

Abstract
The cellular network is highly interconnected. Pathways merge and diverge. They proceed through shared proteins and may change directions. How are cellular pathways controlled and their directions decided, coded, and read? These questions become particularly acute when we consider that a small number of pathways, such as signaling pathways that regulate cell fates, cell proliferation, and cell death in development, are extensively exploited. This review focuses on these signaling questions from the structural standpoint and discusses the literature in this light. All co-occurring allosteric events (including posttranslational modifications, pathogen binding, and gain-of-function mutations) collectively tag the protein functional site with a unique barcode. The barcode shape is read by an interacting molecule, which transmits the signal. A conformational barcode provides an intracellular address label, which selectively favors binding to one partner and quenches binding to others, and, in this way, determines the pathway direction, and, eventually, the cell’s response and fate.
Signaling controls cell actions (Clevers and Nusse, 2012; De Trez, 2012; Herr et al., 2012; Ramos and Camargo, 2012), and signaling cannot take place without allostery (Biter et al., 2012; Chennubhotla et al., 2008; Cui and Karplus, 2008; Dixit and Verkhivker, 2011; Endres et al., 2011; Goodey and Benkovic, 2008; Kenakin and Miller, 2010; Korkmaz et al., 2012; Lesne et al., 2012; Long and Bruschweiler, 2011a; Nussinov etal., 2012; Pandini et al., 2012; Piwonski et al., 2012; Selvaratnam et al., 2011; Tsai et al., 2009a; Tsai and Nussinov, 2011; Tzeng and Kalodimos, 2011; Whitley and Lee, 2009; Zhuravlev and Papoian, 2010). Communications from the extracellular space, via extra-membranous domains, the membrane, and the cytoplasm, to, in, and out of the nucleus are all accomplished with the help of allosteric propagation. Via a conformational biasing mechanism, allostery controls pathway divergence and unification; governs pathway branching and direction in scaffolding proteins, as in the case of the yeast mitogen-activated protein kinase (MAPK) (Zalatan et al., 2012); and coordinates molecular machines and multienzyme complexes (Nussinov et al., 2013a).
How are multiple co-occurring allosteric events acting on a protein (or RNA or DNA) read in the cell? While frequently over looked, this question is of fundamental importance because, at any point in time, multiple co-occurring allosteric events take place (e.g., a protein binds multiple partners in a complex, undergoes allosteric posttranslational modifications [PTMs], and interacts with ions and with the membrane). Collectively, available data suggest that allostery exerts conformational control over cellular pathways and the network to decide cell responses; and it performs these tasks by tagging protein binding sites with barcode labels, where a barcode is a composite of all concomitant allosteric actions on the given protein.
To date, studies of signaling on the conformational level have largely focused on individual molecules and a single allosteric event. However, in living cells, signaling proceeds across pathways and pathway crosstalks, which emanates from and leads to simultaneous allosteric actions (Gunasekaran et al., 2004; Kar et al., 2010; Korcsmáros et al., 2010). The complexity of the cellular network is immense: pathways can integrate and branch and the signal they transmit may strengthen or weaken. In the cell, virtually all pathways are interconnected. Cell responses reflect this network connectivity. Interconnectivity, uniting, and partitioning takes place through proteins that are shared by a number of pathways. These are typically signaling proteins that can bind a large number of partners (Tsai et al., 2009b), which raises questions such as (1) how does a shared protein “know” which partner to bind at any given time? (2) What decides pathway branching, integration, and the consequent change in direction of the signaling pathway? (3) How is the signal coded, and how is it read? (4) Is there a unifying mechanism through which signaling proteins communicate in environment- and cell type-dependent (and ligand-independent) ways? When combined with orthosteric (i.e., binding site) PTMs, mutations, and the physiological environment (i.e., ligand concentration, etc.), barcode labels, which reflect the composite of all concurrent allosteric events, can help in the understanding of physiological function, the mode of action of gain-of-function mutations, and drug discovery. Such a barcode concept is rooted in the free-energy landscape description of proteins (Ferreiro et al., 2011; Frauenfelder et al., 1991).
Here, our major premise is that prior allosteric events may (partially) quench the coupling of the signaling protein to some of its partners and favor others and, by so doing, direct the signals toward specific pathways. The collective effect of all co-occurring allosteric actions (e.g., phosphorylation of a specific set of serine and threonine residues) can provide intracellular address labels, leading to a distinct receptor-binding site shape (and dynamics), which differs from those resulting from other PTMs, noncovalent binding, and allosteric (e.g., gain-of-function) mutational combinations. Simply put, each set of allosteric events provides a distinct conformational barcode. Conformational barcodes may have short residence times; however, their duration should be sufficiently long for recognition and binding for the allosteric signal to go through. A signaling protein may have many barcodes; each spells a specific function. Barcodes are imprinted on receptors, scaffolding, and cytoskeleton proteins, which are also allosteric. Collectively, barcodes can direct signaling in the cell by specifying a distinct allosteric relay under a specific set of conditions and, in this way, switch pathway direction. If allostery is not at play, neither signal propagation nor pathway switching will take place. Cheong et al. (2011) analyzed the information transduction capacity of tumor necrosis factor signaling; it will be interesting to assess such signaling capacity of molecular ensembles and cellular networks for imprinting conformational barcodes.
Conformational barcodes and classical barcode descriptions, such as the DNA barcode encoding genetic information, DNA methylation, and the histone code in epigenetic regulation or the stress history determining subsequent cellular responses (Polman et al., 2012), all integrate changes of a large number of parameters at the molecular and the cellular network levels. However, conformational barcodes are much more general and dynamic than the classical barcodes. A few recent publications describe the barcode of G protein-coupled receptor (GPCR) phosphorylation patterns (Butcher et al., 2011; Liggett, 2011; Nobles et al., 2011; Reiter et al., 2012). Our description generalizes these concepts to all cellular functions and accounts for events other than phosphorylation; all determine the functional response. The conformational barcode concept provides a general view of how the cell decodes multiple co-occurring events.
All components of an allosteric barcode merge and cooperatively determine the shape of a protein functional site. In turn, the functional site selects a partner whose shape is most favorable, thereby biasing the pathway in the direction of this partner, while quenching others. This leads to a population shift in the partner (Ma et al., 1999; Tsai et al., 1999). The subsequent change in the partner’s shape may induce additional allosteric events, such as a distinct phosphorylation pattern and binding, which collectively create a new barcode that stimulates specific downstream signaling through the partner. In vivo, in the absence of an agonist, receptors may oscillate between their inactive and active states. The equilibrium typically (though not always) favors the inactive state, and in the absence of an agonist, the time spent in the active state is short. Binding of the agonist stabilizes the high energy active state, which leads to a population shift toward this conformation. Posttranslational modification enzymes (e.g., kinases, acetylases, methylases, etc.) may recognize this populated conformation. PTMs can be allosteric (i.e., away from the binding site) or at the binding site (Figure 1; Nussinov et al., 2012). Their collective outcome will translate into a specific shape, which will selectively bind another protein, with a subsequent population shift. Each conformation of the receptor can select an agonist that is most favorable to its shape and chemistry. In turn, this more populated receptor conformation, which differs from the first, will lead to an altered pattern of posttranslational modification and binding events and different pathways. Agonists may include proteins, RNA, DNA, lipids, small ligands, such as drugs, ions (Liu et al.,2012), light, and changes in pH that will affect protonation states; all are also allosteric effectors.
Figure 1. Posttranslational Phosphorylation of p53 Takes Place at Numerous Serine and Threonine Residues, Providing Barcodes for p53 Signaling.
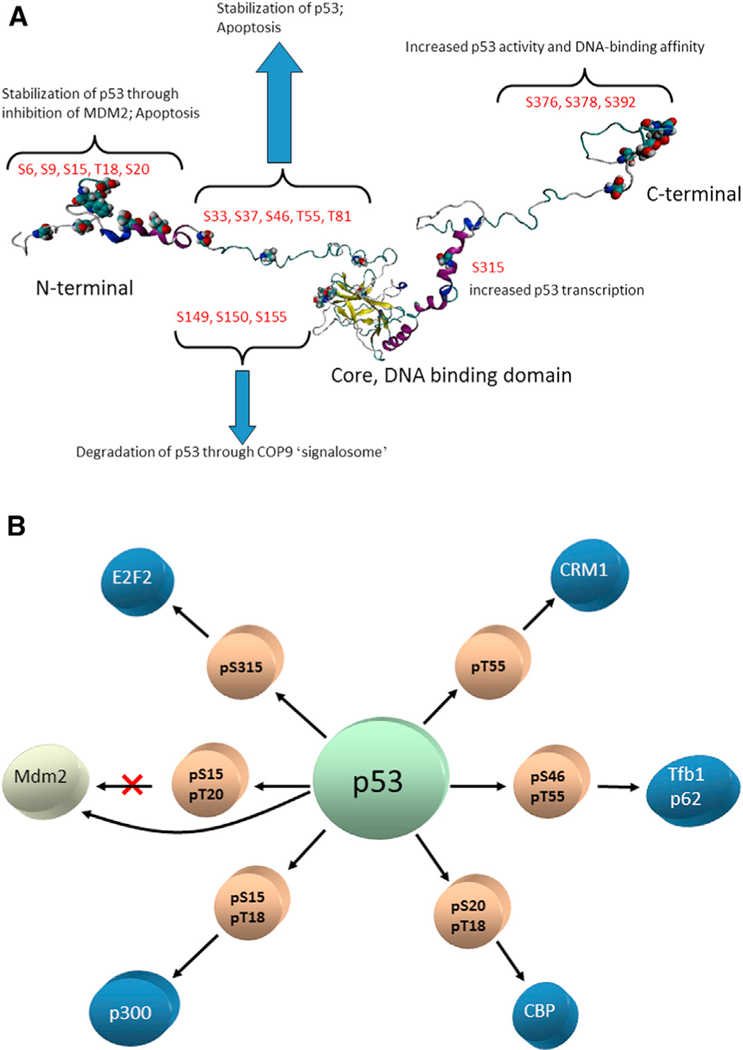
(A) Phosphorylation events in the N-terminal region mostly stabilize the p53 and lead to apoptosis. Several mutations in the core DNA-binding domain can (allosterically, by shifting the equilibrium) destabilize p53, leading to COP9 degradation. Phosphorylation in the C-terminal region can increase p53 activity and p53 DNA-binding affinity. “Signalome” is the cell-signaling network, consisting of highly connected functional modules (Ma’ayan et al., 2005). The COP9 signalosome (CSN) is a conserved protein complex that functions in the ubiquitin-proteasome pathway. It is a multisubunit protease that regulates the activity of cullin-RING ligase. It removes the post-translational modification of ubiquitin-like protein, Nedd8/Rub1, from thecullin component of the ubiquitin E3 ligase (i.e., deneddylation). In addition, it is associated with deubiquitination activity and protein kinase activities capable of phosphorylating important signaling regulators. CSN is a key player in the DNA-damage response, cell-cycle control, and gene expression (Wei and Deng, 2003; Wei et al., 2008). The figure illustrates that PTMs (here, phosphorylation) at the functional sites (i.e., orthosteric PTMs in the body of the text) can serve as barcodes. The N-terminal phosphorylation events are allosteric. In this paper, we focus on allosteric barcodes. (B) p53 is one of the most connected nodes in either the protein-protein interaction network or the gene regulation network. The central circle represents both wild-type p53 and isoforms; the light brown circles are phosphorylation events required for turning on/off specific p53-protein interactions. The blue circles represent proteins interacting with p53. Note that the phosphorylation of Ser15 and Ser20 blocks the p53-MDM2 interaction. The figure is adapted with permission from Tsai et al., (2009b).
Here, we conceptualize allosteric effects as conformational barcode tags along pathways in the cell. A conformational barcode is the result of the unification of all allosteric events acting on a protein at a given time, collectively shifting its conformational ensemble and, by so doing, determining its functional site shape and dynamics. A protein can have many conformational barcodes; these can be the outcome of physiological regulation and allosteric, disease-related events (Nussinov and Tsai, 2013). Below, we relate to and provide examples of both. Conformational barcodes are important because they describe how the combinations of all allosteric events—beyond the PTMs, which to date were credited as defining barcodes—are read by partners and, as such, can explain pathway switching and cell fates.
What Is a Barcode?
The classical definition of a barcode is an optical machine-readable representation of data relating to the object to which it is attached. Linear, or one-dimensional (1D) barcodes represent data by varying the widths of parallel lines and the spaces between them. Geometric patterns in two dimensions (2D), such as rectangles and hexagons, are also increasingly common (Figure 2A). Optical bar code scanners read and interpret the code. Because there are many ways to construct and arrange the shapes and spaces, numerous symbologies are possible; each provides unique information. The scanned data are transmitted and relayed for an appropriate application.
Figure 2. The Concept of a Conformational Barcode.
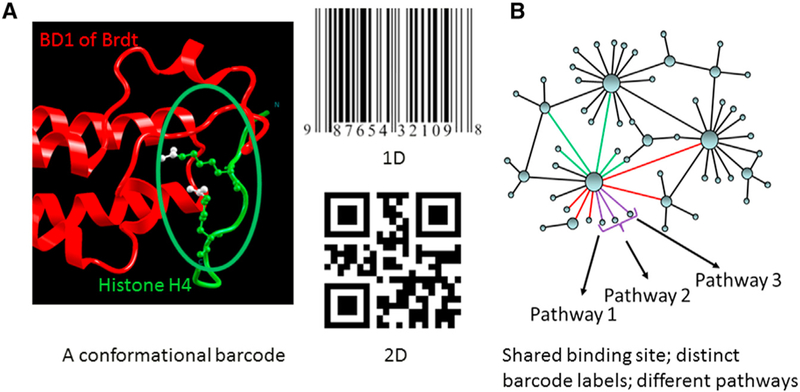
A barcode is a readable representation of data relating to the object to which it is attached. Linear, or 1D, barcodes represent data by varying the widths of parallel lines and the spaces between them; geometric patterns in 2D can employ rectangles and various combinations, including hexagons. Protein-binding site barcodes are high-dimensional. (A) illustrates two acetylation marks (double orthosteric PTMs) on a histone H4 tail (red) recognized by the bromodomain (BD1) of Brdt (green), a testis-specific member of the BET protein family (Protein Data Bank [PDB] 2WP2; Morinière et al., 2009). That BD1 fails to bind monoacetylated H4 tail illustrates the essential role of combinatorial PTM barcode in interaction specificity. The acetylated lysines at positions 5 and 8 are shown. (B) highlights the role of a barcode in the cellular network. Proteins are depicted as nodes and interactions as edges. Some proteins (hubs) can have a large number of partners. A shared binding site of a hub protein can bind multiple partners. Proteins with similar color edges (purple or green) share the same site. Binding of a specific protein determines the signaling pathway. The binding event that takes place at a given time is largely governed by the barcode at the shared binding site.
Translating it to proteins, each functional site shape can be viewed as a barcode, as in the case of the histone recognition code (Musselman et al., 2012); each shape recognizes a specific ligand. The classical barcode concept relates to PTMs that are at the functional site. The histone barcode description captures multiple different PTM combinations in the same (10–15 amino acids) segment of protein sequence. Such a barcode concept applies to direct recognition, where phosphorylation, acetylation, and methylation are all occurring within the same short stretch of sequence. In contrast, allosteric barcode encompasses a much larger part of the protein. For example, β-arrestin 2 sites, which are phosphorylated by GRK2 (i.e., T360, S364, S396, S401, S407, and S411), are responsible for GPCR internalization (Reiter et al., 2012). Thus, while some PTMs may be located in a short sequence stretch and be directly recognized, others are across the molecule and may work via long range conformational effects, and in still other proteins, the effects of both types may combine. Allosteric barcodes may further integrate effectors other than PTMs.
Binding-site shapes are encoded by two major elements: allosteric events away from the binding site and posttranslational modifications at the binding site, which, following the drug terminology, we call “orthosteric PTMs.” Protein recognition domains provide examples where these PTMs are read directly. Figure 1A presents an example relating to serine phosphorylation barcodes. Here, with the exception of the phosphorylation events at the N-terminal, which prevent p53-MDM2 interaction far away, all others are orthosteric and thus recognized directly. However, the consequences of these binding events may also be allosteric. The shape is determined by the collective outcome of all concomitant orthosteric PTMs and allosteric events. Because the number of possible PTMs is large (over 300; Nussinov et al., 2012), and the number of partners binding noncovalently to a given protein, particularly receptors and signaling proteins, can also be large, in principle, the number of possible codes can be very large. Each shape can encode a specific function. The barcode is likely to change in the presence of allosteric gain-of-function mutations. By selecting one specific partner over another (Figure 2B), this variability of the barcodes in a specific protein can control branching and integration of signaling pathways and, in this way, spell cellular response. In the presence of gain-of-function mutations, shifting the pathway may lead to drug resistance.
Functional sites provide conformational barcodes, each of which is an intracellular address label. These conformational barcodes differ from the “conventional” barcodes in four ways: first, in the types of symbology that can be employed (i.e., conformational features instead of combinations of simple geometric shapes); second, rather than a series of symbols that are read and integrated, a conformational barcode and the chemistry that it presents (charged groups, hydrogen bonds, hydrophobic patches, etc.) cannot be separated into distinct components. It is one unit that has to fit well and get selected. Third, the barcodes are sensitive to environmental conditions; fourth, conformational dynamics can be a critical component of the barcode. A conformational barcode does not necessarily consist of a structural change; it can be represented by changes in shape and dynamics or only by changes in the molecular fluctuations. Thus, unlike the 1D (or 2D) conventional barcodes, conformational barcodes are high-dimensional. Mechanistically, they may reflect protein frustration (Zhuravlev and Papoian, 2010), dynamic prestress, and dynamic changes in the rigidity of protein structures (Csermely et al., 2013; Gáspár and Csermely, 2012). All tally to the high dimensionality.
Unlike the “conventional barcodes,” where the optical machine-readable representation of data is translated into a set of numbers, our conformational barcode description lacks quantitation. Quantifying the collective effects of all co-occurring allosteric events, including gain-of-function mutations, is challenging. Nonetheless, allosteric barcodes are useful in providing a comprehensive framework of the cellular network from the functional standpoint, helping to understand how allosteric events can contribute to pathway divergence.
Barcodes Are the Result of Population Shift
In vivo, signaling can initiate or be altered by noncovalent binding and covalent posttranslational modifications. Three theories have been proposed to explain molecular recognition and binding in signaling (Figure 3). The first was the “lock and key” mechanism, which considered the protein and the ligand as rigid molecules that require precise conformational match to form a functional complex. The “lock and key” mechanism is unable to explain the heterogeneity of the recognition where one receptor is able to bind at the same site multiple different ligands, as in the case of the EphA4 receptor and the nine ephrin ligands (Lackmann and Boyd, 2008); it is also unable to explain the modulation of the signaling in the pathway. The second was the “induced fit” hypothesis, which argues that, in complexes, the partners may have different conformations from those observed in their unbound states and that those bound conformations are “induced” by the binding partners. However, a large body of experimental (Boehr et al., 2009) and computational (Long and Bruschweiler, 2011b; Wlodarski and Zagrovic, 2009) data indicate that the bound state conformation already exists before binding. In the late 1990s, we proposed a third theory, that of the “conformational selection and population shift” (Tsai et al., 1999). This theory recognized that biological macromolecules pre-exist in a broad range of conformations, among which are the bound state conformers, and suggested that the most complementary conformer binds, followed by population shift of the ensemble to restore the equilibrium. Conformational transitions between the species determine the time scales of the population shift for the binding reaction. The higher the barriers that need to be overcome in the conformational transitions, the slower the population shift. Conformational selection is followed by induced fit, which involves minor backbone and side-chain conformational changes to optimize the interaction (Csermely et al.,2010). We further proposed (Ma et al., 1999; Tsai et al., 1999) that evolution exploited the pre-existing conformations and optimized and tuned them for cellular life. Thus, the concepts of the pre-existence of all functional conformational states, which allows conformational selection, and of the population shift are powerful in explaining functional mechanisms. They also relate binding to population shift in allosteric transitions. Conformational selection and population shift take place in protein-protein interactions, protein-RNA, and protein-DNA. They are important in protein-small molecule binding, including drugs and metabolites, and in binding of lipids. All can induce allosteric propagation.
Figure 3. Schematic Illustrations of Binding by “Lock and Key,” “Induced Fit,” and “Conformational Selection” Models.
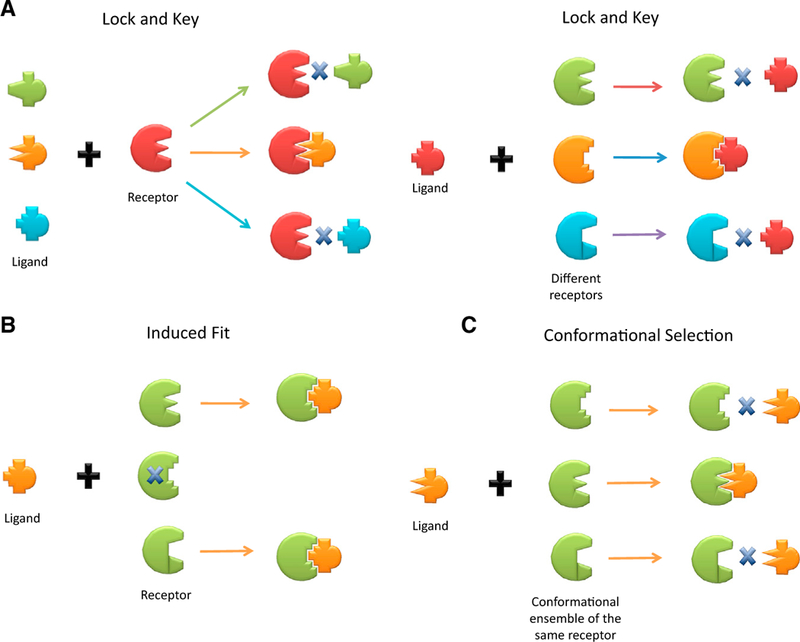
(A) In (A), the drawing emphasizes the fact that the lock and key model only allows a binding to happen with an exact fit between complementary geometric shapes of ligand and receptor. The cross bar drawn in between implies no ligand-receptor binding can occur. Either the case of a receptor selecting among different ligands or the reverse of a ligand selecting among different receptors are depicted, with the binding exclusively determined by the exact lock-and-key fit criterion. (B and C) Different colors imply different receptors. In contrast to the lock and key model, the “induced fit” model highlighted in (B) indicates that there is no exact fit in the prebinding conformation between the ligand and receptor, as shown by the cross bar on the receptor fitted exactly to the ligand, and the consequent binding ends up with the receptor being induced to change the shape to fit into that of ligand. In the “conformational selection” model, the emphasis of the illustration in (C) is placed on the availability of pre-existing, different receptor conformations, and the ligand selects and binds the receptor via the lock and key criterion. Here, different conformations of the same receptor are in the same color (green). The figure emphasizes that the lock-and-key hypothesis considers many different receptor molecules. Each receptor has one specific structure. One of the receptor molecules (lock) fits the ligand (key). In contrast, in conformational selection, the ensemble consists of many distinct conformations of the same receptor molecule.
In a similar manner, signaling may start from a PTM event. PTM is a covalent linkage of a chemical group on the protein surface. The enzymatic reaction can take place during or following protein synthesis. PTMs can be small or large, hydrophobic or charged. They range from phosphate group, acetyl, methyl, to a fatty acid moiety, and to a peptide, such as ubiquitin or ubiquitin-like chains, like Sumo. PTMs can take place at the binding site, in which case they are directly recognized by recognition domains, or elsewhere on the protein surface, in which case they act allosterically. The perturbation to the protein target caused by covalent binding is expected to be at least as large as that incurred by noncovalent binding. PTM events can also take place on nucleic acids, for example, DNA methylation. Population shift explains cooperativity, and barcodes are the result of population shift following such perturbation events.
Allosteric Barcodes: Origins and Consequences
The structural details and the contacts that the partners make, even when binding a shared site, differ; thus, the allosteric consequences vary. Each event initiates a distinct distribution of allosteric pathways and may result in (slightly) different binding site shape and dynamics, which may affect the selection of a binding partner elsewhere and the affinity of that interaction. Since proteins can bind multiple partners simultaneously, as, for example, in the case of transcription factors, a large number of possible combinatorial effects may result. The partners are not necessarily proteins: they can be DNA or RNA; small signaling second messengers, such as cyclic adenosine monophosphate (cAMP); ATP/GTP, nicotinamide adenine dinucleotide (NAD)/ reduced NAD, or flavin adenine dinucleotide; or ions (e.g., Ca2+ and Zn2+) or lipids, etc.; the effects can be compounded by physical environmental factors. Since some (hub) proteins can have tens or hundreds of partners, the number of combinations can be very large.
The multiple PTM types and sites in a protein can further contribute to make the number of possible combinatorial allosteric outcomes vast. There are hundreds of PTM types, some small (e.g., hydroxylation and methylation), some large (ubiquitination), some charged (phosphorylation and acetylation), some hydrophobic (alkylation and myristoylation), and some involving sugars. PTMs are common, and a site may be modified by several PTM types. PTM addition and removal can result in a huge number of possible combinations, which serve to expand the functional breadth of the proteins in pathway modulation. For example, transcription factor p53 contains at least 50 PTM sites (Meek and Anderson, 2009); the FoxO family of Forkhead transcription factors undergoes phosphorylation, acetylation, and ubiquitination, with distinct PTM combinations acting as specific codes. Seventeen potential PTM sites were found in FOXO3a, and it was suggested that single and binary modifications could result in up to 131 072 (217) distinct PTM patterns (Benayoun and Veitia, 2009). Because the perturbation caused by each type of PTM is different and also depends on the protein structural environment, the range of collective variations in the population shift as expressed by the allosteric effect at the functional site is very large. Combined with simultaneous noncovalent binding events, they can code for specific functions. Figure 4 illustrates schematically possible scenarios where PTMs with different volumes (e.g., methylation versus ubiquitination), chemical properties (e.g., the negatively charged phosphate or acetyl group versus palmitoylation, farnesylation, or alkylation), protein structural environments, and combinations, together with non-covalent events, can lead to large variation in a shared binding site, leading it to preferentially select a certain partner over another. These scenarios are described via detailed examples.
Figure 4. Schematic Diagrams of Allosteric PTM Examples.
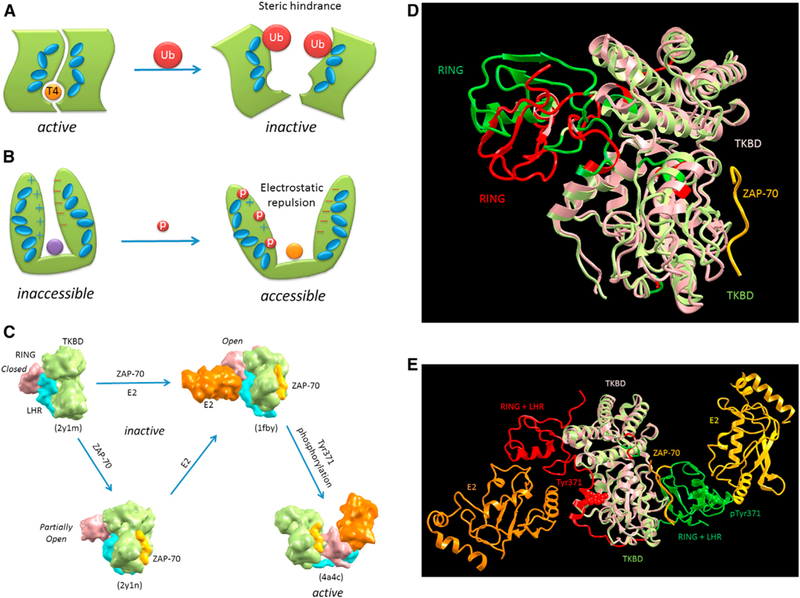
(A) illustratesan activethyroid hormone-activating type 2 deiodinasewith a bound T4 substrate. It istransiently inactivated by ubiquitylation at the dimer interface. The allosteric action of the ubiquitylations is illustrated by the interacting blue ellipsoids, which propagatethe steric hindrance down to the T4 binding site (Sagar et al., 2007). (B) Progressive phosphorylation events up to a certain threshold number at the N-terminal region of circadian clock protein, FRQ, allosterically expose its protected middle region (purple circle) and lead to its own degradation (Querfurth et al., 2011). The allosteric action is illustrated by the interacting blue ellipsoids, which propagate the created electrostatic repulsion between the N-terminal and C-terminal of FRQ down to the exposed middle region (orange circle). (C-E) A phosphorylation-dependent activation of c-Cbl, a RING ubiquitin ligase that attenuate receptortyrosine kinase signal transduction, is illustrated by four captured crystal structures with a combination of two involved allosteric actions. (C) provides a model for the binding of substrate sequences (the ZAP-70 peptide) to the N-terminal SH2-containing tyrosine kinase-binding domain (TKBD) and of ubiquitin-conjugating enzyme (E2) and Tyr371 phosphorylation, together leading toward c-Cbl activation (Dou et al., 2012). In the absence of E2 and the TKBD substrate, the c-CBl (a single-subunit RING E3 that negatively regulates proteins by promoting ubiquitination and subsequent degradation; PDB 2Y1M) adopts a closed conformation, in which the E2-binding surface of RING subdomain associates with the TKBD. The allosteric effect of the ZAP-70 peptide binding partially opens the RING subdomain, making it favorable for E2 binding. This is clearly seen from the superposition of the two crystal structures (PDB 2Y1M and 2Y1N) shown in (D), which indicate the partial RING opening (the RING movement from red to green) due to the TKBD substrate binding (yellow ribbon). The subsequent E2 binding causes the RING subdomain to adopt an open conformation. Tyr371, which mediates the linker helix region (LHR) subdomain binding to the TKBD, secures the c-Cbl in an inactive state. The phosphorylation ofTyr371 activates c-Cbl by releasing LHR from TKBD, which then undergoes a large conformational change that brings the RING subdomain and E2 into proximity of the substrate. The large conformational change due to the pTyr371 is seen from the superposition of two crystal structures (PDB 1FBV and 4A4C) in (E). Both the side chains of Tyr371 (red) and pTyr371 (green) are given in space-fill models. (A) and (B) are adapted with permission from Nussinov et al., (2012)
Taken together, this argues for an allosteric barcode where each combination defines a distinct function. An enormously challenging goal is to spell out the functional outcome of specific combinations. The complexities of the tasks are huge; insights into the allosteric conformational barcode will be extremely useful in forecasting consequences of deleterious mutations and of drug regimens not only on the protein but on the pathway level (Lee et al., 2012). Binding of an allosteric drug similarly defines an allosteric barcode at the active site elsewhere.
Allosteric Effects Propagate Across Protein Interfaces
Population shift occurs across proteins (Lee etal., 2008); it spans their interactions with other proteins (Boehr, 2012; Boehr et al., 2009; Lee etal., 2008; Long and Bruschweiler, 2011a; Mackereth et al., 2011; Moal and Bates, 2012), DNA/RNA (Fuxreiter et al.,2011), and lipids along the pathways (Antal et al., 2009; Nussinov et al., 2011) and throughout the cell (Nussinov and Tsai, 2013; Nussinov et al., 2013b). Propagation takes place across multimolecular complexes, in the membrane, cytoplasm, and organelles, as, for example, in the ligase, proteasome, ribosome, where propagation can span proteins and RNA, and chromatin remodeling complexes. These illustrate how allosteric effects encoded by address labels can relay signals across domain and molecular boundaries.
Propagation across Obligate Interactions Can Be Helped by Molecular Disorder
Obligate protein-protein complexes typically involve interfaces with large buried surface areas and tight binding. Propagation is expected to be efficient when the interfaces are tightly packed. This could be particularly the case if the unbound states of proteins are disordered and couple binding and folding (Tsai and Nussinov, 1997, 2011). Disordered proteins (domains) are frequent components of large multimolecular complexes. One example is the 26 subunit Mediator complex, which consists of three modules: tail, middle, and head. Mediator mediates long range signaling between DNA regulatory elements (REs) bound to transcription factors (TFs), the RNA polymerase II (Pol II), and the basal transcription factors. The TF-bound gene-specific REs can be far from transcription start sites. The DNA is an allosteric effector; its binding to the DNA-binding domain of a TF results in allosteric perturbation that propagates to the activation domain of the TF, which can then bind to the tail/middle or head modules of Mediator. The allosteric propagation pathways proceed through the TF-Mediator interface and across the gigantic Mediator and its multiple subunit interfaces. Cryo-EM images illustrate that the outcome is allosteric “shape-shifting” of subunits (MED17, MED18, and MED20) in the Mediator head module ~400 Ǻ away, leading to a jaw-like opening of the head, which then interacts with the basal transcription factors and RNA Pol II (Cai et al., 2010). “Shape-shifting” is observed when a large conformational change is displayed by a sufficiently high population in the ensemble (Tsai and Nussinov, 1997). Thus, the perturbation by the binding of the TF activation domain causes a significant conformational change that propagates via Mediator head to RNA Pol II subunits Rpb4/Rpb7 and finally to the clamp of Pol II to initiate/activate transcription. Bioinformatics analysis found that: (1) the conformational disorder level of the Mediator exceeds that in other complexes of similar size (Cai et al., 2010); (2) disorder is particularly high in the tail and middle modules, which emerged more recently; (3) the extent of disorder increases through evolution (from yeast to human); and (4) the disordered regions arrangements and interaction sites are similar between yeast and human, which indicates specific recognition at protein-protein interfaces, as further corroborated by available crystal structure data (Seizl et al., 2011). The different DNA sequences of the DNA REs selectively bind the complementary conformation of the functional site on the TF DNA-binding domain. The subsequent population shift allosterically stamps address labels on the TF activation domains (AD). In p53, activating domain (p53AD) and p53 C-terminal domain (p53CTD) interact with MED17 and MED1, respectively, and these interactions affect Mediator structure and Pol II activity (p53AD induces pocket formation in MED17 and this correlates with activation of stalled Pol II, which is not the case for the p53CTD-MED1 interaction). Gain-of-function mutations of p53AD residues L22Q and W23S disrupted the MED17 interaction and thus Pol II binding, apparently due to Mediator’s shape-shifting, which in turn is the outcome of a conformational change in the p53AD binding site (Meyer et al., 2010).
Disorder is frequent in proteins involved in cell signaling (lakoucheva et al., 2002; Zhou, 2012). Analysis of a database of signaling proteins found that there is significantly more predicted disorder in signaling and cancer-associated proteins than in several other categories of protein function, such as metabolism, biosynthesis, and degradation. Because in disordered states the differences in energy among the states can be small and the barriers separating them low, they might be more sensitive to barcode combinations.
Propagation Takes Place across Transient, Dynamic Interactions
Interfaces of short-lived interactions are usually small and often with a limited region of tight packing. This does not hamper allosteric propagation, which can proceed even through small chemical groups; a striking recent example has shown that small variation in a ligand is sufficient to transform an agonist into an antagonist (Sadowsky et al., 2011). That small structural variability can lead to alteration in the allosteric signals can also be seen by mutations. Regulation and crosstalk between pathways largely take place via short-lived interactions. The molecules (or preformed complexes) bind, initiate or transmit the signal, and dissociate, followed by other binding events, as, for example, in the case of Ras protein (a guanosine triphosphatase [GTPase]), which controls the cell growth and differentiation signaling pathways. Ras forms transient complexes with GTPase-activating proteins (GAPs) switching it OFF and with guanine nucleotide-exchange factors switching it ON when the cell is activated or resting in response to stimuli, respectively. The key point is the binding kinetics: so long as the proteinprotein (or ligand) residence time is sufficient for the allosteric propagation wave to go through, the association will be productive in signaling (Stein et al., 2009).
Propagation across Domains
Domain boundaries resemble intermolecular boundaries, and allosteric effects also propagate across domain borders. We cited above the propagation between a TF DNA binding domain and the activation domain; another striking recent example is that of the androgen receptor (AR). There, a study inspired by mutations related to the androgen insensitivity syndromes and prostate cancer observed that AR-ligand-binding domain mutations D695N, R710A, F754S, and P766A induced a decrease in DNA binding but left the ligand binding unaffected; however, DNA-binding domain mutations K590A, K592A, and E621A lowered the ligand binding affinity but not DNA binding (Helsen etal., 2012).
Allosteric Conformational Switches via Phosphorylation Barcodes: The Kinase-Mediated Phosphorylation of GPCRs Example
GPCRs sense extramembranous molecules, such as neurotransmitters, hormones, and pheromones, whose binding activates signaling pathways and cellular responses. GPCRs exist in a broad range of conformational states. In the absence of the ligand agonist, they fluctuate between the active and the more highly populated inactive conformations. Agonist binding selectively stabilizes one of the active GPCR conformations, which leads to a population shift. A different agonist may stabilize another. Heteromeric G protein recognizes a specific agonist-bound state. The dissociation of its α or β and γ subunits signals to effectors such as adenylyl cyclase, leading to a change in the concentration of second messenger cAMP, which affects cellular function. However, with continuous agonist binding, GPCR signaling wanes, becoming desensitized. Desensitization takes place via phosphorylation by GPCR kinases (GRKs), often at the GPCRs’ Ser or Thr residues in the third intracellular loop or in the cytoplasmic tail (there are 11 Ser and Thr residues in the cytoplasmic tail of β2AR), which are at the binding sites of the G proteins. Many possible combinations of Ser and Thr phosphorylation patterns can be obtained, each the result of a different GRK (e.g., GRK2 or GRK6), which likely favor binding to a distinct populated GPCR ligand-bound conformation. The phosphorylated GPCRs become substrates for β-arrestins, which not only partially quench G protein binding but can stimulate signaling through the action of the β-arrestin scaffolds. Distinct phosphorylation patterns may select specific β-arrestin conformations; in turn, each β-arrestin conformation can select its specific partner and, in this way, the effect influences downstream signaling via the β-arrestins. β-arrestins serve as multiprotein scaffolds, regulating a large number of signaling molecules, including the MAPKs ERK, c-Jun N-terminal kinase (JNK), and p38 as well as Akt, PI3 kinase, and RhoA. For example, b-arrestin1 can recruit c-Src, a nonreceptor tyrosine kinase family member, to 7TMRs. Src recruitment to the β2AR leads to ERK activation. β-arrestin2 scaffolds appear to act in ERK activation by mediating binding of ERK1/2 and Raf1 and MEK-1. ERK1/2 and Raf-1 bind β-arrestin2 directly, and the mitogen-activated protein kinase kinase MEK-1 binds indirectly. The β-arrestin scaffold also mediates JNK3 activation. Similar to ERK, JNK3 and apoptosis signaling kinase 1 all bind β-arrestin2 directly, whereas MAP kinase kinase 4 binds indirectly (DeWire et al., 2007). A different conformation of β-arrestin2 is likely to be involved in each pathway (Xiao et al., 2004).
These downstream signals are configured by β-arrestin conformations, which are specified by the GRK barcoding phosphorylation patterns on the receptors (Butcher et al., 2011; Liggett, 2011; Nobles et al., 2011). Each GRK can phosphorylate different serine and threonine residues on a given ligand-bound receptor conformation. Thus, ligand binding biases the GPCR ensemble, which is read by the GRKs. The GRKs redistribute the population of the phosphorylated GPCRs, tagging them by new barcodes. These are read by the ensemble of the β-arrestins. The pattern of GRK phosphorylation sites and the selection of the conformation of β-arrestin can differ substantially, even among closely related GPCRs that are stimulated by the same agonist, which can further diversify its effect on downstream signaling (Nussinov et al., 2013b).
How Diversified Allosteric Barcodes Establish Distinct Types of Molecular Switches: Conformational Switches in the Kinases
Conformational switches do not always involve allosteric effects (Bradshaw, 2010). The Src, Syk, and Tec types of molecular switches provide examples (Figure 5; Bradshaw, 2010; Deindl et al., 2007; Filippakopoulos et al., 2008; Jura et al., 2011; Moarefi et al., 1997; Tsang et al., 2008). Protein kinases, a large family which constitutes almost 2% of the human genome, possess a very similar enzymatic core domain, with the active site located at the cleft between its N-lobe and C-lobe subdomains (Figure 5A). It has been well established that all kinase catalytic activities are regulated through three core actions: (1) orthosteric phosphorylation of key residues located in the activation loop for organizing and/or stabilizing the active conformation; (2) allosteric binding via intra- or inter molecular interactions in the N-lobe (such as the PIF binding pocket) to position the αC-helix in an active configuration; and (3) autoinhibitory binding at various allosteric sites to suppress kinase activity. Figure 5A highlights the orthosteric phosphorylation and the allosteric intramolecular interaction in the Fes kinase. The phosphorylated Tyr (pY713) in the activation loop as well as the tight binding between the SH2 domain and the αC-helix due to electrostatic interactions between Glu (E469 and E472) and Arg (R609) residues are indicated in the figure. Figures 5B and 5C depict crystal structures of suppressed kinase activity, respectively, for the Src kinase and the Syk kinase Zap-70. In Figure 5B, the conformation of inactive Src kinase was superimposed on an active kinase core domain with an orthosteric phosphorylation (pTyr416), revealing a disorientated N-lobe in the inactive kinase. The reason for the distorted kinase domain appears to be due to the intramolecular SH3 binding, mediated by the SH2-kinase linker that interacts with kinase N-lobe. Also highlighted in Figure 5B is the phosphorylated Tyr at the kinase C-terminal (pTyr527) bound to the SH2 domain, which plays a significant role in stabilizing the intramolecular SH3 binding. In Figure 5C, the Syk family kinase Zap-70 is captured in the crystal in an autoinhibitory configuration with a similar organization but different mechanism as compared to the case of Src kinase above. In contrast to Src, not much distortion in the N-lobe is observed based on the superposition to an active Syk kinase with dual orthosteric phosphorylation (Y492 and Y493). Therefore, it has been suggested that rigidification of the SH2-kinase linker, due to the interaction with inter-SH2 linker, is responsible for autoinhibition in Zap-70 by reducing kinase flexibility at the catalytic cleft.
Figure 5. An Illustration of Distinct Molecular Switches that Can Be Decided bythe Diverse Allosteric Barcodes in Cytoplasmic Tyrosine Kinases.
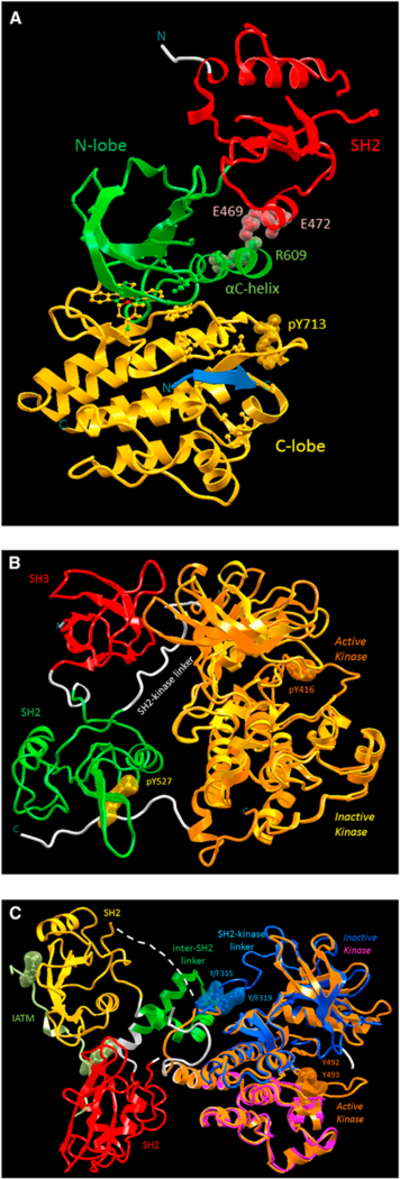
(A) These barcodes are expressed by three catalytic regulation mechanisms. These include (1) orthosteric phosphorylation of Tyr residues in the activation loop; (2) allosteric activation through binding at the N-lobe of kinase core domain; and (3) autoinhibitory binding at various allosteric sites. The first two regulation events are revealed in (A) with the example of active Fes kinase conformation (PDB 3CD3). The active site of Fes kinase is shown in the cleft between N-lobe (green) and C-lobe (yellow) subdomains, with the αC-helix positioned by the SH2 (red) subdomain in an active orientation as well as the activation loop, which is organized by the orthosteric phosphorylation of pY713to an active configuration. The allosteric intramolecular interactions are highlighted by the tight electrostatic interactions between residues Glu (E469 and E472) in SH2 and Arg (R609) in αC-helix. (B and C) The third autoinhibitory regulation event is depicted in (B) and (C) with the examples of Srcand Syk kinase, respectively. In (B), the inactive Src kinase (PDB 1FMK) is superimposed onto an active kinase (PDB 1YI6) with an orthosteric phosphorylation (pTyr416). When compared to the (superimposed) active kinase (orange), the disoriented N-lobe (yellow) from the inactive Src kinase reveals that the autoinhibitory mechanism is through the intramolecular SH3 (red) binding to the kinase N-lobe, mediated by the SH2-kinase linker (white). The autoinhibitory mechanism is also helped by the SH2 (green) binding to the kinase C-terminal through the phosphorylated Tyr (pTyr527). In (C), the inactive Syk kinase Zap-70 (PDB 2OZO) is superimposed on an active kinase (PDB 1U59) with dual orthosteric phosphorylation events (Y492 and Y493) and by an ITAM peptide (PDB 2OQ1) bound to both SH2 domains. The autoinhibitory mechanism suggested that the rigidification of SH2-kinase linker (blue) is due to the interaction with inter-SH2 linker (green), which reduces kinase flexibility at the catalytic cleft. Therefore, the activation of Syk kinase can be achieved either through the phosphorylation within SH2-kinase linker (pY315 and pY319) or binding of phosphorylated ITAM peptide (light green) with the two highlighted phosphorylated Tyr to the tandem SH2 subdomains. The mechanism of the disrupting interactions between inter-SH2 linker and SH2-kinase linker in the former action is orthosteric and is allosteric in the latter. See text for further detailed description of distinct molecular switches observed in cytoplasmic tyrosine kinases.
Cytoplasmic (also known as nonreceptor) tyrosine kinases, the Src, Syk, and Tec kinase families, are essential for signaling of antigen and Fc receptors in hematopoietic cells. Current data suggest that regulation can take place via three types of switches: Src kinase activation functions as a graded switch; Syk kinase activation as an OR-gate switch; and Tec kinase activation as an AND-gate switch. Here, we illustrate how diversified allosteric barcodes establish distinct types of molecular switches. An ideal graded switch can be defined as one where (1) the partial contributions of events toward activation are always additive and (2) full activity requires all contributions from every activation event. The Src kinase has been shown to incrementally increase its enzymatic activity with individual allosteric autoinhibition disruption events, such as dephosphorylation of the C-terminal tail (pY527; Figure 5B) and ligand binding either to the SH2 subdomain or to the SH3 subdomain, as well as orthosteric phosphorylation within the activation loop (pY416; Figure 5B). The definition of an OR-gate switch, as the name suggests, requires only one single activation event to reach full activity. The Syk kinases reach full activity through an allosteric autoinhibition disruption event: either the binding of phosphorylated immunoreceptor tyrosine-based activation motif (ITAM) peptide to the tandem SH2 subdomains (Figure 5C) or phosphorylation within the SH2-kinase linker (pY315 and pY319; Figure 5C). The AND-gate is switched on in an all-or-none fashion only after all activation events. Current data suggest that, to have high activity, the Tec kinase requires both activation loop phosphorylation and docking of the SH2- kinase linker against the kinase (with a similar mechanism to that shown in Figure 5A; Bradshaw, 2010).
Why do they function as distinct types of switches, even though the Src, Syk, and Tec in the cytoplasmic tyrosine kinase family have similar kinase core domain and similar full chain arrangement (with additional SH2 and SH3 subdomains)? Structural insight into the allosteric barcodes may provide answers. First, although the core domains among kinase families are very similar, evolution has ascribed different choices of activation events in different families. For Syk kinase, in addition to the role of organizing the activation loop in the active conformation, orthosteric phosphorylation is also able to propagate local environmental changes to remotely position the αC-helix in an active configuration. Therefore, when the autoinhibition interaction is disrupted either by orthosteric phosphorylation events within the SH2-kinase linker (pY315 and pY319) or by an allosteric ITAM binding, the autophosphorylation (pY491 and pY493) within the activation loop is able to fully activate the Syk kinase. For the AND-gate switch, the activation of the Tec kinase simply requires well-built cooperativity among all activation events in order to shift the dominant kinase population toward the active conformation. Note that no negative autoinhibition disruption action is involved in the Tec kinase case, only positive activation. In a graded switch, a single activation event—whether a disrupted or formed interaction—is unable to shift the active kinase conformation to make it the dominant population in the ensemble. Instead, each activation event adds in a step-by-step manner to shift the most favorable conformation toward the active state.
Conformational Barcodes, Diseases, and Allosteric Drug Therapeutics
Diseases can be caused by external pathogens, such as bacteria, fungi, viruses, protozoa, multicellular organisms, and aberrant proteins, such as prions, or by internal dysfunction. Mutations that lead to failure to produce proteins in correct amounts—too low or too high—can disrupt metabolic or regulatory signaling pathways. Binding of pathogens, gain-of-function mutations, the sun, and irradiation can all have a key role in shaping barcodes, impacting molecular recognition and, thus, cellular pathways (Nussinov and Tsai, 2013).
Binding of Pathogens
Allosteric binding of pathogen proteins may interfere with regulated signaling and lead to disease by similarly causing a population shift. Oncogenic proteins E6 and E7 of human papillomavirus (HPV) provide one example. E6 binds host regulatory proteins, such as p53 and SAP97, and mark these proteins for destruction. E6 binds simultaneously to the three PDZ domains of SAP97 in vitro, leading to their reorganization. Nuclear magnetic resonance experiments detected residues in PDZ2 that are not part of the peptide-binding pocket but experience chemical shift changes upon binding of E6 through an intradomain allosteric effect, which may modulate SAP97 binding to a partner (Chi et al., 2011). A mutant E6 protein lacking the PDZ-binding motif is found at lower levels because of rapid degradation by the proteasome (Nicolaides et al., 2011). Poliovirus (PV) provides an example of a pathogen that initiates allosterically downstream events, leading to cell death. In PV-infected mice, motor neurons undergo apoptosis by PV activation of Bax, a proapoptotic member of the Bcl-2 family, mediated by JNK (Autret et al.,(2007). Bax activation involves a conformational change that exposes the N-terminal of Bax, which leads to its translocation from the cytosol to the mitochondria. Finally, an example is related to apamin, an 18 amino acid peptide neurotoxin in apitoxin (bee venom) whose allosteric effects deregulate channels. Activation of small-conductance calcium Ca2+-dependent potassium channels regulates membrane excitability. There are three subtypes (SK1-SK3) in the nervous system, and apamin distinguishes between them. Apamin binds to a three amino acid motif within the S3 to S4 extracellular loop to produce a high-sensitivity allosteric block by regulating the shape of the outer channel pore. Interestingly, the S3 to S4 loop of one subunit in the heteromeric channel overlaps the outer pore of the adjacent subunit (Weatherall et al., 2011). Heteromeric channels are common, and allosteric effects can also be expected in other cases.
UVB Exposure by Sunlight
Ultraviolet B (UVB)-induced DNA damage is the major agent in neomelanoma skin cancer development, but there is increasing evidence for a role for HPVs, including HPV 5 and HPV 8, in sun-exposed body sites. Following UVB exposure, Bak undergoes a conformational change, exposing the BH3 domain, which is then inserted into a groove of another Bak monomer. This initiates Bak oligomerization, which facilitates pore formation in the outer mitochondrial membrane, leading to the release of proapoptotic factors. The HPV E6 protein affects UVB-induced apoptosis, targeting Bak for proteolytic degradation (Simmonds and Storey,.2008). The mechanism is again via a population shift.
Mutations
Most of the known mutations in proteins that have been associated with disease occur in binding (or active) sites. Active site mutations can abolish, weaken, or alter an interaction or a catalytic activity. There are numerous examples (e.g., the C133W mutation in the active site of serine palmitoyltransferase [Gable et al., 2010], active site mutation Y955C in mitochondrial DNA polymerase γ [Estep and Johnson, 2011], half of the mutations in choline acetyl transferase that cause motor disorders are in the active site [Cai et al., 2004], and Lowe syndrome and Dent disease typically present such mutations in the OCRL protein [Pirruccello et al., 2011]). Point mutations can also take place at a PTM site, abolishing the modification and thus affecting the recognition by a PTM recognition domain, also leading to disease. A recent example is the mutation of H2BK123, which abolishes Dam1 methylation (Latham et al., 2011). Finally, the G201V mutation in the human proteasome subunit β-type 8 gene that encodes the immunoproteasome subunit β5i in patients with Nakajo-Nishimura (NNS) syndrome, a distinct inflammatory and wasting disease, is in close proximity to the threonine and lysine in the catalytic center and causes ubiquitinated proteins to accumulate (Arima et al., 2011).
However, in reality, the majority of the gain-of-function mutations are away from the active (or binding) site. For example, the structure of the enzyme galactocerebrosidase (Deane et al., 2011) illustrates that the Krabbe disease-causing mutations, a devastating neurodegenerative disease, are widely distributed throughout the protein. Dominant mutations in glycyl-transfer RNA (tRNA) synthetase (GlyRS) and tyrosyl-tRNA synthetase cause the Charcot-Marie-Tooth (CMT) disease, the most common heritable disease of the peripheral nervous system. The mutations are scattered in the structure and have no apparent unifying connection (Xie et al., 2007). The structure of CMT-causing mutant (G526R) of homodimeric human GlyRS shows that the mutation is at the site of synthesis of glycyl-adenylate; however, the effects are observed 30 Ǻ away, at the stabilized dimer interface. The choline acetyltransferase structure reveals a broad distribution of mutations that cause motor disorders (Cai et al., 2004). Half of the mutations affect enzyme activity directly; the others are distant from the active site and exert indirect effects. In addition to the OCRL protein mutation at the Rab5 binding site, Lowe syndrome and type 2 Dent disease-associated missense mutations affect the short phenylalanine and histidine motif indirectly by destabilizing the RhoGAP fold (Pirruccello et al., 2011). In the NNS syndrome, the G201V substitution caused conformational changes not only in Thr73 and Lys105 at the catalytic site; additional conformational changes were observed in the S8-H3 loop located at the interface between β4 and β5i, which impaired the assembly of the 20S proteasome (Arima et al., 2011).
In all of these cases, the mutations were away from the functional site; the outcome of all leads to disease. The connection between the mutations and the disease was attributed to different possible causes. However, in all, allostery appears to play a key unifying role by specifying the conformational barcode. Dysfunctional conformational barcodes in disease states can be (partially) restored to their “healthy” barcode ensemble states by allosteric drugs (Csermely et al., 2013; Nussinov et al., 2011). Efficient, nontoxic modification of dysfunctional conformational barcodes is a major challenge.
Conclusions
The cell is complex, and deciphering of its signal transduction is challenging (Kiel and Serrano, 2012). Cellular diagrams illustrate this complexity by dense, interconnected maps. Interconnectivity takes place through shared proteins. Such “hub,” or signaling proteins, may have tens or even hundreds of partners. A key question is then what decides pathway unification and divergence? How does the signaling protein “know” which protein to select at any given time? Such questions are critical to the understanding of cell regulation. Here, our major tenet is that the conformation of a binding site acts like a (high-dimensional) barcode. This barcode is decided by the collective effects of all allosteric events on the protein, including disease-related ones, such as binding of pathogenic proteins, and gain-of-function mutations. From the free-energy landscape standpoint (Frauenfelder et al., 1991), the combined outcome of all is stabilization of a certain conformation; in so doing, the binding site in this conformation becomes an intracellular address label, providing a conformational barcode. The partner selected by this conformational barcode decides the pathway direction, and by not selecting other partners, alternate pathways are quenched. A change in conditions will modify this allosteric conformational barcode, resulting in another pathway taking over. Insight into these questions may help in synthetic biology approaches to signaling (Kiel et al., 2010). Because proteins pre-exist in a broad range of states, all pathways pre-exist in the cell. Those taking place via sparsely populated states do so at the basal, unregulated levels and may or may not be detected. Changes in conformational barcodes induced by extracellular signaling, pathogens, gain-of-function and drug-resistant mutations, and allosteric drugs can all rewire the network, shifting the relative pathway occupancy, and, in this way, transform “dormant” pathways into dominant ones.
This review aims to go beyond aggregating recent developments in the structural biology of allostery. It attempts to provide unifying insights that bring to light emerging trends that may otherwise remain latent and more elusive. In particular, it stresses the importance of collectively evaluating all the multiple co-occurring allosteric and orthosteric perturbations sensed by a given receptor, as opposed to considering each single perturbation in isolation. These perturbations are either covalent (e.g., mutations and/or PTMs) or noncovalent (e.g., effector or drug binding or even UVB radiation). Each unique and time-dependent combination of allosteric and orthosteric perturbations results in a specific “molecular barcode,” defined in the context of the free-energy landscape as a distinct set of populations of protein states with specific structural and dynamical properties. How different sets of perturbations map into different barcodes or sets of populations depends on the type of allosteric switch (i.e., graded switch versus OR- versus AND-gates), as exemplified by the Src, Syk, and Tec cytoplasmic tyrosine kinase families.
The concept of “molecular barcode” has a well-defined combinatorial connotation and, as such, its value is not only semantic but much more substantial, as it may open new perspectives in the field of allostery, explaining the complexity and diversity of signaling networks and stimulating new lines of research. It argues for shifting the focus from single allosteric per-turbations considered in isolation to a more holistic approach, in which multiple allosteric perturbations are simultaneously taken into consideration in an integrative manner. In addition, the concept of “molecular barcode” explains how a single allosteric system serving as hub node in a complex signaling network may initiate, relay, orterminate signal propagation.The molecular barcode treatment helps to explain how select signaling cascades are favored over others, leading toward divergence or convergence and eventually rewiring signaling pathways.
ACKNOWLEDGMENTS
We are grateful to one of our reviewers, who provided a broad, insightful overview of our review, uniquely capturing its significance to the structural and signaling biology community and its implications to the field. We have incorporated it in the Conclusions with only minor modifications. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. It was also supported by research grants from the Hungarian National Science Foundation (OTKA K83314) and by the EU (TÁM0P-4.2.2/B-10/1–2010- 0013). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
REFERENCES
- Antal MA, Bode C, and Csermely P (2009). Perturbation waves in proteins and protein networks: applications of percolation and game theories in signaling and drug design. Curr. Protein Pept. Sci. 10, 161–172. [DOI] [PubMed] [Google Scholar]
- Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, Ichinose K, Nakamura H, Tsujino A, Kawakami A,et al. (2011). Protea- some assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl. Acad. Sci. USA 108, 14914–14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autret A, Martin-Latil S, Mousson L, Wirotius A, Petit F, Arnoult D, Colbère-Garapin F, Estaquier J, and Blondel B (2007). Poliovirus induces Bax- dependent cell death mediated by c-Jun NH2-terminal kinase. J. Virol. 81, 7504–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, and Veitia RA (2009). A post-translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol. 19, 189–197. [DOI] [PubMed] [Google Scholar]
- Biter AB, Lee S, Sung N, and Tsai FT (2012). Structural basis for intersubunit signaling in a protein disaggregating machine. Proc. Natl. Acad. Sci. USA 109,12515–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD (2012). Promiscuity in protein-RNA interactions: conformational ensembles facilitate molecular recognition in the spliceosome: conformational diversity in U2AF65 facilitates binding to diverse RNA sequences. BioEssays 34, 174–180. [DOI] [PubMed] [Google Scholar]
- Boehr DD, Nussinov R, and Wright PE (2009). The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5,789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw JM (2010). The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell. Signal. 22, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Butcher AJ, Prihandoko R, Kong KC, McWilliams P, Edwards JM, Bottrill A, Mistry S, and Tobin AB (2011). Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling barcode. J. Biol. Chem. 286,11506–11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Cronin CN, Engel AG, Ohno K, Hersh LB, and Rodgers DW (2004). Choline acetyltransferase structure reveals distribution of mutations that cause motor disorders. EMBO J. 23, 2047–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G, Imasaki T, Yamada K, Cardelli F, Takagi Y, and Asturias FJ(2010). Mediator head module structure and functional interactions. Nat. Struct. Mol. Biol. 17, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennubhotla C, Yang Z, and Bahar I (2008). Coupling between global dynamics and signal transduction pathways: a mechanism of allostery for chaperonin GroEL. Mol. Biosyst. 4, 287–292. [DOI] [PubMed] [Google Scholar]
- Cheong R, Rhee A, Wang CJ, Nemenman I, and Levchenko A (2011). Information transduction capacity of noisy biochemical signaling networks. Science 334, 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi CN, Bach A, Engstrom A, Stromgaard K, Lundstrom P, Ferguson N, and Jemth P (2011). Biophysical characterization ofthe complex between human papillomavirus E6 protein and synapse-associated protein 97. J. Biol. Chem. 286, 3597–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, and Nusse R (2012). Wnt/p-catenin signaling and disease. Cell 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- Csermely P, Palotai R, and Nussinov R (2010). Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 35, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csermely P, Korcsmaros T, Kiss HJ, London G, and Nussinov R (2013). Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 138, 333–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, and Karplus M (2008). Allostery and cooperativity revisited. Protein Sci. 17, 1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Trez C (2012). Lymphotoxin-beta receptor expression and its related signaling pathways govern dendritic cell homeostasis and function. Immunobiology 217, 1250–1258. [DOI] [PubMed] [Google Scholar]
- Deane JE, Graham SC, Kim NN, Stein PE, McNair R, Cachón-González MB, Cox TM, and Read RJ (2011). Insights into Krabbe disease from structures of galactocerebrosidase. Proc. Natl. Acad. Sci. USA 108, 15169–15173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deindl S, Kadlecek TA, Brdicka T, Cao X, Weiss A, and Kuriyan J (2007). Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell 129, 735–746. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, and Shenoy SK (2007). Beta-arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510. [DOI] [PubMed] [Google Scholar]
- Dixit A, and Verkhivker GM (2011). Computational modeling of allosteric communication reveals organizing principles of mutation-induced signaling in ABL and EGFR kinases. PLoS Comput. Biol. 7, e1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Buetow L, Hock A, Sibbet GJ, Vousden KH, and Huang DT (2012). Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat. Struct. Mol. Biol. 19, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres NF, Engel K, Das R, Kovacs E, and Kuriyan J (2011). Regulation of the catalytic activity of the EGF receptor. Curr. Opin. Struct. Biol. 21, 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estep PA, and Johnson KA (2011). Effect of the Y955C mutation on mitochondrial DNA polymerase nucleotide incorporation efficiency and fidelity. Biochemistry 50, 6376–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro DU, Hegler JA, Komives EA, and Wolynes PG (2011). On the role of frustration in the energy landscapes of allosteric proteins. Proc. Natl. Acad. Sci. USA 108, 3499–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Kofler M, Hantschel O, Gish GD, Grebien F, Salah E, Neudecker P, Kay LE, Turk BE, Superti-Furga G, et al. (2008). Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell 134, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H, Sligar SG, and Wolynes PG (1991). The energy land scapes and motions of proteins. Science 254, 1598–1603. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M, Simon I, and Bondos S (2011). Dynamic protein-DNA recognition: beyond what can be seen. Trends Biochem. Sci. 36, 415–423. [DOI] [PubMed] [Google Scholar]
- Gable K, Gupta SD, Han G, Niranjanakumari S, Harmon JM, and Dunn TM (2010). A disease-causing mutation in the active site of serine palmitoyltransferase causes catalytic promiscuity. J. Biol. Chem. 285, 22846–22852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar ME, and Csermely P (2012). Rigidity and flexibility of biological networks. Brief. Funct. Genomics 11, 443–456. [DOI] [PubMed] [Google Scholar]
- Goodey NM, and Benkovic SJ (2008). Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 4, 474–482. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K, Ma B, and Nussinov R (2004). Isallosteryan intrinsic property of all dynamic proteins? Proteins 57, 433–443. [DOI] [PubMed] [Google Scholar]
- Helsen C, Dubois V, Verfaillie A, Young J,Trekels M, Vancraenenbroeck R, De Maeyer M, and Claessens F (2012). Evidence for DNA-binding domain — ligand-binding domain communications in the androgen receptor. Mol. Cell. Biol. 32, 3033–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr P, Hausmann G, and Basler K (2012). WNT secretion and signalling in human disease. Trends Mol. Med. 18, 483–493. [DOI] [PubMed] [Google Scholar]
- lakoucheva LM, Brown CJ, Lawson JD, Obradović Z, and Dunker AK (2002). Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584. [DOI] [PubMed] [Google Scholar]
- Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, and Kuriyan J(2011). Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar G, Keskin O, Gursoy A, and Nussinov R (2010). Allostery and population shift in drug discovery. Curr. Opin. Pharmacol. 10, 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, and Miller LJ (2010). Seven transmembrane receptors as shape- shifting proteins: the impact ofallosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel C, and Serrano L (2012). Challenges ahead in signal transduction: MAPK as an example. Curr. Opin. Biotechnol. 23, 305–314. [DOI] [PubMed] [Google Scholar]
- Kiel C, Yus E, and Serrano L (2010). Engineering signal transduction pathways. Cell 140, 33–47. [DOI] [PubMed] [Google Scholar]
- Korcsmáros T, Farkas IJ, Szalay MS, Rovo P, Fazekas D, Spiro Z, Böde C, Lenti K, Vellai T, and Csermely P (2010). Uniformly curated signaling pathways reveal tissue-specific cross-talks and support drug target discovery. Bioinformatics 26, 2042–2050. [DOI] [PubMed] [Google Scholar]
- Korkmaz EN, Nussinov R, and Haliloğlu T (2012). Conformational control of the binding of the transactivation domain of the MLL protein and c-Myb to the KIX domain of CREB. PLoS Comput. Biol. 8, e1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackmann M, and Boyd AW (2008). Eph, a protein family coming of age: more confusion, insight, or complexity? Sci. Signal. 1, re2. [DOI] [PubMed] [Google Scholar]
- Latham JA, Chosed RJ, Wang S, and Dent SY (2011). Chromatin signaling to kinetochores: transregulation of Dam1 methylation by histone H2B ubiquitination. Cell 146, 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Natarajan M, Nashine VC, Socolich M, Vo T, Russ WP, Benkovic SJ, and Ranganathan R (2008). Surface sites for engineering allosteric control in proteins. Science 322, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, and Yaffe MB (2012). Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 149, 780–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne A, Becavin C, and Victor JM (2012). The condensed chromatin fiber: an allosteric chemo-mechanical machine for signal transduction and genome processing. Phys. Biol. 9, 013001. [DOI] [PubMed] [Google Scholar]
- Liggett SB (2011). Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci. Signal. 4, pe36. [DOI] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, et al. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long D, and Bruschweiler R (2011a). Atomistic kinetic model for population shift and allostery in biomolecules. J. Am. Chem. Soc. 133, 18999–19005. [DOI] [PubMed] [Google Scholar]
- Long D, and Brüschweiler R (2011b). In silico elucidation of the recognition dynamics of ubiquitin. PLoS Comput. Biol. 7, e1002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma’ayan A, Jenkins SL, Neves S, Hasseldine A, Grace E, Dubin-Thaler B, Eungdamrong NJ, Weng G, Ram PT, Rice JJ, et al. (2005). Formation of regulatory patterns during signal propagation in a Mammalian cellular network. Science 309, 1078–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Kumar S, Tsai CJ, and Nussinov R (1999). Folding funnels and binding mechanisms. Protein Eng. 12, 713–720. [DOI] [PubMed] [Google Scholar]
- Mackereth CD, Madl T, Bonnal S, Simon B, Zanier K, Gasch A, Rybin V, Valcarcel J, and Sattler M (2011). Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature 475, 408–411. [DOI] [PubMed] [Google Scholar]
- Meek DW, and Anderson CW (2009). Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1, a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Lin SC, Bernecky C, Gao Y, and Taatjes DJ (2010). p53 activates transcription by directing structural shifts in Mediator. Nat. Struct. Mol. Biol. 17, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moal IH, and Bates PA (2012). Kinetic rate constant prediction supports the conformational selection mechanism of protein binding. PLoS Comput. Biol. 8, e1002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, and Miller WT (1997). Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 385, 650–653. [DOI] [PubMed] [Google Scholar]
- Morinière J, Rousseaux S, Steuerwald U, Soler-Lopez M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K, Hart DJ, et al. (2009). Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 461,664–668. [DOI] [PubMed] [Google Scholar]
- Musselman CA, Lalonde ME, Cote J, and Kutateladze TG (2012). Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaides L, Davy C, Raj K, Kranjec C, Banks L, and Doorbar J (2011). Stabilization of HPV16 E6 protein by PDZ proteins, and potential implications for genome maintenance. Virology 414, 137–145. [DOI] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, et al. (2011). Distinct phosphorylation sites on the p(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, and Tsai CJ (2013). Allostery in diseaseand in drug discovery. Cell 153, 293–305. [DOI] [PubMed] [Google Scholar]
- Nussinov R,Tsai CJ, and Csermely P (2011).Allo-networkdrugs: harnessing allostery in cellular networks. Trends Pharmacol. Sci. 32, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, Xin F, and Radivojac P (2012). Allosteric post-translational modification codes. Trends Biochem. Sci. 37, 447–455. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Ma B, and Tsai CJ (2013a). A broad view of scaffolding suggests that scaffolding proteins can actively control regulation and signaling of multienzyme complexes through allostery. Biochim. Biophys. Acta 1834, 820–829. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Ma B (2013b). The underappreciated role of allostery in the cellular network. Annu. Rev. Biophys. 42, 169–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandini A, Fornili A, Fraternali F,and Kleinjung J (2012). Detection of allosteric signal transmission by information-theoretic analysis of protein dynamics. FASEB J. 26, 868–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirruccello M, Swan LE, Folta-Stogniew E, and De Camilli P (2011). Recognition of the F&H motif by the Lowe syndrome protein OCRL. Nat. Struct. Mol. Biol. 18, 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwonski HM, Goomanovsky M, Bensimon D, Horovitz A, and Haran G(2012). Allosteric inhibition of individual enzyme molecules trapped in lipid vesicles. Proc. Natl. Acad. Sci. USA 109, E1437–E1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman JA, Hunter RG, Speksnijder N, van den Oever JM, Korobko OB, McEwen BS, de Kloet ER, and Datson NA (2012). Glucocorticoids modulate the mTOR pathway in the hippocampus: differential effects depending on stress history. Endocrinology 153, 4317–4327. [DOI] [PubMed] [Google Scholar]
- Querfurth C, Diernfellner AC, Gin E, Malzahn E, Heifer T, and Brunner M (2011). Circadian conformational change of the Neurospora clock protein FREQUENCY triggered by clustered hyperphosphorylation of a basic domain. Mol.Cell 43,713–722. [DOI] [PubMed] [Google Scholar]
- Ramos A, and Camargo FD (2012). The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 22, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, and Lefkowitz RJ (2012). Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, and Wells JA (2011). Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl. Acad. Sci. USA 108, 6056–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar GD, Gereben B, Callebaut I, Mornon JP, Zeöld A, da Silva WS, Luongo C, Dentice M, Tente SM, Freitas BC, et al. (2007). Ubiquitination-induced conformational change within the deiodinase dimer is a switch regulating enzyme activity. Mol. Cell. Biol. 27, 4774–4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seizl M, Larivière L, Pfaffeneder T, Wenzeck L, and Cramer P (2011). Mediator head subcomplex Med11/22 contains a common helix bundle building block with a specific function in transcription initiation complex stabilization. Nucleic Acids Res. 39, 6291–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam R, Chowdhury S, VanSchouwen B, and Melacini G (2011). Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. USA 108, 6133–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds M, and Storey A (2008). Identification of the regions ofthe HPV5 E6 protein involved in Bak degradation and inhibition of apoptosis. Int. J. Cancer 123, 2260–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Pache RA, Bernado P, Pons M, and Aloy P (2009). Dynamic interactions of proteins in complex networks: a more structured view. FEBS J. 276, 5390–5405. [DOI] [PubMed] [Google Scholar]
- Tsai CJ, and Nussinov R (1997). Hydrophobic folding units at protein-protein interfaces: implications to protein folding and to protein-protein association. Protein Sci. 6, 1426–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, and Nussinov R (2011). Gene-specific transcription activation via long-range allosteric shape-shifting. Biochem. J. 439, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, Kumar S, Ma B, and Nussinov R (1999). Folding funnels, binding funnels, and protein function. Protein Sci. 8, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, Del Sol A, and Nussinov R (2009a). Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Mol. Biosyst. 5, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CJ, Ma B, and Nussinov R (2009b). Protein-protein interaction networks: how can a hub protein bind so many different partners? Trends Biochem. Sci. 34, 594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang E, Giannetti AM, Shaw D, Dinh M, Tse JK, Gandhi S, Ho H, Wang S, Papp E, and Bradshaw JM (2008). Molecular mechanism of the Syk activation switch. J. Biol. Chem. 283, 32650–32659. [DOI] [PubMed] [Google Scholar]
- Tzeng SR, and Kalodimos CG (2011). Protein dynamics and allostery: an NMR view. Curr. Opin. Struct. Biol. 21, 62–67. [DOI] [PubMed] [Google Scholar]
- Weatherall KL, Seutin V, Liegeois JF, and Marrion NV (2011). Crucial role of a shared extracellular loop in apamin sensitivity and maintenance of pore shape of small-conductance calcium-activated potassium (SK) channels. Proc. Natl. Acad. Sci. USA 108, 18494–18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N, and Deng XW (2003). The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 19, 261–286. [DOI] [PubMed] [Google Scholar]
- Wei N, Serino G, and Deng XW (2008). The COP9 signalosome: more than a protease. Trends Biochem. Sci. 33, 592–600. [DOI] [PubMed] [Google Scholar]
- Whitley MJ, and Lee AL (2009). Frameworks for understanding long-range intra-protein communication. Curr. Protein Pept. Sci. 10, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarski T, and Zagrovic B (2009). Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc. Natl. Acad. Sci. USA 106, 19346–19351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Shenoy SK, Nobles K, and Lefkowitz RJ (2004). Activation-dependent conformational changes in beta-arrestin 2. J. Biol. Chem. 279, 55744–55753. [DOI] [PubMed] [Google Scholar]
- Xie W, Nangle LA, Zhang W, Schimmel P, and Yang XL (2007). Long- range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNAsynthetase. Proc. Natl. Acad. Sci. USA 104, 9976–9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalatan JG, Coyle SM, Rajan S, Sidhu SS, and Lim WA (2012). Conformational control of the Ste5 scaffold protein insulates against MAP kinase misactivation. Science 337, 1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX (2012). Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem. Sci. 37, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravlev PI, and Papoian GA (2010). Protein functional landscapes, dynamics, allostery: a tortuous path towards a universal theoretical framework. Q. Rev. Biophys. 43, 295–332. [DOI] [PubMed] [Google Scholar]