

Abstract
ZNF335 plays an essential role in neurogenesis and biallelic variants in ZNF335 have been identified as the cause of severe primary autosomal recessive microcephaly in two unrelated families. We describe herein two additional affected individuals with biallelic ZNF335 variants, one individual with a homozygous c.1399T>C, p.(Cys467Arg) variant, and a second individual with compound heterozygous c.2171_2173delTCT, p.(Phe724del) and c.3998A>G, p.(Glu1333Gly) variants in ZNF335; with the latter variant predicted to affect splicing. Whereas the first case presented with early death and a severe phenotype characterized by anterior agyria with prominent extra-axial spaces, absent basal ganglia, and hypoplasia of the brainstem and cerebellum, the second case had a milder clinical presentation with hypomyelination and otherwise preserved brain structures on MRI. Our findings expand the clinical spectrum of ZNF335 associated microcephaly.
Keywords: ZNF335, microcephaly, basal ganglia, neurogenesis, neurodegeneration
Graphical abstract
Introduction
ZNF335 is a zinc finger protein that acts as an essential link between H3K4 complexes and REST/NRSF and is involved in regulating neuronal gene expression and cell fate during brain development in human and mice1. Mouse models with Znf335 null mutations are embryonically lethal while a conditional knockout of Znf335 leads to severely reduced cortical size1. Biallelic variants in ZNF335 have been reported as the cause of autosomal recessive congenital or acquired microcephaly in two independent families1,2, with severe spastic quadriplegia and increased extra-axial spaces, absence of the basal ganglia, absent or hypoplastic corpus callosum, enlarged ventricles, markedly reduced white matter volume, delayed myelination, hypoplasia of the brainstem, cerebellar hemispheres and vermis on brain imaging. Variants in ZNF335 in the previously reported families included a homozygous c.3332G>A, p.(Arg1111His) variant that effects the 5’ splice site of exon 201, as well as compound heterozygous missense variants c.1399T>C, p.(Cys467Arg) and c.1505A>G, p.(Tyr502Cys)2. Here we report one individual with homozygous c.1399T>C, p.(Cys467Arg) variant, and a second individual with compound heterozygous c.2171_2173delTCT, p.(Phe724del) and c.3998A>G, p.(Glu1333Gly) variants in ZNF335 and further delineate the associated clinical spectrum of ZNF335 related severe primary autosomal recessive microcephaly.
Material and methods
Written informed consent, approved by the Research Ethics Boards of UZ Brussel and UZ Leuven respectively, was obtained from both families.
For family A, genomic DNA was extracted from peripheral blood samples of the affected child, parents and two brothers. Samples were processed using a multi-gene panel analysis including 193 genes involved in Malformations of Cortical Development (http://www.brightcore.be/mcd). Raw data were quality-controlled by use of FastQC and mapped to the human reference genome with BWA (0.7.10). Mapping qualities were assessed via overall coverage analysis by an in-house designed script. Mapped reads were processed using the GATK (2.7.2) pipeline and detected variants were annotated by Annovar or Alamut Batch. The identification of potentially causal variants was done with use of Highlander, an in-house developed software program for variant classification. Variants were filtered over several variant databases (dbSNP, ExAC), and using tools predicting splice effects (MaxEntScan, SpliceSiteFinder). Synonymous variants not affecting splicing and those with an allele frequency >2% in one or more populations were excluded. Variant validation was performed by PCR and Sanger sequencing according to standard procedures. For family B, whole exome sequencing on the proband and both parents were performed by GeneDx (Gaithersburg, MD) using standard procedures (https://www.genedx.com/). The RefSeq used for ZNF335 was: NM_022095.3.
Results
Clinical features
Patient A
Patient A is the third child of consanguineous parents of North-African origin. Family history was negative for microcephaly, developmental delay or other neurological problems. He was born at term after an uneventful pregnancy. Birth weight was 3.450 kg (43rd percentile; −0.18 SD), length was 49 cm (33rd percentile; −0.44 SD), and occipitofrontal circumference was 28.5 cm (<1st percentile; −3.24 SD). Physical examination at birth revealed a low sloping forehead, flat occiput and generalized hypotonia (Table 1). He developed seizures with bradycardia and apnea from the first hours of life that were refractory to treatment and led to death at 5 days of age. Brain MRI revealed large extra-axial spaces, anterior agyria and a posterior simplified gyral pattern, enlarged ventricles, absent basal ganglia, thin corpus callosum, and hypoplasia of brainstem and cerebellum (Figure 1).
Table 1.
Clinical and Imaging features of individuals with biallelic ZNF335 variants
| CLINICAL FEATURES | |||||
|---|---|---|---|---|---|
| Family 1 | Family 2 | Family 3 | Family 4 | ||
| Reference | This report, Patient A | This report, Patient B | Sato et al., 2016 | Yang et al., 2012, Individual 8 | Yang et al., 2012, Individual 9 |
| Sex | M | M | F | M | M |
| Nucleotide variant(s) (NM_022095.3) | homozygous c.1399T>C | Compund heterozygous c.2171_2173delTCT and c.3998A>G | Compound heterozygous c.1399T>C and c.1505A>G | homozygous c.3332G>A | homozygous c.3332G>A |
| Protein sequence variations(s) | p.(Cys467Arg) | p.(Phe724del) and p.(Glu1333Gly) | p.(Cys467Arg) and p.(Tyr502Cys) | p.(Arg1111His) | p.(Arg1111His) |
| Age at examination [age at death] | 5 days [5 days] | 3 months | 33 months | 3 months [unknown] | 8 months [unknown] |
| Birth head circumference (SD) | 28.5 cm (−3.24 SD) | 36.8 cm (+0.55 SD) | 32 cm (−0.6 SD) | ND | 26.5 cm (−4.1 SD) |
| Head circumference on examination (standard deviations) | 28 cm (−3.7 SD) | 40 cm (−1.22 SD) | 41 cm (−5.1 SD) | 27.5 cm (−9 SD; −7.1 SD corrected for prematurity) | 32 cm (−9.9 SD) |
| Neurological features | Severe hypotonia | Hypertonia, spasticity | Spastic paralysis, hypertonia, moderate sensorineural hearing loss | Hypertonia, spasticity | ND |
| Epilepsy | Yes | No | Yes | Yes | Yes |
| Age at seizure onset | Birth | N/A | 3 months | Birth | ND |
| Seizure type | Focal | N/A | Afebrile seizures, partial seizures, focal | Paroxysmal myoclonic jerks | Paroxysmal myoclonic jerks |
| Refractory | Yes | N/A | No, well controlled on monotherapy | ND | ND |
| Growth and development | NA | Normal growth with mild delay in voluntary movements | Severe motor delay: no voluntary movement; language delay: absent speech | Short stature: 51 cm at 3 months (−3 SD, corrected for prematurity) | Low weight: 5.2 kg at 8 months (−4 SD) |
| Dysmorphic features | Low sloping forehead, flat occiput | Prominent nasal bridge | Low sloping forehead, micrognathia | Low sloping forehead, prominent nasal bridge, micrognathia, prominent ear helices, bilateral simian creases | ND |
| CLINICAL FEATURES | |||||
| Other | Bradycardia, apnea | Gastroesophageal reflex | Systolic murmur, ventricular septal defect, bradycardia, entropium ciliarum, gastroesophageal reflux | Twin, born at 35 weeks gestation, IUGR, choanal atresia, bilateral cataracts, bilateral flexion contractures of thumbs and hands with overriding fingers, bilateral dorsiflexion of feet with overring toes | Joint contractures |
| BRAIN IMAGING FEATURES | |||||
| Age at MRI | 4 days | 3 months | 3, 5, and 16 months | 3 months | NA |
| Subarachnoid spaces | Enlarged, most pronounced in the temporal fossa bilaterally | Normal | Enlarged | Enlarged | |
| Gyral pattern | Frontal agyria, posterior simplified gyral pattern | Normal | Progressive corticosubcortical atrophy | Markedly simplified gyral pattern | |
| White matter | Posterior limb of the internal capsule present, little to no myelinated white matter | Hypomyelination | Hypomyelination | Severely reduced white matter with delayed myelination | |
| Lateral Ventricles | Enlarged lateral ventricles, most pronounced over the occipital and hippocampal horns | Normal | Enlarged | Enlarged relative to hemispheres | |
| Corpus callosum | Thin, prominent column of the fornix | Normal | Thin | Absent | |
| Basal Ganglia | Absent | Present | Absent | Absent | |
| Thalami | Present | Present | Present | ND | |
| Hippocampus | Malformed | Present | ND | ND | |
| Brainstem | Hypoplasia | Present | Hypoplasia | Hypoplasia | |
| Cerebellar hemispheres | Hypoplasia | Present | Progressive atrophy | Hypoplasia | |
| Cerebellar vermis | Hypoplasia | Present | Progressive atrophy | Hypoplasia | |
SD standard deviations; ND not described; NA not applicable; IUGR intra-uterine growth retardation
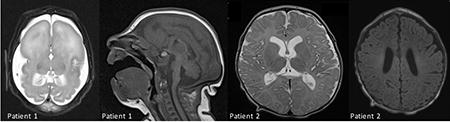
Figure 1.

(A–C) Brain MRI at age 4 days of life for patient A. A. Axial T2 shows enlarged subarachnoid spaces, anterior agyria and posterior simplified gyral pattern, enlarged lateral ventricles, absent basal ganglia and white matter hypomyelination. B. Sagital T1 shows hypoplasia of corpus callosum, brainstem and cerebellum. C. Coronal T2 shows severe hypoplasia of the cerebellar vermis and hemispheres. (D–E) Brain MRI at 3 months of life for patient B. D. Axial T1 showing hypomyelination of perirolandic white matter and corona radiata. E. Axial T2 showing age appropriate myelination of posterior limb of the internal capsule as well as heterogeneous signal in thalami and otherwise preserved brain structures. F. Axial T1 showing heterogeneous signal in thalami and otherwise preserved brain structures.
Hypomyelination was suspected but was difficult to accurately assess given his young age and severe brain malformations. Array-CGH showed a paternally inherited 257–513 kb deletion of 2q36.3, which is unlikely related to the patient’s phenotype since the father is asymptomatic and deletions of 2q36.3 have not previously been associated with microcephaly. Analysis of the MCD gene panel revealed a homozygous c.1399T>C, p.(Cys467Arg) variant in ZNF335. Both parents and one brother were heterozygous carriers, while the variant was absent in the other sibling.
Patient B
Patient B is the second child of non-consanguineous parents of Caucasian European descent. Family history is negative for microcephaly, and positive for mother with miscarriage at 8 weeks, maternal uncle with a fatty acid oxidation defect, and paternal cousin with autism. Patient B was born at 39 weeks after an uncomplicated pregnancy. Vaginal delivery was complicated by nuchal cord with Apgar scores of 8 and 8 at 1 and 5 minutes respectively. Birth weight was 4.054 kg (85th percentile; +1.03 SD), length 53.3 cm (90th percentile; +1.26 SD) and head circumference 36.8 cm (71st percentile; +0.55 SD). After birth he was noted to have body tremors. Workup at that time demonstrated normal glucose as well as a normal cranial ultrasound. After discharge he developed progressive feeding difficulties, gastroesophageal reflux disease, frequent spasms and arching of his back and was given a clinical diagnosis of Sandifer Syndrome. At the 2-month clinic visit head circumference was 38.1 cm (19th percentile; −0.87 SD). Growth parameters at three months of age were: weight 6.25 kg (41st percentile; −0.22 SD), length 63.3 cm (67th percentile; +0.44 SD), and head circumference 40 cm (11th percentile; −1.22 SD). Physical exam was notable for a coronal suture ridge, a small anterior fontanelle, prominent nasal bridge, truncal and axial hypertonia at rest that was increased with activity, spasticity, frequent tongue thrusts, and hyperreflexia with up to 4–6 beats of ankle clonus. Brain MRI revealed hypomyelination as well as heterogenous signal within both thalami with otherwise preserved brain structures. Notably, his basal ganglia appeared intact, unlike previously reported patients with biallelic ZNF335 variants. Laboratory testing included a normal lactate, ammonia, urine organic acids, acylcarnitine profile, VLCFAs, CSF amino acids, and lysosomal storage disorder enzyme activity panel. A chromosomal microarray was normal. Trio whole exome sequencing revealed a paternally inherited ZNF335 c.2171_2173delTCT, p.(Phe724del) and maternally inherited ZNF335 c.3998A>G, p.(Glu1333Gly) variant of uncertain significance (VUS) in addition to a paternally inherited OPA1 c.1712G>A, p.(Arg571His) VUS.
Variant Analysis
The homozygous p.(Cys467Arg) variant in patient A is located at a highly conserved zinc finger domain, and is predicted to be pathogenic by SIFT, PolyPhen-2 and MutationTaster. The cysteine to arginine substitution is likely to be deleterious because it changes a thiol (uncharged residue) to a positively charged residue. Moreover, the p.(Cys467Arg) allele is absent from more than 138,000 individuals in the gnomAD Consortium3. The heterozygous c.3998A>G, p.(Glu1333Gly) variant in patient B is located at an amino acid conserved throughout mammals in the C-terminal portion of ZNF335 that is integral for interacting with DBC-14. This variant is predicted to be damaging (SIFT), probably damaging (Polyphen-2) or Disease Causing (MutationTaster) and is rare, seen in a heterozygous state in 31 individuals in gnomAD consortium, with an allele frequency of 0.01119% and no homozygotes3. Moreover, this variant is predicted to strongly activate a cryptic donor site. It currently remains uncertain how this possible splice variant affects the mRNA transcript and the protein sequence (Supplementary Figure 1). The heterozygote c.2171_2173delTCT, p.(Phe724del) variant in individual B is located at an amino acid conserved throughout vertebrates. This variant is predicted to be Disease Causing by MutationTaster, is absent from over 138,000 individuals in gnomAD3 and is classified as a VUS in ClinVar based on one prior independent unpublished case.
Discussion
This article reports two additional patients with biallelic variants in ZNF335 and expands the clinical spectrum of biallelic ZNF335 related microcephaly. Specifically, compound heterozygous ZNF335 p.(Phe724del) and p.(Glu1333Gly) variants result in an acquired microcephaly with hypomyelination, spasticity and hypertonia with otherwise preserved brain structures and relatively normal growth by 3 months of life. In contrast, homozygosity of the ZNF335 p.(Cys467Arg) variant results in severe congenital microcephaly, generalized hypotonia, and refractory seizures leading to death at 5 days of age with multiple structural brain anomalies. The c.1399T>C, p.(Cys467Arg) variant described in patient A was also previously reported in a compound heterozygous state with c.1505A>G, p.(Tyr502Cys) in a child with a milder clinical phenotype characterized by a less severely affected cerebral cortex as seen on MRI and survival beyond 33 months2. It might be hypothesized that the homozygous p.(Cys467Arg) missense mutation detected in patient A is affecting the structure of the ZNF335 protein and consequently results in a more severe phenotype. In patient B, both detected variants might have a less severe impact on the protein function. The p.(Phe724del) variant is removing a single amino acid from the protein, while the missense variant p.(Glu1333Gly) might have an impact on splicing. It remains, however, unsure to what degree splicing is affected.
All affected individuals surviving beyond 3 months of life developed hypomyelination and severe spasticity, implicating these as core features of this condition. Severe cases of biallelic ZNF335 variants also appear to be associated with a variable degree of cortical malformation (including a simplified gyral pattern or agyria), progressive cerebral and cerebellar atrophy, large extra-axial spaces, enlarged lateral ventricles, thin to absent corpus callosum, absent basal ganglia, and hypoplasia of brainstem and cerebellum (Table 1). The progressive cerebral and cerebellar atrophy highlights the ZNF335-associated neurodegeneration. Microcephaly in combination with absent basal ganglia should orient the clinician towards biallelic variants in ZNF335, but does not exclude this condition, as is highlighted in patient B. However, the differential diagnosis of microcephaly and hypomyelination is quite broad, necessitating a comprehensive environmental, metabolic and genetic evaluation. The presence of epilepsy was variable in the four families, with patient A demonstrating refractory seizures leading to death at age 5 days, whereas the proband in Sato et al. had rare focal seizures controlled by treatment2 and patient B lacked any clinical seizures, but has frequent spasms that have yet to be recorded by EEG. Although not noted by Yang et al.1, affected individuals in that family had seizures described as paroxysmal myoclonic jerks (personal communication). Including this report, of the ten patients with biallelic ZNF335 variants reported to date (7 males, 3 females), only two girls survived beyond the age of 18 months. In all cases, death was likely secondary to respiratory complications or seizures (Current report, Yang et al. 20121, Sato et al. 20162).
The degree of microcephaly at birth was variable between patients (Table 1), with severe microcephaly in patient A and the family reported by Yang et al. compared to a normal head circumference in patient B and the proband reported by Sato et al.2. It is notable that both patient B and the proband in Sato et al. developed an acquired microcephaly, indicating a role of ZNF335 in both prenatal and postnatal brain development, consistent with histopathological changes in patients and Znf335 null mice, which suggest that variants in ZNF335 cause both a loss of progenitor cells as well as neurodegeneration1.
The presence of the ZNF335 p.(Cys467Arg) variant in both patient A in this report, who is of North African descent, as well as the proband in Sato et al., who is of Japanese descent, suggests that there may be only a limited number of ZNF335 alleles that alter function enough to show a severe phenotype, but are still incompletely disabling so as to permit survival past birth. So far, no nonsense or frameshift mutations have been described in the ZNF335 gene. Possible, these severe mutations might be lethal. Furthermore, the identification of 31 individuals heterozygous for the c.3998A>G, p.(Glu1333Gly) variant in gnomAD suggests that milder cases of this condition caused by compound heterozygotes with this variant, as seen in patient B, may be more prevalent than previously recognized.
Supplementary Material
Supplementary Figure 1: Prediction of splice site effects adapted for alteration c.3998A>G, p.(Glu1333Gly) using Alamut Visual. The substitution possibly causes the activation of a cryptic donor site. The normal stop codon will not be present in the exon. It is difficult to predict the sequence of the altered transcript.
Acknowledgments
AJ was supported by a Senior Clinical Investigator Fellowship from the Research Foundation Flanders (FWO) and has received funding from the Scientific Fund Willy Gepts and the VUB Wetenschappelijk Steunfonds. LVDV received funding from the Foundation Marguerite-Marie Delacroix and an FWO Travelgrant. AD received an EPNS research fellowship. DJ was supported by the National Institutes of Health (NIH) MSTP training grant (NIGMS T32GM007753). CAW was supported by grant R01NS035129 from the NINDS and is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Conflict of interest: none
References
- 1.Yang YJ, Baltus AE, Mathew RS, et al. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell. 2012;151:1097–1112. doi: 10.1016/j.cell.2012.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sato R, Takanashi J, Tsuyusaki Y, et al. Association Between Invisible Basal Ganglia and ZNF335 Mutations: A Case Report. Pediatrics. 2016;138 doi: 10.1542/peds.2016-0897. [DOI] [PubMed] [Google Scholar]
- 3.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garapaty S, Xu CF, Trojer P, Mahajan MA, Neubert TA, Samuels HH. Identification and characterization of a novel nuclear protein complex involved in nuclear hormone receptor-mediated gene regulation. J Biol Chem. 2009;284:7542–7552. doi: 10.1074/jbc.M805872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Prediction of splice site effects adapted for alteration c.3998A>G, p.(Glu1333Gly) using Alamut Visual. The substitution possibly causes the activation of a cryptic donor site. The normal stop codon will not be present in the exon. It is difficult to predict the sequence of the altered transcript.