from the PLGA SMPs caused a roughly 50% decrease in MMP-2 expression relative to the control. There was no significant difference (p > 0.05) in MMP-2 expression between cultures receiving conjugated or unconjugated DOX SMPs. In all cases, the expression of MMP-9 was too low to quantify via densitometry [8, 25].
Figure 8.
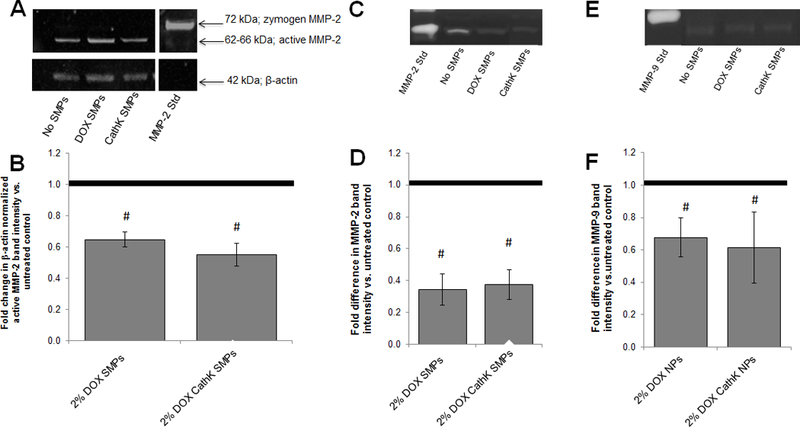
Panels 8A and 8B show effects of co-culture with unconjugated and cathepsin K antibody-conjugated DOX-SMPs on MMP-2 protein synthesis in TNF-α-activated EaRASMCs cultures, as analyzed by western blots. Panel A shows a representative blot. Panel B shows fold difference in β-actin normalized band intensity for active MMP-2 protein in DOX-SMP-treated EaRASMC layers, relative to control cultures cultured with no SMPs (mean ± SD; n = 3 cultures/condition). # indicates significant differences versus controls (assigned a value of 1.0) deemed for a p value < 0.05. Panels 8C-F show effects of co-culture with unconjugated and cathepsin K antibody-conjugated DOX-SMPs on MMP-2 and MMP-9 activity in TNF-α-activated EaRASMCs cultures, as analyzed by gel zymography. Panel C shows representative image of gel zymogram for MMP-2. Panel D shows fold difference in β-actin-normalized MMP2 band intensities compared to SMP-free control cultures (assigned value of 1.0; dotted line). Panel E shows representative gel zymogram for MMP-9. Panel F shows the fold difference in β-actin-normalized MMP9 band intensities versus SMP-free control cultures (assigned value of 1.0) Values shown indicate mean ± SD based on analysis of n = 3 cultures per condition. # denotes significance of differences versus control cultures, deemed for p < 0.05.
Figures 8C and 8E shows a representative image of the gel zymography for MMP-2 and −9 activity, respectively, in all three test cases. The control-normalized band intensities between EaRASMC cultures receiving conjugated or unconjugated DOX SMPs showed significantly reduced (p < 0.05) MMP-2 and −9 activity as evidenced in Figures 8D and 8F, with ~3-fold attenuation in MMP-2 activity and about 40% attenuation in MMP-9 activity. There was no significant difference in activity between cultures receiving either conjugated or unconjugated DOX SMPs.
4. Discussion
SMP-based delivery of DOX is attractive from the standpoint of a) avoiding potential side effects and body wide inhibition of MMPs due to systemically delivered DOX, and b) enabling controlled and sustained release of DOX within AAA tissue at doses significantly below the systemic doses, at which we have shown the drug to not only continue to inhibit MMPs but also uniquely stimulate elastin regeneration [11]. In prior articles, we described a novel formulation of cationic amphiphile-surface modified DOX- PLGA SMPs that provide, independent of the released DOX, both pro-elastogenic and anti-proteolytic effects [11, 17, 25]. The goal of this study is to develop a mechanism to provide greater specificity of targeting DOX-loaded PLGA SMPs to AAA tissue for localized and sustained release of active agents to augment regenerative repair of elastic matrix towards arresting or regressing AAA growth. An added interest is to assess the implications of improved binding and retention of the modified SMPs within cell layers to possibly enhance their pro-regenerative and anti-proteolytic effects on the cells. In this study, we sought to improve specificity of targeting of our SMPs by conjugating them with a cathepsin K Ab. This has been based on recent findings that cathepsin K is chronically overexpressed in AAA tissues [14, 15]. This has been attributed to a reduction in cystatin, a competitive cysteine protease inhibitor in the inflamed AAA environment [15, 16], which results in increased elastolytic activity of cathepsin K, and through a positive feedback mechanism triggered by matrix breakdown, further increases in cathepsin K expression [4, 33].
For this study, we continued to study PLGA SMPs since the polymer is FDA approved for clinical drug delivery applications due to their biodegradability and high biocompatibility [27, 28]. PLGA containing a 50:50 ratio of lactide:glycolide content has a molecular weight equivalent to 117 kDa, and because its biodegradation occurs over several months, it is ideal for our proposed application [21, 28]. SMP sizes between 200 and 500 nm were targeted since we have shown SMPs larger than 200 nm to be mostly excluded in the extracellular space, where matrix assembly occurs [11] and SMPs > 500 nm exhibit increased uptake by phagocytes [33]. Using DMAB, as an emulsion stabilizer during SMP formulation resulted in functionalization of the cationic amphiphile on the SMP surface to impart a cationic charge. In our prior study [11], we showed cationic moieties on the SMP surface to interact electrostatically with the active site of MMPs to reduce their proteolytic activity [34, 56]. Differently, hydrocarbon chains present on the cationic amphiphiles bind highly hydrophobic elastin substrates, and induce conformational changes that expose lysine side chains on the elastin molecule for crosslinking, while the cationic elastin-surfactant complex attracts anionic LOX enzyme to increase crosslinking of the elastin [57, 58]. We also showed the magnitude of these effects to be dependent on the choice of cationic amphiphile and the imparted surface charge [11].
We investigated two different methods for conjugating cathepsin K Abs to the SMP surface, namely, adsorption and covalent reaction, which are frequently used to conjugate biomolecules to surfaces [22, 35–40]. With each method we also investigated the impact of conjugation time on the efficiency of Ab conjugation onto the SMPs. While adsorption uses weak van Der Waals forces to hold the Abs to the SMP surface [37, 38], covalent conjugation involves the use of carbodiimide reaction chemistry to generate strong covalent bonds via a defined chemical reaction stoichiometry [39]. As a result of the much stronger and resilient covalent bonding (Figure 1C, 1D), in the latter case, the conjugated Abs are more resistant to displacement by the vastly more abundant serum proteins over at least 2 weeks [37, 38]. Our results in Figure 1A shows, while increasing conjugation time to 5 hours resulted in significant increases in Ab conjugation, especially for the adsorption method, in both methods, only very modest increases in Ab conjugation were observed at longer conjugation times (up to 24 hours; not shown), suggesting that at 5 hours of incubation, saturation of the SMP surface has already been likely achieved; in any case, extending the incubation time is also undesirable due to increased hydrolytic degradation of the PLGA matrix. No significant differences in Ab conjugation normalized to SMP quantity were observed between for 5 h covalent binding and adsorption conditions (Figure 1B). In an early experiment (not shown), we determined that blocking (saturating) non-specific binding sites on the SMPs with serum (5% v/v; 1 h) prior to secondary Ab incubation had no effect in reducing the relatively high non-specific binding of the fluorophore, the absorbance ratio for which was however significantly lower than the antibody-modified SMPs. However, likely because of the defined reaction chemistry and strong covalent linkages, SMPs conjugated via the covalent method were found to exhibit more uniform (Figure 1E), and resilient (Figure 1C, 1D) Ab-binding. On the basis of these findings, we deemed that covalent conjugation of the cathepsin K Abs over a 5 hour period is most appropriate, due to a) stronger bonds formed which resist displacement by serum proteins, and b) likely saturation of the SMP surface with Abs.
Cathepsin K overexpression in the AAA wall, has been observed both in humans [14, 15] and in rat models [41, 55]. In vivo, cathepsin K is generated by vascular SMCs only upon stimulation by macrophage-derived inflammatory cytokines (e.g., intereukin 1-β, interferon-γ, and TNF-α) [14]. As a result, constitutive expression of cathepsin K by unstimulated SMCs (aneurysmal and healthy) in culture and in non-injured arteries is minimal as our results in Figures 2 and 3 indicate. Activating cultured EaRASMCs with TNF-α, as has been performed by others to stimulate an inflammatory response [15, 16], was found to significantly increase cathepsin K expression (Figure 2). Similarly, injuring arteries with elastase significantly increased cathepsin K expression (Figure 3). Cathepsin K expression was found to be localized in both the intra- and extra-cellular domains, agreeing well with published findings that the protease is localized both within lysosomes and endosomes in the cytoplasm and also in the extracellular space [42, 43]. The extracellular presence of cathepsin K at the cell surface presents a significant advantage in our efforts to target our SMPs to this compartment for augmenting regenerative elastic matrix repair.
Confocal imaging (Figure 4) showed co-localization and hence likely binding of cathepsin K Ab conjugated SMPs to sites of cathepsin K expression within cell layers, mostly on the cell surface, but also intracellularly. These results are consistent with our earlier published findings that our SMPs of size 300–400 nm are mostly excluded to the cell exterior. SMP binding also correlated positively to the expression of cathepsin K, being significantly higher in cultures activated with TNF-α, than in unstimulated EaRASMC and RASMC controls. Similarly, as expected, uptake and retention of cathepsin K Ab-conjugated SMPs was significantly greater within the walls of matrix injured arteries expressing higher levels of cathepsin K (Figure 5A1 vs. 5A4); SMPs bound with a non-specific IgG (Figure 5A3) showed significantly lower binding to the vessel wall, to the extent of unmodified SMPs (Figure 5A2). Since our earlier published findings that our SMPs of size 300–400 nm are mostly excluded to the cell exterior, it is highly likely that our cathepsin K Ab-conjugated SMPs bind to cathepsin K on the cell surface/ extracellular space.
Release of DOX from the Ab-conjugated SMPs was not significantly different from unconjugated SMPs (Figure 6), which was an expected outcome since the size and surface charge of both sets of SMPs were identical. With both preparations, the total amount of DOX cumulatively released over the 45 days was only ~ 20% of the theoretical DOX loading, suggesting the potential for sustaining DOX release from the SMPs over a significantly longer period. The near-steady state levels of DOX released were much lower than the typical DOX dosing levels (16–54 μg/ml) [8, 11, 44] above which the drug was found to inhibit proliferation of aneurysmal SMCs and their deposition of crosslinked elastic matrix [45, 46], outcomes we have confirmed in recent culture studies (unpublished results). Differences in proliferation between EaRASMCs cultured with DOX-loaded SMPs (both Ab-conjugated and unconjugated) or without SMPs (controls) were insignificant (Figure 7A), confirming our prior findings that the SMPs and DOX released therefrom at the doses observed, do not impact EaRASMC proliferation. That said the proliferation levels observed in cultures supplemented with the cathepsin K-Ab-conjugated SMPs was found to be modestly lower, and statistically different from that in cultures supplemented with the unconjugated SMPs. Although this is highly likely to be related to intrinsic experimental errors in cell enumeration, especially since the cell proliferation in both cases was not different from the SMP-free controls, there are other possible explanations. One possibility is that the more intimate binding and retention of the Ab-conjugated SMPs to the cells results in greater and more sustained depolarization of the cell membrane due to the cationic charge on the SMPs. This has been shown by other groups to trigger increases in intracellular calcium ion (Ca2+) concentrations, and to in turn inhibit proliferation in cultures of normal cells [47]. Alternately, more intimate binding of the cathepsin K Ab-conjugated SMPs to the cell surface results in more effective MMP inhibition due to the action of both DOX and the surface cationic amphiphiles, as we have previously reported [11]. There are several published studies that have shown MMP inhibition to enhance SMC proliferation and migration [59].
As expected, based on our published findings as to the pro-elastogenic effects of a) DOX at the currently released dose range, and b) our cationic amphiphile-surface functionalized PLGA SMPs, both sets of DOX-SMPs (cathepsin K Ab-conjugated and unconjugated) stimulated synthesis of elastic matrix (on a per cell basis) by EaRASMCs over SMP-free cell cultures (Figure 7B). The lack of any differences in elastic matrix synthesis on a per cell basis, between the two DOX-SMP-supplemented culture groups suggests a) no differences in DOX release between the DOX-SMP preparations, which we have confirmed (Figure 6), and b) no adverse impact of the Ab-conjugated DOX-SMPs on, or significant benefit to cell elastogenicity as a result of their improved binding and/or retention within the cell layers. While our TEM images shown in Figure 7C do mirror the results of the Fastin assay in showing greater elastic matrix deposition in SMP-treated EaRASMC cultures relative to SMP-untreated but TNF-α-activated EaRASMC controls, no mature fibers were seen in the SMP-treated cultures. Rather, elastic fiber structures in the process of forming were observed, with a visibly greater number of amorphous elastin deposits seen associated with the microfibrillar components and elastin fibrils in the cathepsin K Ab-conjugated SMP-treated cultures. While the poor elastin deposits and fiber formation in control cultures can be related to both intrinsically poor ability of EaRASMCs to synthesize tropoelastin and assemble elastic fibers on one hand and increased proteolytic activity resulting from overexpression of cathepsin K and other proteases, the improved outcomes in the SMP-treated cultures may be attributed to pro-elastogenic and elastic matrix crosslinking-promoting effects of our DMAB-functionalized polymer carriers and released DOX. It is also quite possible that improved binding of the cathepsin K conjugated SMPs to the cell layers contributed to localized anti-proteolytic effect, which might account for the greater number of amorphous elastin deposits within the forming elastic fibers in these cultures versus those in cultures treated with unconjugated SMPs. Further investigation is required in this regard.
Chronic overexpression of the elastolytic gelatinases MMP-2 and MMP-9 have been shown to drive AAA growth [10, 48–51], and oral DOX therapy inhibit these enzymes [10, 48] to slow AAA growth in both animal models [5, 44, 51] and humans [10]. The mechanism behind this inhibition is described in our lab’s previous work [11], and briefly, is based on ability of DOX to chelate Zn2+ and Ca2+ in the catalytically active domain of the MMPs-2 and −9 [52], which leads to enzyme denaturation and degradation [53, 54]. In our previous publication [11] we also showed that imparting a positive surface charge on the SMPs via use of DMAB as an emulsion stabilizer during SMP formulation, enables them to bind and anionic glutamic acid residues in the catalytically active domain of MMPs, leading to a decrease in their proteolytic activity. It is also possible that the two dodecyl chains presented by DMAB may block the active site of MMP-2 via steric hindrance to exert its inhibitory effect. Thus, the MMP inhibitory effects of our DOX SMPs may be attributed to both the drug and to the DMAB-surface modified polymer SMPs. Our present results (Figure 8) confirm our prior reported findings on the significant MMP inhibitory effects of our DOX-SMPs, and showed that cathepsin K antibody conjugation does not alter the aggregate MMP inhibitory effects due to DOX and the polymer nanocarriers. This outcome suggests that likely neither the covalent reaction chemistry for antibody conjugation nor the presence of the cathepsin K antibody on the SMP surface interferes with DOX release or MMP interactions with the pendant cationic amphiphiles on the SMP surface.
5. Conclusions
This study has shown that cathepsin K Ab conjugation is a useful approach for targeted binding of therapeutic DOX-SMPs to cathepsin K overexpressing, cytokine-activated SMCs typical of those within the aneurysmal wall. We have demonstrated that covalent conjugation is a more reliable and efficient method over physical adsorption, for incorporating cathepsin K Abs on the DOX-SMP surface. Our results have confirmed that cathepsin K Ab conjugation does not alter DOX release profiles and the pro-elastogenic and anti-proteolytic effects of the SMPs, attributable to both the released DOX and pendant cationic amphiphiles on the SMP surface. Future studies will investigate the ability of cathepsin K Ab-conjugated SMPs to penetrate the disrupted endothelium of the AAA wall to localize and be retained in the AAA wall in rat models following intravenous or catheter-based infusion to a flow occluded, matrix-disrupted aorta and their efficacy in stimulating regenerative matrix repair towards achieving AAA growth arrest.
Acknowledgements
The authors acknowledge research funding for this project from the National Institutes of Health (HL132856), American Heart Association (16IRG27250113) and National Science Foundation (BME Division; 1508642) awarded to AR. The PerkinElmer IVIS Spectrum CT In Vivo Imager purchased with funding from the National Institutes of Health SIG grant 1S10OD018205–01A1 was used in this work as well as the FEI Tecnai G2 Spirit transmission electron microscope purchased using funds from the National Institutes of Health SIG grant 1S10RR031536–01.
References
- [1].Lederle FA, Johnson GR, Wilson SE, Chute EP, Littooy FN, Bandyk D, Krupski WC, Barone GW, Acher CW, Ballard DJ. Prevalence and associations of abdominal aortic aneurysm detection through screening. Ann Intern Med 1997;126;441–9. [DOI] [PubMed] [Google Scholar]
- [2].Tanaka A, Hasegawa T, Chen Z, Okita Y, Okada K. A novel rat model of abdominal aortic aneurysm using a combination of intraluminal elastase infusion and extraluminal calcium chloride exposure. Journal for Vascular Surgery 2009;50;6;1423–32. [DOI] [PubMed] [Google Scholar]
- [3].Thompson SG, Ashton HA, Gao L, Scott RAP. Screening men for abdominal aortic aneurysm: 10 year mortality and cost effectiveness results from the randomized multicenter aneurysm screening study. BMJ 2009;338;2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Upchurch GR Jr, Schaub TA. Abdominal Aortic Aneurysm. Am Fam Physician 2006;73;1198–204. [PubMed] [Google Scholar]
- [5].Bartoli MA, Parodi FE, Chu J, Pagano MB, Mao DL, Baxter BT, Buckley C, Ennis TL, Thompson RW. Localized administration of doxycycline suppresses aortic dilation in an experimental mouse model of abdominal aortic aneurysm. Ann Vasc Surg 2006;20;228–36. [DOI] [PubMed] [Google Scholar]
- [6].Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. [DOI] [PubMed] [Google Scholar]
- [7].Chang WYC, Clements D, Johnson SR. Effect of doxycycline on proliferation, MMP production, and adhesion in LAM-related cells. Am J Physiol Lung Cell Mol Physiol 2010;299;L393–400. [DOI] [PubMed] [Google Scholar]
- [8].Franco C, Ho B, Mulholland D, Hou GP, Islam M, Donaldson K, Bendeck MP. Doxycycline alters vascular smooth muscle cell adhesion, migration, and reorganization of fibrillar collagen matrices. Am J Pathol 2006;168;1697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Baxter BT, Pearce WH, Waltke EA, Littooy FN, Hallett JW, Kent KC, Upchurch GR Jr, Chaikof EL, Mills JL, Fleckten B, Longo GM, Lee JK, Thompson RW. Prolonged administration of doxycycline in patients with small asymptomatic abdominal aortic aneurysms: report of a prospective (Phase II) multicenter study. J Vasc Surg 2002;36;1–12. [DOI] [PubMed] [Google Scholar]
- [10].Curci JA, Mao DL, Bohner DG, Allen BT, Rubin BG, Reilly JM, Sicard GA, Thompson RW. Preoperative treatment with doxycycline reduces aortic wall-expression and activation of matrix metalloproteinases in patients with abdominal aortic aneurysms. J Vasc. Surg. 2000;31:325–341. [DOI] [PubMed] [Google Scholar]
- [11].Sivaraman B, Ramamurthi A. Multifunctional nanoparticles for doxycycline delivery towards localized elastic matrix stabilization and regenerative repair . Acta Biomaterialia 2013;9;6511–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sylvester A, Sivaraman B, Deb P, Ramamurthi A. Nanoparticles for localized delivery of hyaluronan oligomers towards regenerative repair of elastic matrix. Acta Biomaterialia 2013;9;12;9292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, Moritz JD, Schu P, von Figura K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K deficient mice. Proc Natl Acad Sci USA 1998;95;13453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest 1998;102;576–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sukhova GK, Shi GP. Do cathepsins play a role in Abdominal Aortic Aneurysm Pathogenesis? Annals of the New York Academy of Sciences 2006;1085;161–9. [DOI] [PubMed] [Google Scholar]
- [16].Keegan P, Wilder C, Platt M. Tumor necrosis factor alpha stimulates cathepsin K and V activity via juxtacrine monocyte – endothelial cell signaling and JNK activation. Mol Cell Biochem 2012;367;65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gacchina CE, Deb PP, Bartha JL, Ramamurthi A. Elastogenic inductability of smooth muscle cells from a rat model of late stage abdominal aortic aneurysms. Tissue Eng 2011;17;1699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Anidjar S, Salzmann JL, Gentric D, Lagneau P, Camilleri JP, Michael JB. Elastase induced experimental aneurysms in rats. Circulation 1990;82;973–81. [DOI] [PubMed] [Google Scholar]
- [19].Guzman LA, Labhasetwar V, Song C, Jang Y, Lincoff AM, Levy R, Topol EJ. Local intraluminal infusion of biodegradable polymeric nanoparticles. A novel approach for prolonged drug delivery after balloon angioplasty. Circulation 1996;94;1441–8. [DOI] [PubMed] [Google Scholar]
- [20].Labhasetwar V, Song C, Humphrey W, Shebuski R, Levy RJ. Arterial uptake of biodegradable nanoparticles: effects of surface modifications. J Pharm Sci 1998;87;1229–34. [DOI] [PubMed] [Google Scholar]
- [21].Song CX, Labhasetwar V, Murphy H, Qu X, Humphrey WR, Shebuski RJ, Levy RJ. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. J Control Release 1997;43;197–212. [Google Scholar]
- [22].Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Con Rel 2007;120;18–26. [DOI] [PubMed] [Google Scholar]
- [23].Wissink M, Beernink R, Pieper J, Poot A, Engbers G, Beugling T, van Aken WG, Feijen J. Immobilization of heparin to EDC/NHS-crosslinked collagen. Characterization and in-vitro evaluation. Biomaterials 2001;22;151–163. [DOI] [PubMed] [Google Scholar]
- [24].Pieper J, Hafmans T, Veerkamp J, van Kuppevelt T. Development of tailor-made collagen-glycosaminoglycan matrices: EDC/NHS crosslinking, and ultrastructural aspects. Biomaterials 2000;21;581–593. [DOI] [PubMed] [Google Scholar]
- [25].Venkataraman L, Ramamurthi A. Induced elastin matrix generation within 3-dimensional collagen scaffolds. Tissue Eng Part A 2011;17;2879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay procedure. Anal Biochem 1980;102;344–52. [DOI] [PubMed] [Google Scholar]
- [27].Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polym Ed 2006;17;247–289. [DOI] [PubMed] [Google Scholar]
- [28].Lue J-M, Wang X, Marin-Muller C, Wang H, Lin PH, Yao Q, Chen C. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn 2009;9;325–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Panyam J, Dali MA, Sahoo SK, Ma WX, Chakravarthi SS, Amidon GL, Levy RJ, Labhasetwar V. Polymer degradation and in vitro release of a model protein from poly(D,L-lactide-co-glycolide) nano and microparticles. J Control Release 2003;92;173–87. [DOI] [PubMed] [Google Scholar]
- [30].Wang G, Uludag H. Recent developments in nanoparticle-based drug delivery and targeting systems with emphasis on protein-based nanoparticles. Expert Opin Drug Deliv 2008;5;499–515. [DOI] [PubMed] [Google Scholar]
- [31].Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly(D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release 2002;82;105–14. [DOI] [PubMed] [Google Scholar]
- [32].Reeps C, Lohöfer F, Eckstein HH. Rudelius M, Pelisek J. Cellular expression of cathepsin proteases in symptomatic and asymptomatic AAA. Springer; Berlin Heidelberg: 2009;38;313–15. [Google Scholar]
- [33].Nguyen KT, Shukla KP, Moctezuma M, Braden ARC, Zhou J, Hu ZB, Tang L. Studies of the cellular uptake of hydrogel nanospheres and microspheres by phagocytes, vascular endothelial cells, and smooth muscle cells. J Biomed Mater Res 2009;88A;1022–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases – structure, function, and biochemistry. Circ Res 2003;92;827–39. [DOI] [PubMed] [Google Scholar]
- [35].Chau Y, Tan FE, Langer R. Synthesis and characterization of dextran-peptide-methotrexate conjugates for tumor targeting via mediation by matrix metalloproteinases II and matrix metalloproteinases IX. Bioconjug Chem 2004;15;931–41. [DOI] [PubMed] [Google Scholar]
- [36].Shamsipour F, Zarnani AH, Ghods R, Chamankhah M, Forouzesh F, Vafaei S, Bayat AA, Akhondi MM, Ali Oghabian M, Jeddi-Tehrani M. Conjugation of Monoclonal Antibodies to Super Paramagnetic Iron Oxide Nanoparticles for Detection of her2/neu Antigen on Breast Cancer Cell Lines. Avicenna Journal of Medical Biotechnology. 2009;1;27–31. [PMC free article] [PubMed] [Google Scholar]
- [37].Gruttnera C, Mullera K, Tellera J, Westphala F, Foremanb A, Ivkovb R. Synthesis and antibody conjugation of magnetic nanoparticles with improved specific power absorption rates for alter-nating magnetic field cancer therapy. J Magn Mater 2007;311:181–186. [Google Scholar]
- [38].Illum L, Jones PDE, Kreuter J, Baldwin RW, Davis SS. Adsorption of monoclonal antibodies to polyhexylcyanoacrylate nanoparticles and subsequent immunospecific binding to tumor cells in vitro. Int J Pharm 1983;17;65–76. [Google Scholar]
- [39].Barbet J, Machy P, Leserman L. Monoclonal covalently coupled to liposomes: specific targeting to cells. J Supramol Struct Cell Biochem 1981;16;243–258. [DOI] [PubMed] [Google Scholar]
- [40].Hermanson GT. Bioconjugated techniques. Academic Press, London: 1996;169–172. [Google Scholar]
- [41].Miyake T, Aoki M, Osako MK, Shimamura M, Nakagami H, Morishita R. Systemic administration of ribbon-type decoy oligodeoxynucleotide against nuclear factor κB and ets prevents abdominal aortic aneurysm in rat model. Mol Ther. 2011. January;19(1):181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sage J, Leblanc-Noblesse E, Nizard C, Sasaki T, Schnebert S, Perrier E, Kurfurst R, Bromme D, Lalmanach G, Lecaille F. Cleavage of Nidogen-1 by Cathepsin S Impairs Its Binding to Basement Membrane Partners. PLoS ONE. 2012;7;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lindeman J, Hanemaaijer R, Mulder A, Dijkstra PD, Szuhai K, Bromme D, Verheijen JH, Hogendoorn PC. Cathepsin K is the principal protease in giant cell tumor of bone. American J Path. 2004;165;593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Curci JA, Petrinec D, Liao SX, Golub LM, Thompson RW. Pharmacologic suppression of experimental abdominal aortic aneurysms : a comparison of doxycycline and four chemically modified tetracyclines. J Vasc Surg 1998;28;1082–93. [DOI] [PubMed] [Google Scholar]
- [45].Prall AK, Longo GM, Mayhan WG, Waltke EA, Fleckten B, Thompson RW, Baxter BT. Doxycycline in patients with abdominal aortic aneurysms and in mice: Comparison of serum levels and effect on aneurysm growth in mice. J Vasc Surg 2002;35:923–928. [DOI] [PubMed] [Google Scholar]
- [46].Ding R, McGuinness CL, Burnand KG, Sullivan E, Smith A. Matrix Metalloproteinases in the Aneurysm Wall of Patients Treated with Low-Dose Doxycycline. Vascular. 2005;13:290–297. [DOI] [PubMed] [Google Scholar]
- [47].Arvizo RR, Miranda OR, Thompson MA, Pabelick CM, Bhattacharya R, Robertson JD, Rotello VM, Prakash YS, Mukherjee P. Effect of nanoparticle surface charge at the plasma membrane and beyond. Nano Lett. 2010. July 14;10(7):2543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu J, Xiong WF, Baca-Regen L, Nagase H, Baxter BT. Mechanism of inhibition of matrix metalloproteinase-2 expression by doxycycline in human aortic smooth muscle cells. J Vasc Surf 2003;38;1376–83. [DOI] [PubMed] [Google Scholar]
- [49].Longo GM, Xiong WF, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest 2002;110;625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Davis V, Persidskaia R, Baca-Regen L, Itoh Y, Nagase H, Persidky Y, Ghorpade A, Baxter BT. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Throm Vasc Biol 1998;18;1625–33. [DOI] [PubMed] [Google Scholar]
- [51].Petrinec D, Liao SX, Holmes DR, Reilly JM, Parks WC, Thompson RW. Doxycycline inhibition of aneurysmal degeneration in an elastase-induced rat model of abdominal aortic aneurysm: preservation of aortic elastin associated with suppressed production of 92 kDa gelatinase. J Vasc Surg 1996;23;336–46. [DOI] [PubMed] [Google Scholar]
- [52].Galindo-Rodriguez S, Allemann E, Fessi H, Doelker E. Physiochemical parameters associated with nanoparticle formation in the salting-out, emulsification-diffusion, and nanoprecipitation methods. Pharm Res 2004;21;1428–39. [DOI] [PubMed] [Google Scholar]
- [53].Seftor REB, Seftor EA, De Larco JE, Kleiner DE, Leferson J, Stetler-Stevenson WG, McNamara TF, Golub LM, Hendrix MJ. Chemically modified tetracyclines inhibit human melanoma cell invasion and metastasis. Clin Exp Metastasis 1998;16;217–25. [DOI] [PubMed] [Google Scholar]
- [54].Garcia RA, Pantazatos DP, Gessner CR, Go KV, Woods VL, Villarreal FJ. Molecular interactions between matrilysin and the matrix metalloproteinase inhibitor doxycycline investigated by deuterium exchange mass spectrometry. Mol Pharmacol 2005;67;1128–36. [DOI] [PubMed] [Google Scholar]
- [55].Sun J, Sukhova G, Zhang J, Chen H, Sjoberg S, Libby P, Xia M, Xiong N, Gelb, BD, Shi G. Cathepsin K deficiency reduces elastase perfusion-induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol 2012:32;15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tezvergil-Mutluay A, Agee KA, Uchiyama T, Imazato S, Mutluay MM, Cadenaro M, Breschi L, Nishitani Y, Tay FR, Pashley DH. The Inhibitory Effects of Quaternary Ammonium Methacrylates on Soluble and Matrix-bound MMPs. J Dent Res 2011;90:535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kagan HM, Tseng L, Simpson DE. Control of elastin metabolism by elastin ligands. Reciprocal effects on lysyl oxidase activity. J Biol Chem 1981;256:5417–5421. [PubMed] [Google Scholar]
- [58].Kagan HM, Sullivan KA, Olsson TA, Cronlund AL. Purification and properties of four species of lysyl oxidase from bovine aorta. Biochem J 1979;177:203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zempo N, Koyama N, Kenagy RD, Lea HJ, Clowes AW. Regulation of vascular smooth muscle cell migration and proliferation in vitro and in injured rat arteries by a synthetic matrix metalloproteinase inhibitor. Arterioscler Thromb Vasc Biol. 1996;1;28–33 [DOI] [PubMed] [Google Scholar]