

Abstract
Background
Monoclonal antibodies inhibiting tumour necrosis factor-α (TNFα) signalling pathway (anti-TNFα) have been widely used in Crohn’s disease (CD). However, treatment response varies among patients with CD and the clinical outcome is dependent on single nucleotide polymorphisms (SNP) in TNFα receptor superfamily 1A and 1B (TNFRSF1A/1B).
Methods
We tested nine SNPs in TNFα, TNFRSF1A and TNFRSF1B by TaqMan genotyping from peripheral blood samples of 104 subjects. Additionally, we quantified the effects of these SNPs on their corresponding gene expression by RT-PCR and susceptibility to Mycobacterium avium subsp paratuberculosis (MAP) infection by IS900 nested PCR.
Results
Four SNPs (TNFα:rs1800629, TNFRSF1A:rs767455, TNFRSF1B:rs1061624 and TNFRSF1B:rs3397) were over-represented significantly (p<0.05) among patients with CD compared with healthy controls. The TNFRSF1A:rs767455 GG genotype was found in 15/54 patients with CD (28%), while it was only found in 2/50 healthy controls (4%) (OR 9.2, 95% CI 1.98 to 42.83). The TNFRSF1B:rs3397 TT genotype was found in 15/54 patients with CD (28%) compared with (4/50) healthy controls (8%) (OR 4.4, 95% CI 1.36 to 14.14). Furthermore, the SNPs TNFRSF1A:rs767455 and TNFRSF1B:rs3397 were associated with downregulating their corresponding genes significantly (p<0.05). MAP infection was predominantly found among patients with CD in comparison to healthy controls (57% vs 8%, respectively), which was also dependent on the SNPs TNFRSF1A:rs767455 and TNFRSF1B:rs3397. Our SNP haplotype analysis of TNFRSF1A:rs767455 and TNFRSF1B:rs3397 indicates that the G–T haplotype is significantly distributed among patients with CD (46%) and MAP infection susceptibility is also associated with this specific haplotype (31%).
Conclusion
The SNPs TNFRSF1A:rs767455 and TNFRSF1B:rs3397, which are known to affect anti-TNFα clinical outcome in CD, were associated with lower corresponding gene expression and higher MAP infection susceptibility.
Keywords: Crohn’s disease, tumour necrosis factor-α, TNFα, anti-TNFα, adalimumab, infliximab, TNFRSF1A, TNFRSF1B, SNPs, MAP, mycobacteria paratuberculosis
Summary box.
What is already known about this subject?
There is a significant variable response among patients with Crohn’s disease (CD) receiving anti-tumour necrosis factor-α (TNFα).
About 10%–30% of patients with inflammatory bowel disease have no initial response to anti-TNFα treatment.
Over 50% of the anti-TNFα initial responders have lost response to treatment over time.
Genetic polymorphisms in TNFRSF1A and TNFRSF1B have been shown to affect anti-TNFα treatment response significantly among patients with CD.
The risk for tuberculosis infection has substantially increased in patients receiving anti-TNFα.
Mycobacterium avium subsp paratuberculosis (MAP) is the most investigated suspect to be a causative pathogen in a subset of patients with CD.
What are the new findings?
The single nucleotide polymorphisms (SNP) TNFRSF1A:rs767455 and TNFRSF1B:rs3397 were both associated with lower expression of their corresponding genes.
MAP infection susceptibility is associated with the SNPs TNFRSF1A:rs767455 and TNFRSF1B:rs3397.
SNP haplotype analysis of TNFRSF1A:rs767455 and TNFRSF1B:rs3397 indicates that the G–T haplotype has a significant difference in frequency among patients with CD, and MAP infection susceptibility is also associated with this specific haplotype.
How might it impact on clinical practice in the foreseeable future?
Genetic testing for TNFRSF1A:rs767455 and TNFRSF1B:rs3397, in addition to MAP infection screening methods, should be considered before initiation of anti-TNFα treatment in CD for a better clinical outcome.
Introduction
Tumour necrosis factor-α (TNFα) is a proinflammatory cytokine that has been found to be dysregulated in numerous inflammatory disorders including rheumatoid arthritis, psoriasis and Crohn’s disease (CD).1 2 It is secreted primarily by macrophages, but it can also be produced by lymphocytes, natural killer cells and mast cells.3 In order to exert its biological activity, TNFα binds to two different cell-surface receptors: TNFα receptor superfamily 1A (TNFRSF1A), which is expressed in most tissues, and TNFα receptor superfamily 1B (TNFRSF1B), which is typically found in immune cells.4
Targeting TNFα by monoclonal antibodies (anti-TNFα) such as adalimumab (Humira) and infliximab (Remicade) has shown that blocking this cytokine signalling pathway may control the symptoms of hyperactive immune response in CD initially.5 However, there is a significant variable response to anti-TNFα therapeutics among patients receiving this treatment.6 Additionally, clinical observations have reported that 10%–30% of patients with inflammatory bowel disease have no initial response to anti-TNFα treatment, and over 50% of the initial responders have lost response to treatment over time.7
Clinical studies have shown that anti-TNFα therapeutics have many deleterious side effects, such as malignancy, neurologic disorders, heart failure and, more importantly, multiple infections.8 Since TNFα plays a critical role in the immune defence against infections, it was unsurprising to notice that TNFα-deficient animal models are more susceptible to develop mycobacterial infections compared with wild-type controls.9 On the other hand, patients receiving anti-TNFα treatment are at higher risk for meningitis, sepsis, histoplasmosis, and pneumonia.10–12 Moreover, the risk for tuberculosis development has substantially increased in patients receiving anti-TNFα, which might raise a question about their effect on Mycobacterium avium subsp paratuberculosis (MAP) infection in a subset of patients with CD.13–16 Thus, prescribing anti-TNFα to patients with CD without considering MAP infection could worsen disease condition eventually.
Genetic polymorphisms are associated with the overall risk of developing an inflammatory disorder, and they play a role in the treatment outcome. For instance, polymorphisms in TNFRSF1A and TNFRSF1B have been shown to affect anti-TNFα treatment response significantly among patients with CD.17–19 However, the mechanism of those effects remains unknown. Predicting the efficacy of anti-TNFα treatment based on patient’s genetics and MAP infection status would be more effective and beneficial to patients by reducing the risk for adverse effects and treatment cost.
In this study, we investigated the frequency of single nucleotide polymorphisms (SNP) in TNFα, TNFRSF1A and TNFRSF1B among patients with CD in comparison to healthy controls, in addition to their effect on gene expression and susceptibility to MAP intracellular infection. Finally, we further linked the effect of SNPs in these genes and presence of MAP to anti-TNFα treatment response in patients with CD.
Materials and methods
Clinical samples
Peripheral blood from a total of 104 subjects (54 patients with CD and 50 healthy controls) was included in this study. All participants provided written informed consent prior to enrolment.
Each subject provided two 4.0 mL K2-EDTA coded blood tubes, where one tube was processed for detection of MAP infection, and the second tube was processed for gene expression analysis and genotyping of nine SNPs in TNFα, TNFRSF1A and TNFRSF1B. There was no significant difference in average age or gender between the two groups (CD vs healthy controls). All subject demographics are listed in table 1.
Table 1.
Demographics of study participants
| Diagnosis | n | Age range | Average age | Gender ratio (Male:female %) |
| All subjects | 104 | 20–66 | 35 | 53:47 |
| Crohn’s disease | 54 | 21–66 | 39 | 48:52 |
| Healthy controls | 50 | 20–63 | 31 | 58:42 |
Extraction of DNA and detection of MAP by IS900 nested PCR
Purified white blood cells separated from blood samples were cultured in BD Bactec MGIT Para-TB Medium for 6 months of incubation at 37°C. Mycobacterial growth was measured initially using the ultraviolet illuminator and 1.0 mL was used for DNA extraction and nested PCR (nPCR) analysis as described earlier.15 20 Briefly, MAP infection was detected using MAP-specific IS900 derived oligonucleotide primers. Round 1 of amplification was performed using P90 (GTTCGGGGCCGTCGCTTAGG) and P91 (GAGGTCGATCGCCCACGTGA) primers following these conditions: 95°C for 5 min, then 34 cycles of 95°C for 1 min, 58°C for 1.5 min, 72°C for 1.5 min. Final extension of 10 min at 72°C. The amplified product from this round was 398 bp.
A second round of amplification involved using AV1 (ATGTGGTTGCTGTGTTGGATGG) and AV2 (CCGCCGCAATCAACTCCAG) primers, which was performed following these conditions: 95°C for 5 min, then 34 cycles of 95°C for 1 min, 58°C for 1.5 min, 72°C for 1.5 min. Final extension of 10 min at 72°C. The final product following this round was 298 bp, which was identified on 2% agarose gel.
Identification of SNPs in TNFα, TNFRSF1A and TNFRSF1B genes
Genomic DNA was purified from peripheral blood leucocytes using QIAamp DNA Blood Mini Kit (Qiagen) following manufacturer’s instructions. TaqMan genotyping assays (Applied Biosystems) for TNFα (rs1800629, rs1799964, and rs1799724), TNFRSF1A (rs4149584, rs767455, and rs4149570) and TNFRSF1B (rs1061624, rs1061622, and rs3397) were performed on isolated DNA samples following manufacturer protocol at the University of Florida Pharmacotherapy and Translational Research Department (Gainesville, FL). Briefly, the reaction mixture had 2× TaqMan master mix and 20× assay working stock (primers with VIC and FAM fluorophore attachment). Isolated DNA samples along with reaction mixture and controls were added into a 384-well microtitre plate, following RT-PCR assay (one cycle at 95°C for 10 min, 92°C for 15 s and 50 cycles at 58°C for 1 min) using Applied Biosystems QuantStudio RT-PCR System. The plate was analysed for VIC and FAM fluorophores at 551 and 517 nm, respectively. Fluorescence of VIC or FAM alone determined that the sample had allele 1 or allele 2, while VIC and FAM together determined that the sample was heterozygous for each allele. Gene mutation, location and phenotype for SNPs used in this study are summarised in table 2.21–27
Table 2.
Gene mutations, locations and mutation phenotypes of SNPs selected for this study
| Gene | Reference SNP | Gene mutation* | Location and AA change* | Mutation phenotype | Reference |
| TNF | rs1800629 | G→A | Promoter | Higher susceptibility to CD | 22 |
| rs1799964 | T→C | Promoter | Associated with IBD in general | 23 | |
| rs1799724 | C→T | Promoter | Linked to ankylosing spondylitis | 24 | |
| TNFRSF1A | rs4149584 | C→T | Exon 4 (R→Q) | Higher susceptibility to MS | 25 |
| rs767455 | A→G | Exon 1: No AA change | Used to predict anti-TNFα response in CD | 17 | |
| rs4149570 | G→T | 320 bp upstream of gene | Used to predict anti-TNFα response in RA | 26 | |
| TNFRSF1B | rs1061622 | T→G | Exon 6 (M→R) | Higher susceptibility to IBD | 27 |
| rs1061624 | G→A | Exon 10 | Higher susceptibility to IBD | 27 | |
| rs3397 | C→T | Exon 10 | Used to predict anti-TNFα response in CD | 18 |
*Gene mutation and location data were obtained from the National Center for Biotechnology Information (NCBI).21
AA, amino acid; CD, Crohn’s disease; IBD, inflammatory bowel disease; MS, multiple sclerosis; Q, glutamine; R, arginine; RA, rheumatoid arthritis; SNP, single nucleotide polymorphism; TNFα, tumour necrosis factor-α.
Gene expression of TNFα, TNFRSF1A, and TNFRSF1B by RT-PCR
RNA was isolated from 1.0 mL of whole blood samples, and used for cDNA synthesis in order to analyse gene expression of TNFα, TNFRSF1A, and TNFRSF1B via RT-PCR. Briefly, 1.0 mL of blood was transferred into a 2.0 mL RNase-free microcentrifuge tube, and then centrifuged at 1000 rpm for 5 min. Pellets containing leucocytes were collected and suspended in 1.0 mL of TRIzol reagent (Invitrogen) for 15 min of gentle shaking. A total volume of 0.2 mL of chloroform was added to each tube, and the mixture was then incubated at room temperature for 3 min. All tubes were then centrifuged at 11 400 RPM for 15 min at 4°C. The upper aqueous colourless phase containing RNA was transferred into a new 2.0 mL RNase-free microcentrifuge tube. A total volume of 0.5 mL of 100% isopropanol was then added following 10 min of incubation at room temperature. Each tube was then centrifuged at 11 400 RPM for 10 min at 4°C. A total volume of 1.0 mL of 75% ethanol was used to wash RNA pellets and then centrifuged at 8700 RPM for 5 min at 4°C. RNA pellets were air-dried for 15–30 min, suspended in 20 μL of RNase-free H2O, and finally heated at 60°C for 10 min.
For cDNA synthesis, 600 ng of RNA from each sample was added to 0.25 mL tubes containing 0.2 mL of PCR reaction, 4 μL of iScript Reverse Transcription (Bio-Rad), and up to 20 μL RNase-free H2O. All tubes were then transferred to thermal cycler (MyGene Series Peltier Thermal Cycler) and ran for 5 min at 25°C, 20 min at 46°C and 1 min at 95°C. The final concentration of cDNA synthesised for each sample was 30 ng/μL. A total volume of 1 μL of cDNA was mixed with 10 μL of Fast SYBR Green Mastermix (Thermo Fisher Scientific), 1 μL of either TNFa, TNFRSF1A, or TNFRSF1B PrimePCR SYBER Green Assay mix (Bio-Rad) and 8 μL of molecular biological grade sterile H2O in a 96-well microamp RT-PCR reaction plate. Controls of 18s RNA gene oligonucleotide primers (forward primer: 5′-GTA ACC CGT TGA ACC CCA TT-3′; reverse primer: 5′-CCA TCC AAT CGG TAG TAG CG-3′) were used in order to obtain baseline CT readings. The 7500 Fast Real-Time PCR System (Applied Biosystems) was used to perform the RT-PCR reaction. Relative mRNA expression levels were calculated using the equation (2^(-∆CT) × 1000), where ∆CT=Sample RT-PCR CT reading − 18s CT baseline.
Statistical analysis
All statistical tests were performed using GraphPad Prism V.7.02. MAP infection incidence was used to estimate the power of this study. Since patients with CD have MAP infection incidence proportion of nearly 40%, while in healthy controls MAP infection incidence proportion is about 10%,13 14 at 90% power and alpha (type I error) of 0.05, our calculated sample size was 84 samples (42 CD and 42 healthy controls). We also aimed for a similar number of Matsukura et al’s anti-TNFα treatment response study which included 81 participants.17 For genotype frequency, we used two-tailed Z test and OR analysis to compare between the presence of SNPs in patients with CD versus healthy controls. At each locus examined, SNP genotypes were subcategorised into four groups (major, heterozygous, homozygous and both heterozygous+homozygous), then tested for significance within each subcategory at p<0.05 and a 95% CI. For haplotype analysis, we used Fisher’s exact test since we had a smaller number of samples. Patients with CD were subcategorised into four haplotype groups and then tested for significance in-between MAP infection groups. A p value <0.05 was considered significant. For gene expression analysis, first, we compared the average gene expression in CD versus healthy control for each gene regardless of their genotype using unpaired two-tailed t-test at p<0.05 and a 95% CI, then we compared individuals who carried two major alleles with others for each SNP tested by using one-way analysis of variance, where Newman-Keuls post-test was selected for multiple comparisons. For MAP infection susceptibility, we compared infection proportions between SNP genotypes and major alleles in CD group and healthy controls separately using two-tailed Z test at p<0.05. Age and gender were not included as covariates as for all data sets no age or gender effects were observed.
Results
Frequency of SNPs in TNFα, TNFRSF1A, and TNFRSF1B among patients with CD
We have assessed 104 subjects (54 patients with CD and 50 healthy controls) for three SNPs in TNFα (rs1800629, rs1799964, and rs1799724), three SNPs in TNFRSF1A (rs4149584, rs767455, and rs4149570) and three SNPs in TNFRSF1B (rs1061624, rs1061622, and rs3397). Genotype distribution of these SNPs conveyed the Hardy-Weinberg equilibrium.
We identified one SNP in TNFα (TNFα:rs1800629) with a significant difference in frequency in patients with CD compared with healthy controls (p<0.05). The TNFα:rs1800629 GA genotype was found in 20/54 patients with CD (37%) compared with 7/50 healthy controls (14%) (OR 3.6, 95% CI 1.37 to 9.54). As shown in table 3, the other two SNPs (TNFα:rs1799964 and TNFα:rs1799724) have shown no significance among patients with CD compared with healthy controls (p>0.05). Similarly, there was a significant difference in frequency of one SNP in TNFRSF1A (TNFRSF1A:rs767455) among patients with CD compared with healthy controls (p<0.05). The TNFRSF1A:rs767455 GG genotype was found in 15/54 patients with CD (28%), while it was only found in 2/50 healthy controls (4%) (OR 9.2, 95% CI 1.98 to 42.83). The other two SNPs (TNFRSF1A:rs4149584 and TNFRSF1A:rs4149570) have shown no significance (p>0.05).
Table 3.
Genotype frequencies of selected SNPs for patients with CD and healthy controls
| Genotype | Patients with CD (n=54) | Healthy controls (n=50) | P value* OR (95% CI) |
| TNFα rs1800629 | |||
| GG (reference allele) | 34 (64%) | 41 (82%) | 0.03 |
| GA | 20 (37%) | 7 (14%) |
0.01
3.6 (1.37 to 9.54) |
| AA | 0 (0%) | 2 (4%) | 0.13 0.2 (0.01 to 5.20) |
| GA+AA | 20 (37%) | 9 (18%) |
0.03
2.7 (1.08 to 6.64) |
| TNFα rs1799964 | |||
| TT (reference allele) | 33 (61%) | 34 (68%) | 0.75 |
| TC | 18 (33%) | 16 (32%) | 0.89 1.2 (0.51 to 2.64) |
| CC | 3 (6%) | 0 (0%) | 0.09 7.2 (0.35 to 144.9) |
| TC+CC | 21 (39%) | 16 (32%) | 0.73 1.4 (0.60 to 3.00) |
| TNFα rs1799724 | |||
| CC (reference allele) | 45 (83%) | 43 (86%) | 0.7 |
| CT | 9 (16%) | 7 (14%) | 0.38 1.2 (0.42 to 3.59) |
| TT | 0 (0%) | 0 (0%) | NA |
| CT+TT | 9 (16%) | 7 (14%) | 0.70 1.2 (0.42 to 3.59) |
| TNFRSF1A rs4149584 | |||
| CC (reference allele) | 52 (96%) | 49 (98%) | 0.6 |
| CT | 2 (4%) | 1 (2%) | 0.60 1.9 (0.17 to 21.40) |
| TT | 0 (0%) | 0 (0%) | NA |
| CT+TT | 2 (4%) | 1 (0%) | 0.60 1.9 (0.17 to 21.40) |
| TNFRSF1A rs767455 | |||
| AA (reference allele) | 17 (31%) | 29 (58%) | 0.01 |
| AG | 22 (41%) | 19 (38%) | 0.29 2.0 (0.83 to 4.65) |
| GG | 15 (28%) | 2 (4%) |
0.01
9.2 (1.98 to 42.83) |
| AG+ GG | 37 (68%) | 21 (42%) |
0.01
3.0 (1.34 to 6.71) |
| TNFRSF1A rs4149570 | |||
| GG (reference allele) | 25 (46%) | 26 (52%) | 0.56 |
| GT | 13 (24%) | 18 (36%) | 0.18 0.75 (0.31 to 1.84) |
| TT | 16 (30%) | 7 (14%) | 0.06 2.4 (0.83 to 6.75) |
| GT+TT | 29 (54%) | 25 (50%) | 0.70 1.2 (0.56 to 2.59) |
| TNFRSF1B rs1061624 | |||
| AA (reference allele) | 13 (24%) | 27 (54%) | 0.01 |
| AG | 27 (50%) | 15 (30%) |
0.04
2.3 (1.04 to 5.22) |
| GG | 14 (26%) | 8 (16%) |
0.02
3.6 (1.21 to 10.83) |
| AG+ GG | 41 (76%) | 23 (46%) |
0.01
3.7 (1.61 to 8.53) |
| TNFRSF1B rs1061622 | |||
| TT (reference allele) | 24 (44%) | 31 (62%) | 0.07 |
| TG | 22 (41%) | 16 (32%) | 0.36 1.8 (0.77 to 4.1) |
| GG | 8 (15%) | 3 (6%) | 0.14 3.44 (0.82 to 14.4) |
| TG+GG | 30 (%) | 19 (38%) | 0.07 2.03 (0.93 to 4.47) |
| TNFRSF1B rs3397 | |||
| CC (reference allele) | 12 (22%) | 31 (62%) | 0.01 |
| CT | 31 (57%) | 15 (30%) |
0.01
3.1 (1.4 to 7.07) |
| TT | 15 (28%) | 4 (8%) |
0.01
4.4 (1.36 to 14.14) |
| CT+TT | 46 (85%) | 19 (38%) |
0.01
6.25 (2.66 to 14.69) |
Two-tailed Z test and OR analysis were used to compare between the presence of SNPs in patients with CD versus healthy controls.
*P<0.05 was considered as significance threshold.
CD, Crohn’s disease; NA, unknown residue change; SNP, single nucleotide polymorphism; TNFα, tumour necrosis factor-α.
Additionally, two SNPs in TNFRSF1B (TNFRSF1B:rs1061624 and TNFRSF1B:rs3397) were found to be significant in patients with CD compared with healthy controls (p<0.05). The TNFRSF1B:rs1061624 AG genotype was found in 27/54 patients with CD (50%) compared with 15/50 healthy controls (30%) (OR 2.3, 95% CI 1.04 to 5.22). The TNFRSF1B:rs3397 CT genotype was found in 31/54 patients with CD (57%) compared with (15/50) healthy controls (30%) (OR 3.1, 95% CI 1.40 to 7.07). Besides, the TNFRSF1B:rs3397 TT genotype was also significantly found in 15/54 patients with CD (28%) compared with (4/50) healthy controls (8%) (OR 4.4, 95% CI 1.36 to 14.14). However, TNFRSF1B:rs1061622 was not found to be significant among patients with CD in comparison to healthy controls (p>0.05). A complete list of SNPs and genotyping frequency is shown in table 3.
SNPs downregulate TNFRSF1A and TNFRSF1B gene expression in CD
We quantified relative gene expression level of TNFα, TNFRSF1A, and TNFRSF1B in all 104 study participants. Then, we further analysed the data according to each SNP present, in order to find out if SNPs are associated with gene expression level. In general, the relative expression of TNFα was more than three times higher in patients with CD (2.44±0.30), in comparison to healthy subjects (0.72±0.20), regardless of SNPs involved (p<0.5). However, none of TNFα SNPs (rs1800629, rs1799964, and rs1799724) were associated with gene expression level significantly (p>0.05) (figure 1A–C).
Figure 1.

Gene expression level of TNFα, TNFRSF1A and TNFRSF1B according to each allele type in selected single nucleotide polymorphisms (SNP) (A–I) among patients with Crohn’s disease (CD) (n=54) and healthy controls (n=50). Unpaired two-tailed t-test at p<0.05 and a 95% CI was used to test gene expression significance in patients with CD versus healthy controls, then one-way analysis of variance (ANOVA), where Newman-Keuls post-test was selected for multiple comparisons, was used to test individuals who carried two major alleles with others for each SNP. TNFα, tumour necrosis factor.
Overall, the expression level of TNFRSF1A and TNFRSF1B was significantly downregulated in patients with CD (0.38±0.15; p<0.5 and 0.34±0.13; p<0.05, respectively), compared with healthy subjects (0.79±0.24 and 0.66±0.17, respectively) (figure 1D–I). Nevertheless, patients with CD with the SNP TNFRSF1A:rs767455 who had GG genotype, had a significantly lower relative gene expression level compared with patients without SNP who had AA genotype (0.26±0.09 vs 0.49±0.12; p<0.5) (figure 1E).
Similarly, the expression level of TNFRSF1B was significantly lower in patients with CD with the SNP TNFRSF1B:rs3397 who had either CT (0.32±0.11) or TT (0.2±0.09) genotype, compared with patients without SNP (0.59±0.10) who had CC genotype (p<0.5) (figure 1I).
SNPs in TNFRSF1A and TNFRSF1B induce susceptibility to MAP infection
We evaluated MAP infection status in all 104 study participants by IS900 nPCR analysis, which is very sensitive and specific for MAP. Overall, 31/54 patients with CD were infected with MAP compared with only 4/50 healthy controls (OR 15.5, 95% CI 4.88 to 49.22, p<0.05).
We further looked for any association between a specific genotype in all SNPs we tested and the presence of MAP infection. Interestingly, patients with CD with the SNP TNFRSF1A:rs767455 who had either AG or GG genotype were more susceptible to MAP infection (63% and 66.6%, respectively), while patients with CD without SNP (AA genotype) were 41% infected with MAP. However, this result was not statistically significant (p>0.05) (figure 2A). On the other hand, patients with CD with the SNP TNFRSF1B:rs3397 who had either CT or TT genotype were significantly susceptible to MAP infection (58% and 70.5%, respectively), compared with patients with CD without SNP (CC genotype), who were only 17% infected with MAP (p<0.05) (figure 2B). None of the additional SNPs we tested (rs1800629, rs1799964, and rs1799724 of TNFα, rs4149584 and rs4149570 of TNFRSF1A, rs1061624 and rs1061624 of TNFRSF1B) were found to be significantly associated with higher susceptibility to MAP infection.
Figure 2.
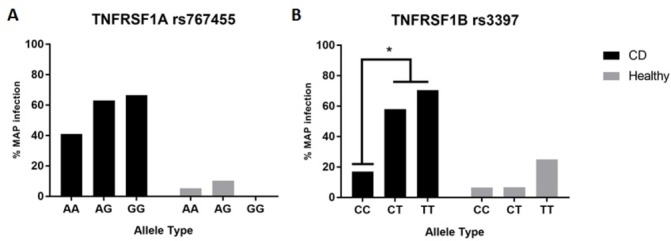
Influence of (A) TNFRSF1A (rs767455) and (B) TNFRSF1B (rs3397) single nucleotide polymorphisms (SNP) on MAP infection susceptibility in patients with CD (n=54) and healthy subjects (n=50). Infection proportions were compared between SNP genotypes and major alleles in patients with CD and healthy controls separately using two-tailed Z test at p<0.05. CD, Crohn’s disease; MAP, Mycobacterium avium subsp paratuberculosis.
SNP haplotypes distribution and MAP infection susceptibility
After we have identified TNFRSF1A:rs767455 and TNFRSF1B:rs3397 as SNPs associated with lower gene expression and higher MAP infection susceptibility in patients with CD, we further performed haplotype analysis of these two SNPs. The haplotype frequencies among patients with CD and healthy controls showed a significant difference for the G–T haplotype (p<0.05), which was found in 25/54 patients with CD (46%), while in healthy controls it was 7/50 (14%) (figure 3). Consequently, MAP infection was significantly higher among patients with CD who had the G–T haplotype (17/25), which contributes to 31% of patients with CD overall, while none of the healthy controls had the G–T haplotype and MAP infection at the same time. As described in table 4, none of the other SNP haplotypes inferred from TNFRSF1A and TNFRSF1B (A–C, G–C and AT) were found to be associated with susceptibility to MAP infection significantly (p>0.05).
Figure 3.
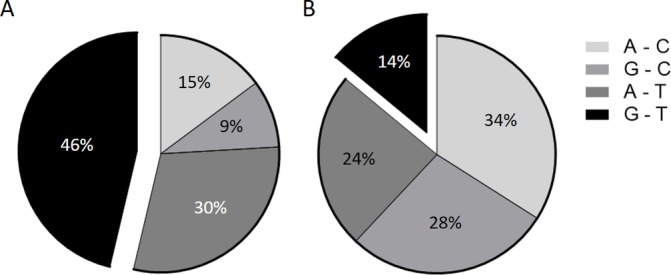
Haplotypes inferred from rs767455 (A/G) and rs3397 (C/T), and their distributions in patients with Crohn’s disease (CD) (A) and healthy subjects (B).
Table 4.
Haplotypes inferred from TNFRSF1A rs767455 (A/G)–TNFRSF1B rs3397 (C/T) and MAP infection distributions among patients with CD
| Haplotype* (rs767455–rs3397) |
CD MAP (+) (n=31) |
CD MAP (−) (n=23) |
Overall | P value |
| A–C | 2 (4%) | 6 (11%) | 8 (15%) | 0.14 |
| G–C | 3 (5%) | 2 (4%) | 5 (9%) | 0.64 |
| A–T | 9 (17%) | 7 (13%) | 16 (30%) | 0.59 |
| G–T | 17 (31%) | 8 (15%) | 25 (46%) | 0.01 |
Fisher’s exact test was used to test group significance at p<0.05.
CD, Crohn’s disease; MAP, Mycobacterium avium subsp paratuberculosis.
Discussion
Increased level of TNFα has been found in the inflamed intestinal mucosa of patients with CD, which is considered an essential mediator of immunologic response for this inflammatory disease.28 Consequently, TNFα plays a role in increasing intestinal permeability, granuloma formation and coagulation pathways.3 Most recently, we elucidated the relationship between MAP infection and the upregulation of TNFα expression in vitro, which has shown that both MAP and M. tuberculosis induce gene expression significantly in comparison to other mycobacteria and non-mycobacteria strains.29 Although inhibition of TNFα has shown a positive clinical outcome in some patients with CD, poor response was also found in others, which leaves those patients with severe side effects and higher susceptibility to infections.13 Therefore, several pharmacogenetic studies have evaluated the variation of anti-TNFα treatment response among patients with CD in order to predict treatment response ultimately.17–19
Genetic polymorphisms in TNFRSF1A and TNFRSF1B were found to be correlated with anti-TNFα treatment response in patients with CD. Specifically, the TNFRSF1A:rs767455 AG and GG genotype has a significant difference in frequency among non-responders to anti-TNFα treatment compared with the majority of drug responders who had the AA genotype.16 Similarly, the TNFRSF1B:rs3397 CT and TT also have a significant difference in frequency among anti-TNFα non-responders in comparison to patients with CD who were classified as drug responders (CC genotype).18 30 However, these observations were limited to drug response only, while the molecular effects of these SNPs were not discussed.
Since MAP is a suspected microbial causative agent of CD,14 15 31 32 it was worthy to find out if genetic polymorphisms are inducing susceptibility to MAP infection in patients with CD, which will ultimately influence anti-TNFα treatment outcome. Therefore, we tested 104 subjects (54 patients with CD and 50 healthy controls) for selective SNPs in TNFα (rs1800629, rs1799964, and rs1799724), TNFRSF1A (rs4149584, rs767455, and rs4149570) and TNFRSF1B (rs1061624, rs1061622, and rs3397). First, four out of these nine SNPs were found to have a significant difference in frequency among patients with CD compared with healthy controls (TNFα:rs1800629, TNFRSF1A:rs767455, TNFRSF1B:rs1061624 and TNFRSF1B:rs3397), which also correlates with previous reports.33–35 Second, we quantified gene expression level of TNFα, TNFRSF1A and TNFRSF1B in all study participants. We found that TNFα relative expression level is about 3.4-fold higher in patients with CD compared with healthy controls overall. However, none of TNFα-associated SNPs had a significant association with TNFα gene expression level. The expression of TNFRSF1A and TNFRSF1B was significantly downregulated (~2-fold) in patients with CD compared with healthy controls regardless of SNPs involved. Furthermore, the SNPs TNFRSF1A:rs767455 and TNFRSF1B:rs3397 were both found to be associated with a significant lower gene expression. Additionally, MAP infection was predominantly found among patients with CD in comparison to healthy controls (57% vs 8%, respectively). Interestingly, MAP infection was also associated with the SNPs TNFRSF1A:rs767455 and TNFRSF1B:rs3397. Furthermore, our SNP haplotype analysis of TNFRSF1A:rs767455 and TNFRSF1B:rs3397 indicates that the G–T haplotype has a significant difference in frequency among patients with CD (46%) and MAP infection susceptibility is also associated with this specific haplotype (31%).
It is relevant to compare between MAP and M. tuberculosis infection since mycobacterial protein tyrosine phosphatase (PtpA) in MAP shares 90% homology to PtpA in M. tuberculosis.36 This protein inhibits phagosome–lysosome fusion in the macrophage by dephosphorylating the host vacuolar protein sorting 33B. Consequently, the pathogen will be able to avoid containment eradication and it establishes a successful intracellular infection by preventing fusion of lysosome and phagosome into the phagolysosomal complex.37 38 Therefore, the primary mechanism for MAP eradication is by inducing apoptosis of the infected macrophage through TNFα-dependent mechanism.39 40 This is a critical point when we consider anti-TNFα treatment for patients with CD who are infected with MAP. In addition to genetic testing of selective SNPs we discussed, MAP infection screening method should be implemented before initiation of therapy. Finally, in figure 4, we suggest following a treatment algorithm for CD, based on patient’s haplotype inferred from TNFRSF1A rs767455 (A/G) and TNFRSF1B rs3397 (C/T).
Figure 4.

Recommended Crohn’s disease (CD) treatment algorithm based on haplotypes inferred from TNFRSF1A rs767455 (A/G)–TNFRSF1B rs3397 (C/T). MAP, Mycobacterium avium subsp paratuberculosis; SNP, single nucleotide polymorphism; TNFα, tumour necrosis factor.
Acknowledgments
Our thanks are due to all members of Dr Naser’s lab and to Brent Cao for his valuable assistance in the statistical analysis. A sincere acknowledgement to all the subjects whose clinical samples were included in this study.
Footnotes
Contributors: AQ is the primary author who performed all experiments, collected data and participated in writing the manuscript. SR is our collaborator gastroenterologist at the Digestive and Liver Center of Florida, who provided us with clinical samples. SAN is the leading investigator in the lab and has supervised all aspects of the study including writing and editing of the manuscript.
Funding: This study was funded, in part, by the Florida Legislative Grant and UCF Doctoral Research Support Award.
Competing interests: None declared.
Patient consent for publication: Obtained.
Ethics approval: University of Central Florida Institutional Review Board (IRB00001138).
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: The data sets supporting the conclusions of this article are included in the article.
References
- 1. Victor FC, Gottlieb AB. TNF-alpha and apoptosis: implications for the pathogenesis and treatment of psoriasis. J Drugs Dermatol 2002;1:264–75. [PubMed] [Google Scholar]
- 2. Brynskov J, Foegh P, Pedersen G, et al. . Tumour necrosis factor alpha converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut 2002;51:37–43. 10.1136/gut.51.1.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Furst DE, Wallis R, Broder M, et al. . Tumor necrosis factor antagonists: different kinetics and/or mechanisms of action may explain differences in the risk for developing granulomatous infection. Semin Arthritis Rheum 2006;36:159–67. 10.1016/j.semarthrit.2006.02.001 [DOI] [PubMed] [Google Scholar]
- 4. Papadakis KA, Targan SR. Tumor necrosis factor: biology and therapeutic inhibitors. Gastroenterology 2000;119:1148–57. 10.1053/gast.2000.18160 [DOI] [PubMed] [Google Scholar]
- 5. Hyrich KL, Watson KD, Silman AJ, et al. . Predictors of response to anti-TNF-alpha therapy among patients with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register. Rheumatology 2006;45:1558–65. 10.1093/rheumatology/kel149 [DOI] [PubMed] [Google Scholar]
- 6. Tracey D, Klareskog L, Sasso EH, et al. . Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008;117:244–79. 10.1016/j.pharmthera.2007.10.001 [DOI] [PubMed] [Google Scholar]
- 7. Roda G, Jharap B, Neeraj N, et al. . Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin Transl Gastroenterol 2016;7:e135 10.1038/ctg.2015.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Antoni C, Braun J. Side effects of anti-TNF therapy: current knowledge. Clin Exp Rheumatol 2002;20(6 Suppl 28):S152–7. [PubMed] [Google Scholar]
- 9. Botha T, Ryffel B. Reactivation of latent tuberculosis infection in TNF-deficient mice. J Immunol 2003;171:3110–8. 10.4049/jimmunol.171.6.3110 [DOI] [PubMed] [Google Scholar]
- 10. Marotte H, Charrin JE, Miossec P. Infliximab-induced aseptic meningitis. The Lancet 2001;358:1784 10.1016/S0140-6736(01)06810-6 [DOI] [PubMed] [Google Scholar]
- 11. Baghai M, Osmon DR, Wolk DM, et al. . Fatal sepsis in a patient with rheumatoid arthritis treated with etanercept. Mayo Clin Proc 2001;76:653–6. 10.4065/76.6.653 [DOI] [PubMed] [Google Scholar]
- 12. Ritz MA, Jost R. Severe pneumococcal pneumonia following treatment with infliximab for Crohn's disease. Inflamm Bowel Dis 2001;7:327 10.1097/00054725-200111000-00009 [DOI] [PubMed] [Google Scholar]
- 13. Qasem A, Naser AE, Naser SA. The alternate effects of anti-TNFα therapeutics and their role in mycobacterial granulomatous infection in Crohn's disease. Expert Rev Anti Infect Ther 2017;15:637–43. 10.1080/14787210.2017.1328276 [DOI] [PubMed] [Google Scholar]
- 14. Chamberlin WM, Naser SA. Integrating theories of the etiology of Crohn's disease. On the etiology of Crohn's disease: questioning the hypotheses. Med Sci Monit 2006;12:RA27–RA33. [PubMed] [Google Scholar]
- 15. Qasem A, Abdel-Aty A, Abu-Suwa H, et al. . Oxidative stress due to Mycobacterium avium subspecies paratuberculosis (MAP) infection upregulates selenium-dependent GPx activity. Gut Pathog 2016;8:12 10.1186/s13099-016-0090-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cao BL, Qasem A, Sharp RC, et al. . Systematic review and meta-analysis on the association of tuberculosis in Crohn's disease patients treated with tumor necrosis factor-α inhibitors (Anti-TNFα). World J Gastroenterol 2018;24:2764–75. 10.3748/wjg.v24.i25.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matsukura H, Ikeda S, Yoshimura N, et al. . Genetic polymorphisms of tumour necrosis factor receptor superfamily 1A and 1B affect responses to infliximab in Japanese patients with Crohn's disease. Aliment Pharmacol Ther 2008;27:765–70. 10.1111/j.1365-2036.2008.03630.x [DOI] [PubMed] [Google Scholar]
- 18. Medrano LM, Taxonera C, Márquez A, et al. . Role of TNFRSF1B polymorphisms in the response of Crohn's disease patients to infliximab. Hum Immunol 2014;75:71–5. 10.1016/j.humimm.2013.09.017 [DOI] [PubMed] [Google Scholar]
- 19. Steenholdt C, Enevold C, Ainsworth MA, et al. . Genetic polymorphisms of tumour necrosis factor receptor superfamily 1b and fas ligand are associated with clinical efficacy and/or acute severe infusion reactions to infliximab in Crohn's disease. Aliment Pharmacol Ther 2012;36:650–9. 10.1111/apt.12010 [DOI] [PubMed] [Google Scholar]
- 20. Naser SA, Ghobrial G, Romero C, et al. . Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn's disease. The Lancet 2004;364:1039–44. 10.1016/S0140-6736(04)17058-X [DOI] [PubMed] [Google Scholar]
- 21. NCBI , 2018. National Center for Biotechnology Information. Available from: ncbi.nlm.nih.gov
- 22. Ferreira AC, Almeida S, Tavares M, et al. . NOD2/CARD15 and TNFA, but not IL1B and IL1RN, are associated with Crohn's disease. Inflamm Bowel Dis 2005;11:331–9. 10.1097/01.MIB.0000158153.71579.b4 [DOI] [PubMed] [Google Scholar]
- 23. Cooke J, Zhang H, Greger L, et al. . Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm Bowel Dis 2012;18:2128–37. 10.1002/ibd.22942 [DOI] [PubMed] [Google Scholar]
- 24. Zhu X, Wang Y, Sun L, et al. . A novel gene variation of TNFalpha associated with ankylosing spondylitis: a reconfirmed study. Ann Rheum Dis 2007;66:1419–22. 10.1136/ard.2006.068528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Comabella M, Caminero AB, Malhotra S, et al. . TNFRSF1A polymorphisms rs1800693 and rs4149584 in patients with multiple sclerosis. Neurology 2013;80:2010–6. 10.1212/WNL.0b013e318294b2d6 [DOI] [PubMed] [Google Scholar]
- 26. Sode J, Vogel U, Bank S, et al. . Anti-TNF treatment response in rheumatoid arthritis patients is associated with genetic variation in the NLRP3-inflammasome. PLoS One 2014;9:e100361 10.1371/journal.pone.0100361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferguson LR, Han DY, Huebner C, et al. . Tumor necrosis factor receptor superfamily, member 1B haplotypes increase or decrease the risk of inflammatory bowel diseases in a New Zealand caucasian population. Gastroenterol Res Pract 2009;2009:1–9. 10.1155/2009/591704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. MacDonald TT, Hutchings P, Choy MY, et al. . Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol 1990;81:301–5. 10.1111/j.1365-2249.1990.tb03334.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qasem A, Naser SA. TNFα inhibitors exacerbate Mycobacterium paratuberculosis infection in tissue culture: a rationale for poor response of patients with Crohn's disease to current approved therapy. BMJ Open Gastroenterol 2018;5:e000216 10.1136/bmjgast-2018-000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen W, Xu H, Wang X, et al. . The tumor necrosis factor receptor superfamily member 1B polymorphisms predict response to anti-TNF therapy in patients with autoimmune disease: A meta-analysis. Int Immunopharmacol 2015;28:146–53. 10.1016/j.intimp.2015.05.049 [DOI] [PubMed] [Google Scholar]
- 31. Naser A, Qasem A, Naser SA. Mycobacterial infection influences bone biomarker levels in patients with Crohn's disease. Can J Physiol Pharmacol 2018;96:662–7. 10.1139/cjpp-2017-0700 [DOI] [PubMed] [Google Scholar]
- 32. Qasem A, Safavikhasraghi M, Naser SA. A single capsule formulation of RHB-104 demonstrates higher anti-microbial growth potency for effective treatment of Crohn's disease associated with Mycobacterium avium subspecies paratuberculosis. Gut Pathog 2016;8:45 10.1186/s13099-016-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Waschke KA, Villani AC, Vermeire S, et al. . Tumor necrosis factor receptor gene polymorphisms in Crohn's disease: association with clinical phenotypes. Am J Gastroenterol 2005;100:1126–33. 10.1111/j.1572-0241.2005.40534.x [DOI] [PubMed] [Google Scholar]
- 34. Kawasaki A, Tsuchiya N, Hagiwara K, et al. . Independent contribution of HLA-DRB1 and TNF alpha promoter polymorphisms to the susceptibility to Crohn's disease. Genes Immun 2000;1:351–7. 10.1038/sj.gene.6363689 [DOI] [PubMed] [Google Scholar]
- 35. Bouma G, Xia B, Crusius JB, et al. . Distribution of four polymorphisms in the tumour necrosis factor (TNF) genes in patients with inflammatory bowel disease (IBD). Clin Exp Immunol 1996;103:391–6. 10.1111/j.1365-2249.1996.tb08292.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bach H, Sun J, Hmama Z, et al. . Mycobacterium avium subsp. paratuberculosis PtpA is an endogenous tyrosine phosphatase secreted during infection. Infect Immun 2006;74:6540–6. 10.1128/IAI.01106-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crowle AJ, Dahl R, Ross E, et al. . Evidence that vesicles containing living, virulent Mycobacterium tuberculosis or Mycobacterium avium in cultured human macrophages are not acidic. Infect Immun 1991;59:1823–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frehel C, de Chastellier C, Lang T, et al. . Evidence for inhibition of fusion of lysosomal and prelysosomal compartments with phagosomes in macrophages infected with pathogenic Mycobacterium avium. Infect Immun 1986;52:252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fratazzi C, Arbeit RD, Carini C, et al. . Macrophage apoptosis in mycobacterial infections. J Leukoc Biol 1999;66:763–4. 10.1002/jlb.66.5.763 [DOI] [PubMed] [Google Scholar]
- 40. Fratazzi C, Arbeit RD, Carini C, et al. . Programmed cell death of Mycobacterium avium serovar 4-infected human macrophages prevents the mycobacteria from spreading and induces mycobacterial growth inhibition by freshly added, uninfected macrophages. J Immunol 1997;158:4320–7. [PubMed] [Google Scholar]