

Abstract
We present a general strategy for determining the cholesterol-binding site of eukaryotic membrane proteins in native-like lipid membranes by NMR spectroscopy. The strategy combines yeast biosynthetic 13C enrichment of cholesterol with detection of protein-cholesterol 13C-13C cross peaks in 2D correlation NMR spectra under the dynamic nuclear polarization (DNP) condition. Low-temperature DNP not only allows high-sensitivity detection of weak protein-cholesterol cross peaks in 2D spectra but also immobilizes cholesterol and protein to enable intermolecular distance measurements. We demonstrate this approach on the influenza M2 protein, which utilizes cholesterol to conduct membrane scission in the last step of virus budding and release from the host cell plasma membrane. A 13C-13C double-quantum filter was employed to significantly simplify the 2D 13C-13C correlation spectra and facilitate the identification of protein-cholesterol cross peaks. A number of cross peaks between the M2 transmembrane residues’ sidechains and the cholesterol sterol group were detected, which complement recently measured protein contacts to the isooctyl tail of cholesterol to define an extended binding interface. These results provide atomic-level evidence of M2-cholesterol interaction to cause membrane curvature and scission, and the approach is generally applicable to other eukaryotic membrane proteins for understanding the influence of cholesterol on membrane protein function.
Graphical Abstract
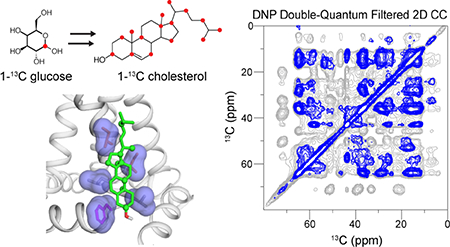
Introduction
Cholesterol is an essential component of mammalian cell membranes 1. It consists of four fused rings bonded to a polar hydroxyl group at one end and an isooctyl tail at the other end. In phospholipid bilayers, cholesterol is oriented with the long axis of the sterol rings approximately parallel to the bilayer normal; the polar OH moiety resides at the membrane-water interface whereas the aliphatic tail inserts into the bilayer center 2,3. In mammalian membranes, cholesterol is present at 20–30 mol%, while in enveloped viruses cholesterol is enriched to ~45 mol% in the viral lipid envelope 4,5. The rigid sterol plane of cholesterol renders phospholipid bilayers more viscous at high temperature but more dynamic at low temperature compared to cholesterol-free membranes, which has profound consequences to membrane physical properties such as viscosity, bilayer thickness, domain formation, and curvature stress 6.
Cholesterol is known to regulate a diverse range of membrane protein functions. For example, it modulates the conformational equilibria, stability, and ligand-binding affinities of G-protein coupled receptors to affect signal transduction 7–11. It is implicated in Alzheimer’s disease and binds the amyloid precursor protein 12,13. Cholesterol also changes the conformation and localization of viral fusion proteins 14–16. Cholesterol can modulate membrane protein function by specific binding to proteins or by non-specific effects on the membrane physical properties. However, the precise mechanism of cholesterol’s influence on membrane proteins is so far poorly understood, and likely varies from protein to protein. The lack of information is largely due to the small size and complex dynamics of cholesterol in lipid bilayers, and the incompatibility of cholesterol with the detergent micelles and small isotropic bicelles that are commonly used for membrane protein reconstitution before solution NMR studies 17. In comparison, cholesterol can be advantageous for protein crystallization when used in concert with curvature lipids such as monoolein 18. Of the approximately dozen high-resolution (< 3 Å) cholesterol-bound membrane protein structures that have been deposited in the Protein Databank so far, most were solved using X-ray crystallography. However, in most these cases, the bound cholesterol in these structures results from co-crystallization from monoolein-cholesterol lipid cubic phases, and has no known function. The β2 adrenergic receptor and the glutamate receptor structures are examples where the cholesterol-binding sites have partly understood functions 7,19. Therefore, a strategy for determining functionally important protein-cholesterol binding sites in lipid bilayers is important for elucidating the biological roles of cholesterol in lipid membranes.
Magic-angle-spinning (MAS) solid-state NMR (SSNMR) is ideally suited to the investigation of protein-lipid interactions in lipid bilayers 20–22. Solid-state NMR spectroscopy can detect angstrom-to-nanometer intermolecular contacts 23,24 under a wide range of temperature, bilayer composition, and membrane viscosities, and thus can potentially provide detailed structural information about cholesterol-protein binding. A number of recent solution NMR studies have reported cholesterol interactions with membrane proteins by monitoring protein chemical shift perturbations 12,25,26. However, it is recognized that chemical shift perturbations can occur through allosteric effects, so that residues far from the bound cholesterol may exhibit significant chemical shift changes. Therefore, to unambiguously determine the location of cholesterol relative to the protein, distance measurements are necessary.
Recently, we showed that aliphatic tail-fluorinated cholesterol provides an effective probe for determining the cholesterol-binding site to proteins using solid-state NMR of bilayer-bound proteins 27. By combining 13C-labeled protein with aliphatic-heptafluorinated cholesterol, we determined the binding site of cholesterol to the influenza M2 protein. However, current commercially available 13C-labeled cholesterol compounds place 13C-enriched sites either at the sterol polar end or at the nonpolar aliphatic tail, whereas the vast central region of cholesterol is NMR-silent, thus restricting the detectable region of cholesterol for protein binding to the two ends of the molecule.
In this work we demonstrate biosynthetic 13C-labeling of cholesterol from yeast, together with sensitivity-enhancing dynamic nuclear polarization (DNP) NMR, for determining the cholesterol-binding site in membrane proteins with high sensitivity. Yeast normally produce the sterol ergosterol, which shares common biosynthetic steps with cholesterol until the final transformations, where three enzymes, Erg4p, Erg5p, and Erg6p catalyze methyltransferase and reductase activity at C24 and desaturase activity at C22 (Fig. 1a). A genetically engineered Saccharomyces cerevisiae strain, RH6829, has been developed in which the Erg5p and Erg6p enzymes were replaced with two dehydrocholesterol reductases for C24 and C7, thus producing cholesterol 28,29. Using this metabolically engineered yeast strain, we show that cholesterol can be 13C skip-labeled at alternate carbons throughout the molecule 28–30, which dramatically increases the detectable intermolecular interface with the influenza virus M2 protein.
Figure 1.

Biosynthetic 13C labeling of cholesterol in yeast for investigating protein-cholesterol interactions. (a) Key steps in the Saccharomyces cerevisiae biosynthetic pathway for ergosterol. Replacement of Erg6p and Erg5p enzymes by DHCR7 and DHCR24 enzymes enables efficient production of cholesterol. The 1-13C and 2-13C glucose labeled carbons are colored in red and blue, respectively. (b) 1D and 2D 13C CP spectra of 1-13C CHOL in POPC/POPG (4 : 1) bilayers, measured at 275 K. The sterol ring naming convention is indicated on the chemical structure. Assignment of the cross peaks confirm the 13C labeled positions.
The influenza M2 protein is a tetrameric membrane protein 31 that conducts protons to initiate viral uncoating 32–35 as well as mediating membrane scission of the budding virus 36. The membrane scission function has been shown to require cholesterol 37,38 and an amphipathic helix (AH) that is C-terminal to the proton-conducting transmembrane (TM) helix 39,40. Direct interaction between cholesterol and M2 was first suggested based on biochemical assays 41,42. We recently measured M2-cholesterol 13C-19F distances to show that cholesterol binds to two of the four TM helices from the vertices of the tetramer 27, and binding does not require a putative cholesterol-recognition amino acid sequence 43,44 but requires the L-shape formed by the TM helix and the AH. Based on the measured distances between the TM residues and cholesterol tail and the measured sterol plane orientation 27, we proposed a structure of the bound complex. However, this previous study did not have any distance restraints for the extended sterol of cholesterol due to the lack of suitable 13C-labeled cholesterol. We now show 2D 13C-13C correlation spectra with multiple cross peaks between the protein and yeast-labeled cholesterol, which allow us to determine an extensive binding interface between M2 and cholesterol.
Materials and Methods
Biosynthesis of 13C-labeled cholesterol from yeast
Saccharomyces cerevisiae strain RH6829 28,29, was a gift from Professor Riezman (University of Geneva). The yeast was plated and inoculated into starter culture following established protocols 45,46. The 500 mL production medium consisted of 3.34 g (0.67%) yeast nitrogen base with ammonium sulfate but without amino acids (VWR AMRESCO), 695 mg (0.14%) yeast dropout medium supplements without His, Leu, Trp, and uracil (Sigma), 190 mg Leu, 76 mg uracil, and 7.5 g (15%) of 1-13C glucose (Cambridge Isotope Laboratories). This medium was filter-sterilized before use. The production medium was split into two 250 mL portions, each inoculated with 25 μL (1 : 10,000) RH6829 starter culture, and incubated for 68 h at 30°C while shaking at 220 rpm. The cells were harvested and washed with trichloroacetic acid 47. The cell pellets were lyophilized and transferred to 20 mL glass scintillation vials for sterol extraction.
The cells were resuspended in an extraction cocktail consisting of 3 mL 60% (w/v) KOH in water, 3 mL 0.5% (w/v) pyrogallol in methanol, and an additional 4.5 mL methanol; 10.5 mL was used per 250 mL culture. The cells were heated at 80°C for 2 h with occasional mixing, and then extracted three times with ~4 mL petroleum ether. The ether phase was pooled into 20 mL scintillation vials and dried by nitrogen gas. The dried material was redissolved in acetone and passed through a 0.22 μm syringe filter to prepare for HPLC purification. The concentration of the extracted material was estimated by comparing the extract solution against commercial cholesterol solutions spotted on a TLC plate and visualized with an acidic FeCl3 stain (50% w/v FeCl3 in water with 5% v/v glacial acetic acid and 5% v/v sulfuric acid, Fig. S1a).
The extracted fraction was purified by HPLC using a Vydac C18 reversed-phase column (4.6 mm i.d. × 100 mm, 5 μm particle size) under isocratic conditions of 30% solvent A (90% ethanol, 5% isopropanol, 4.6% methanol) and 70% solvent B (acetonitrile) and a 1.5 mL/min flow rate. The extract solution (~1.5 mg/mL) was purified over 14 injections of 300 μl; the main peak eluted at 9 minutes (Fig. S1b). Approximately 8 mL of eluant was collected per injection. The total mass of purified 1-13C CHOL from the 500 mL production culture was 9 mg.
Expression of recombinant M2(21–97) protein
An Udorn 1972/H3N2 M2 construct (residues 21–97) spanning the TM domain and the full cytoplasmic domain was expressed in E. coli as described previously 48,49. The construct, termed M2T below, contains an N-terminal His6 tag and a TEV protease cleavage site for purification. The protein was 13C, 15N-labeled at Ile, Phe, Gly, and Ala (IFGA-M2T) by adding 150 mg/L of each 13C, 15N-labeled amino acid to the M9 growth media (48 mM Na2HPO4, 22 mM KH2PO4, 8.55 mM NaCl, 1 g/L NH4Cl, 2 g/L D-glucose, 2 mM MgSO4, 0.1 mM CaCl2, 100 mg/mL ampicillin). In addition, 50 mg/L unlabeled Ser was added to prevent Ser labeling due to scrambling from Gly 50,51.
Cells were grown in 2 L of Luria broth media to an OD600 of 0.8, then pelleted and resuspended in 1 L M9 medium. About 40 minutes after suspension, protein expression was induced by adding isopropyl-β-D-thiogalactopyranoside (IPTG) to a concentration of 0.4 mM. After 4 hours, cells were pelleted and resuspended in lysis buffer (50 mM Tris, 50 mM NaCl, pH 7.8) and sonicated on ice for 30 min in cycles of 30 s on, 30 s off. M2T was purified from both the lysate supernatant and inclusion bodies by Ni-NTA affinity chromatography, concentrated and washed, and incubated with ~1 mg TEV protease for 48 h at room temperature. Cleaved M2T was purified by HPLC on a Vydac C4 reversed-phase column (22 mm × 250 mm, 10 μm particle size) using a mobile phase of water/acetonitrile containing 0.1% trifluoroacetic acid. Purification was conducted with a gradient of 30–99% acetonitrile over 70 minutes at a flow rate of 10 mL/min. About 12 mg of M2T was purified from 1 L of M9 medium and was lyophilized to dryness before use.
Synthesis of M2(22–61) peptides
A 13C, 15N-labeled M2 Weybridge peptide (residues 22–61) (NH2-SSDPLVVAASII-GILHLILWILDRLFFKSIYRRLKYGLKR-amide) was synthesized using a custom-designed fast-flow peptide synthesizer 27,52. The peptide was 13C, 15N-labeled at G34 and I39 (henceforth referred to as GI-M2W) to allow site-specific verification of the M2-cholesterol contacts. Amino acids were coupled using HATU (O-7-(azabenzotriazol)-1-yl-N,N,N’,N’-tetramethyluronium hexafluorophosphate) at 70°C under flow rates of 2.0 or 3.1 mL/min. Coupling times were 60 s for 13C, 15N-labeled residues and 80 s for unlabeled residues. Ten equivalents of unlabeled amino acids and four equivalents of labeled amino acids were used for each coupling. Resin cleavage and sidechain deprotection were conducted using 95% trifluoroacetic acid (TFA), 2.5% triisopropyl silane (TIPS), and 2.5% (H2O) at room temperature for 2–3 h. The cleaved peptide was precipitated with cold diethyl ether. The crude peptide was purified by reverse-phase HPLC on a C4 column (22 mm × 250 mm) with an acetonitrile-water gradient of 30–70%. All mobile phases contained 0.1% TFA. Purified peptide masses were verified on an Agilent 6520 LC-MS equipped with a Zorbax 300SB-C3 column. Pure peptide fractions were pooled and lyophilized for later reconstitution into membrane bilayers. About 14 mg purified peptide was obtained from a 0.06 mmol synthesis scale.
Membrane sample preparation
IFGA-M2T and GI-M2W were reconstituted into lipid membranes containing 1-13C CHOL by organic-phase mixing. Two membrane mixtures were used: a POPC/POPG/CHOL membrane with a molar ratio of 8 : 2 : 2 or 9.8 : 3.3 : 2, and a virus-mimetic membrane (VMS) containing POPC, POPE, POPS, sphingomyelin, and CHOL at a molar ratio of 36 : 18 : 18 : 11 : 17 53. Thus, the cholesterol mole fractions in these membranes are 13–17 mol%. The protein-lipid molar ratios are 1 : 20 to 1 : 25, while the protein-cholesterol molar ratio was 1 : 4.5 for all samples. All lipids and cholesterol were dissolved in chloroform, with up to 10% methanol for POPS, while the M2 protein was dissolved in trifluoroethanol. The solutions were combined, dried with a stream of nitrogen gas, and placed under vacuum overnight to remove residual organic solvent. The samples were rehydrated in ~3 mL Tris buffer (20 mM, pH 7.5) and subjected to 6–7 freeze/thaw cycles between liquid nitrogen temperature and a 37°C water bath. The liposome suspension was pelleted by ultracentrifugation at 160,000 × g and 4°C for 4–16 h, and the supernatant was decanted to obtain the proteoliposomes pellet with ~70 μL associated buffer.
Membrane samples for DNP experiments were prepared by adding D2O to the centrifuged membrane pellets to create a 3 : 1 D2O/H2O composition. This mixture was vortexed to obtain a uniform suspension, then the sample volume was reduced to ~100 μL by repeated lyophilization of 10–20 minutes at a time, with vortexing in between to maintain a uniform suspension and prevent sample losses along the walls of the vessel. An equivalent mass of “DNP juice”, which consists of 15 mM AMUPOL in 60% (v/v) 13C-depleted glycerol (Cambridge Isotope Laboratories), 30% D2O, and 10% H2O, was added to the membrane suspensions, and vortexed to mix the radicals and the membrane uniformly. The mixture was dried to ~50% hydration by brief lyophilization, and centrifuged (3000 × g) into 3.2 mm MAS rotors.
Solid-state NMR experiments
Non-DNP 1D and 2D 13C spectra at 293 – 255 K were measured on a 600 MHz (14.1 T) Bruker AVANCE III HD spectrometer using a 3.2 mm magic-angle-spinning (MAS) probe. Radiofrequency (rf) field strengths were 71–85 kHz for 1H and 31–50 kHz for 13C. 13C chemical shifts were externally referenced to the CH2 resonance of adamantane at 38.48 ppm on the tetramethylsilane scale. 13C cross-polarization (CP) spectra were measured under 9–13 kHz MAS from 293 K to 255 K, which spans both the liquid-crystalline phase and gel phase of the POPC/POPG membrane.
DNP sensitivity-enhanced 2D 13C-13C correlation spectra at ~110 K were measured on a 600 MHz NMR spectrometer equipped with a 7.2 T gyrotron operating at a Larmor frequency of 395 GHz (Bruker BioSpin, Billerica, MA). The samples were spun at 12 kHz or 13.33 kHz using a 3.2 mm MAS probe. Typical rf field strengths were 100 kHz for 1H and 60–67 kHz for 13C. 1H-13C CP used a tangent shape on H and contact times of 0.5 – 1.0 ms. Sample temperatures were ~98 K and ~110 K without and with microwave irradiation. The 13C sensitivity enhancement factors were 28–42, as evaluated from 1D 13C CP spectra measured with a 10 s recycle delay. 2D 13C-13C correlation spectra were measured using C CORD spin diffusion mixing times of 30 ms or 100–500 ms (Table S1) 54. To simplify the ω1 dimension of the 2D spectra, we applied a 13C-13C double-quantum filter (DQF) before the t1 evolution period in many experiments, so that cholesterol 13C peaks that do not have directly bonded 13C sites are suppressed. This DQF strategy in 2D correlation experiments has been shown before in the absence of DNP for removing natural abundance 13C signals 55. A 2D dipolar INADEQUATE spectrum of POPC/POPG bound IFGA-M2T was measured to assign the protein resonances. The 2D DQF-CC and INADEQUATE experiments used the SPC5 sequence 56 for 13C dipolar recoupling with a combined DQ excitation and reconversion time of 0.6–0.9 ms 57.
Molecular docking with distance constraints
To model the M2-CHOL complex using the measured distance and orientation constraints, we used the docking program HADDOCK 58,59. The input restraints include the 13C-13C distance contacts reported in this work, 13C-19F and 13C-13C distances previously reported on M2 peptides, and cholesterol orientation relative to the bilayer normal measured using 2H NMR 27. HADDOCK allows designation of ambiguous and unambiguous interaction restraints to guide the docking: all unambiguous restraints were maintained throughout the docking process, while 50% of ambiguous restraints were discarded at the beginning of each run 60. M2-CHOL contacts with specific residue and atom assignments, obtained from the site-specifically labeled peptide M2(22–61), were input as unambiguous restraints, while assignments obtained from M2T samples, which can correspond to multiple residues (e.g. I39 or I42), were input as ambiguous restraints (Table S3). Inter-vector angular restraints 61 were defined between the bilayer normal, which is equivalent to the channel axis, and a molecule-fixed cholesterol coordinate system 27. Previously, we defined the channel axis as the average vector of the four TM helices where the TM Cα atoms define each helix axis, and the cholesterol coordinate system was defined with respect to the C3 moiety 27,62. For HADDOCK, addition of artificial vectors interferes with docking, thus we adopted an alternative approach of selecting proxy vectors in the protein and cholesterol to represent the same coordinate axes. The I35 Cα–W41 Cα vector of helix D is nearly parallel to the channel axis; the Ch13–Ch3 vector of cholesterol defines the Z-axis of the cholesterol molecular frame, while the Ch10–Ch19 vector is approximately coincident with the X-axis of the cholesterol molecular frame (Table S4). All these vectors lie within 5° of the previously established bilayer normal and cholesterol coordinate axes, and were used for implementing orientation restraints in HADDOCK.
A number of changes were made to the default HADDOCK settings as recommended for protein-ligand binding. The mode for clustering analysis was changed to RMSD with a 2.5 Å cutoff and a minimum cluster size of 6. The weighting factors for scoring during rigid-body docking and solvent refinement were altered, starting temperatures for the second and third temperature-accelerated dynamics (TAD) steps were reduced to 500 and 300°C, respectively, and the number of MD steps for rigid high-temperature TAD and first rigid-body cooling were set to 0. Dimethylsulfoxide was used during the final explicit solvent refinement stage as a membrane mimic. These settings are modifiable with “guru” access to the HADDOCK webserver. The default settings of 1000 initial structures for rigid body docking and 200 structures each for semi-flexible refinement and explicit solvent refinement were used. The input M2 structure used for the docking was the first model of the SSNMR structure ensemble (PDB accession code: 2L0J) 40. The residues were renumbered to avoid non-uniqueness to conform to HADDOCK guidelines. The input cholesterol structure was accessed from the ZINC ligand database 63. The full input parameter file (except for the PDB coordinate file), is given in Appendix A.
To evaluate and compare different docked structures, we first used the HADDOCK cluster and ranking analysis based on the HADDOCK scoring function. However, the output score does not perform as well when ranking protein-ligand complexes 64, thus we additionally analyzed the docking structures using the PRODIGY-LIGAND contact-based binding energy predictor 64–67. In addition, we compared the output cholesterol β and γ orientation angles to the experimentally measured angles from 2H NMR spectra 27. To quantify the docked cholesterol’s orientational deviations from the measured orientations, we calculated the residual sum-of-squares (RSS) for each structure using the equation:
where βo and γo are the output angles of the docked structure while βi and γi are the experimentally measured input angles of 11° and 10°, respectively. The top 10 structures by the RSS ranking are shown in Fig. S9 and the RSS, PRODIGY, and HADDOCK scores are summarized in Table S5. The docked structures were visualized in PyMOL and the orientation angles and RSS were calculated in Matlab.
Results
Yeast biosynthesis of 1-13C CHOL
Yeast-based 13C enrichment of sterols has recently been extended beyond ergosterol by metabolic engineering of S. cerevisiae 29. The biosynthetic pathways of fungal ergosterol, vertebrate cholesterol, and plant campesterol, diverge near the end. This allowed the replacement of two ergosterol-synthesis enzymes by the corresponding cholesterol-synthesis enzymes, DHCR7 and DHCR24, to produce cholesterol (Fig. 1a). Supplying 1-13C labeled glucose labels 15 carbons in cholesterol, 12 of which do not have directly bonded carbons 28,30. This skip labeling facilitates NMR structural studies by removing 13C-13C J-couplings and dipolar couplings, narrowing the linewidths and increasing spectral sensitivity. Compared to 2-13C glucose, 1-13C glucose labels all five methyl carbons in cholesterol - Ch19, Ch18, Ch21, Ch26 and Ch27 - which may have a higher propensity to form specific interactions with proteins. Moreover, the 1-13C labeled sites have larger chemical diversity and hence larger chemical shift dispersion (Fig. S2), making 1-13C labeling the more promising choice for studying M2-cholesterol interactions 68.
We obtained 9.0 mg of purified and skip-labeled 1-13C CHOL from 500 mL of yeast medium containing 7.5 g of 1-13C glucose. One-dimensional 13C CP spectra of 1-13C CHOL reconstituted into POPC/POPG bilayers show linewidths of ~0.2 ppm in the liquid-crystalline phase of the membrane at 275 K. Two-dimensional 13C-13C correlation spectra measured with 300 ms mixing display up to four-bond correlations. These cross peaks are consistent with the spectra of cholesterol obtained with 2-13C sodium acetate, which produces the same labeling pattern as 1-13C glucose (Fig. 1b) 30. The cholesterol 13C chemical shifts were assigned based on the connectivity patterns in the spectra and verified by comparison with literature chemical shifts 69
Identifying protein-cholesterol contacts using 1-13C CHOL and 13C-labeled M2
Labeling with 1-13C glucose produced a large number of NMR reporters throughout cholesterol, which dramatically facilitated the search for the M2-CHOL interface and allowed a more detailed description of the binding site obtained from previous distance measurements, which involved only the cholesterol tail 27. We reconstituted 1-13C CHOL with 13C-labeled M2T in POPC/POPG bilayers to measure cholesterol-protein 13C-13C cross peaks in 2D correlation spectra. To immobilize cholesterol in the membrane, we first conducted the 2D experiments at ~253 K, in the gel phase of the membrane. However, at this temperature the cholesterol 13C signals broaden significantly, to ~2 ppm, compared to the liquid-crystalline phase (Fig. S3), with the concomitant drop in sensitivity. Indeed, 2D 13C-13C correlation spectra measured at 250 K show relatively low sensitivity and no clear intramolecular long-range cross peaks for cholesterol are observed. For example, the cholesterol Ch3 cross section shows no cross peaks with any other carbons, even though the two-bond Ch3-Ch1 and Ch3-Ch5 correlations are fully expected (Fig. S4a). Under these conditions, M2 signals show up relatively well, and a potential Ile Cα-Ch18 cross peak was observed. However, no other intermolecular cross peaks are detected. Therefore, at this moderate low temperature regime, the dynamic properties of M2 and cholesterol are not conducive for detecting intermolecular cross peaks. We thus turned to DNP-based 2D experiments at ~110 K, where both the protein and cholesterol are well immobilized in the membrane. We routinely obtained sensitivity enhancement factors of 28–42. The cholesterol ring 13C signals do not broaden noticeably from 255 K to 110 K. facilitating spectral assignment. In addition, the cholesterol 13C chemical shifts are unperturbed between the high and low temperatures, thus allowing assignments made at 273 K to be transferred to the DNP spectra at 110 K (Fig. S3, S5a). The methyl 13C signals exhibit the expected line broadening due to the well known methyl rotation at this temperature 70.
The 2D DNP 13C-13C correlation spectra are shown in Fig. 2–5 and Fig. S4b-S7. The 30 ms spectrum (Fig. 2) shows predominantly intramolecular correlations. In the 300 ms CHOL-only spectrum, Ch3-Ch17 and Ch3-Ch18 cross peaks are observed (Fig. 3a). These carbons reside on opposite ends of the sterol rings at distances of 8–9 Å 71, thus cross peaks for very long distances can be detected with DNP sensitivity enhancement. However, for the mixed M2T and CHOL sample, the 2D spectra shows significant congestion (Fig. 4a): for example, between 12 and 45 ppm, many CHOL resonances overlap with the 13C signals of the labeled Ile, Phe, Gly and Ala.
Figure 2.

30 ms 2D CC spectrum of IFGA-M2T and 1-13C CHOL in POPC/POPG membranes, showing full resonance assignment. The spectrum was measured at ~110 K under 13 kHz MAS with sensitivity enhancement by DNP. Significant spectral congestion is seen due to the large number of CHOL cross peaks in addition to the protein cross peaks. Cholesterol cross peaks are assigned in orange, protein cross peaks in black, and CHOL-M2 cross peaks in red.
Figure 5.

Regions of 2D 13C-13C correlation spectra of IFGA-M2T and 1-13C CHOL in POPC/POPG membranes, showing protein-CHOL cross peaks. All spectra were measured at ~110 K under DNP condition. Left column shows 30 ms spectra (purple) where only short-distance contacts are detected. Middle column (blue) shows 300 ms spectra where longer-range cross peaks are detected. Right column (black) shows 300 ms spectra of the protein-free control membrane with 1-13C CHOL. (a) Aromatic region of the 2D spectra, showing a Phe Cγ-Ch15 cross peak. (b) Ile regions of the 2D spectra, showing cross peaks between Ile sidechain carbons and CHOL Ch13, Ch17, Ch19 and Ch21.
Figure 3.

300 ms (a) 2D CC spectrum and (b) 2D DQF-CC spectrum of 1-13C CHOL in POPC/POPG membranes, showing full assignment of the intramolecular CHOL cross peaks. The spectra were measured at ~110 K with DNP.
Figure 4.

Simplification of 2D CC correlation spectra by DQF. All 2D spectra were measured at ~110 K with DNP. (a) 300 ms 2D CC spectrum of IFGA-M2T and 1-13C CHOL in POPC/POPG membranes measured without the DQF. Representative protein (blue) and CHOL (orange) 13C chemical shifts in the ω1 dimension are shaded. (b) 300 ms 2D DQF-CC spectrum of protein-free POPC/POPG membranes with 1-13C CHOL. The DQF suppresses all 13C signals without a directly bonded 13C in the ω1 dimension, leaving only Ch18, Ch13, and Ch17 signals. (c, d) 2D DQF-CC spectra of IFGA-M2T and 1-13C CHOL in POPC/POPG membranes measured with 13C spin diffusion mixing times of (c) 30 ms and (d) 300 ms. DQF significantly simplifies the ω1 dimension of the spectra, facilitating assignment of intermolecular protein-CHOL cross peaks.
To simplify the spectra and allow assignment, we took advantage of the uniform 13C-labeling of the selected protein residues, which contrasts with the mostly skip-labeled 1-13C CHOL, by adding a 13C-13C double-quantum filter (DQF) before t1 evolution 55. This 2D DQF-CC experiment removes most cholesterol peaks from the ω1 dimension except for the directly bonded Ch17, Ch13, and Ch18 carbons. As a result, roughly 75% cholesterol cross peaks are removed in the 2D DQF-CC spectrum (Fig. 3b), while those that remain can be easily assigned from the CHOL-only spectra with or without DQF (Fig. S5). In the protein-containing sample, the 30 ms DQF-CC spectrum emphasized the protein signals, and enabled assignment of the protein 13C peaks (Fig. 4c). A 2D dipolar INADEQUATE spectrum (Fig. S6) was also measured to confirm the assignment. The protein-cholesterol cross peaks of interest can thus be identified in long-mixing-time 2D DQF spectra (Fig. 4d, Fig. 5a, Fig. S7b, S8).
The 2D DQF-CC spectra shows multiple Ile cross peaks with cholesterol. Ile Cγ1 cross peaks with Ch17 (Fig. 5a) and with Ch24 (Fig. 2) are already observed at 30 ms mixing. The cross peak with Ch17 remains in the 300 ms DQF-CC spectrum, while the cross peak with Ch24 is below detection. With DQF, a 36.9 ppm peak in the ω1 dimension exhibits cross peaks with CHOL methyl carbons Ch19 (19.8 ppm) and Ch21 (20.6 ppm) and D ring carbons Ch13 and Ch17 (Fig. 5a). The 36.9 ppm chemical shift can in principle be assigned to either Ile Cβ or Phe Cβ; however, the dipolar INADQUATE spectrum (Fig. S6) shows cross peaks between Ile Cα (64.1 ppm) and Ch13 (43.1 ppm), thus supporting the assignment of these cross peaks in the DQF-CC spectra as Ile Cβ to cholesterol contacts.
For the aromatic region of the 2D spectra, the spectra without DQF already resolve several cross peaks between Phe sidechain and CHOL (Fig. 5b). Specifically, cross peaks between Phe Cδ/Cε and CHOL Ch3 and Ch9 are observed, thus establishing protein contacts with the sterol A and B rings. The Phe-C3 cross peak agrees with a previous report of Phe Cγ − Ch3 contact in DMPC bilayers by SSNMR at 262 K 72. These Phe-CHOL contacts were not observed in the DQF-CC spectra, due to the removal of Ch3 and Ch9 signals in the ω1 dimension and the low overall sensitivity in the aromatic spectral region.
Protein-cholesterol contacts from site-specifically labeled M2 peptide
Since the M2T sample labels all Ile, Phe, Gly and Ala residues, sequence-specific assignment cannot be made. We thus sought to provide some residue-specific information about the cholesterol-protein cross peaks using a G34, I39-labeled M2W(22–61) peptide. The M2Wamino acid sequence was taken from the chicken/Weybridge H7N7 strain, which contains a putative cholesterol-recognition motif 43. However, our recent study 27 has shown that this motif is not required for cholesterol binding. Indeed, the Udorn M2 sequence used in the previous experiments 27 and in the current IFGA-M2T sample both give significant protein-cholesterol 13C-19F dipolar couplings and 13C-13C cross peaks, despite the fact that Udorn M2 does not have a cationic residue thought to constitute part of the cholesterol-recognition motif. We reconstituted GI-M2W into a POPC/POPG membrane and a virus-mimetic (VMS) membrane, to investigate whether M2-cholesterol binding depends on the membrane environment.
Although only a single Ile was labeled in the peptide, the unfiltered 2D CC spectra remain sufficiently congested. We therefore conducted the same 2D DQF-CC experiments to measure I39-cholesterol cross peaks. Fig. 6 shows selected regions of the 2D spectra. The two membrane samples show very similar protein and cholesterol chemical shifts, with at most a 0.3 ppm deviation. I39 Cβ and Cγ2 cross peaks with cholesterol Ch13, Ch15, Ch17, and Ch21 are observed. These intermolecular contacts again involve the cholesterol D ring and the nearby methyl carbon, as was the case for the M2T sample (Fig. 5a). The similarity of the observed cholesterol cross peaks with the protein in both the cytoplasmic-containing M2 and the site-specifically labeled TM-AH peptide indicates that I39 is a major contributor of the Ile-cholesterol cross peaks observed in the M2T spectra, although it does not rule out the possibility that other Ile residues such as I42 may also interact with cholesterol. It also means that the M2-cholesterol binding is unaffected by the presence or absence of the cytoplasmic tail or the cholesterol-recognition sequence motif. Interestingly, the two lipid membranes do not give an identical set of M2-cholesterol cross peaks. For example, the I39 Cγ2 cross peak to Ch17 is detected only in the VMS membrane and not in the POPC/POPG membrane (Fig 4c, d). This difference suggests that the M2-cholesterol binding interactions may be slightly different between the POPC/POPG bilayer and the VMS membrane. This difference may be a result of changes in the TM helix tilt angle in the VMS membrane, whose rigidity and larger thickness can affect the TM helix orientation 73–75.
Figure 6.

Protein-cholesterol correlations observed in DNP 2D DQF-CC spectra of GI-M2W(22–61) peptide in two lipid membranes. (a) Regions of the 400 ms 2D DQF-CC spectrum of GI-M2W in POPC/POPG membranes. (b) Regions of the 400 ms 2D DQF-CC spectrum of GI-M2W in the VMS membranes. I39 cross peaks with several CHOL carbons are detected. (c) 1D cross sections extracted from the I39 Cβ chemical shift, showing similar cross peaks in the two lipid membranes. (d) 1D cross sections extracted from the CHOL Ch17 chemical shift, showing a protein-cholesterol cross peak in the VMS membrane but not the POPC/POPG membrane.
13C-13C distances constrain cholesterol docking onto the M2 protein
The many intermolecular 13C-13C cross peaks observed in these 2D spectra allow us to establish distance restraints between M2 and the sterol rings of cholesterol, which were previously inaccessible in commercially available terminally labeled cholesterol. These 13C-13C distance restraints were input into the HADDOCK program for molecular docking 58,59. In addition, the previously measured 13C-19F distances and cholesterol orientation angles 27 were also supplied as restraints. HADDOCK uses both ambiguous and unambiguous distance restraints (Table S3), of which the unambiguous restraints are always applied, while half of the ambiguous restraints were used for each of the 1000 initial runs. The ambiguous restraint list consisted of contacts obtained from the M2T sample for which sequence-specific assignment cannot be made. The three lipid-facing Ile residues of the M2 TM domain are I35, I39, and I42, where I39 and I42 reside at a similar depth while I35 lies in the middle of the bilayer. Thus, many of the observed Ile-cholesterol cross peaks were assigned to either I39 or I42. Of the four Phe residues of the amphipathic helix, only F47 and F54 face lipids. Based on our previous docking model, we had assigned the Phe–Ch3 contact observed by Ekanayake et al. to F47 27,72; nevertheless, we submitted this restraint at first as ambiguous. Preliminary docking simulations ruled out F48 and F54 as possible residues to contact cholesterol, since structures with these contacts strongly violated the submitted distance or angular restraints for cholesterol (Tables S2 and S3). Thus, we re-categorized the F47- cholesterol cross peaks as unambiguous, and the Phe/Ile Cβ cross peaks to cholesterol were assigned to I39 or I42 based on closer relative distances compared to F47. Additional unambiguous restraints included 13C-13C contacts determined using the G34, I39-M2W peptide, and distances from 13C-19F experiments and 13C-13C correlations with previously reported M2W and M2 Udorn peptides 27.
We used a combination of metrics to critique the 200 HADDOCK output structures, including the HADDOCK score, PRODIGY score, and residual sum of squares (RSS) of the cholesterol β and γ orientation angles. These metrics and the rankings for the top 10 structures according to RSS are listed in Table S5. The HADDOCK score is applied both to whole clusters and to individual structures. However, in a recent protein-ligand binding challenge, ranking by HADDOCK score alone produced less than 20% good matches 64. Consistently, we observed that similar M2-cholesterol docked structures can show large discrepancies in the HADDOCK scores. In comparison, the PRODIGY-LIGAND method identifies and categorizes atomic contacts to the ligand within 10.5 Å and assigns an energy value based on the atom pairs, resulting in more accurate structures and binding affinities 64. The PRODIGY scores for the top 100 PRODIGY-ranked structures differ only by ~0.3 kcal/mol. Thus, we used RSS as the primary metric for ranking the docking results and at the same time monitored the PRODIGY scores for red flags when comparing structures. Of the top 10 structures ranked by RSS (with the lowest RSS values), the two poorest ranking structures by PRODIGY show cholesterol ring orientations that differ substantially from the rest of the ensemble (Fig. S9). We consider these structures as not accurately representing M2-bound cholesterol, despite the good RSS rankings. The remaining eight structures show consistent docking poses for the bound cholesterol. For the subsequent discussion, we examine the top-ranking structure by RSS that also ranked favorably by PRODIGY.
The HADDOCK docked M2-cholesterol structural model is shown in Fig. 7, while the measured distances and orientations are given in Table S3 and S4. Two cholesterol molecules are docked onto the four-helix bundle (Fig. 7a), in accordance with the previous finding that cholesterol binds M2 with a stoichiometry of 2 per tetramer. However only one cholesterol molecule was used during the simulation 27. In the docked model, cholesterol is positioned on the outer face of the TM helix, with the sterol rings aligned with a groove formed by the lipid-facing sidechains from I39 to F47. The rough, methyl-studded β-face of cholesterol is flush with the TM helix, allowing the Ch18 and Ch19 methyl groups to form van der Waals contacts with the protein. Overall, Ile sidechains interact with the aliphatic tail and the D ring. Residue F47 is in contact with the A ring and likely forms CH-π interactions with the cholesterol hydrogens 76. Meanwhile L43 and L46, which were not labeled in this study, constitute the lipid-facing residues in the final turn of the TM helix and are located at the same depth as the cholesterol A and B rings. The position of L43 suggests the presence of CH-π interaction with the Ch5-Ch6 double bond as another means of stabilizing the bound complex. Other stabilizing factors may stem from interactions between the distal cholesterol tail and more deeply embedded sidechains such as I35, but the principal forces stabilizing the bound complex likely originates from the interactions between M2 and the sterol ring system.
Figure 7.

Cholesterol docking onto M2 (PDB code: 2L0J) using HADDOCK. Distance restraints obtained from the 2D CC spectra were used to constrain the binding site. Cholesterol carbons that exhibit cross peaks with the M2 protein are shown as spheres. Key Ile and Phe residues at the binding interface are indicated, along with the distances to the cholesterol carbons.
Discussion
Implication of the M2-cholesterol complex structure for membrane scission
The current data demonstrate the feasibility and utility of using biosynthetic 13C-labeled cholesterol to determine protein-cholesterol binding structures in lipid bilayers. Out of a total of 27 cholesterol carbons, 15 were 13C-enriched by 1-13C glucose, and among these 9 carbons, distributed on all four sterol rings as well as in the aliphatic tail, showed correlations peaks with the M2 protein (Table S3). Importantly, detecting these protein-cholesterol correlation peaks required a careful choice of the experimental temperature at which both the protein and cholesterol are immobilized in the lipid membrane. In the liquid-crystalline phase of the membrane, cholesterol is highly dynamic 30,77, giving well-resolved spectra but reducing the efficiency of 13C spin diffusion for measuring long-range correlation peaks. Cooling the membrane to moderate low temperatures of about 253 K improves 13C spin diffusion, but the spectra show heterogeneously broadened lines for the protein and cholesterol (Fig. S3), which reduce both the resolution and the sensitivity of the spectra. Use of DNP NMR experiments at cryogenic temperatures circumvent these problems and allow high-sensitivity detection of intermolecular correlation peaks that are inherently much weaker than one- or two-bond cross peaks.
The measured 13C-13C restraints support our previous finding that M2 binds cholesterol at an annular binding site on the lipid-facing face of the TM helix, as opposed to a buried or internal binding site. The cholesterol-binding interface consists mostly of Ile and Leu sidechains, with F47 at the polar head of cholesterol. The observed contacts between the Ile sidechains and cholesterol methyl groups suggest that the methyl-studded β face of cholesterol is in direct contact with M2, as is supported by the HADDOCK model (Fig. 7b). Sidechain-methyl interactions provide key stabilization of the complex in a manner similar to models of α-synuclein 78, the human type 3 somatostatin receptor 79, and the human β2-adrenergic receptor 19. The M2-cholesterol interface shows both methyl-methyl interactions and CH-π interactions, includes at least five TM residues, and spans a region that is larger than the 9 Å length of the cholesterol rings. These results indicate an extended TM binding interface with cholesterol. Interestingly, previous biochemical data suggest that perturbation of some of these TM residues may not abolish M2-cholesterol interaction. For example, single alanine mutation at L46 did not affect the formation of Udorn viral filaments 37, and triple alanine mutations of L46, F47, and F48 showed only a minor effect on viral infectivity 80. In comparison, a penta-Ala mutation of five aromatic residues in the AH abolished cholesterol-dependent membrane scission 36,81,82. This is consistent with our observation that the TM-only peptide, lacking the AH, does not bind cholesterol 27. Moreover, TM and AH conformation and dynamics of the penta-Ala mutant was shown to be insensitive to the presence or absence of cholesterol in the membrane 82. Therefore, our M2-cholesterol complex structure and the mutagenesis data together indicate that the amphipathic helix is necessary for keeping the TM helix in the correct conformation to interact with cholesterol. Furthermore, cholesterol binding to the neck of the TM and AH may control the insertion depth of the AH in the bilayer, as indicated by recent EPR studies of spin-labeled M2 in the presence and absence of cholesterol 38,82. This effect represents a mechanism for regulating AH-mediated membrane curvature induction through cholesterol binding to M2.
Binding cholesterol at two of the four vertices of the four-helix TM bundle provides a means to target M2 to the site of virus budding. This is consistent with M2’s preferential localization to the boundary of the raft and non-raft domains 4,36,42,83,84. A previously suggested binding site in the groove between two TM helices 72 does not agree with our measured restraints. In addition to cholesterol, other factors that have been proposed to drive M2 to the neck of the budding virus including M2’s preference for high curvature regions of the membrane 36,83 and for negative-curvature phosphatidylethanolamine 85, S-palmitoylation at Cys50 86, and M2’s interaction with other influenza matrix proteins 87,88. Cholesterol is also known to promote the formation of lipid stalks in membrane fusion 14, thus the M2-cholesterol interaction represents a confluence of several physiological mechanisms to form the high-curvature neck of the budding virus to induce the eventual membrane scission.
High-sensitivity studies of cholesterol-protein interactions by biosynthetic 13C labeling of cholesterol
In this study, we used 1-13C glucose to preferentially label methyl carbons in cholesterol, since we hypothesized that they are an important element for stabilizing M2-cholesterol binding. However, the similar 13C chemical shifts between the cholesterol and protein methyl signals and the line broadening at the ~110 K temperature used for the DNP experiments complicates resonance assignment. Use of 2-13C glucose labeled cholesterol, termed 2-13C CHOL, is a promising alternative 28. 2-13C CHOL contains no 13C chemical shifts upfield of 20 ppm, and only one 13C resonance between 20 and 25 ppm (Fig. S2b), which is a region densely populated by Leu, Val and Ile methyl 13C signals. Thus, 2-13C CHOL will be especially useful for measuring protein methyl-cholesterol cross peaks. Although the 13C chemical shift dispersion of 2-13C CHOL is smaller than that of 1-13C CHOL, only 12 carbons are 13C-enriched, thus resonance overlap should be reduced. 2-13C CHOL can be prepared both from 2-13C glucose and 1-13C acetate. Likewise, it has been shown that 2-13C-acetate, through the mevalonate pathway, produces the same 1-13C CHOL labeling pattern as 1-13C glucose, but with higher yields per mass of 13C starting material and greater than 80% 13C incorporation 30. Thus, acetate labeling has the benefits of higher sensitivity, increased statistical probability for protein-cholesterol cross peaks, and increased efficiency for spectral editing techniques such as the DQF experiment used here. The higher 13C labeling level also gives more freedom in preparing membrane samples with appropriate compositions. For the membrane samples used in this study, the mass ratio of 1-13C CHOL is 1 mg per 20 mg total dry mass, including lipids and protein. Given the 30 μL volume of the 3.2 mm MAS rotors, it is impractical to add more sample for boosting sensitivity. The membrane lipid molar ratio is dictated by the need for sufficient lipids to solvate the protein and the restriction to less than ~17 mol% cholesterol for studying curvature-inducing M2-cholesterol interactions 36. Thus, with the limited mass of cholesterol, doubling the 13C enrichment level will facilitate NMR studies of protein-cholesterol binding.
This work focuses on the use of biosynthetic 13C-cholesterol in conjunction with NMR to determine the M2-cholesterol binding interface structure. The 13C-labeled cholesterol involves natural isotopes without the potential for structural perturbation, which makes it a superior approach to fluorescent labels, spin-labeled cholesterol derivatives, and even fluorinated cholesterol. While these non-natural molecules can be used to detect much longer distances to map protein-cholesterol binding sites, they have the potential to behave non-natively and perturb protein structure 89. 13C-labeled cholesterol can also be used to study cholesterol dynamics in the membrane and when associated with proteins 3,30,77. Beyond NMR, biosynthetically labeled cholesterol finds various other uses. For example, it has been exploited for mass spectrometric studies of membrane domain formation and lateral organization 68, and perdeuterated cholesterol has been used for Raman spectroscopy and neutron scattering studies of membrane physical properties and intracellular cholesterol storage 90–92. Therefore, biosynthetic cholesterol labeling opens many avenues for investigating biologically important interaction of this ubiquitous component of mammalian membranes with proteins and lipids.
Supplementary Material
Acknowledgments
The authors thank Prof. Howard Riezman and Brigitte Bernadets for providing S. cerevisiae strain RH6829 and recommendations for storage and production culture composition. M. R. E. thanks V. S. Mandala and Dr. S. Y. Liao for assistance in the expression of M2T, and Prof. Alexandre Bonvin for advice about using HADDOCK. This work is supported by NIH grant GM088204 to M. H.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Supporting Information Available
- Experimental parameters for 2D 13C correlation spectra
- Summary of observed M2-cholesterol contacts and distance and angular restraints used as input to HADDOCK
- Analysis and ranking of the top HADDOCK outputs
- Analytical data for preparation of 1-13C CHOL, including HPLC and NMR.
- Temperature-dependent 1D and 2D 13C spectra of membranes containing 1-13C CHOL and M2 and 1-13C CHOL
- Fully assigned 2D spectra of samples containing IFGA-M2T and 1-13C CHOL
- Extracted 1D cross sections showing M2 – cholesterol cross peaks
- Ensemble of 10 best docked M2-cholesterol complexes
- Parameter file for HADDOCK docking
This material is available free of charge via the Internet at http://pubs.acs.org
References
- (1).Gennis RB Biomembranes: Molecular Structure and Function; Springer: New York, 1989. [Google Scholar]
- (2).Dufourc EJ; Smith IC, A detailed analysis of the motions of cholesterol in biological membranes by 2H-NMR relaxation, Chem. Phys. Lipids 1986, 41, 123–135. [DOI] [PubMed] [Google Scholar]
- (3).Marsan MP; Muller I; Ramos C; Rodriguez F; Dufourc EJ; Czaplicki J; Milon A, Cholesterol orientation and dynamics in dimyristoylphosphatidylcholine bilayers: a solid state deuterium NMR analysis, Biophys. J 1999, 76, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gerl MJ; Sampaio JL; Urban S; Kalvodova L; Verbavatz JM; Binnington B; Lindemann D; Lingwood CA; Shevchenko A; Schroeder C; Simons K, Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane, J. Cell. Biol 2012, 196, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Brügger B; Glass B; Haberkant P; Leibrecht I; Wieland FT; Kräusslich HG, The HIV lipidome: a raft with an unusual composition, Proc. Natl. Acad. Sci. U. S. A 2006, 103, 2641–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Brown DA; London E, Functions of lipid rafts in biological membranes, Annu. Rev. Cell Dev. Biology 1998, 14, 111–136 [DOI] [PubMed] [Google Scholar]
- (7).Wu H; Wang C; Gregory KJ; Han GW; Cho HP; Xia Y; Niswender CM; Katritch V; Meiler J; Cherezov V; Conn PJ; Stevens RC, Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator, Science 2014, 344, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Katritch V; Cherezov V; Stevens RC, Structure-function of the G protein-coupled receptor superfamily, Annu. Rev. Pharmacol. Toxicol 2013, 53, 531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zocher M; Zhang C; Rasmussen SG; Kobilka BK; Muller DJ, Cholesterol increases kinetic, energetic, and mechanical stability of the human beta2-adrenergic receptor, Proc. Natl. Acad. Sci. U. S. A 2012, 109, E3463–E3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ruprecht JJ; Mielke T; Vogel R; Villa C; Schertler GF, Electron crystallography reveals the structure of metarhodopsin I, EMBO J. 2004, 23, 3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mitchell DC; Straume M; Miller JL; Litman BJ, Modulation of metarhodopsin formation by cholesterol-induced ordering of bilayer lipids, Biochemistry 1990, 29, 9143–9149. [DOI] [PubMed] [Google Scholar]
- (12).Barrett PJ; Song YL; Van Horn WD; Hustedt EJ; Schafer JM; Hadziselimovic A; Beel AJ; Sanders CR, The Amyloid Precursor Protein Has a Flexible Transmembrane Domain and Binds Cholesterol, Science 2012, 336, 1168–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Beel AJ; Sakakura M; Barrett PJ; Sanders CR, Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships?, Biochim. Biophys. Acta 2010, 1801, 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yang ST; Kreutzberger AJB; Lee J; Kiessling V; Tamm LK, The role of cholesterol in membrane fusion, Chem. Phys. Lipids 2016, 199, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Qiang W; Sun Y; Weliky DP, A strong correlation between fusogenicity and membrane insertion depth of the HIV fusion peptide, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 15314–15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yang ST; Kiessling V; Simmons JA; White JM; Tamm LK, HIV gp41-mediated membrane fusion occurs at edges of cholesterol-rich lipid domains, Nat. Chem. Biol 2015, 11, 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Howell SC; Mittal R; Huang L; Travis B; Breyer RM; Sanders CR, CHOBIMALT: a cholesterol-based detergent, Biochemistry 2010, 49, 9572–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Caffrey M; Cherezov V, Crystallizing membrane proteins using lipidic mesophases, Nat. Protoc 2009, 4, 706–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hanson MA; Cherezov V; Griffith MT; Roth CB; Jaakola VP; Chien EYT; Velasquez J; Kuhn P; Stevens RC, A specific cholesterol binding site is established by the 2.8 angstrom structure of the human beta(2)-adrenergic receptor, Structure 2008, 16, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Mandala VS; Williams JK; Hong M, Structure and Dynamics of Membrane Proteins from Solid-State NMR, Ann. Rev. Biophys 2018, 47, 201–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hong M; Zhang Y; Hu F, Membrane Protein Structure and Dynamics from NMR Spectroscopy, Annu. Rev. Phys. Chem 2012, 63, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hong M; Su Y, Structure and dynamics of cationic membrane peptides and proteins: Insights from solid-state NMR, Protein Sci. 2011, 20, 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Roos M; Wang T; Shcherbakov AA; Hong M, Fast Magic-Angle-Spinning 19F Spin Exchange NMR for Determining Nanometer 19F–19F Distances in Proteins and Phamaceutical Compounds, J. Phys. Chem. B 2018, 122, 2900–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shcherbakov AA; Hong M, Rapid measurement of long-range distances in proteins by multidimensional 13C-19F REDOR NMR under fast magic-angle spinning, J. Biomol. NMR 2018, 71, 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jaipuria G; Leonov A; Giller K; Vasa SK; Jaremko L; Jaremko M; Linser R; Becker S; Zweckstetter M, Cholesterol-mediated allosteric regulation of the mitochondrial translocator protein structure, Nat. Commun 2017, 8, 14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jaipuria G; Ukmar-Godec T; Zweckstetter M, Challenges and approaches to understand cholesterol-binding impact on membrane protein function: an NMR view, Cell. Mol. Life Sci 2018, 75, 2137–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Elkins MR; Williams JK; Gelenter MD; Dai P; Kwon B; Sergeyev IV; Pentelute BL; Hong M, Cholesterol-binding site of the influenza M2 protein in lipid bilayers from solid-state NMR, Proc. Natl. Acad. Sci. U. S. A 2017, 114, 12946–12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shivapurkar R; Souza CM; Jeannerat D; Riezman H, An efficient method for the production of isotopically enriched cholesterol for NMR, J. Lipid Res 2011, 52, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Souza CM; Schwabe TM; Pichler H; Ploier B; Leitner E; Guan XL; Wenk MR; Riezman I; Riezman H, A stable yeast strain efficiently producing cholesterol instead of ergosterol is functional for tryptophan uptake, but not weak organic acid resistance, Metab. Eng 2011, 13, 555–569. [DOI] [PubMed] [Google Scholar]
- (30).Della Ripa LA; Petros ZA; Cioffi AG; Piehl DW; Courtney JM; Burke MD; Rienstra CM, Solid-State NMR of highly (13)C-enriched cholesterol in lipid bilayers, Methods 2018, 138–139, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Holsinger LJ; Lamb RA, Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds, Virology 1991, 183, 32–43. [DOI] [PubMed] [Google Scholar]
- (32).Hong M; DeGrado WF, Structural basis for proton conduction and inhibition by the influenza M2 protein, Protein Sci. 2012, 21, 1620–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cady SD; Luo WB; Hu F; Hong M, Structure and function of the influenza M2 proton channel, Biochemistry 2009, 48, 7356–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Pinto LH; Holsinger LJ; Lamb RA, Influenza-Virus M2 Protein Has Ion Channel Activity, Cell 1992, 69, 517–528. [DOI] [PubMed] [Google Scholar]
- (35).Pinto LH; Lamb RA, The M2 proton channels of influenza A and B viruses, J. Biol. Chem 2006, 281, 8997–9000. [DOI] [PubMed] [Google Scholar]
- (36).Rossman JS; Jing XH; Leser GP; Lamb RA, Influenza Virus M2 Protein Mediates ESCRT-Independent Membrane Scission, Cell 2010, 142, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rossman JS; Jing XH; Leser GP; Balannik V; Pinto LH; Lamb RA, Influenza Virus M2 Ion Channel Protein Is Necessary for Filamentous Virion Formation, J. Virol 2010, 84, 5078–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kim SS; Upshur MA; Saotome K; Sahu ID; McCarrick RM; Feix JB; Lorigan GA; Howard KP, Cholesterol-Dependent Conformational Exchange of the C-Terminal Domain of the Influenza A M2 Protein, Biochemistry 2015, 54, 7157–7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Nguyen PA; Soto CS; Polishchuk A; Caputo GA; Tatko CD; Ma C; Ohigashi Y; Pinto LH; DeGrado WF; Howard KP, pH-induced conformational change of the influenza M2 protein C-terminal domain, Biochemistry 2008, 47, 9934–9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sharma M; Yi MG; Dong H; Qin HJ; Peterson E; Busath DD; Zhou HX; Cross TA, Insight into the Mechanism of the Influenza A Proton Channel from a Structure in a Lipid Bilayer, Science 2010, 330, 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Schroeder C; Heider H; Moncke-Buchner E; Lin TI, The influenza virus ion channel and maturation cofactor M2 is a cholesterol-binding protein, Eur. Biophys. J 2005, 34, 52–66. [DOI] [PubMed] [Google Scholar]
- (42).Thaa B; Levental I; Herrmann A; Veit M, Intrinsic membrane association of the cytoplasmic tail of influenza virus M2 protein and lateral membrane sorting regulated by cholesterol binding and palmitoylation, Biochem. J 2011, 437, 389–397. [DOI] [PubMed] [Google Scholar]
- (43).Li H; Papadopoulos V, Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus patterns, Endocrinology 1998, 139, 4991–4997. [DOI] [PubMed] [Google Scholar]
- (44).Epand RM, Cholesterol and the interaction of proteins with membrane domains, Prog. Lipid Res 2006, 45, 279–294. [DOI] [PubMed] [Google Scholar]
- (45).Treco DA; Lundblad V, Preparation of Yeast Media, Curr. Protoc. Mol. Biol 1993, 23, 13.11.11–13.11.17 [DOI] [PubMed] [Google Scholar]
- (46).Treco DA; Winston F, Growth and Manipulation of Yeast, Curr. Protoc. Mol. Biol 2008, 82, 13.12.11–13.12.12. [DOI] [PubMed] [Google Scholar]
- (47).Guan XL; Riezman I; Wenk MR; Riezman H, Yeast lipid analysis and quantification by mass spectrometry, Methods. Enzymol 2010, 470, 369–391. [DOI] [PubMed] [Google Scholar]
- (48).Liao SY; Yang Y; Tietze D; Hong M, The Influenza M2 Cytoplasmic Tail Changes the Proton-Exchange Equilibria and the Backbone Conformation of the Transmembrane Histidine Residue to Facilitate Proton Conduction, J. Am. Chem. Soc 2015, 137, 6067–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liao SY; Fritzsching KJ; Hong M, Conformational analysis of the full-length M2 protein of the influenza A virus using solid-state NMR, Protein Sci. 2013, 22, 1623–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hong M, Resonance Assignment of 13C/15N Labeled Proteins by Two- and Three-Dimensional Magic-Angle-Spinning NMR, J. Biomol. NMR 1999, 15, 1–14. [DOI] [PubMed] [Google Scholar]
- (51).Hong M; Jakes K, Selective and Extensive 13C Labeling of a Membrane Protein for Solid-State NMR Investigation, J. Biomol. NMR 1999, 14, 71–74. [DOI] [PubMed] [Google Scholar]
- (52).Simon MD; Heider PL; Adamo A; Vinogradov AA; Mong SK; Li X; Berger T; Policarpo RL; Zhang C; Zou Y; Liao X; Spokoyny AM; Jensen KF; Pentelute BL, Rapid flow-based peptide synthesis, ChemBioChem 2014, 15, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kwon B; Lee M; Waring AJ; Hong M, Oligomeric Structure and Three-Dimensional Fold of the HIV gp41 Membrane-Proximal External Region and Transmembrane Domain in Phospholipid Bilayers, J. Am. Chem. Soc 2018, 140, 8246–8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Hou GJ; Yan S; Trebosc J; Amoureux JP; Polenova T, Broadband homonuclear correlation spectroscopy driven by combined R2(n)(v) sequences under fast magic angle spinning for NMR structural analysis of organic and biological solids, J. Magn. Reson 2013, 232, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lopez JJ; Kaiser C; Shastri S; Glaubitz C, Double quantum filtering homonuclear MAS NMR correlation spectra: a tool for membrane protein studies, J. Biomol. NMR 2008, 41, 97–104. [DOI] [PubMed] [Google Scholar]
- (56).Hohwy M; Rienstra CM; Jaroniec CP; Griffin RG, Fivefold symmetric homonuclear dipolar recoupling in rotating solids: Application to double quantum spectroscopy, J. Chem. Phys 1999, 110, 7983–7992. [Google Scholar]
- (57).Hong M, Solid-state dipolar INADEQUATE NMR spectroscopy with a large double-quantum spectral width, J. Magn. Reson 1999, 136, 86–91. [DOI] [PubMed] [Google Scholar]
- (58).Dominguez C; Boelens R; Bonvin AMJJ, HADDOCK: a protein-protein docking approach based on biochemical or biophysical information, J. Am. Chem. Soc 2003, 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- (59).van Zundert GCP; Rodrigues JPGLM; Trellet M; Schmitz C; Kastritis PL; Karaca E; Melquiond ASJ; van Dijk M; de Vries SJ; Bonvin AMJJ, The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes, J. Mol. Biol 2016, 428, 720–725. [DOI] [PubMed] [Google Scholar]
- (60).de Vries SJ; van Dijk M; Bonvin AMJJ, The HADDOCK web server for data-driven biomolecular docking, Nat. Protoc 2010, 5, 883–897. [DOI] [PubMed] [Google Scholar]
- (61).Meiler J; Blomberg N; Nilges M; Griesinger C, A new approach for applying residual dipolar couplings as restraints in structure elucidation, J. Biomol. NMR 2000, 16, 245–252. [DOI] [PubMed] [Google Scholar]
- (62).Dufourc EJ; Parish EJ; Chitrakorn S; Smith ICP, Structural and Dynamical Details of Cholesterol Lipid Interaction as Revealed by Deuterium NMR, Biochemistry 1984, 23, 6062–6071. [Google Scholar]
- (63).Irwin JJ; Sterling T; Mysinger MM; Bolstad ES; Coleman RG, ZINC: a free tool to discover chemistry for biology, J. Chem. Inf. Model 2012, 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Kurkcuoglu Z; Koukos PI; Citro N; Trellet ME; Rodrigues J; Moreira IS; Roel-Touris J; Melquiond ASJ; Geng C; Schaarschmidt J; Xue LC; Vangone A; Bonvin A, Performance of HADDOCK and a simple contact-based protein-ligand binding affinity predictor in the D3R Grand Challenge 2, J. Comput. Aided Mol. Des 2018, 32, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Xue LC; Rodrigues JP; Kastritis PL; Bonvin AMJJ; Vangone A, PRODIGY: a web server for predicting the binding affinity of protein-protein complexes, Bioinformatics 2016, 32, 3676–3678. [DOI] [PubMed] [Google Scholar]
- (66).Vangone A; Bonvin AMJJ, Contacts-based prediction of binding affinity in protein-protein complexes, eLife 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Vangone A; Schaarschmidt J; Koukos P; Geng C; Citro N; Trellet ME; Xue LC; Bonvin AMJJ, Large-scale prediction of binding affinity in protein small-ligand complexes: the PRODIGY-LIG web server, Bioinformatics 2018. DOI: 10.1093/bioinformatics/bty816 [DOI] [PubMed] [Google Scholar]
- (68).Lozano MM; Hovis JS; Moss FR 3rd; Boxer SG, Dynamic Reorganization and Correlation among Lipid Raft Components, J. Am. Chem. Soc 2016, 138, 9996–10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Kalinowski H-O; Berger S; Braun S 13C-NMR-Spektroskopie; Thieme-Verlag: Stuttgart, 1984. [Google Scholar]
- (70).Ni QZ; Markhasin E; Can TV; Corzilius B; Tan KO; Barnes AB; Daviso E; Su Y; Herzfeld J; Griffin RG, Peptide and Protein Dynamics and Low-Temperature/DNP Magic Angle Spinning NMR, J. Phys. Chem. B 2017, 121, 4997–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Mcmullan RK; Koetzle TF; Fronckowiak MD, Structure of [20-Ch3],[20-Cd3]-Methylpregnene-3,20-Diol Methanolate from Neutron-Diffraction at 123-K, Acta Crystallogr. C 1992, 48, 1509–1512. [Google Scholar]
- (72).Ekanayake EV; Fu R; Cross TA, Structural Influences: Cholesterol, Drug, and Proton Binding to Full-Length Influenza A M2 Protein, Biophys. J 2016, 110, 1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Luo W; Cady SD; Hong M, Immobilization of the influenza A M2 transmembrane peptide in virus envelope-mimetic lipid membranes: a solid-state NMR investigation, Biochemistry 2009, 48, 6361–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Duong-Ly KC; Nanda V; DeGrado WF; Howard KP, The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment, Protein Sci. 2005, 14, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Cady SD; Goodman C; Tatko C; DeGrado WF; Hong M, Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: a 2H, 13C, and 15N solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle, J. Am. Chem. Soc 2007, 129, 5719–5729. [DOI] [PubMed] [Google Scholar]
- (76).Nishio M; Umezawa Y; Hirota M; Takeuchi Y, The Ch/Pi Interaction - Significance in Molecular Recognition, Tetrahedron 1995, 51, 8665–8701. [Google Scholar]
- (77).Ferreira TM; Coreta-Gomes F; Ollila OH; Moreno MJ; Vaz WL; Topgaard D, Cholesterol and POPC segmental order parameters in lipid membranes: solid state 1H-13C NMR and MD simulation studies, Phys. Chem. Chem. Phys 2013, 15, 1976–1989. [DOI] [PubMed] [Google Scholar]
- (78).Fantini J; Carlus D; Yahi N, The fusogenic tilted peptide (67–78) of alpha-synuclein is a cholesterol binding domain, Biochim. Biophys. Acta 2011, 1808, 2343–2351. [DOI] [PubMed] [Google Scholar]
- (79).Fantini J; Barrantes FJ, How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains, Front. Physiol 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Stewart SM; Pekosz A, Mutations in the membrane-proximal region of the influenza A virus M2 protein cytoplasmic tail have modest effects on virus replication, J. Virol 2011, 85, 12179–12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Martyna A; Bahsoun B; Badham MD; Srinivasan S; Howard MJ; Rossman JS, Membrane remodeling by the M2 amphipathic helix drives influenza virus membrane scission, Sci. Rep 2017, 7, 44695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Herneisen AL; Sahu ID; McCarrick RM; Feix JB; Lorigan GA; Howard KP, A Budding-Defective M2 Mutant Exhibits Reduced Membrane Interaction, Insensitivity to Cholesterol, and Perturbed Interdomain Coupling, Biochemistry 2017, 56, 5955–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Madsen JJ; MGrime JMA; Rossman JS; Voth GA, Entropic Forces Drive Clustering and Spatial Localization of Influenza A M2 During Viral Budding, Proc. Natl. Acad. Sci. U. S. A 2018, 115, E8595–E8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Ho CS; Khadka NK; She F; Cai J; Pan J, Influenza M2 Transmembrane Domain Senses Membrane Heterogeneity and Enhances Membrane Curvature, Langmuir 2016, 32, 6730–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Schmidt NW; Mishra A; Wang J; DeGrado WF; Wong GC, Influenza virus A M2 protein generates negative Gaussian membrane curvature necessary for budding and scission, J. Am. Chem. Soc 2013, 135, 13710–13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Thaa B; Siche S; Herrmann A; Veit M, Acylation and cholesterol binding are not required for targeting of influenza A virus M2 protein to the hemagglutinin-defined budozone, FEBS Lett. 2014, 588, 1031–1036. [DOI] [PubMed] [Google Scholar]
- (87).Chen BJ; Leser GP; Jackson D; Lamb RA, The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding, J. Virol 2008, 82, 10059–10070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Leser GP; Lamb RA, Lateral Organization of Influenza Virus Proteins in the Budozone Region of the Plasma Membrane, J. Virol 2017, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Scheidt HA; Muller P; Herrmann A; Huster D, The potential of fluorescent and spin-labeled steroid analogs to mimic natural cholesterol, J. Biol. Chem 2003, 278, 45563–45569. [DOI] [PubMed] [Google Scholar]
- (90).Nickels JD; Cheng X; Mostofian B; Stanley C; Lindner B; Heberle FA; Perticaroli S; Feygenson M; Egami T; Standaert RF; Smith JC; Myles DA; Ohl M; Katsaras J, Mechanical Properties of Nanoscopic Lipid Domains, J. Am. Chem. Soc 2015, 137, 15772–15780. [DOI] [PubMed] [Google Scholar]
- (91).Marquardt D; Heberle FA; Greathouse DV; Koeppe RE; Standaert RF; Van Oosten BJ; Harroun TA; Kinnun JJ; Williams JA; Wassall SR; Katsaras J, Lipid bilayer thickness determines cholesterol’s location in model membranes, Soft Matter 2016, 12, 9417–9428. [DOI] [PubMed] [Google Scholar]
- (92).Alfonso-Garcia A; Pfisterer SG; Riezman H; Ikonen E; Potma EO, D38-cholesterol as a Raman active probe for imaging intracellular cholesterol storage, J. Biomed. Opt 2016, 21, 61003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.