

Abstract
Background:
Elevated uptake of the [18F]AV-1451 tau-PET ligand has been observed cross-sectionally in subjects with progressive supranuclear palsy (PSP). However, it is unknown how the ligand performs longitudinally in PSP. We aimed to determine how regional measures of change on [18F]AV-1451 PET perform as longitudinal biomarkers of PSP compared to the more established biomarker of rate of midbrain atrophy.
Methods:
Sixteen subjects with PSP underwent two serial [18F]AV-1451 tau-PET and 3 Tesla MRI over 12 months, and were age and gender-matched to 39 healthy controls with longitudinal [18F]AV-1451 PET. Median [18F]AV-1451 uptake was calculated for each scan for regions-of-interest across the brain and divided by uptake in cerebellar crus to create standard uptake value ratios. Midbrain volume on MRI was also calculated for each scan. Sample sizes required to power placebo-controlled treatment trials were calculated.
Results:
Rate of midbrain atrophy was significantly increased in PSP compared to controls. [18F]AV-1451 regional change measures were significantly increased in PSP compared to controls in the pallidum, precentral cortex, dentate nucleus of the cerebellum and midbrain. Change over time in the PSP Rating Scale correlated with change in midbrain volume but did not correlate with change in the [18F]AV-1451 measures. Smallest sample size estimates were obtained with rate of midbrain atrophy, followed by the PSP Rating Scale, with both outperforming [18F]AV-1451 measures.
Conclusions:
[18F]AV-1451 tau-PET measures increase over time in subjects with PSP, but longitudinal [18F]AV-1451 measures may not perform as well as rate of midbrain atrophy as biomarkers for PSP clinical trials.
Keywords: Tau, positron emission tomography, Richardson’s syndrome, serial, rates
Introduction
Progressive supranuclear palsy (PSP) is a devastating neurodegenerative disease. Patients with PSP typically have difficulty with gait and posture, frequently fall, and have vertical supranuclear gaze palsy on neurological examination1, 2. At autopsy, the vast majority of patients clinically diagnosed with PSP have deposition of 4-repeat tau3–5; hence, tau is a major target for future treatments of PSP, and PSP subjects are being targeted for clinical treatment trials for tau-based therapies. Hence, biomarkers that track disease progression in PSP are extremely valuable. At present, the most studied and validated biomarkers come from MRI6, with rate of midbrain atrophy confirmed as a reliable biomarker in PSP, which allows small sample size estimates for clinical treatment trials7–9. However, MRI only provides a surrogate biomarker of underlying pathology. No disease-specific biomarker currently exists that can track progression of tau pathology during life that could be used as an outcome measure in treatment trials.
Positron emission tomography (PET) imaging using recently developed tau ligands, such as [18F]AV-145110, 11, are therefore potentially important and may provide disease-specific in vivo biomarkers. Cross-sectional studies in PSP patients have demonstrated elevated uptake of [18F]AV-1451, predominantly in subcortical regions, including the cerebellar dentate nucleus, midbrain, pallidum, thalamus, subthalamic nucleus and striatum compared to controls, although it is unclear whether [18F]AV-1451 uptake in these patients really reflects 4R tau pathology12–15. The majority of groups, with one exception16, have also found that [18F]AV-1451 uptake in PSP does not correlate with clinical disease severity16–21.
[18F]AV-1451 tau-PET could provide a biologically plausible biomarker of PSP disease progression. Obviously, it would be important to determine whether [18F]AV-1451 tau-PET signal changes over time in PSP and correlates with clinical disease progression. Currently, it is unknown how [18F]AV-1451 performs longitudinally in this disorder. Therefore, we aimed to assess longitudinal tau-PET using the [18F]AV-1451 ligand in PSP subjects who had undergone serial scanning and compare the performance of [18F]AV-1451 PET to the MRI-based biomarker of rate of midbrain atrophy. We hypothesized that [18F]AV-1451 uptake in subcortical regions would increase over time in PSP, and that [18F]AV-1451 would perform comparably to midbrain atrophy in differentiating PSP subjects from controls.
Methods
Subjects
Sixteen subjects that fulfilled clinical criteria for probable PSP2 underwent two serial [18F]AV-1451 PET scans and accompanying 3T MRI with follow-up intervals of approximately 12 months. All subjects had been identified from the Department of Neurology, Mayo Clinic, and consecutively recruited into an NIH-funded prospective longitudinal PSP study by a neurodegenerative specialist and PSP expert (KAJ). All baseline evaluations occurred between February 18th 2015 and December 6th 2016 and follow-ups were completed between March 1st 2016 and December 5th 2017. Subjects were included in the study if they met criteria for probable PSP, had symptoms for ≤5 years and were able to ambulate at least short distances independently. Of the 16 subjects, 12 fulfilled clinical criteria for Level 1 PSP-Richardson’s syndrome2, one met criteria for PSP-parkinsonism2, two met criteria for PSP-speech/language, and one met criteria for PSP-progressive gait freezing. One subject has died and had pathologically-confirmed PSP.
All subjects underwent clinical and neurological examinations and completed standardized and validated testing, including the PSP Rating Scale22 as a measure of clinical disease severity, the PSP Saccadic Impairment Scale23 as a measure of eye movement abnormalities, the Montreal Cognitive Assessment (MoCA)24 as a measure of global cognitive function, and the Frontal Assessment Battery25 as a measure of executive function. Demographic and clinical features of the nine PSP subjects are shown in Table 1.
Table 1:
Demographic and clinical features of the cohort
| PSP (N = 16) | |
|---|---|
| Demographic data | |
| No. Female (%) | 6 (40%) |
| Age at baseline, years | 68 (6) [59, 83] |
| Age at onset, years | 63 (6) [54, 80] |
| Scan interval, years | 0.98 (0.10) [0.8, 1.2] |
| Disease duration at baseline, years | 4.4 (1.7) [2.3,8.9] |
| Clinical data | |
| PSP Rating Scale | |
| Baseline | 36 (14) [9–60] |
| Change/yr | 11.7 (6.5) [1.9, 23.2] |
| PSP Rating Scale - gait/midline | |
| Baseline | 9 (6) [0–15] |
| Change/yr | 3.1 (2.4) [0, 9.1] |
| PSP Saccadic Impairment Scale | |
| Baseline | 2.6 (0.96) [1, 4] |
| Change/yr | 0.7 (0.75) [0, 2] |
| Montreal Cognitive Assessment Battery | |
| Baseline | 26 (3) [19–30] |
| Change/yr | −1.6 (2.8) [3.5, −5.9] |
| Frontal Assessment Battery | |
| Baseline | 15 (2) [12, 18] |
| Change/yr | −0.8 (1.4) [1.2, −4] |
Data is shown as mean (standard deviation) [min, max]
The 16 PSP subjects were matched by age to 39 healthy control subjects that had undergone serial [18F]AV-1451 imaging in the Mayo Clinic Study of Aging. The control cohort consisted of 18 (46%) female subjects with mean education of 15 years (standard deviation=2), mean age at tau-PET of 68 years (8), mean follow-up interval of 1.29 (0.13) years and mean MoCA of 26 (2). All control subjects were negative for beta-amyloid deposition on Pittsburgh Compound B PET26. The study was approved by the Mayo IRB. All subjects consented to research.
Neuroimaging analyses
Tau-PET imaging was performed using the [18F]AV-1451 ligand and a PET/CT scanner (GE Healthcare, Milwaukee, Wisconsin). An intravenous bolus injection of approximately 370MBq (range 333–407 MBq) of [18F]AV-1451 was administered, followed by a 20 minute PET acquisition performed 80 minutes after injection. Partial volume correction (PVC) of PET data was performed using a two-compartment model27. All subjects had a 3T MPRAGE sequence performed at the time of the tau-PET (TR/TE/T1, 2300/3/900 ms; flip angle 8°, 26-cm field of view; 256 × 256 in-plane matrix with a phase FOV of 0.94, slice thickness of 1.2 mm, in-plane resolution 1mm).
MPRAGE images were each segmented and corrected for intensity inhomogeneity using SPM12 Unified Segmentation28 with modifications and tissue priors from the Mayo Clinic Adult Lifespan Template (MCALT) (https://www.nitrc.org/projects/mcalt/)29. Midbrain volume was measured for each scan by propagating a template-drawn midbrain volume mask into the native space of each patients scan and summing segmented per-voxel gray and white matter probabilities30. The template-drawn midbrain volume mask was traced using Analyze software (Biomedical Imaging Resource, Mayo Clinic, Rochester, MN) according to previously published guidelines31.
[18F]AV-1451 images were each co-registered and resampled to the subjects corresponding MPRAGE MRI using SPM12 6-degree-of-freedom rigid body registration and B-spline interpolation. All regions of interest were defined in MCALT space. The dentate nucleus of the cerebellum was defined as previously described20 on MCALT, and voxels containing the dentate nucleus of the cerebellum were removed from all other cerebellar regions-of-interest. The midbrain volume mask described above31 was transformed to MCALT. The Deep Brain Stimulation Intrinsic Template Atlas (DISTAL)32 was transformed into MCALT space and used to define regions for the subthalamic nucleus, red nucleus and substantia nigra. All other regions of interest were defined using the MCALT_ADIR122 atlas. For each subject MPRAGE, a nonlinear mapping to MCALT was created using Advanced Normaliation Tools (ANTs)33, and this mapping was used to automatically localize all regions on the native MPRAGE image using GenericLabel interpolation. Each region was then masked to include only gray or white matter-segmented voxels, and PET regional values were measured as the median image intensity across these voxels. Regional median values were divided by the median uptake in cerebellar crus grey matter to create standard uptake ratios (SUVRs). The crus region was selected to provide cerebellar grey matter in relative isolation from CSF spaces and to avoid adjacency to parahippocampal, fusiform and lingual gyri to avoid bleed-in signal from tau-pathology34. Left and right hemisphere structures were combined for each region-of-interest. Manual quality control checks were performed on the [18F]AV-1451 and MRAGE scans, as well as on the segmentations and registrations of the atlas regions-of-interest onto the [18F]AV-1451 scans. Two subjects were excluded from the MRI analysis because of motion artifacts on one of their MRI scans. However, all 16 subjects were included in the tau-PET analysis since the atlas to [18F]AV-1451 registrations were acceptable
An initial assessment of the longitudinal data was performed using 52 regions-of-interest across all cortical and subcortical structures. This allowed us to investigate general patterns in the data in an unbiased manner. A smaller more specific set of regions was then used to assess rates of change in [18F]AV-1451 uptake over time. The smaller set of 15 regions included nine subcortical and brainstem regions that have been shown to have tau uptake in cross-sectional studies (midbrain, thalamus, dentate nucleus of the cerebellum, globus pallidus, caudate, putamen, subthalamic nucleus, substantia nigra, red nucleus), four cortical regions that show high tau deposition at autopsy35,36 (superior frontal, precentral, postcentral, supplementary motor area), as well as two cortical regions that are typically relatively unaffected pathologically in PSP (entorhinal cortex and occipital lobe).
Statistical analysis
For each subject, we calculated the annualized change of tau-PET, defined as follow-up SUVR minus baseline SUVR divided by time in years. We summarized the annual change measures by reporting the mean and 95% CI. To investigate the differences in annualized change between PSP and controls subjects across the 15 regions-of-interest, we calculated the area under the receiver operating characteristic curve (AUROC), a nonparametric effect size measure that is independent of the underlying scales of the data37. In general, the higher the AUROC estimates, the better the discrimination between groups. In order to correct for age, for each ROI, we fitted a linear regression between Tau SUVR and age. The residual values from the regression were used for the AUROC analysis. P-values and 95% confidence intervals for the regions-of-interest were based on the link between AUROC and the Mann-Whitney U statistic38. The p-values were also adjusted for multiple comparisons using false discovery rate correction. A non-parametric analysis approach was appropriate for our data because the [18F]AV-1451 estimates were not normally distributed. However, for comparison purposes, we also provide the parametric Cohen’s D effect size metric. The Cohen’s d estimate was the difference in means scaled by residual standard deviation from an age adjusted linear regression model. We investigated differences in annualized midbrain volume change between PSP and controls using this same analytical approach. Spearman’s correlations were calculated between annualized change in the PSP Rating Scale and annualized change in imaging metrics, and between baseline and annualized change measures for [18F]AV-1451. Sample size requirements per treatment arm for a placebo-controlled treatment trial were estimated for rates of midbrain atrophy, change in PSPRS over time, and change in the top [18F]AV-1451 measures. Estimated sample sizes needed for PSP subjects to detect a 20% reduction in the rate of change based on 80% and 90% power, two-sided two-sample t-test with an alpha level 0.05 were calculated. Statistical analyses were performed using PET scans that had undergone PVC, and also repeated using PET scans without PVC. The statistical analyses were performed using R version 3.4.1.
Results
Evaluation of subject-level [18F]AV-1451 change
Tau-PET SUVRs across all regions are shown for each scan within each subject in Figure 1. The SUVR profile across regions remains relatively consistent over time in all subjects. However, the regional patterns of change over time varied across subjects. Subject 13 (Supplemental Figure 1) showed increases in uptake over time across nearly all cortical and subcortical regions. Patterns of increased uptake predominantly in the dentate nucleus of the cerebellum and brainstem regions were observed in subjects 1, 2, 4, 8 and 15, with increases observed in frontal regions in subjects 3, 10, 11 and 12. Subjects 6 and 16 (Supplemental Figure 1) showed subtle decreases in uptake in the midbrain, dentate nucleus of the cerebellum and putamen, with subject 6 also showing decreases in frontotemporal regions, similar to subject 7. Regional profiles were very similar when using PET scans without PVC.
Figure 1:
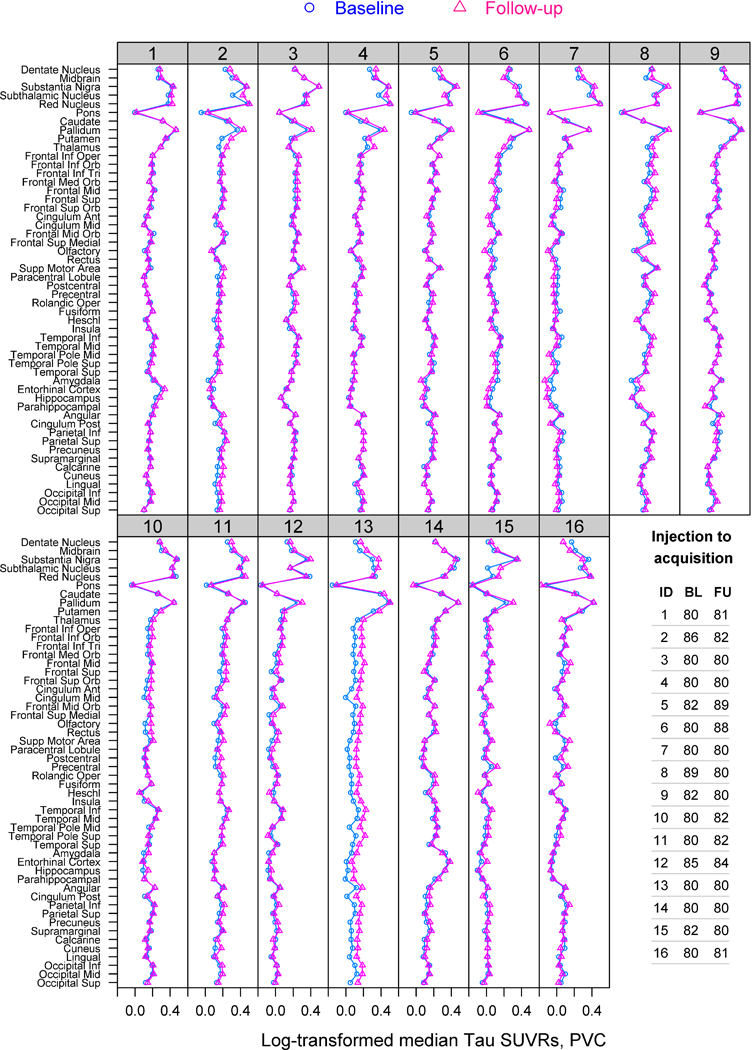
[18F]AV-1451 SUVR regional profiles for baseline and follow-up scans of the 16 PSP subjects. Time from injection of [18F]AV-1451 to PET acquisition (minutes) is shown for each scan for each subject.
Regional comparisons of PSP and controls
Rate of midbrain atrophy on 3T MR imaging was significantly increased in the PSP subjects compared to controls, with an AUROC estimate of 0.93 to differentiate the two groups (Table 2). In the [18F]AV-1451 analysis, the midbrain, pallidum and precentral cortex showed significantly increased rates of uptake in PSP compared to controls, with AUROCs ranging 0.75–0.79 (Table 2). The dentate nucleus of the cerebellum and subthalamic nucleus also showed trends for increased rates of uptake in PSP compared to controls (AUROCs 0.71 and 0.70), but these results did not survive correction for multiple comparisons. AUROC estimates were very similar when age effects were removed. In the analysis without PVC, the pallidum, precentral cortex, dentate nucleus of the cerebellum and midbrain showed significantly increased rates of [18F]AV-1451 uptake in PSP compared to controls, with AUROCs ranging from 0.71–0.76, with trends for increased rates of uptake in the subthalamic nucleus and substantia nigra (AUROCs 0.69 and 0.68) (Supplemental Table 1).
Table 2:
Comparisons of change measures in PSP and controls
| Annualized change (Mean (95% CI)) |
AUROC | Cohen’s d* | ||||
|---|---|---|---|---|---|---|
| PSP (N = 16) |
Control (N = 39) |
Est (95% CI) | P | Est (95% CI) removed age effect |
Est (95% CI) | |
| MRI volume | ||||||
| Midbrain | −0.25 (−0.32, −0.18) | −0.03(−0.07, 0.003) | 0.93 (0.79, 0.98) | <0.001a | 0.92 (0.78, 0.97) | −1.87 (1.33, 2.71) |
|
[18F]AV-1451
uptake | ||||||
| Midbrain | 0.04 (0.01, 0.06) | −0.009 (−0.02, 0.003) | 0.79 (0.62, 0.89) | <0.001a | 0.79 (0.62, 0.89) | 1.07 (0.40, 1.97) |
| Pallidum | 0.07 (0.03, 0.11) | −0.004 (−0.02, 0.01) | 0.76 (0.60, 0.87) | 0.002a | 0.76 (0.60, 0.87) | 1.10 (0.48, 1.92) |
| Precentral | 0.03 (0.009, 0.05) | −0.001 (−0.009, 0.008) | 0.75 (0.58, 0.86) | 0.003a | 0.74 (0.57, 0.85) | 0.98 (0.32, 1.85) |
| Dentate Nucleus | 0.03 (0.003, 0.06) | −0.003 (−0.02, 0.01) | 0.71 (0.54, 0.83) | 0.01 | 0.70 (0.53, 0.83) | 0.65 (0.05, 1.39) |
| Subthalamic Nucleus | 0.05 (0.001, 0.11) | −0.01 (−0.03, 0.009) | 0.70 (0.53, 0.83) | 0.02 | 0.70 (0.53, 0.82) | 0.86 (0.17, 1.65) |
| Supp Motor Area | 0.02 (0.002, 0.05) | 0.0004 (−0.009, 0.01) | 0.67 (0.50, 0.80) | 0.05 | 0.67 (0.50, 0.80) | 0.71 (0.05, 1.56) |
| Thalamus | 0.02 (−0.006, 0.05) | −0.004 (−0.02, 0.007) | 0.67 (0.50, 0.80) | 0.06 | 0.67 (0.50, 0.80) | 0.66 (−0.04, 1.47) |
| Occipital | 0.01 (−0.001, 0.03) | −0.001 (−0.009, 0.007) | 0.66 (0.49, 0.79) | 0.06 | 0.66 (0.49, 0.79) | 0.55 (−0.03, 1.23) |
| Substantia Nigra | 0.02 (−0.03, 0.070) | −0.02 (−0.04, 0.005) | 0.66 (0.49, 0.79) | 0.07 | 0.66 (0.49, 0.79) | 0.51 (−0.13, 1.25) |
| Putamen | 0.03 (0.002, 0.06) | 0.005 (−0.009, 0.02) | 0.66 (0.49, 0.79) | 0.07 | 0.64 (0.47, 0.78) | 0.59 (−0.06, 1.30) |
| Caudate | 0.02 (−0.01, 0.04) | −0.004 (−0.02, 0.01) | 0.63 (0.46, 0.77) | 0.14 | 0.63 (0.47, 0.77) | 0.45 (−0.16, 1.11) |
| Postcentral | 0.02(−0.003, 0.03) | 0.001 (−0.008, 0.009) | 0.62 (0.45, 0.76) | 0.16 | 0.61 (0.44, 0.75) | 0.53 (−0.10, 1.22) |
| Red Nucleus | 0.01 (−0.03, 0.06) | −0.02(−0.04, 0.002) | 0.61 (0.44, 0.75) | 0.21 | 0.61 (0.44, 0.75) | 0.45 (−0.21, 1.16) |
| Entorhinal Cortex | 0.007 (−0.02, 0.03) | −0.004 (−0.02, 0.007) | 0.58 (0.42, 0.73) | 0.34 | 0.59 (0.42, 0.73) | 0.31 (−0.33, 0.98) |
| Frontal Sup | 0.02(−0.006, 0.04) | −0.001 (−0.01, 0.009) | 0.58 (0.42, 0.73) | 0.34 | 0.60 (0.43, 0.74) | 0.54 (−0.13, 1.27) |
Difference in means scaled by residual standard deviation from an age adjusted linear regression model
Still statistically significant (adjusted p-value or q-value < 0.05) after controlling for false discovery rate
Correlations with baseline uptake and disease severity in PSP
Change in midbrain volume showed a reasonably strong correlation with change in PSP Rating Scale over the 12 month interval (r=−0.48, p=0.09). However, we did not observe any significant correlations between change in [18F]AV-1451 uptake over time for the midbrain, precentral cortex, dentate nucleus of the cerebellum or pallidum and change in the PSP Rating Scale (Figure 2). We also did not see any significant associations between baseline [18F]AV-1451 uptake and change in [18F]AV-1451 uptake over time for these regions (Figure 2). These [18F]AV-1451 results remained the same in the analysis without PVC.
Figure 2:
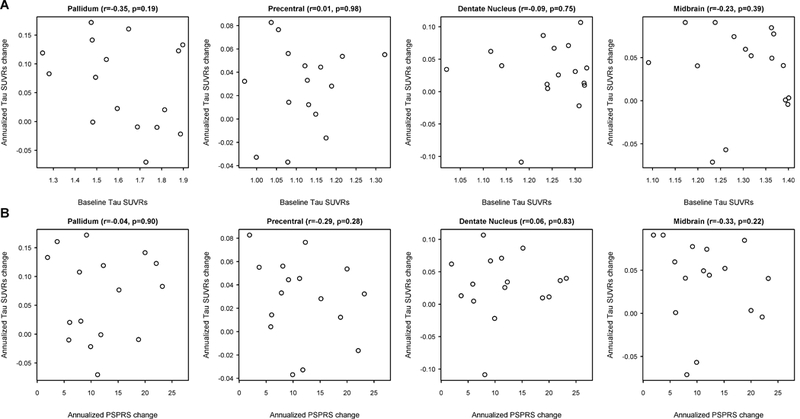
Scatter-plots showing the relationship between (A) annualized change in [18F]AV-1451 measures versus annualized change in PSP Rating Scale (PSPRS), and (B) baseline [18F]AV-1451 uptake versus annualized change in [18F]AV-1451.
Sample size estimates
The smallest sample size estimates were achieved with rate of midbrain volume followed by rate of change in the PSPRS (Table 3). Both of these two metrics provided smaller sample size estimates than the [18F]AV-1451 measures. Within the [18F]AV-1451 measures, sample sizes for the top four regions were smaller with PVC compared to without PVC, with the smallest achieved with change in the pallidum.
Table 3:
Estimated sample size estimates per treatment arm in a clinical trial to detect a reduction in each longitudinal outcome variable in PSP. Data is shown at 80% and 90% power for a potential 20% reduction in each outcome. [18F]AV-1451 measures are shown both without and with partial volume correction (PVC).
| Outcome variable | PVC |
No PVC |
||
|---|---|---|---|---|
| 80% Power | 90% Power | 80% Power | 90% Power | |
| Midbrain volume | 94 | 125 | NA | NA |
| PSP rating scale | 165 | 220 | NA | NA |
| Pallidum [18F]AV-1451 | 521 | 697 | 551 | 737 |
| Precentral [18F]AV-1451 | 626 | 837 | 867 | 1160 |
| Midbrain [18F]AV-1451 | 741 | 992 | 2105 | 2817 |
| Dentate Nucleus of the cerebellum [18F]AV-1451 | 1126 | 1507 | 1094 | 1464 |
Discussion
This regional longitudinal analysis of [18F]AV-1451 tau-PET binding showed evidence that [18F]AV-1451 uptake increased over time in the pallidum, precentral cortex, dentate nucleus of the cerebellum and midbrain in PSP. However, the effect sizes for these measures were smaller than those observed for rate of midbrain atrophy; only midbrain atrophy, and not [18F]AV-1451 uptake, correlated with disease severity; and midbrain atrophy provided smaller sample size estimates for clinical treatment trials. This suggests that rate of midbrain atrophy would be a more clinically relevant measure of PSP progression than [18F]AV-1451, and thus more useful as a longitudinal biomarker for clinical treatment trials in PSP.
The four key regions that showed the most robust evidence for increased uptake of [18F]AV-1451 over time in PSP are typically affected by tau pathology and neurodegeneration in PSP39–41. The pallidum, midbrain and dentate nucleus of the cerebellum have also consistently shown elevated [18F]AV-1451 in PSP compared to controls in cross-sectional studies16–18, 20, 21. Elevated uptake in cortical regions, including the precentral cortex, has been observed in some cross-sectional studies20, 21, although not all17, 18, and uptake was typically not as striking as that observed in the subcortical structures20, 21. Our findings suggest that the precentral cortex may be actively accumulating tau at this point in the disease, perhaps reflecting disease spread from subcortical structures to the cortex. The amount a subject changes over time in these regions is, however, not related to the amount of uptake at baseline, perhaps telling us something biological about disease spread or reflecting noise in the measurements. The regions-of-interest placed on the subthalamic nucleus and substantia nigra also performed reasonably well in our longitudinal analysis likely reflecting the fact that they are both capturing the elevated signal observed in the midbrain in PSP subjects. However, neither performed as well as the midbrain region-of-interest that is larger and subsumes both of these brainstem nuclei.
Individual patterns of change in uptake did show variability, however, with some subjects showing evidence for increased [18F]AV-1451 uptake, but others showing decreased uptake over time. We also had a subject which stood out because increased uptake was observed across all regions of the brain. This variability likely reduced power in the group-level results. Of course, the difficulty in interpreting these longitudinal tau-PET results is that we have no biologic marker documenting how tau burden changes over the course of this disease. Pathological studies can only sample one time point at the end of the disease. While one might assume that tau pathology gradually accumulates over the course of the disease, it has been found that some oligodendroglial tau pathology actually decreases with disease duration in PSP42. In addition, the accumulated tau burden at any point in time may be partially offset by physiological degradation of tau aggregates43. The variability we observed across subjects could reflect this biological heterogeneity. Furthermore, we have no current assurances that [18F]AV-1451 tau-PET is providing an accurate measure of the burden of underlying 4R tau in PSP. It may instead reflect off-target binding to something other than 4R tau. If this is the case then our data suggests that whatever the off-target binding site is, it does increase over time in PSP.
Despite the fact that we observed regions with increasing [18F]AV-1451 uptake over time, it was clear that [18F]AV-1451 did not perform as well as midbrain volume as a disease biomarker in PSP. An important aspect of a disease biomarker is that it should relate to progression of the disease. This is the case for rate of midbrain atrophy which we found to correlate to change over time on the PSPRS in our cohort. Others have similarly observed good correlations between midbrain measures and disease severity in PSP9, 44–46. However, we did not observe any correlation between decline in disease severity and longitudinal measures of [18F]AV-1451. Furthermore, sample size estimates for clinical trials were much smaller for rate of midbrain volume compared to the [18F]AV-1451 measures, with only 94 subjects required per treatment arm with rate of midbrain volume loss for a trial with 80% power to detect a 20% treatment effect. These estimates are roughly in line with those obtained in previous studies for rate of midbrain volume change7–9. In comparison, 521 subjects per treatment arm would be required when using the pallidum which was the best of the [18F]AV-1451 measures. Of note, we found that the sample size estimates were the smallest and the effect sizes of the difference between PSP and controls were the largest, when using PVC data compared to estimates without PVC. There is a current lack of consensus in the field concerning the value of PVC in [18F]AV-1451 analysis, but in this situation it does increase power slightly. Sample size estimates from the PSP Rating Scale were also smaller than those from the [18F]AV-1451 measures, although it did not perform as well as rate of midbrain volume, consistent with previous findings8, 9.
Strengths of our study include the fact that our cohort consisted of three different clinical variants of PSP which may increase generalizability to other PSP cohorts and our findings were very similar in the analysis with and without PVC, with the same top set of regions identified in both analyses. However, our findings may not generalize to other variant syndromes of PSP that we did not include, and the number of subjects was too small to allow us to perform analyses across the different clinical variants. Future studies with larger numbers of subjects will need to determine whether patterns of change in [18F]AV-1451 differ across the clinical variants of PSP. It is important to also acknowledge that our results may not generalize to patients earlier in the disease course. The longitudinal trajectory of [18F]AV-1451 changes has not yet been established in PSP and it is quite possible that rates of change may be greatest in earlier disease stages and could perform better than MRI measures in this phase of the disease. In addition, a limitation of the study is our necessary use of late-uptake SUVR images. SUVR images are less reliable than PET quantification methods using full-dynamic acquisitions, and longitudinal studies using SUVR can be confounded by differences in brain perfusion47. However, late-uptake scans with SUVR remain the most common method for PSP studies due to the large increase in cost, patient burden, and scanner access required for full-dynamic acquisitions. Performing full-dynamic scans for AV-1451 would require over 100 minutes of scan time, rather than 20 minutes, and this was not feasible for our study.
Our study provides some evidence that [18F]AV-1451 uptake changes over time in subjects with PSP. However, given the variability we observed in longitudinal change patterns in [18F]AV-1451, and issues with how our results generalize to other disease stages, it will be important for in vivo [18F]AV-1451 findings to be further validated at autopsy and in other PSP cohorts, particularly in a range of subjects at different stages of the disease.
Supplementary Material
Acknowledgements
This study was funded by R01-NS89757, U01-AG006786 and R01-AG34676. We would like to acknowledge AVID Radiopharmaceuticals for provision of AV-1451 precursor, chemistry production advice and oversight, and FDA regulatory cross-filing permission and documentation needed for this work.
Funding sources: This study was funded by R01-NS89757, U01-AG006786 and R01-AG34676.
Grants: JW was supported by grants from the NIH. DK was supported by grants from the NIH. RP was supported by grants from the NIH. CJ was supported by grants from the NIH and the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Foundation. VL was supported by grants from the NIH, GE Healthcare, Siemens Molecular Imaging, and AVID Radiopharmaceuticals. KJ was supported by grants from the NIH.
Footnotes
Conflicts of interest: The authors have no conflicts of interest
Author roles
Conception and design of the study: JW, KJ
Acquisition and analysis of data: JLW, NT, SDW, CGS, MLS, AJS, JEA, DSK, RCP, CRJ, VJL, KAJ
Drafting a significant portion of the manuscript or figures: JW, KJ
Financial Disclosures of all authors: Stock Ownership in medically-related fields: MLS owns stock in Align Technology, Inc., Gilead Sciences, Inc., Globus Medical Inc., Inovio Biomedical Corp., Johnson & Johnson, LHC Group, Inc., 2017, Medtronic, Inc., Mesa Laboratories, Inc., Natus Medical Incorporated, Parexel International Corporation, and Varex Imaging Corporation. CJ owns stock in Johnson & Johnson.
Intellectual Property Rights: None
Consultancies: DK is a consultant for TauRX, Lundbeck Pharmaceuticals, Elan Pharmaceuticals, Lilly Pharmaceuticals, and Baxter Pharmaceuticals. RP is a consultant for Roche, Inc., Merck, Inc., Genentech, Inc., Biogen, Inc., and Eli Lilly and Co. CJ has provided consulting services for Eli Lilly.
Expert Testimony: None
Advisory Boards: VL serves on scientific advisory boards for Bayer Schering Pharma and Piramal Life Science. RP serves on data monitoring committees for Pfizer, Inc. and Janssen Alzheimer Immunotherapy.
Employment: None
Partnerships: None
Contracts: None
Honoraria: None
Royalties: RP receives publishing royalties from Mild Cognitive Impairment (Oxford University Press, 2003)
REFERENCES
- 1.Steele JC, Richardson JC, Olszewski J. Progressive Supranuclear Palsy. a Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy, Nuchal Dystonia and Dementia. Arch Neurol 1964;10:333–359. [DOI] [PubMed] [Google Scholar]
- 2.Hoglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Josephs KA, Hodges JR, Snowden J, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122(2):137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osaki Y, Ben-Shlomo Y, Lees AJ, et al. Accuracy of clinical diagnosis of progressive supranuclear palsy. Mov Disord 2004;19(2):181–189. [DOI] [PubMed] [Google Scholar]
- 5.Respondek G, Roeber S, Kretzschmar H, et al. Accuracy of the National Institute for Neurological Disorders and Stroke/Society for Progressive Supranuclear Palsy and neuroprotection and natural history in Parkinson plus syndromes criteria for the diagnosis of progressive supranuclear palsy. Mov Disord 2013;28(4):504–509. [DOI] [PubMed] [Google Scholar]
- 6.Whitwell JL, Hoglinger GU, Antonini A, et al. Radiological biomarkers for diagnosis in PSP: Where are we and where do we need to be? Mov Disord 2017;32(7):955–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paviour DC, Price SL, Lees AJ, Fox NC. MRI derived brain atrophy in PSP and MSA-P. Determining sample size to detect treatment effects. J Neurol 2007;254(4):478–481. [DOI] [PubMed] [Google Scholar]
- 8.Whitwell JL, Xu J, Mandrekar JN, Gunter JL, Jack CR, Josephs KA. Rates of brain atrophy and clinical decline over 6 and 12-month intervals in PSP: determining sample size for treatment trials. Parkinsonism Relat Disord 2012;18(3):252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoglinger GU, Schope J, Stamelou M, et al. Longitudinal magnetic resonance imaging in progressive supranuclear palsy: A new combined score for clinical trials. Mov Disord 2017;32(6):842–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis 2013;34(2):457–468. [DOI] [PubMed] [Google Scholar]
- 11.Xia CF, Arteaga J, Chen G, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement 2013;9(6):666–676. [DOI] [PubMed] [Google Scholar]
- 12.Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun 2016;4(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 2015;78(5):787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sander K, Lashley T, Gami P, et al. Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimers Dement 2016;12(11):1116–1124. [DOI] [PubMed] [Google Scholar]
- 15.Smith R, Scholl M, Honer M, Nilsson CF, Englund E, Hansson O. Tau neuropathology correlates with FDG-PET, but not AV-1451-PET, in progressive supranuclear palsy. Acta Neuropathol 2017;133(1):149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith R, Schain M, Nilsson C, et al. Increased basal ganglia binding of 18 F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord 2017;32(1):108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H, Choi JY, Hwang MS, et al. Subcortical 18 F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord 2017;32(1):134–140. [DOI] [PubMed] [Google Scholar]
- 18.Passamonti L, Vazquez Rodriguez P, Hong YT, et al. 18F-AV-1451 positron emission tomography in Alzheimer’s disease and progressive supranuclear palsy. Brain 2017;140(3):781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitwell JL, Ahlskog JE, Tosakulwong N, et al. Pittsburgh Compound B and AV-1451 positron emission tomography assessment of molecular pathologies of Alzheimer’s disease in progressive supranuclear palsy. Parkinsonism Relat Disord 2018;48:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitwell JL, Lowe VJ, Tosakulwong N, et al. [18 F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord 2017;32(1):124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schonhaut DR, McMillan CT, Spina S, et al. (18) F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: A multicenter study. Ann Neurol 2017;82(4):622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain 2007;130(Pt 6):1552–1565. [DOI] [PubMed] [Google Scholar]
- 23.Whitwell JL, Master AV, Avula R, et al. Clinical correlates of white matter tract degeneration in progressive supranuclear palsy. Arch Neurol 2011;68(6):753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53(4):695–699. [DOI] [PubMed] [Google Scholar]
- 25.Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a Frontal Assessment Battery at bedside. Neurology 2000;55(11):1621–1626. [DOI] [PubMed] [Google Scholar]
- 26.Jack CR Jr, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement 2017;13(3):205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meltzer CC, Kinahan PE, Greer PJ, et al. Comparative evaluation of MR-based partial-volume correction schemes for PET. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 1999;40(12):2053–2065. [PubMed] [Google Scholar]
- 28.Ashburner J, Friston KJ. Unified segmentation. Neuroimage 2005;26(3):839–851. [DOI] [PubMed] [Google Scholar]
- 29.Schwarz CG, Gunter JL, Ward CP, et al. THE MAYO CLINIC ADULT LIFESPAN TEMPLATE: BETTER QUANTIFICATION ACROSS THE LIFESPAN. Alzheimer’s & Dementia 2017;13(7, Supplement):P792. [Google Scholar]
- 30.Botha H, Whitwell JL, Madhaven A, Senjem ML, Lowe V, Josephs KA. The pimple sign of progressive supranuclear palsy syndrome. Parkinsonism Relat Disord 2014;20(2):180–185. [DOI] [PubMed] [Google Scholar]
- 31.Fujiwara A, Yoshida T, Otsuka T, et al. Midbrain volume increase in patients with panic disorder. Psychiatry Clin Neurosci 2011;65(4):365–373. [DOI] [PubMed] [Google Scholar]
- 32.Ewert S, Plettig P, Li N, et al. Toward defining deep brain stimulation targets in MNI space: A subcortical atlas based on multimodal MRI, histology and structural connectivity. Neuroimage 2018;170:271–282. [DOI] [PubMed] [Google Scholar]
- 33.Avants BB, Epstein CL, Grossman M, Gee JC. Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal 2008;12(1):26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowe VJ, Wiste HJ, Senjem ML, et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 2018;141(1):271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dickson DW, Hauw JJ, Agid Y, Litvan I. Progressive Supranuclear Palsy and Corticobasal Degeneration In: Dickson D, Weller RO, eds. Neurodegeneration: The molecular pathology of dementia and movement disorders. 2nd edition ed. Chichester, UK: Wiley-Blackwell, 2011. [Google Scholar]
- 36.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 1994;44(11):2015–2019. [DOI] [PubMed] [Google Scholar]
- 37.Acion L, Peterson JJ, Temple S, Arndt S. Probabilistic index: an intuitive non-parametric approach to measuring the size of treatment effects. Stat Med 2006;25(4):591–602. [DOI] [PubMed] [Google Scholar]
- 38.Newcombe RG. Confidence intervals for an effect size measure based on the Mann-Whitney statistic. Part 1: general issues and tail-area-based methods. Stat Med 2006;25(4):543–557. [DOI] [PubMed] [Google Scholar]
- 39.Josephs KA, Whitwell JL, Dickson DW, et al. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging 2008;29(2):280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 2010;23(4):394–400. [DOI] [PubMed] [Google Scholar]
- 41.Whitwell JL, Avula R, Master A, et al. Disrupted thalamocortical connectivity in PSP: a resting-state fMRI, DTI, and VBM study. Parkinsonism Relat Disord 2011;17(8):599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Josephs KA, Mandrekar JN, Dickson DW. The relationship between histopathological features of progressive supranuclear palsy and disease duration. Parkinsonism Relat Disord 2006;12(2):109–112. [DOI] [PubMed] [Google Scholar]
- 43.Sato C, Barthelemy NR, Mawuenyega KG, et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018;97(6):1284–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitwell JL, Xu J, Mandrekar J, Gunter JL, Jack CR, Josephs KA. Imaging measures predict progression in progressive supranuclear palsy. Mov Disord 2012;27(14):1801–1804. [DOI] [PubMed] [Google Scholar]
- 45.Dutt S, Binney RJ, Heuer HW, et al. Progression of brain atrophy in PSP and CBS over 6 months and 1 year. Neurology 2016;87(19):2016–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paviour DC, Price SL, Jahanshahi M, Lees AJ, Fox NC. Longitudinal MRI in progressive supranuclear palsy and multiple system atrophy: rates and regions of atrophy. Brain 2006;129(Pt 4):1040–1049. [DOI] [PubMed] [Google Scholar]
- 47.van Berckel BN, Ossenkoppele R, Tolboom N, et al. Longitudinal amyloid imaging using 11C-PiB: methodologic considerations. J Nucl Med 2013;54(9):1570–1576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.