

INTRODUCTION
The complement system is traditionally considered a component of the innate immune system. In the context of transplantation, evidence published since the late 1990s has expanded our understanding of links between complement and adaptive immunity, including alloimmune responses and transplant injury. Among many pathogenic functions in transplantation, complement participates in ischemia-reperfusion (I/R) injury, controls the strength of the donor-reactive T cell immune response, and acts as an effector for antibody-initiated allograft injury. Herein, we will review the biology of the complement system and discuss current concepts regarding how complement contributes to the pathogenesis of transplant injury. We will also discuss how the discovery of these fundamental mechanisms has led to development and testing of a) novel treatment strategies that could limit allograft injury and improve the health of transplant recipients and b) complement-derived biomarkers for post-transplant risk stratification.
BIOLOGY OF THE COMPLEMENT SYSTEM
The complement system is comprised of >30 soluble and membrane-bound proteins, including zymogens, receptors, and regulators. The individual complement components can be categorized by their ability to initiate, amplify, or regulate the complement cascade and/or perform effector functions (Figure 1).
Figure 1. Overview of the complement cascade and its regulators.
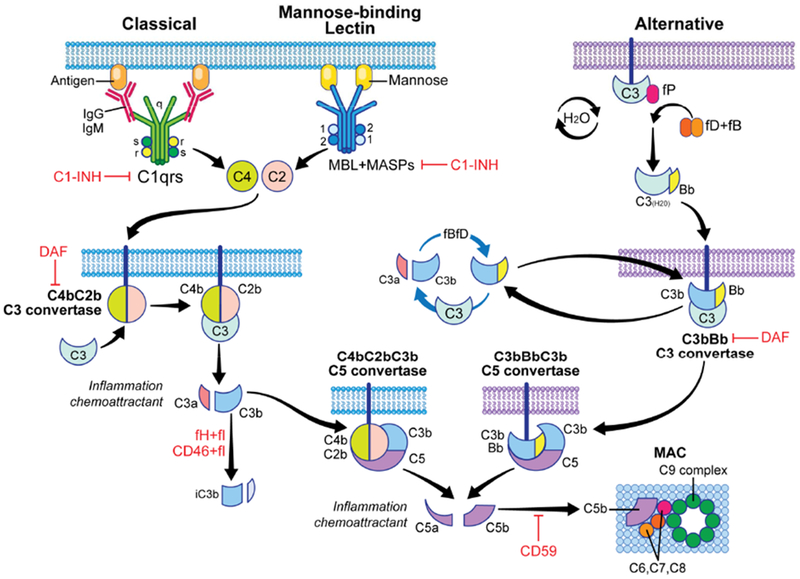
Complement activation can be initiated by the classical pathway triggered by cross-linking cell-bound subclasses of IgG and IgM antibodies, the mannose binding lectin (MBL) pathway triggered by carbohydrates present on bacteria surface, and the alternative pathway that undergoes spontaneous activation on cell surfaces. All three pathways converge into one key amplification step to form multimeric C3 convertases which cleave C3 to C3a and C3b, the latter forming additional C3 convertases (amplification) and then initiating formation of the C5 convertase. Subsequently, C5 cleavage yields C5a and C5b, ultimately forming the membrane attack complex (MAC, C5b-9) on the target cells. Complement activation/amplification is restrained on self-cells by several membrane-bound and soluble regulatory proteins (shown in red). See text for further details. Surface-expressed regulators include decay accelerating factor (DAF or CD55, accelerates the decay of cell-surface assembled C3 convertases), membrane cofactor protein [MCP or CD46, co-factor for factor I (fI) that inactivates C3b to iC3b], and CD59 (protectin, inhibits formation of the MAC). Factor H (fH) is a soluble complement regulator that exhibits both decay accelerating and co-factor activity. C1 inhibitor (C1-INH) inhibits C1qrs and MBL-MASP complexes, limiting classical pathway and MBL pathway activation, respectively. MASP: mannose-associated serine protease.
Complement activation can be initiated via the classical pathway, the lectin pathway, and the alternative pathway 1. The classical pathway is activated when C1q, as part of the C1qrs complex, binds to the Fc regions of IgG or IgM. The activated C1qrs complex then cleaves C4 and C2, forming the membrane bound C4bC2b C3 convertase, which enzymatically cleaves C3 into C3a (an anaphylatoxin) and C3b. In the lectin pathway, mannose-binding lectins (MBLs), as well as collectins and ficolins, function as pattern recognition receptors and bind bacterial carbohydrate and/or lipid motifs. MBL can also directly recognize neoantigens expressed by injured or apoptotic cells 2, a mechanism pathogenically linked to ischemia reperfusion injury (see discussion below). Once bound, the lectin binding proteins function as opsonins and also interact with MBL-associated serine proteases (MASPs). Upon binding to MBL, MASP-1 and MASP-2 act analogously to the C1qrs complex; the MBL/MASP-1/2 complex that cleaves C4 and C2 to generate C4bC2b C3 convertases. In the alternative pathway, complement activation occurs continuously and spontaneously at low levels, through a process commonly referred to as “tickover.” This process is initiated by spontaneous hydrolysis of C3, forming C3(H2O), which permits binding of Factor B (fB). Factor D (fD)-mediated cleavage of C3(H2O) bound fB forms the initial C3 convertase, C3(H2O)Bb, which cleaves C3 into C3a and C3b. C3b then associates with Bb to form the membrane bound C3bBb alternative pathway C3 convertase. Once C3 convertases are formed by any of the 3 activation pathways, the alternative pathway amplifies complement activation by utilizing the locally produced C3b molecules to form more C3 convertases.
The surface bound, multimeric, C3 convertases can bind additional C3b molecules to yield the C5 convertases, C4bC2bC3b and C3bBbC3b (Figure 1). These enzymes cleave C5 into C5a (another anaphylatoxin) and C5b, the latter initiating the formation of the C5b-9 membrane attack complex (MAC) 3. The MAC forms a pore in cell membranes, which promotes cytolysis in non-nucleated cells (including bacteria and red blood cells). MACs inserted into nucleated cells generally induce cellular activation 4, rather than lysis, and/or promote tissue injury 5. Soluble and surface bound split products, including C3a, C3b, iC3b, C3dg and C5a mediate separate but overlapping effector functions 6. C3a and C5a ligate their respective receptors C3aR1 and C5aR1, 7-transmembrane-spanning, G-protein coupled receptors (GPCR) expressed on various cell types to mediate chemotaxis, cytokine release, antigen-presenting cell (APC) activation, and T-cell activation and expansion (see below) 7–10. C3b, iC3b, and C3dg bind to various complement receptors, functioning as opsonins.
Under physiological conditions, multiple regulatory mechanisms throughout the cascade prevent injury to self-cells but permit complement activation on pathogens that do not express complement regulators 6 (Figure 1). The C1-inhibitor (C1-INH) inactivates the C1qrs complex, as well as the MBL/MASP-1/MASP-2 complex, to prevent classical and lectin pathway activation. Decay accelerating factor (DAF; CD55), a glycophosphatidylinositol-(GPI)-anchored, cell surface expressed protein, accelerates the decay of the C3 convertases, thereby limiting amplification of the complement cascade. Soluble factor H and surface-expressed membrane cofactor protein (MCP, CD46) are cofactors for factor I that cleave C3b into iC3b, a mechanism that irreversibly disassociates C3 convertases. Downstream, ubiquitously expressed carboxypeptidases rapidly inactivate the C3a and C5a anaphylatoxins while the surface expressed, GPI-anchored protein CD59 (protectin) prevents the formation of MACs.
While the liver produces the majority of the circulating (plasma) complement components, complement proteins are locally produced by multiple other cell types including endothelial cells, parenchymal cells [e.g. tubular cells in the kidney 11] and immune cells, including T cells and antigen presenting cells (APCs) 12.
COMPLEMENT AND ISCHEMIA-REPERFUSION INJURY
Static cold storage of donor organs induces tissue hypoxia, mitochondrial damage, and ATP depletion, which, upon reperfusion, results in the generation of free oxygen radicals and organ damage commonly referred to as ischemia reperfusion (I/R) injury 13. In kidney transplantation, I/R injury presents clinically as delayed graft function (DGF). Extensive experimental evidence now supports a crucial role for complement in this process (see Figure 2). I/R-induced injury to endothelial cells results in surface expression of neoantigens that can be recognized by MBL 14–16 or collectins 17. Subsequent complement activation yields C3a and C5a that bind to their respective receptors on endothelial cells (among other cell types). These ligations crucially mediate I/R injury 18–20 (Figure 2). Data from animal models also suggests that donor brain death upregulates complement activation in renal allografts prior to procurement, which provides the dominant source of complement driving I/R injury 21,22. The elucidation of these mechanisms has resulted in novel strategies aimed at preventing I/R injury and DGF. Chun et al showed in 2018 that peri-transplant treatment with C1-INH (to block MBL and classical pathway complement activation) overcame prolonged cold ischemia-induced, complement-dependent, I/R injury and cardiac allograft rejection in a mouse model 23. Subsequent work by Jordan and colleagues translationally showed that C1-INH administration can limit DGF and improve kidney function in human recipients of deceased donor kidneys 24 C1-INH also improved lung function in a small cohort of lung transplant patients with early, primary graft dysfunction (a form of I/R injury) 25, further highlighting the utility of this treatment strategy.
Figure 2. Mechanisms through which complement mediates ischemia-reperfusion injury.
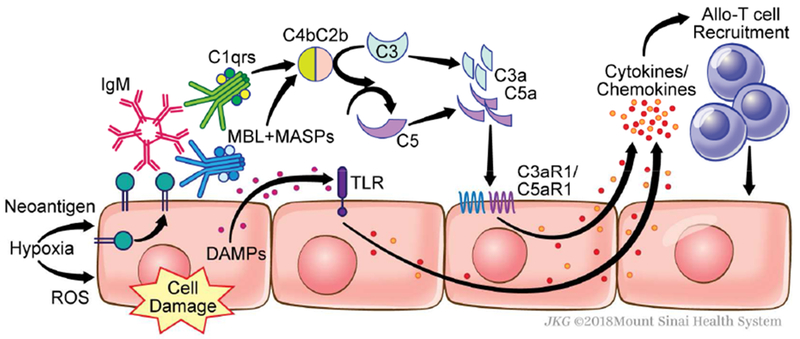
Hypoxia induces surface expression of neoantigens that are recognized by natural, pre-formed IgM and MBL (and/or collectins), which then initiate complement activation. Following reperfusion, the generation of reactive oxygen species (ROS) is associated with graft-derived complement production and activation as well as the release of damage-associated molecular patterns (DAMPs). Subsequent Toll-like receptor (TLR) signaling synergizes with and amplifies complement activation, together yielding C3a and C5a. These anaphylatoxins signal through their 7-transmembrane spanning G-protein coupled receptors on endothelial cells (among other cellular targets) inducing cytokine/chemokine release and facilitating T cell infiltration into the allograft.
In another approach targeting complement-dependent injury, investigators treated donor lungs with a nebulized C3a receptor (C3aR1) antagonist to ameliorate brain death-associated I/R injury in a murine lung transplant model 26. Additionally, mirococept (APT070), a lipid-tailed C3 convertase inhibitor that can be perfused into donor organs and inserts into the endothelial cell membranes within the allograft has been shown to prevent kidney I/R injury in rodents 27 and to improve early islet allograft inflammation in a humanized mouse model 28. Studies testing efficacy of mirococept to prevent DGF in human recipients of deceased donor kidneys are ongoing 29. In contrast to the positive effects of C1-INH alluded to above, a recent prospective, randomized trial testing anti-C5 mAb, eculizumab, in kidney transplantation showed no effect in preventing DGF 30, suggesting that prevention of DGF requires proximal complement inhibition (at or prior to the C3 convertase step).
COMPLEMENT AND T CELL- MEDIATED REJECTION
Building upon the paradigm-shifting observation that C3-deficiency prolongs murine renal allograft survival 31, work from several groups revealed an unanticipated role for immune cell-derived complement as a regulator of T cells, including T cells reactive to transplant antigens (Figure 3). These studies showed that during cognate interactions between CD4+ T cells and APCs, both cell types downregulate surface expression of DAF and secrete alternative pathway complement components which together result in local production of C3a and C5a. The interacting partners also upregulate surface expression C3aR1 and C5aR1 during activation 32. Subsequent autocrine and paracrine C3a/C3aR1 and C5a/C5aR1 signaling via the phosphatidylinositol-3 kinase-gamma (PI3Kγ) pathway induces alloreactive, effector T cell proliferation/differentiation and inhibits T cell apoptosis. Simultaneously, immune cell-derived C3a/C5a ligations with their receptors on APCs induce upregulation of costimulatory molecules and cytokines 32 to further amplify the effector T cell response. Remarkably, Sheen et al showed in 2017 that Toll-like receptor-induced APC activation, a process thought to be crucial for protective immune responses against pathogens and for pathogenic immune responses directed at transplanted organs, requires immune cell production of complement and subsequent C3a/C3aR1 and C5a/C5aR1 ligations on the APC 33.
Figure 3. Mechanisms through which complement modulates T cell-mediated rejection.
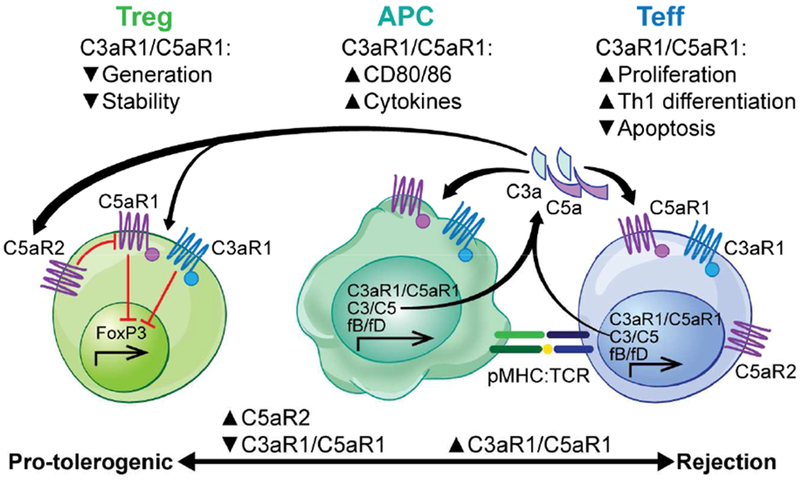
Cognate interactions between antigen presenting cells (APC) and T cells yield immune cell-derived complement production, which activates through the alternative pathway to yield C3a and C5a. The anaphylatoxins bind to their respective receptors (C3aR1 and C5aR1 respectively) on the interacting partners to induce APC maturation and effector T cell proliferation/expansion, survival, and differentiation. The same signals simultaneously inhibit the generation, stability, and function of regulatory T cells (Treg) in part by inhibiting Foxp3 expression. As C5aR2 binds C5a but lacks a GPCR signaling motif, C5aR2 expression on T cells limits the ability of free C5a to bind to C5aR1 and thus facilitates Treg induction by limiting C5a/C5aR1 ligations.
Analogous processes apply to human T cell/APC interactions. C3a and C5a are locally produced during in vitro cultures of human T cells and allogeneic dendritic cells (DCs), resulting in alloreactive T cell activation and expansion 34,35. Separate work by Kemper and colleagues showed a distinct role for complement in human T cell immunity by demonstrating that the intracellular production of complement cleavage products, including C5a, is crucially involved in human T cell activation, in part via activation of the inflammasome 36. Consistent with a role for complement as a modulator of human T cells, patients genetically deficient in C3 have impaired Th1 differentiation 37.
In contrast to complement-dependent promotion of effector T cell responses, immune cell-derived, locally produced C3a/C5a inhibits the generation and stability of regulatory T cells (Treg) in mice and humans 9,10,38 (Figure 3), which contributes to augmenting the antigen-specific effector T cell response. The inhibitory effects of C3aR1/C5aR1 signaling on Treg generation are modulated by T cell expressed C5aR2 that binds C5a but lacks the intracellular signaling domain to initiate G-protein coupled receptor signaling 39. Evidence suggests that in T cells, C5aR2 functions to bind C5a and prevent C5a from binding to and activating C5aR1. Verghese et al showed in 2018 that absence of C5aR2 limits, while transgenic overexpression of C5aR2 enhances, in vivo Treg generation, independent of C5aR1 expression on the T cells 40
Mechanistic experiments showed that C3aR1/C5aR1-initiated signaling, which is prevented by overexpression of C5aR2 3, results in AKT-dependent phosphorylation of the transcription factor Foxo1, preventing its nuclear translocation and subsequently limiting FoxP3 transcription 10. Conversely, disrupting C3aR1/C5aR1 signaling on T cells genetically or pharmacologically 10,38 augments Treg generation, stabilizes FoxP3 expression and is protolerogenic.
Complement regulation of T cell immunity is broadly relevant and applies in the context of infectious pathogens 41, autoimmune diseases 42, and T cell driven transplant rejection. As examples of the latter, absence of DAF (lifts restraint on production of C3a and C5a 43) accelerates cardiac allograft rejection 44 while anti-C5 mAb 45, C5aR1 antagonism 46, or absence of C3aR1 signaling 47 reduces T cell priming and improves allograft survival in mice. Similarly, Treg-dependent allograft survival/tolerance is facilitated by blocking C3aR1/C5aR1 signaling 9 or augmenting C5aR2 expression on immune cells 40. Together with the observations that these mechanisms apply to human T cells 34, the data support the need to test the clinical impact of complement inhibition on T-cell-mediated allograft rejection in humans.
COMPLEMENT AND ANTIBODY-MEDIATED TRANSPLANT INJURY
Complement has established effects on antibody production and function. Complement receptor 1 (CR1, CD35) and complement receptor 2 (CR2, CD21) bind to antigen-bound C3b and iC3b, respectively, the latter lowering the threshold for B cell activation and promoting antigen retention by follicular dendritic cells 48,49. C3-deficient mice fail to produce high-affinity IgG responses against major histocompatibility antigens in skin grafts 50, confirming its relevance to transplantation.
In addition to complement’s role in donor specific antibody (DSA) production, complement activation has been shown to be an important effector mechanism of antibody-initiated allograft injury in murine models 51 and human transplant recipients 52,53 (Figure 4). DSA can activate complement via the classical pathway, ultimately inducing graft inflammation and injury through C3a/C5a production and MAC formation 52. While MAC formation mediates lysis in non-nucleated cells, terminal complement activation on donor human endothelial cells induces non-canonical NF-κB signaling, initiating a pro-inflammatory gene program that facilitates recruitment of alloreactive T cells required for the development of allograft injury 54
Figure 4. Mechanisms through which complement modulates antibody-mediated rejection.
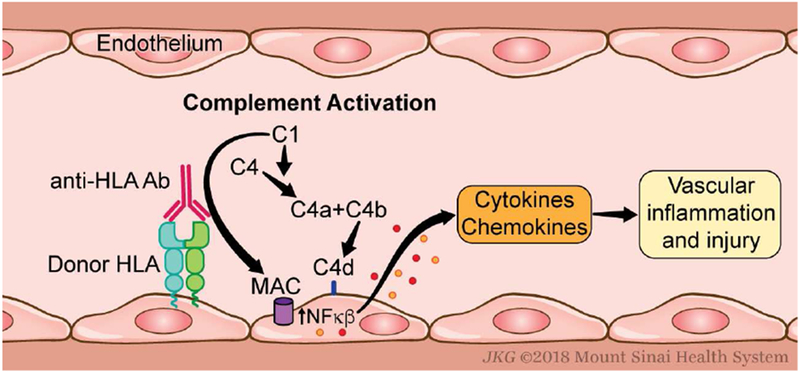
Donor specific antibodies (DSA) bind to donor human leukocyte antigen (HLA) on endothelial cells and initiate complement activation via the classical pathway, leading to complement deposition, local C3a/C5a production, and MAC formation. The latter causes non-canonical NF-κB signaling that initiates a pro-inflammatory gene program, enhancing recruitment of alloreactive T cells and development of vascular injury.
Building upon these fundamental observations, clinicians have studied the efficacy of eculizumab, an anti-human C5 mAb, to prevent and/or treat antibody-mediated rejection (ABMR) in human transplant recipients. Eculizumab plus plasma exchange reduced the incidence of ABMR in 26 sensitized kidney transplant recipients compared to a control group 55, and it successfully treated ABMR in a small cohort 56, although it failed to demonstrate efficacy in another case series 57. Notably, ABMR can be resistant to eculizumab 55, suggesting that complement components proximal to the C5 convertase and/or non-complement-(e.g. Fc receptor)-dependent effector mechanisms can contribute to the antibody-initiated pathology. A pilot study of C1-INH, which blocks activation of the classical and MBL pathways (Figure 1), suggests efficacy for treatment of ABMR 58, but larger scale studies are needed.
COMPLEMENT AND TRANSPLANT REJECTION BIOMARKERS
With the recognition that complement activation is a critical mediator of I/R injury, T-cell mediated rejection, and ABMR, investigators have begun to test whether complement-derived biomarkers could be useful as prognostic risk-assessment tools in transplantation. To this end, research on potential biomarkers has focused on three broad areas: single nucleotide polymorphisms (SNPs) in complement genes, measures of complement-binding capacity of DSA, and detection of serum-based complement components.
Both functional and non-functional SNPs have been identified in complement components throughout the cascade, including within the complement regulators. Their functional effects and relevance to renal transplant outcomes are summarized in a 2017 review 59. Recipient SNPs in C5 (rs17611, increases serum C5a levels) and C5aR1 (rs4804049, nonfunctional) have been linked to worse renal allograft function at 1 year post-transplant and allograft failure, respectively. Similarly, two C3 SNPs (rs10411506 and rs2230205) correlate with ABMR in renal transplant recipients 60. As donor-expressed complement regulatory proteins could confer protection from DSA 61, investigators have hypothesized that donor SNPs within regulators could also be used to assess post-transplant risk. Indeed, a donor CD59 promoter polymorphism (rs147788946) is associated with an increased rate of early acute rejection in renal allografts 62 and a higher incidence of chronic rejection among lung transplant recipients 63. Going forward, sufficiently powered validation studies will be crucial in ultimately determining whether treatment strategies should be individualized based on expression of the various SNPs.
DSA-associated allograft injury is in part dependent on complement-mediated effector functions, so the prognostic value of the complement-binding capacity of anti-HLA antibodies has become an important question in transplantation. A 2013 cohort study of 1016 kidney transplant recipients suggested that patients with C1q-binding (C1q+) DSA have elevated rates of ABMR compared to C1q− DSA+ patients (48% vs 16%) 64. Similar associations were documented in heart 65 and in lung 66 transplant recipients. In another cohort study of 70 renal transplant patients that developed de novo DSA (dnDSA), C1q-binding was correlated with post-dnDSA graft loss 67. However, the authors of this study also demonstrated that C1q positivity correlated with DSA titer and mean fluorescence intensity (MFI), suggesting that C1q binding may simply reflect the strength of the serum DSA and may not add significant prognostic information beyond standard DSA testing.
A non-invasive, serum-based biomarker for allograft rejection could help avoid unnecessary biopsies and lead to earlier treatment, potentially improving transplant outcomes. While no such validated biomarker currently exists in transplantation, there are complement-related candidates. Soluble CD59 (sCD59), which is associated with cellular damage, including post- myocardial infarction 68, was found to be elevated in a cohort of patients who developed bronchiolitis obliterans syndrome (BOS) after lung transplantation 69. Importantly, serum sCD59 concentrations were elevated prior to the clinical diagnosis of BOS in these patients, and sCD59 was determined to be an independent predictor for the development of BOS. In another study, investigators demonstrated a correlation between serum C4d+ (a marker of classical pathway complement activation) microvesicles and ABMR in a cohort of kidney transplant patients 70; treatment of their ABMR was associated with an improvement in estimated glomerular filtration rate and a reduction in C4d+ microvesicle concentration. With regard to acute cellular rejection (ACR), a 2018 study found an association between circulating immune complexes, specific for complement factor H, and ACR in liver transplant recipients 71. These intriguing findings support the need for further testing of complement biomarkers and/or complement biomarker profiles aimed at individualizing risk stratification and treatment algorithms for transplant patients.
CONCLUDING REMARKS
The complement system, traditionally considered a component of innate immunity, is now recognized as a crucial mediator of the adaptive immune response in solid organ transplantation. Pre-clinical and early human trials have begun to elucidate how specific complement components modulate the various forms of post-transplant anti-graft immune responses (e.g. I/R injury, T-cell mediated cellular rejection, ABMR). These data, together with the pharmaceutical industry’s increasing interest in developing complement inhibitors 72, support the need to test whether complement inhibition, potentially guided by complement-derived risk assessing biomarkers, can improve allograft survival and patient-centered outcomes in transplantation.
KEY POINTS.
Complement activation, via the lectin and classical pathways, is an important mediator of ischemia-reperfusion injury.
Local production of complement by immune cells induces antigen-presenting cell maturation and effector T cell expansion, while inhibiting regulatory T cell generation.
Donor specific antibody (DSA)-mediated complement activation initiates a pro-inflammatory gene program in donor endothelial cells.
Serum-based complement components have the potential to serve as biomarkers for allograft rejection.
SYNOPSIS.
The complement system, traditionally considered a component of innate immunity, is now recognized as a crucial mediator of the adaptive immune response in solid organ transplantation. Pre-clinical and early human trials have demonstrated the importance of complement effector mechanisms in driving allograft injury during specific anti-graft immune responses, including ischemia-reperfusion injury, T cell-mediated rejection, and antibody-mediated rejection, as well as a potential role for complement-derived risk stratification biomarkers. These data support the need for further testing of complement inhibitors in solid organ transplant recipients.
Funding/Acknowledgments:
The work was supported by NIH grants R01 AI071185 and AI132405 awarded to PSH and K08 AI135101 to NHC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have nothing to disclose.
REFERENCES
- 1.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–1066. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Ip WE, Michelow IC, Ezekowitz RA. The mannose-binding lectin: a prototypic pattern recognition molecule. Curr Opin Immunol. 2006;18(1): 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bamberg CE, Mackay CR, Lee H, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J Biol Chem. 2010;285(10):7633–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park P, Haas M, Cunningham PN, et al. Inhibiting the complement system does not reduce injury in renal ischemia reperfusion. J Am Soc Nephrol. 2001;12(7):1383–1390. [DOI] [PubMed] [Google Scholar]
- 5.Adler S, Baker PJ, Johnson RJ, Ochi RF, Pritzl P, Couser WG. Complement membrane attack complex stimulates production of reactive oxygen metabolites by cultured rat mesangial cells. J Clin Invest. 1986;77(3):762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. [DOI] [PubMed] [Google Scholar]
- 8.Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009;46(14):2753–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med. 2013;210(2):257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, Heeger PS. Cutting edge: Receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol. 2013;190(12):5921–5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peake PW, O’Grady S, Pussell BA, Charlesworth JA. C3a is made by proximal tubular HK-2 cells and activates them via the C3a receptor. Kidney Int. 1999;56(5):1729–1736. [DOI] [PubMed] [Google Scholar]
- 12.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112(5):1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant. 2011;11(11):2279–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang M, Carroll MC. Natural IgM-mediated innate autoimmunity: a new target for early intervention of ischemia-reperfusion injury. Expert Opin Biol Ther. 2007;7(10):1575–1582. [DOI] [PubMed] [Google Scholar]
- 15.Haas MS, Alicot EM, Schuerpf F, et al. Blockade of self-reactive IgM significantly reduces injury in a murine model of acute myocardial infarction. Cardiovasc Res. 2010;87(4):618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am J Physiol Heart Circ Physiol. 2009;297(5):H1853–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nauser CL, Farrar CA, Sacks SH. Complement Recognition Pathways in Renal Transplantation. J Am Soc Nephrol. 2017;28(9):2571–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thurman JM, Ljubanovic D, Edelstein CL, Gilkeson GS, Holers VM. Lack of a functional alternative complement pathway ameliorates ischemic acute renal failure in mice. J Immunol. 2003;170(3):1517–1523. [DOI] [PubMed] [Google Scholar]
- 19.De Vries B, Matthijsen RA, Wolfs TG, Van Bijnen AA, Heeringa P, Buurman WA. Inhibition of complement factor C5 protects against renal ischemia-reperfusion injury: inhibition of late apoptosis and inflammation. Transplantation. 2003;75(3):375–382. [DOI] [PubMed] [Google Scholar]
- 20.Peng Q, Li K, Smyth LA, et al. C3a and C5a promote renal ischemia-reperfusion injury. J Am Soc Nephrol. 2012;23(9):1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farrar CA, Zhou W, Lin T, Sacks SH. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. FASEB J. 2006;20(2):217–226. [DOI] [PubMed] [Google Scholar]
- 22.Atkinson C, Floerchinger B, Qiao F, et al. Donor brain death exacerbates complement-dependent ischemia/reperfusion injury in transplanted hearts. Circulation. 2013;127(12):1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chun N, Fairchild RL, Li Y, et al. Complement Dependence of Murine Costimulatory Blockade-Resistant Cellular Cardiac Allograft Rejection. Am J Transplant. 2017; 17(11):2810–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jordan SC, Choi J, Aubert O, et al. A phase I/II, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Am J Transplant. 2018. [DOI] [PubMed] [Google Scholar]
- 25.Sommer W, Tudorache I, Kuhn C, et al. C1-esterase-inhibitor for primary graft dysfunction in lung transplantation. Transplantation. 2014;97(11):1185–1191. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Q, Patel K, Lei B, et al. Donor pretreatment with nebulized complement C3a receptor antagonist mitigates brain-death induced immunological injury post-lung transplant. Am J Transplant. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel H, Smith RA, Sacks SH, Zhou W. Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage. J Am Soc Nephrol. 2006; 17(4): 1102–1111. [DOI] [PubMed] [Google Scholar]
- 28.Xiao F, Ma L, Zhao M, et al. APT070 (mirococept), a membrane-localizing C3 convertase inhibitor, attenuates early human islet allograft damage in vitro and in vivo in a humanized mouse model. Br J Pharmacol. 2016;173(3):575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kassimatis T, Qasem A, Douiri A, et al. A double-blind randomised controlled investigation into the efficacy of Mirococept (APT070) for preventing ischaemia reperfusion injury in the kidney allograft (EMPIRIKAL): study protocol for a randomised controlled trial. Trials. 2017;18(1):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heeger P, Akalin E, Baweja M, et al. Lack of Efficacy of Eculizumab for Prevention of Delayed Graft Function (DGF) in Deceased Donor Kidney Transplant Recipients. Am J Transplant. 2018;18(S4). [Google Scholar]
- 31.Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8(6):582–587. [DOI] [PubMed] [Google Scholar]
- 32.Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28(3):425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheen JH, Strainic MG, Liu J, et al. TLR-Induced Murine Dendritic Cell (DC) Activation Requires DC-Intrinsic Complement. J Immunol. 2017;199(1):278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant. 2013;13(10):2530–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li K, Fazekasova H, Wang N, et al. Expression of complement components, receptors and regulators by human dendritic cells. Mol Immunol. 2011. ;48(9–10):1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liszewski MK, Kolev M, Le Friec G, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):1143–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghannam A, Fauquert JL, Thomas C, Kemper C, Drouet C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol Immunol. 2014;58(1):98–107. [DOI] [PubMed] [Google Scholar]
- 38.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14(2):162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okinaga S, Slattery D, Humbles A, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry. 2003;42(31):9406–9415. [DOI] [PubMed] [Google Scholar]
- 40.D AV, Demir M, Chun N, et al. T Cell Expression of C5a Receptor 2 Augments Murine Regulatory T Cell (TREG) Generation and TREG-Dependent Cardiac Allograft Survival. J Immunol. 2018;200(6):2186–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoermer KA, Morrison TE. Complement and viral pathogenesis. Virology. 2011. ;411 (2):362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190(8):3831–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lublin DM, Atkinson JP. Decay-accelerating factor: biochemistry, molecular biology, and function. Annu Rev Immunol. 1989;7:35–58. [DOI] [PubMed] [Google Scholar]
- 44.Pavlov V, Raedler H, Yuan S, et al. Donor deficiency of decay-accelerating factor accelerates murine T cell-mediated cardiac allograft rejection. J Immunol. 2008;181 (7):4580–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raedler H, Vieyra MB, Leisman S, et al. Anti-complement component C5 mAb synergizes with CTLA4Ig to inhibit alloreactive T cells and prolong cardiac allograft survival in mice. Am J Transplant. 2011;11 (7): 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gueler F, Rong S, Gwinner W, et al. Complement 5a receptor inhibition improves renal allograft survival. J Am Soc Nephrol. 2008;19(12):2302–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horwitz J, Mathern D, Heeger P. C3a Receptor Regulates the CD8 T-Cell Alloresponse via Intrinsic and Extrinsic Mechanisms Am J Transplant. 2018;18(S4). [Google Scholar]
- 48.Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol. 1998;160(11):5273–5279. [PubMed] [Google Scholar]
- 49.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271 (5247):348–350. [DOI] [PubMed] [Google Scholar]
- 50.Marsh JE, Farmer CK, Jurcevic S, Wang Y, Carroll MC, Sacks SH. The allogeneic T and B cell response is strongly dependent on complement components C3 and C4. Transplantation. 2001;72(7):1310–1318. [DOI] [PubMed] [Google Scholar]
- 51.Wang H, Arp J, Liu W, et al. Inhibition of terminal complement components in presensitized transplant recipients prevents antibody-mediated rejection leading to longterm graft survival and accommodation. J Immunol. 2007;179(7):4451–4463. [DOI] [PubMed] [Google Scholar]
- 52.Valenzuela NM, McNamara JT, Reed EF. Antibody-mediated graft injury: complement-dependent and complement-independent mechanisms. Curr Opin Organ Transplant. 2014;19(1):33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat Rev Nephrol. 2012;8(11):670–678. [DOI] [PubMed] [Google Scholar]
- 54.Jane-Wit D, Manes TD, Yi T, et al. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-kappaB signaling in endothelial cells. Circulation. 2013;128(23):2504–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stegall MD, Diwan T, Raghavaiah S, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011; 11(11):2405–2413. [DOI] [PubMed] [Google Scholar]
- 56.Locke JE, Magro CM, Singer AL, et al. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant. 2009;9(1):231–235. [DOI] [PubMed] [Google Scholar]
- 57.Burbach M, Suberbielle C, Brocheriou I, et al. Report of the inefficacy of eculizumab in two cases of severe antibody-mediated rejection of renal grafts. Transplantation. 2014;98(10):1056–1059. [DOI] [PubMed] [Google Scholar]
- 58.Montgomery RA, Orandi BJ, Racusen L, et al. Plasma-Derived C1 Esterase Inhibitor for Acute Antibody-Mediated Rejection Following Kidney Transplantation: Results of a Randomized Double-Blind Placebo-Controlled Pilot Study. Am J Transplant. 2016;16(12):3468–3478. [DOI] [PubMed] [Google Scholar]
- 59.Michielsen LA, van Zuilen AD, Muskens IS, Verhaar MC, Otten HG. Complement Polymorphisms in Kidney Transplantation: Critical in Graft Rejection? Am J Transplant. 2017;17(8):2000–2007. [DOI] [PubMed] [Google Scholar]
- 60.Wang Z, Yang H, Guo M, et al. Impact of complement component 3/4/5 single nucleotide polymorphisms on renal transplant recipients with antibody-mediated rejection. Oncotarget. 2017;8(55):94539–94553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griesemer AD, Okumi M, Shimizu A, et al. Upregulation of CD59: potential mechanism of accommodation in a large animal model. Transplantation. 2009;87(9):1308–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michielsen LA, van Zuilen AD, Kardol-Hoefnagel T, Verhaar MC, Otten HG. Association Between Promoter Polymorphisms in CD46 and CD59 in Kidney Donors and Transplant Outcome. Front Immunol. 2018;9:972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Budding K, van de Graaf EA, Kardol-Hoefnagel T, et al. A Promoter Polymorphism in the CD59 Complement Regulatory Protein Gene in Donor Lungs Correlates With a Higher Risk for Chronic Rejection After Lung Transplantation. Am J Transplant. 2016;16(3):987–998. [DOI] [PubMed] [Google Scholar]
- 64.Loupy A, Lefaucheur C, Vernerey D, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med. 2013;369(13):1215–1226. [DOI] [PubMed] [Google Scholar]
- 65.Zeevi A, Lunz J, Feingold B, et al. Persistent strong anti-HLA antibody at high titer is complement binding and associated with increased risk of antibody-mediated rejection in heart transplant recipients. J Heart Lung Transplant. 2013;32(1):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Witt CA, Gaut JP, Yusen RD, et al. Acute antibody-mediated rejection after lung transplantation. J Heart Lung Transplant. 2013;32(10):1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wiebe C, Gareau AJ, Pochinco D, et al. Evaluation of C1q Status and Titer of De Novo Donor-Specific Antibodies as Predictors of Allograft Survival. Am J Transplant. 2017;17(3):703–711. [DOI] [PubMed] [Google Scholar]
- 68.Vakeva A, Lehto T, Takala A, Meri S. Detection of a soluble form of the complement membrane attack complex inhibitor CD59 in plasma after acute myocardial infarction. Scand J Immunol. 2000;52(4):411–414. [DOI] [PubMed] [Google Scholar]
- 69.Budding K, van de Graaf EA, Kardol-Hoefnagel T, et al. Soluble CD59 is a Novel Biomarker for the Prediction of Obstructive Chronic Lung Allograft Dysfunction After Lung Transplantation. Sci Rep. 2016;6:26274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tower CM, Reyes M, Nelson K, et al. Plasma C4d+ Endothelial Microvesicles Increase in Acute Antibody-Mediated Rejection. Transplantation. 2017;101(9):2235–2243. [DOI] [PubMed] [Google Scholar]
- 71.Aibara N, Ohyama K, Hidaka M, et al. Immune complexome analysis of antigens in circulating immune complexes from patients with acute cellular rejection after living donor liver transplantation. Transpl Immunol. 2018;48:60–64. [DOI] [PubMed] [Google Scholar]
- 72.Thurman JM, Le Quintrec M. Targeting the complement cascade: novel treatments coming down the pike. Kidney Int. 2016;90(4):746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]