

Abstract
Nuclear receptor corepressor 1 (NCOR1) and NCOR2 (also known as SMRT) regulate gene expression by activating histone deacetylase 3 through their Deacetylase Activation Domain (DAD). We show that mice with DAD knock-in mutations have memory deficits, reduced anxiety levels, and reduced social interactions. Mice with NCOR1/2 depletion specifically in GABAergic neurons (NS-V mice) recapitulated the memory deficits and had reduced GABRA2 expression in lateral hypothalamus GABAergic neurons (LHGABA). This was associated with LHGABA neuron hyperexcitability and impaired hippocampal long-term potentiation, through a monosynaptic LHGABA to CA3GABA projection. Optogenetic activation of this projection caused memory deficits, while targeted manipulation of LHGABA or CA3GABA neuron activity reversed memory deficits in NS-V mice. We describe de novo variants in NCOR1, NCOR2 or HDAC3 in patients with intellectual disability or neurodevelopmental defects. These findings identify a hypothalamus–hippocampus projection that may link endocrine signals with synaptic plasticity through NCOR-mediated regulation of GABA signaling.
INTRODUCTION
Regulation of gene expression is a key component of intracellular signaling and confers long-lasting effects that are particularly relevant to the memory formation1. Many endocrine factors use nuclear receptors (NRs) to regulate gene expression. NRs recruit nuclear receptor coactivators (NCOAs) or nuclear receptor corepressor (NCORs) in a ligand-dependent manner, which alters epigenome modifications such as histone acetylation, remodels chromatin architecture, and regulates gene transcription2. NCOR1 and its homolog NCOR2 (also known as SMRT, silencing mediator for retinoid and thyroid hormone receptors) are the fundamental scaffold proteins of the NCOR corepressor complex in mammals3,4. NCORs regulate gene expression by recruiting and activating histone deacetylase 3 (HDAC3)5–8.
Our previous work has elucidated the metabolic functions of NCORs and HDAC3 in multiple tissues9–11. NCORs have been shown to interact with methylated CpG binding protein 2 (MeCP2) that binds methylated DNA12. Mutations in the human MECP2 gene are known to cause Rett syndrome13, a neurodevelopmental disorder characterized by intellectual disability, developmental regression, autism spectrum disorders, seizures, and acquired microcephaly. It was shown that MeCP2 missense mutations affecting MeCP2-NCORs interactions can cause Rett syndrome-like phenotype in mice14,15. NCORs can also form a stable protein complex with transducin beta like 1 X-linked receptor 1 (TBL1XR1 or TBLR1)16, another protein that is associated with neurocognitive disorders presenting with variable phenotypes including autism, intellectual disability, and multiple congenital anomalies in humans17. However, whether and how NCORs regulate cognitive functions has not been directly studied.
RESULTS
Disruption of NCORs function by NS-DADm mutations causes cognitive deficits
NCORs regulate gene expression by recruiting and activating histone deacetylase 3 (HDAC3)5–8. The Deacetylase Activation Domain (DAD) on the N-terminal region of NCORs is responsible for activating HDAC3 enzyme activity (Fig 1a). HDAC3 can be inactivated both in vitro and in vivo by missense mutations in either DAD or HDAC3 that abolishes the DAD-HDAC3 interaction8,10,18,19. Conversely, HDAC3 is the only HDAC that confers deacetylase enzymatic activity to the NCOR complex20,21. Therefore, inactivating HDAC3 renders the NCOR complex deficient in the deacetylase activity. We have previously constructed an NS-DADm whole-body knock-in mouse model that contains NCOR1-Y478A and NCOR2-Y470A19. NS-DADm mice do not have HDAC3 enzymatic activity but display normal development and appearance19.
Figure 1. NS-DADm mice display cognitive dysfunction, altered anxiety, and social avoidance.
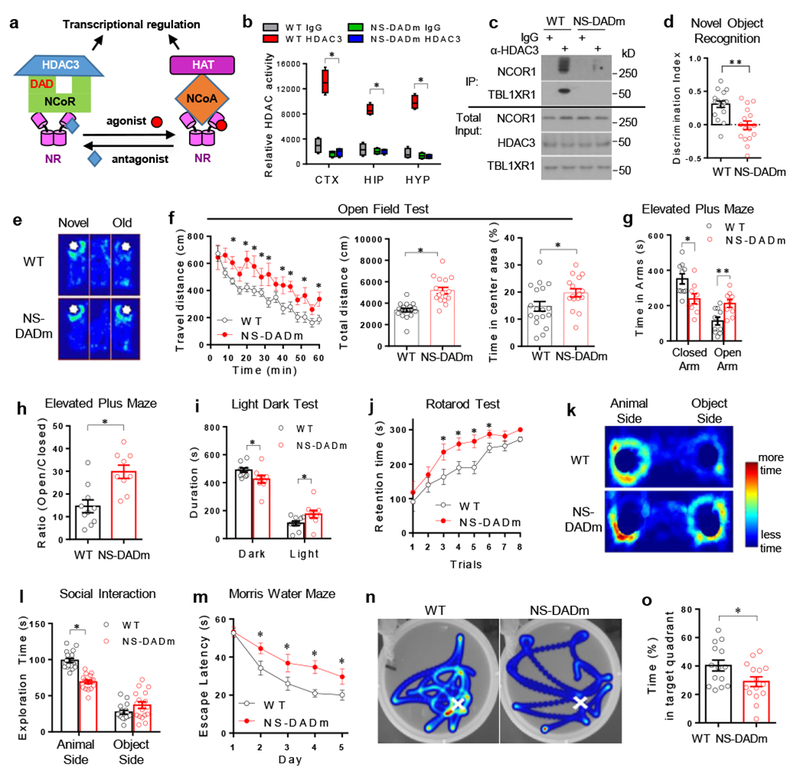
. (a) The Deacetylase Activation Domain (DAD) in NCOR mediates transcriptional regulation by nuclear receptors (NR), which counteracts with coactivators (NCOA) and histone acetyltransferases (HAT). (b) Fluorescence-based HDAC enzyme assay using lysates of mouse brain cortex (CTX), hippocampus (HIP), and hypothalamus (HYP) after immunoprecipitation with HDAC3 antibody or normal IgG. Box plots center line, median; box limits, upper and lower quartiles; whiskers, minimal and maximum values. 2-way ANOVA analysis was used followed by post hoc Tukey test. n=4 mice for each group. (c) Western blot of proteins associated with HDAC3 in HDAC3 immunoprecipitates (IP). The experiement was repeated independently once with similar results. n=4 mice per group. Male 4 months-old mice were used. The blot images have been cropped. (d) Discrimination index in Novel Object Recognition (NOR) test. Data was analyzed by two-tailed unpaired t test. nWT=18, nNS-DADm=16 mice. Male 4 months-old mice were used. (e) Representative heat map of NOR test. White spots indicate novel or old objects in a 3-chamber box. Time duration that a mouse spent in exploring each object was denoted by color from dark blue (less time) to light blue (more time). (f) Open field test. Data was analyzed by one-way repeated ANOVA and two-tailed unpaired t test. nWT=18, nNS-DADm=16 mice. Male 4 months-old mice were used. (g) Elevated plus maze test. Data was analyzed by two-tailed unpaired t test. nWT=10, nNS-DADm=9 mice. Male 4 months-old mice were used. (h) Time ratio in Elevated plus maze test. Data was analyzed by two-tailed unpaired t test. nWT=10, nNS-DADm=9 mice. Male 4 months-old mice were used. (i) Light-dark test. Data was analyzed by two-tailed unpaired t test. nWT=10, nNS-DADm=9 male 4 months-old mice. (j) Rotarod test. Data was analyzed by one-way repeated ANOVA. nWT=18, nNS-DADm=16 male 4 months-old mice. (k-l) Social interaction test representative heat map and statistics. 2-way ANOVA analysis was used followed by post hoc Tukey test. nWT=17, nNS-DADm=17 male 4months-old mice (m) Time spent before reaching the hidden platform in Morris Water Maze test (MWM). One-way repeated ANOVA analysis was used. nWT=13, nNS-DADm=14 male 4 months-old mice. (n-o) Representative heat map and time spent in the target quadrant during MWM test after the hidden platform was removed from the original location (indicated by white crosses). Data was analyzed by two-tailed unpaired t test. nWT=13, nNS-DADm=14 male 4 months-old mice. Data are expressed as mean ± S.E.M. For detailed statistics results, see Supplementary Table 1. * P ≤ 0.05 is set as significance.
In the whole-body knock-in NS-DADm mouse line, HDAC3 enzyme activity was undetectable in all brain regions tested, including cortex, hippocampus, and hypothalamus (Fig 1b). The interaction between HDAC3 and NCOR or TBLR1 was also reduced in the hypothalamus compared to littermate control (referred to as wild-type, WT), although not completely abolished (Fig 1c). The HDAC1 enzyme activity remained unaltered in the NS-DADm brain (Supplemental Fig S1a). NS-DADm mice were born at the Mendelian ratio and had slightly lower body weight than WT, with normal brain weight, breathing pattern, hindlimb activity, circadian locomotor activity, and gross histological morphology in various brain regions (Supplemental Fig S1b-i). Novel Object Recognition (NOR) test revealed a robust deficit in recognition memory in NS-DADm mice (Fig 1d-e, Fig S1j-k). This was not confounded by a lack of locomotor activity or more susceptibility to stress during the test, because NS-DADm mice actually showed hyperactivity and less anxiety compared to control in the Open Field test (Fig 1f), Elevated Plus Maze test (Fig 1g-h), and Light-Dark test (Fig 1i). The recognition memory deficit was not associated with defective motor learning, because NS-DADm mice had enhanced motor coordination and motor learning in the Rotarod test (Fig 1j) even when compared with body weight-matched control mice (Supplemental Fig S1d). Importantly, NS-DADm mice were less interested in social interaction with other animals (Fig 1k, l) and displayed spatial memory deficits in the Morris Water Maze (MWM) test (Fig 1m-o). These findings demonstrated that NCOR loss-of-function causes cognitive deficits, which can be independent of general developmental defects.
Depletion of HDAC3 in the LH impairs memory
To explore which brain regions are susceptible to the loss of HDAC3 enzyme activity in the context of memory deficits, we stereotaxically injected HDAC3loxP/loxP mice with adeno-associated virus (AAV) expressing Cre recombinase from a neuron-specific promoter using AAV-GFP as a control. Depletion of HDAC3 in the hippocampal CA1 region was shown to enhance memory22, suggesting that CA1 is unlikely the region mediating the memory deficit due to the loss of HDAC3 enzyme activity. We started with the hippocampus CA3 region. After injecting AAV-Cre into the CA3, we did not find altered learning behaviors or memory deficit in the NOR or MWM tests (Supplemental Fig S2a-i). This suggests that the CA3 region unlikely mediates the memory deficit due to the loss of HDAC3 enzyme activity, although we cannot rule out the possibility that the absence of behavioral changes could be due to non-complete HDAC3 depletion (Supplemental Fig S2c). The lateral hypothalamus is another brain region involved in motivated behaviors. We found that depletion of HDAC3 at the lateral hypothalamus resulted in robust memory deficit in both NOR and MWM tests (Supplemental Fig S2j-r), suggesting that the NCOR/HDAC3 function in the lateral hypothalamus is required for learning and memory. These findings do not exclude the possibility that other brain regions could also be important in the memory deficit in NS-DADm mice, especially considering that NS-DADm mice were deficient in the HDAC3 enzyme activity while HDAC3 regional knockout mice had disrupted HDAC3 protein levels.
Downregulation of GABAA receptor subunits in the NS-DADm hypothalamus
To address how NCORs regulate gene expression in the hypothalamus, we performed RNA-seq analysis using total RNAs extracted from the hypothalamus of NS-DADm and their WT littermates. There were more genes downregulated than upregulated in NS-DADm compared to control (Fig 2a). The differentially expressed genes were highly enriched in pathways involved in synapse function and cell junction (Fig 2b), with multiple GABAA receptor subunits downregulated in NS-DADm compared to WT (Fig 2c). The downregulation of the GABAA receptors was further confirmed by RT-qPCR and immunostaining using antibodies against α2 subunit (GABRA2) in the NS-DADm hypothalamus, especially in the lateral hypothalamus (LH) (Fig 2d-e, Supplemental Fig S3a). HDAC3 chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) revealed several possible HDAC3 binding sites at the genes of GABAA receptors. ChIP-qPCR analysis further confirmed that HDAC3 indeed bound to those regions and the binding intensity was lower in NS-DADm mice compared to WT (Fig 2f). The residual binding in NS-DADm mice was presumably due to the interaction of HDAC3 with the NCORs C-terminal region6,7. Of note, GABAA receptors gene expression remained unaltered in the NS-DADm hippocampus (Supplemental Fig S3b-c), suggesting that NCORs-mediated transcription regulation is highly region-specific. To address the functional significance of NCORs-mediated regulation of GABA signaling, we used diazepam, a positive modulator of GABAA receptor, at a dosage lower than what was commonly shown to be amnesic23. Diazepam rescued the recognition memory deficit of NS-DADm mice in the NOR test (Fig 2g), suggesting a causative role of disrupted GABA signaling in the memory deficit of NS-DADm mice.
Figure 2. NCORs regulate GABAA receptor gene expression in the hypothalamus.
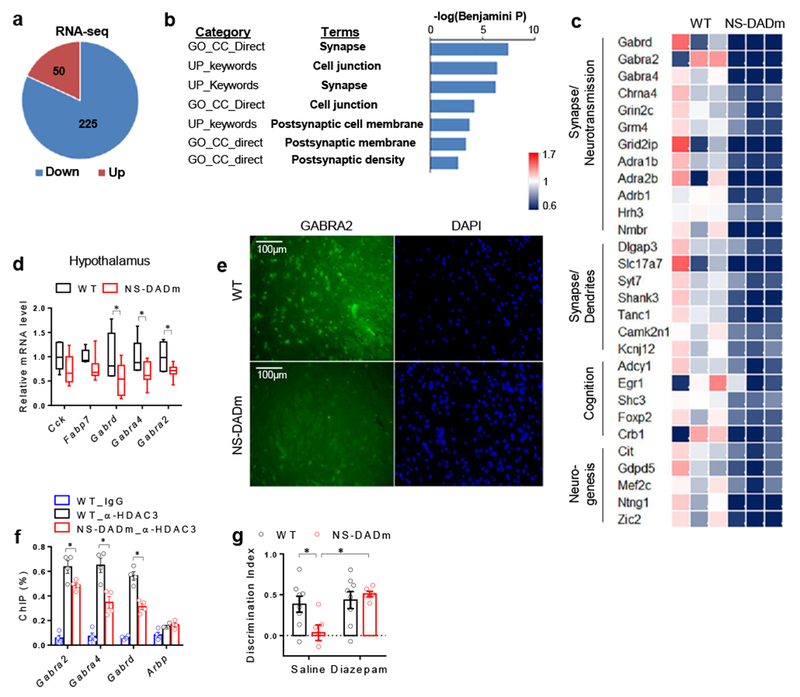
. (a) Upregulated (Up) or downregulated (Down) genes in the hypothalamus of the NS-DADm mice compared to control littermate, as identified by RNA-seq analysis. 3 biological replicates in each group. DESeq2 (version 1.8.2) was used to normalized the read counts and perform differential gene analysis with a threshold of 5% false discovery rate (FDR). (b) Gene ontology (GO) analysis of differentially expressed genes, showing top enriched pathway clusters. DESeq2 (version 1.8.2) was used to normalized the read counts and perform differential gene analysis with a threshold of 5% FDR. n = 3 mice for each group. (c) Heat map of top differentially expressed gene. (d) RT-qPCR analysis of mRNA of hypothalamus from WT and NS-DADm mice. Box plots center line, median; box limits, upper and lower quartiles; whiskers, minimal and maximum values. Data was analyzed by two-tailed unpaired t test. nWT=6, nNS-DADm=8 male 4 months-old mice. (e) Immunostaining of GABAA receptor α2 subunit (GABRA2) in the lateral hypothalamus. The experiment was repeated independently once with similar results. n=4 mice for each group. (f) ChIP-qPCR analysis. The experiment was repeated independently once with similar results. Data was analyzed by two-tailed unpaired t test. n=4 samples for each group, with 3 male 4 months-old mice pooled in each sample. (g) Novel Object Recognition (NOR) test in NS-DADm mice after i.c.v. administration of diazepam, a positive modulator of GABAA receptors. Data was analyzed by 2-way ANOVA followed by post hoc LSD test. nWT_Vehicle=8, nNS-DADm_Vehicle=6, nWT_Diazepam=8, nNS-DADm_Diazepam=6 female 4 months-old mice. Data is expressed as mean ± S.E.M. For detailed statistics results, see Supplementary Table 1. * P ≤ 0.05 is set as significance.
GABA neuron-specific depletion of NCORs impairs memory
We found that NCOR1 was ubiquitously expressed across different brain regions, including those that are highly populated with GABAergic neurons (Supplemental Fig S3d). To address the function of NCORs specifically in GABAergic neurons, we crossbred NCOR1loxP/loxP, NCOR2loxP/loxP mice10 with Vgat-Cre mice24 that expresses Cre recombinase only in GABAergic neurons. The NCOR1loxP/loxP/NCOR2loxP/loxP/Vgat-Cre (NS-V) mice were born at the Mendelian ratio and showed no obvious abnormality from littermates NCOR1loxP/loxP/NCOR2loxP/loxP or Vgat-Cre controls (referred as wild-type, WT). In situ hybridization confirmed deletion of NCOR1 in GABAergic neurons in multiple brain regions (Supplemental Fig 3e-f). Unlike NS-DADm mice, NS-V mice maintained normal locomotor activity and anxiety level in the Open Field test and Elevated Plus Maze test (Fig 3a-b). However, NS-V mice displayed memory and learning deficits in both the NOR test (Fig 3c-d) and the MWM test (Fig 3e-g) to a similar degree as the whole-body knock-in NS-DADm mice, demonstrating that NCORs loss-of-function specifically in GABAergic neurons is sufficient to cause cognitive deficits. These data do not necessarily suggest that NCORs dysfunction in GABAergic neurons fully account for the memory deficit in NS-DADm mice, considering that NCORs have functions other than activation of the HDAC3 enzyme activity.
Figure 3. Depletion of NCORs specifically in GABAergic neurons causes memory deficits.
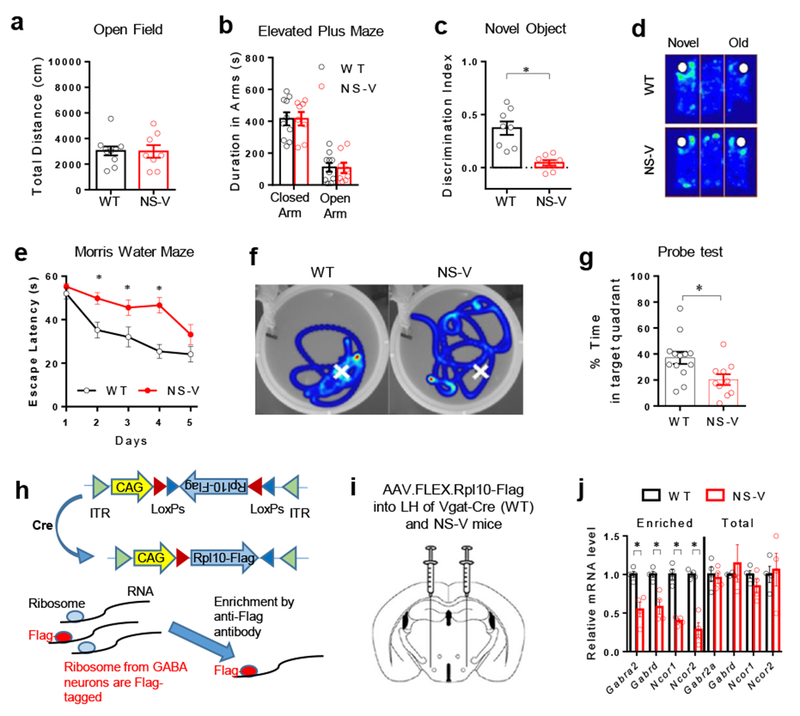
. (a) Open field test and elevated plus maze test of NS-V mice and their littermate WT control mice. Data were analyzed by two-tailed unpaired t test. nWT=10, nNS-V=8 male 4 months-old mice. (b) Elevated plus maze test. Data were analyzed by two-tailed unpaired t test. nWT=10, nNS-V=8 male 4 months-old mice. (c-d) NOR test and representative heat map. White spots indicate novel or old objects. Time duration spent in exploring each object was denoted by color from dark blue (less time) to light blue (more time). Data were analyzed by two-tailed unpaired t test. nWT=8, nNS-V=8 male 4 months-old mice. (e-f) Morris water maze test and representative heat map after the hidden platform was removed from the original location (indicated by white crosses). Data were analyzed by one-way repeated ANOVA. nWT=13, nNS-V=10 male 4 months-old mice. (g) Probe test in the water maze. Data were analyzed by two-tailed unpaired t test. nWT=13, nNS-V=10 male 4 months-old mice. (h) Ribosomal protein L10 (Rpl10) is expressed in the AAV vector with the flip-excision (FLEX) system. ITR, inverted terminal repeats. CAG, promoter. (i) AAV injection scheme. (j) RT-qPCR analysis of total and enriched mRNA (before and after immunoprecipitation, respectively). Hypothalami from 6 male 4 months-old mice were pooled for each sample in RT-qPCR analysis. n=4 samples. Data were analyzed by two-tailed unpaired t test. Data is expressed as mean ± S.E.M. For detailed statistics results, see Supplementary Table 1. * P ≤ 0.05 is set as significance.
NCORs regulate GABAA receptor expression in LHGABA neurons
Does the GABAA receptor downregulation occur in GABAergic neurons? NS-V mice and control Vgat-Cre mice were stereotaxically injected at the LH with AAV expressing a Flag-tagged 60S ribosomal protein L10a (Rpl10a) using the flip-excision (FLEX) system25 to ensure Cre-dependent expression of Rpl10a-Flag (Fig 3h-i). Subsequent immunoprecipitation with anti-Flag antibodies enriched ribosome-bound mRNAs that were only expressed in LHGABA neurons (Fig 3h). The expression of GABAA receptors was downregulated in LHGABA neurons upon NCORs depletion (Fig 3j), which confirmed that NCORs regulate expression of GABAA receptors in LHGABA neurons.
NCORs depletion causes LHGABA hyperexcitability
How do NCORs regulate firing activities of GABAergic neurons? After crossbreeding NS-V mice or Vgat-Cre mice (referred to as WT control) with Rosa26-tdTomato mice that express the red fluorescent protein tdTomato in a Cre-dependent manner26, we used electrophysiology patch clamp to survey tdTomato-labeled GABAergic neurons in different brain regions. LH was the region showing the most prominent change (Fig 4a-j). NCORs-depleted LHGABA neurons displayed higher spontaneous firing rate and more depolarized than control (Fig 4a-c), while hippocampal CA3GABA neurons showed a hyperpolarized resting membrane potential (Fig 4c). These results were further confirmed in perforated clamp analysis (Supplemental Fig S4a). The amplitude of mIPSCs was drastically reduced by more than 50% in LHGABA neurons after NCORs depletion (Fig 4h-i). As a result, the excitation/inhibition (E/I) ratio in LHGABA neurons was higher in the absence of NCORs compared to control (Fig 4j). The frequency of mIPSC in LHGABA neurons remained unchanged (Fig 4g), suggesting a postsynaptic mechanism for the hyperexcitability in LHGABA neurons. This could be explained by the lower GABAA receptor expression in these neurons.
Figure 4. NCORs control the excitation/ inhibition (E/I) balance of LH GABAergic neurons.
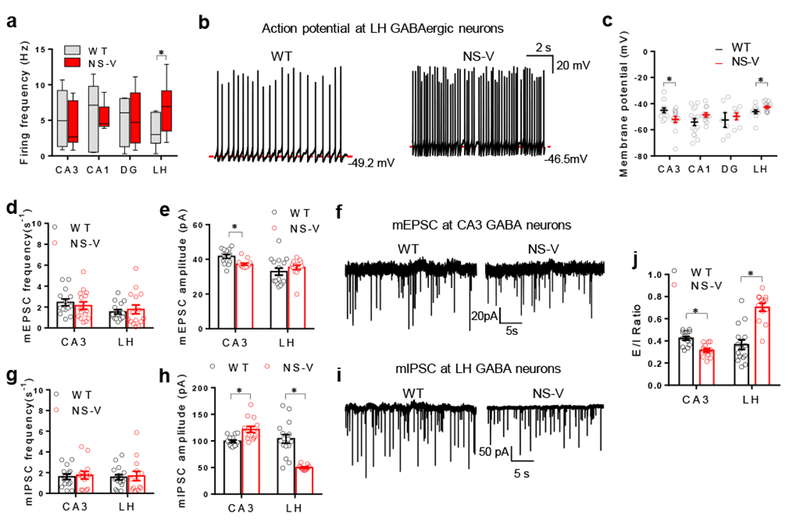
. (a-c) Spontaneous firing frequency, representative traces, and resting membrane potential of GABAergic neurons in the hippocampus CA3, CA1, dentate gyrus (DG), and the lateral hypothalamus (LH) of the NS-V mice and Vgat-Cre control (referred to as wild-type, WT). Box plots center line, median; box limits, upper and lower quartiles; whiskers, minimal and maximum values. The experiment was repeated independently once with similar results. 2 months-old male mice were used. 2-way ANOVA analysis was used. Firing Frequency: CA3: nWT=8 neurons/2 mice, nNS-V= 6 neurons/2 mice; CA1: nWT=7 neurons/2 mice, nNS-V=7 neurons/2 mice; DG: nWT=4 neurons/2 mice, nNS-V=5 neurons/2 mice; LH: nWT=7 neurons/2 mice, nNS-V=14 neurons/2 mice. Membrane Potential: CA3: nWT=12 neurons/2 mice, nNS-V=13neurons /2 mice; CA1: nWT=15 neurons/2 mice, nNS-V=12 neurons/2 mice; DG: nWT=6 neurons/2 mice, nNS-V=7 neurons/2 mice; LH: nWT=11 neurons/2 mice, nNS-V=15 neurons/2 mice. (d-f) Frequency, amplitude, and representative traces of mEPSCs of GABAergic neurons in CA3 and LH of the NS-V mice and Vgat-Cre control mice. 2months-old male mice were used. 2-way ANOVA analysis was used. mEPSC Frequency: CA3: nWT=14 neurons/2 mice, nNS-V=15 neurons/2 mice; LH: nWT=15 neurons/2 mice, nNS-V=15 neurons/2 mice; mEPSC amplitude: CA3: nWT=14 neurons/2 mice, nNS-V=15 neurons/2 mice; LH: nWT=15 neurons/2 mice, nNS-V=15 neurons/2 mice. (g-i) Frequency, amplitude, and representative traces of mIPSCs of GABAergic neurons in CA3 and LH of the NS-V mice and Vgat-Cre control. Male 2 months-old mice were used. 2-way ANOVA analysis was used. mIPSC Frequency: CA3: nWT=14 neurons/2 mice, nNS-V=13 neurons/2 mice; LH: nWT=16 neurons/2 mice, nNS-V=13 neurons/2 mice; mIPSC amplitude: CA3: nWT=14 neurons/2 mice, nNS-V=13 neurons/2 mice; LH: nWT=16 neurons/2 mice, nNS-V=13 neurons/2 mice. (j) Excitation/Inhibition (E/I) ratio (ratio of mEPSC amplitude versus mIPSC amplitude). Male 2 months-old mice were used. 2-way ANOVA analysis was used. E/I ratio: CA3: nWT=14 neurons/2 mice, nNS-V=13 neurons/2 mice; LH: nWT=16 neurons/2 mice, nNS-V=13 neurons/2 mice; mIPSC amplitude: CA3: nWT=14 neurons/2 mice, nNS-V=13 neurons/2 mice; LH: nWT=15 neurons/2 mice, nNS-V=13 neurons/2 mice. Data is expressed as mean ± S.E.M. For detailed statistics results, see Supplementary Table 1. * P ≤ 0.05 is set as significance. Independently, the electrophysiological data was re-analyzed using the linear mixed-effects models (Supplementary Table S2).
LHGABA sends monosynaptic projections to CA3GABA
The concurrence of the LHGABA hyperexcitability and the seemingly CA3GABA hyperpolarization prompted us to check the neural connection between LHGABA and CA3GABA using wheat germ agglutinin (WGA)-mediated anterograde tracing27. WGA travels across the axon terminals and labels downstream neurons. Vgat-Cre/tdTomato mice were stereotaxically injected at LH with adenovirus expressing a WGA-GFP fusion protein in a Cre-dependent manner (Ad-iN/WGA-GFP)27 (Fig 5a). Robust co-labeling of WGA-GFP (green) and tdTomato (red) at CA3 demonstrated an anatomical LHGABA > CA3GABA projection (Fig 5b-c). To further characterize the LHGABA > CA3GABA projection, we used channelrhodopsin-2 (ChR2)-assisted circuit mapping (CRACM). ChR2 is an excitatory light-gated ion channel. Its expression was restricted in LHGABA neurons by injecting an AAV vector expressing ChR2 in a Cre-dependent manner (AAV-FLEX-hChR2-EYFP) into the LH of Vgat-Cre mice (Fig 5d). Ad-iN/WGA-GFP was co-injected to label CA3GABA neurons that receive LHGABA projections. Upon excitation of LHGABA neurons with blue light shed on the CA3 region, evoked IPSCs (eIPSCs) were detected within 10 ms in GFP-labeled CA3GABA neurons (Fig 5e-g). The eIPSCs were insensitive to potassium channel blocker 4-aminopyridine (4-AP) and sodium channel blocker tetrodotoxin (TTX), but could be blocked by GABAA receptor antagonist bicuculline (Bic) (Fig 5e), suggesting a monosynaptic GABA input from LH to CA3. Similar findings were made in NS-V mice (Supplemental Fig S4b-d), suggesting that the projection is intact in the NS-V mice. In comparison to CA3GABA neurons, CA3Glutamate neurons do not receive as many projections from LHGABA in WGA-based antegrade tracing (Supplemental Fig S4e-f). As a negative control, contralateral CA3 did not show WGA-GFP signals (Supplemental Fig S4g). In addition, when a GFP-tagged synaptophysin, a synaptic vesicle glycoprotein, was expressed in the LH through injection of Vgat-Cre mice with AAV-FLEX-Synaptophysin-GFP28, GFP-labeled neuron axons were observed in CA3 (Supplemental Fig S4h), further confirming the monosynaptic projections from LH to CA3. Taken together, the data suggest that the hyperexcitability of LHGABA neurons in NS-V mice could inhibit CA3GABA neurons through the monosynaptic GABA input.
Figure 5. The LHGABA to CA3GABA projection regulates memory and learning behaviors.
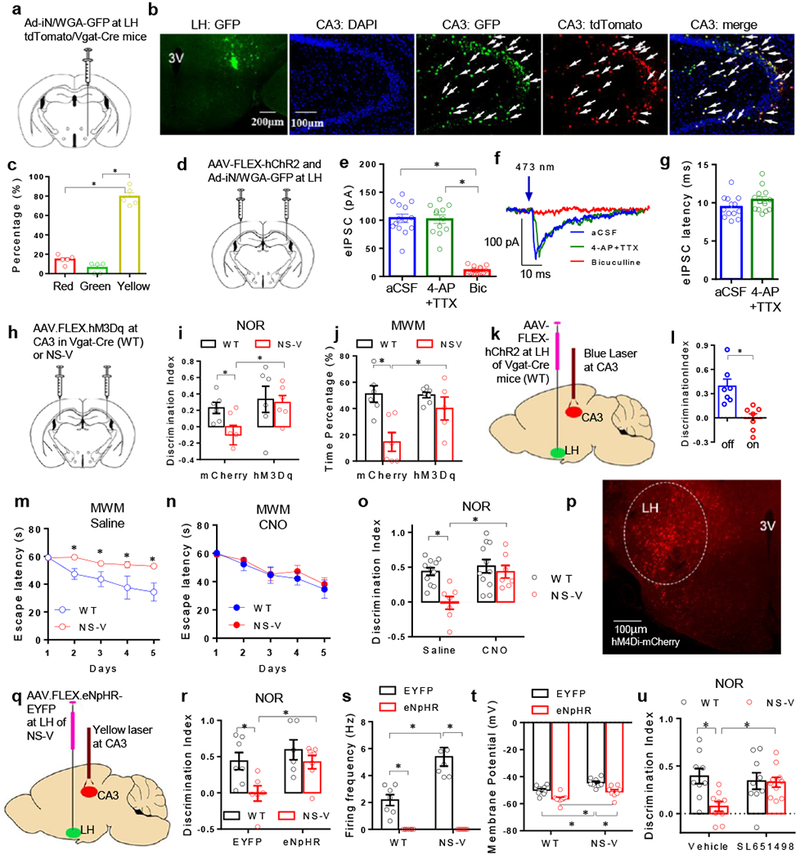
. (a) Anterograde tracing scheme. (b) Microscopy analysis of WGA-GFP at the primary injection site (LH) or projected site (CA3) in WT mice. 3V, 3rd ventricle. Green: WGA-GFP, Blue: DAPI, Red: Vgat positive neurons. Arrows indicate cells positive for both WGA-GFP and tdTomato. The experiment was repeated independently once with similar results. n=5 mice. Male 4 months-old mice were used. (c) Quantification of GFP-positive (green), tdTomato-positive (red), and double-positive (yellow) cells in CA3. One-way ANOVA analysis was used followed by post hoc Tukey test. n=5 mice. Male 4 months-old mice were used. (d) Scheme of intracranial injection of AAV-ChR2 in the LH. (e) The amplitude of eIPSCs of CA3GABA neurons in WT upon blue light stimulation of LHGABA neurons. Brain slices were recorded in pure artificial CSF (aCSF), or in the presence of 4-aminopyridine (4-AP, a potassium channel blocker) plus tetrodotoxin (TTX, a sodium channel blocker), or GABAA Receptor antagonist bicuculline (Bic). 2-way ANOVA analysis was used. Male 4 months-old mice were used. n=15 neurons/3 mice. (f) Representative traces of eIPSCs of CA3GABA neurons. (g) Latency of eIPSCs of CA3GABA neurons. 2-way ANOVA analysis was used. Male 4 months-old mice were used. n=13 neurons/3 mice. (h) Scheme of intracranial injection of AAV-FLEX-hM3Dq at CA3. (i) NOR test in hM3Dq-injected or mCherry-injected NS-V or WT (Vgat-Cre) mice with CNO treatment prior to the test. 2-way ANOVA analysis was used followed by post hoc LSD test. nWT_mCherry=6, nWT_hM3Dq=6, nNS-V_mCherry=6, nNS-V_hM3Dq=6 mice. Female 4 months-old mice were used. (j) MWM Probe test in hM3Dq-injected or mCherry-injected NS-V or WT (Vgat-Cre) mice with CNO treatment prior to the test. 2-way ANOVA analysis was used followed by post hoc LSD test. nWT_mCherry=6, nWT_hM3Dq=6, nNS-V_mCherry=6, nNS-V_hM3Dq=6 mice. Female 4 months-old mice were used. (k) AAV injection and laser probe implant scheme for optogenetic manipulation. (l) Novel Object Recognition (NOR) test with a blue laser stimulation in CA3 on mice infected with AAV-ChR2 at LH. Data were analyzed by two-tailed unpaired t test. nWT_light off=7, nWT_light on=7 mice. Male 4 months-old mice were used. (m-n) MWM test in the presence of CNO or saline treatment in NS-V or WT (Vgat-Cre) mice injected with AAV.FLEX.hM4Di at the LH. 2-way repeated ANOVA analysis was used followed by post hoc LSD test. nWT_Saline=7, nWT_CNO=7, nNS-V_Saline=8, nNS-V_CNO=7 mice. Male 4 months-old mice were used. (o) NOR test in hM4Di-injected NS-V or WT (Vgat-Cre) mice with or without CNO treatment. 2-way ANOVA analysis was used followed by post hoc LSD test. nWT_Saline=11, nWT_CNO=7, nNS-V_Saline=11, nNS-V_CNO=7 mice. Male 4 months-old mice were used. (p) Microscopy analysis of hM4Di-mCherry at the primary injection site. Male 4 months-old mice were used. The experiment was repeated independently once with similar results. (q) Scheme of AAV injection and laser probe implant for optogenetic manipulation. (r) NOR test of NS-V or WT (Vgat-Cre) mice with AAV.FLEX.EYFP or AAV.FLEX.eNpHR-EYFP injected at LH with yellow light on for all mice during the test. (Output power at the probe tip is around 7mW). 2-way ANOVA analysis was used followed by post hoc LSD test. nWT_EYFP=7, nWT_eNpHR=7, nNS-V_EYFP=6, nNS-V_eNpHR=7 mice. Male 4 months-old mice were used. (s-t) Spontaneous firing frequency and resting membrane potential of EYFP-infected or eNpHR-infected LHGABA neurons in response to yellow light in brain slices. 2-way ANOVA analysis was used. Male 4 months-old mice were used. Firing Frequency: nWT_EYFP=7 neurons/2 mice, nNS-V_EYFP=7 neurons/2 mice, nWT_eNpHR=8 neurons/2 mice, nNS-V_eNpHR=7 neurons/2 mice; Membrane Potential: nWT_EYFP=9 neurons/2 mice, nNS-V_EYFP=8 neurons/2 mice, nWT_eNpHR=9 neurons/2 mice, nNS-V_eNpHR=8 neurons/2 mice. (u) Novel Object Recognition (NOR) test after LH-specific infusion of a selective GABAA agonist SL651498. 2-way ANOVA analysis was used followed by post hoc LSD test. nWT_Vehicle=9 mice, nNS-V_Vehicle=9 mice, nWT_SL651498=9 mice, nNS-V_SL651498=10 mice. Male 4 months-old mice were used. Data is expressed as mean ± S.E.M. For detailed statistics results, see Supplementary Table 1. * P ≤ 0.05 is set as significance. Independently, the electrophysiological data was re-analyzed using the linear mixed-effects models (Supplementary Table S2).
The LHGABA to CA3GABA projection regulates memory and learning behaviors
Could the altered excitability in LHGABA and CA3GABA neurons explain the cognitive deficit in NS-V mice? DREADD (Designer Receptor Exclusively Activated by Designer Drugs) uses engineered G-protein coupled receptors to activate or silence specific neurons in response to clozapine-N-oxide (CNO)29. We first used an activating DREADD receptor hM3Dq to address the role of CA3GABA in the memory deficit of the NS-V mice. NS-V mice and control Vgat-Cre mice were injected at the CA3 with AAV-FLEX-hM3Dq-mCherry or a control virus AAV-FLEX-mCherry (Fig 5h). All mice were subjected to behavior tests after administration of CNO to control for potential direct pharmacological effects of CNO itself30. NOR and MWM tests showed that NS-V mice showed improved spatial memory and recognition memory after the DREADD-mediated activation of CA3 neural activities (Fig 5i-j, Supplemental Fig S5a-d). To specifically activate the LHGABA-projecting CA3 neurons, we used an optogenetic approach by expressing ChR2 in LHGABA neurons in Vgat-Cre mice and shedding blue light at CA3 (Fig 5k). Such stimulation during the NOR test reduced the discrimination index (Fig 5l), without affecting the total exploration time (Supplemental Fig S5e). This finding demonstrates that the targeted activation of the LHGABA–CA3 circuit is sufficient to cause memory deficits.
We then address whether the hyperexcitability of LHGABA neurons is necessary for the memory deficits in NS-V mice. We used hM4Di, a silencing DREADD29. AAV-FLEX-hM4Di was injected into the LH of the NS-V mice and the control Vgat-Cre mice (Fig 5m-p). DREADD-mediated suppression of LHGABA neurons in NS-V mice rescued their spatial memory (Fig 5m-n) and recognition memory (Fig 5o) without affecting explorative or locomotor activity (Supplemental Fig S5f-g). To further address whether the LHGABA -CA3 projection per se mediates such effect, we used an enhanced natronomonas halorhodopsin (eNpHR), a suppressing optogenetic receptor. NS-V mice and control Vgat-Cre mice were injected at the LH with AAV-FLEX-eNpHR-EYFP or AAV-FLEX-EYFP, and implanted with an optic fiber at CA3 (Fig 5q). The NS-V mice showed improved recognition memory in the NOR test when all 4 groups of mice received photosuppression (Fig 5r, Supplemental Fig S5h-i). The efficient photosuppression of CA3GABA-innervating LHGABA neurons in NS-V mice was validated in patch clamp analysis in brain slices after shedding a yellow light on the CA3 (Fig 5s-t). LHGABA hypoexcitability did not cause memory deficits, suggesting that the relationship between the LHGABA excitability and the cognitive function is not linear on different sides of the normal range. These data demonstrated that the LHGABA hyperexcitability contributes to the memory deficits in NS-V mice.
In addition to chemogenetic and optogenetic rescue, we sought to treat the cognitive defects in NS-V mice by pharmaceutical manipulation of LH neurons. We found that the recognition memory deficit in the NS-V mice was ameliorated by infusion of SL651498, a selective positive modulator GABAA receptors31, specifically into the LH via an implanted cannula (Fig 5u; Supplemental Fig S5j-k). SL651498 did not change total travel distance or exploration time (Supplemental Fig S5l). These data demonstrated an indispensable role of the LHGABA neurons in regulating memory formation, which underlies the cognitive deficit observed in the NS-V mice.
LHGABA hyperexcitability impairs hippocampal synaptic plasticity
Synaptic plasticity in the hippocampus is considered as a major cellular mechanism for memory formation. Long-term potential (LTP) in hippocampal interneurons is a form of synaptic plasticity involved in memory32–34. We found that hippocampal CA3GABA neurons in NS-V mice displayed impaired LTP when induced by high-frequency stimulation (HFS) at the dentate gyrus (Fig 6a). We used DREADD to address whether the hyperexcitability of LHGABA neurons plays a role in impaired LTP formation in CA3GABA neurons. AAV-FLEX-hM4Di was co-injected with Ad-iN/WGA-GFP into the LH of the NS-V mice and the control Vgat-Cre mice (Fig 6b-j). Administration of CNO silenced the spontaneous firing of LHGABA neurons in both WT and NS-V to a comparable level (Fig 6c). CNO also abolished the difference of LHGABA resting membrane potential between WT and NS-V mice (Fig 6d), validating efficient suppression of LHGABA neurons. Remarkably, CNO completely rescued LTP at LHGABA-innervated CA3GABA neurons (co-labeled by WGA-GFP and tdTomato) (Fig 6e-f). These data demonstrated that the LHGABA hyperexcitability contributes to the impaired hippocampal synaptic plasticity in NS-V mice. We then used optogenetics to address the role of the LHGABA-CA3GABA projection. NS-V mice and control Vgat-Cre mice were injected at the LH with AAV-FLEX-eNpHR-EYFP or AAV-FLEX-EYFP and LTP was measured after shedding yellow light on CA3 in brain slices (Fig 6g). Such photosupression rescued LTP at LHGABA-innervated CA3GABA neurons (Fig 6h-i). These data demonstrated that LHGABA hyperexcitability impaired hippocampal synaptic plasticity through the LHGABA to CA3GABA projection.
Figure 6. The LHGABA to CA3GABA projection regulates hippocampal synaptic plasticity in NS-V mice.
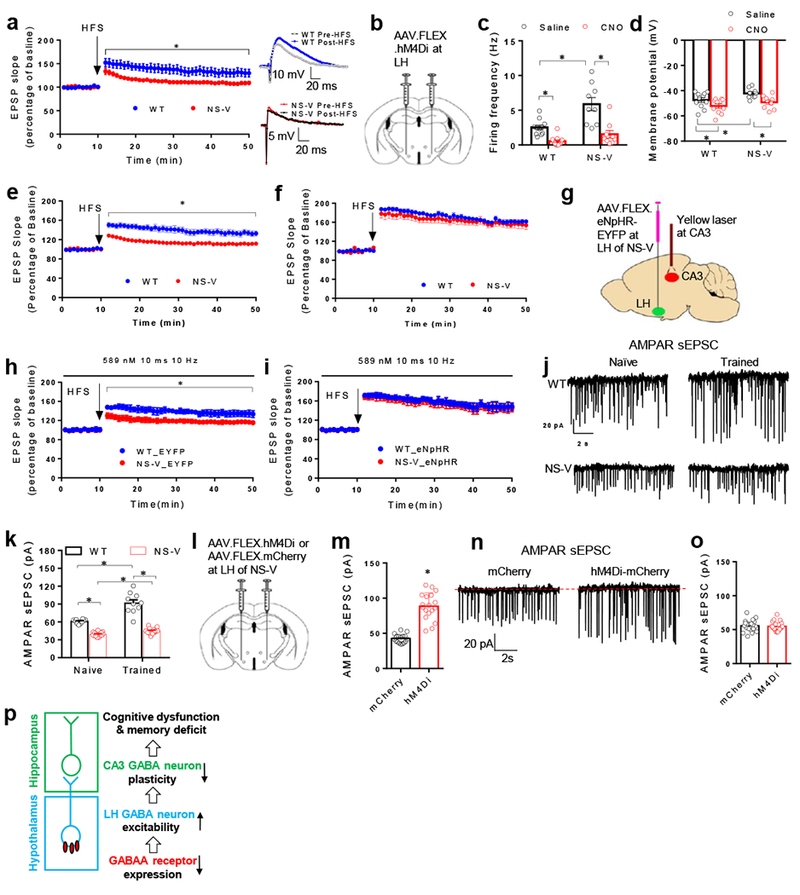
. (a) Long term potentiation (LTP) formation in CA3GABA neurons upon high frequency stimulation (HFS) at dentate gyrus in brain slices. Data were analyzed by one-way repeated ANOVA. nWT=7 mice, nNS-V=5 mice. Male 3 months-old mice were used. (b) Injection scheme for DREADD. (c) Spontaneous firing frequency of LHGABA neurons with or without CNO. Data were analyzed by 2-Way ANOVA. nWT_saline=11 neurons/2 mice, nNS-V_Saline=9 neurons/2 mice, nWT_CNO=11 neurons/2 mice, nNS-V_CNO=9 neurons/2 mice. Male 3 months-old mice were used. (d) Resting membrane potential of LHGABA neurons in response to CNO. Data were analyzed by 2-Way ANOVA. nWT_saline=11 neurons/2 mice, nNS-V_Saline=9 neurons/2 mice, nWT_CNO=11 neurons/2 mice, nNS-V_CNO=9 neurons/2 mice. Male 3 months-old mice were used. (e-f) LTP formation in LHGABA-innervated CA3GABA neurons without or in the presence of CNO. Data were analyzed by 2-Way repeated ANOVA. nWT_saline=6 mice, nNS-V_Saline=6 mice, nWT_CNO=6 mice, nNS-V_CNO=7 mice. Male 3 months-old mice were used. (g) Scheme of AAV injection and laser probe implant for optogenetic manipulation. (h) LTP formation with yellow light on in LHGABA-innervated CA3GABA neurons in brain slices from EYFP-infected mice. (i) LTP formation with yellow light on in LHGABA-innervated CA3GABA neurons in brain slices from eNpHR-infected mice. Data were analyzed by 2-Way repeated ANOVA. nWT_EYFP=6 mice, nNS-V_EYFP=6 mice, nWT_eNpHR=9 mice, nNS-V_CNO=9 mice. Male 3 months-old mice were used. (j-k) Representative traces and the amplitude of AMPAR-dependent sEPSC in LHGABA-innervated CA3GABA neurons in mice either trained during the MWM test or left untrained (naive). Data were analyzed by 2-Way ANOVA. nWT_Naive=10 neurons/2 mice, nNS-V_Naive=13 neurons/2 mice, nWT_Train=10 neurons/2 mice, nNS-V_Train=14 neurons/2 mice. Male 3 months-old mice were used. (l) Scheme of intracranial injection of AAV.FLEX.hM4Di-mCherry or AAV.FLEX.mCherry in the LH of NS-V mice. (m) Injected NS-V mice were trained in the MWM test for 3 days with CNO administration (1mg/kg, i.p.) before each training session. The amplitude of AMPAR-dependent sEPSC in the LHGABA-innervated CA3GABA neurons was measured in brain slices at the 5th day. Data were analyzed by 2-Way ANOVA. nmCherry=17 neurons/3 mice, nhM4Di=18 neurons/3 mice. Male 3 months-old mice were used. (n) Representative traces of AMPAR sEPSC. (o) Measurement of AMPAR-dependent sEPSC in naive NS-V mice without MWM training. Data were analyzed by 2-Way ANOVA. nmCherry=20 neurons/3 mice, nhM4Di=20 neurons/3 mice. Male 3 months-old mice were used. (p) A model. Data is expressed as mean ± S.E.M. For detailed statistics results, see Supplementary Table 1. * P ≤ 0.05 is set as significance. Independently, the electrophysiological data was re-analyzed using the linear mixed-effects models (Supplementary Table S2).
The changes in AMPA receptors (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, AMPAR) and NMDA receptors (N-methyl-D-aspartate receptor, NMDAR) constitute a molecular basis of synaptic plasticity. Using WGA-GFP/tdTomato to label LHGABA-innervated CA3GABA neurons, we recorded AMPAR- and NMDAR-dependent sEPSC as a surrogate for learning-induced synaptic plasticity in vivo in this specific neuron population. Mice were either left untrained (naive) or trained for 3 days to memorize the location of a hidden platform in the MWM test, and were then allowed to rest for a day before recording. Training increased the amplitude of AMPAR-dependent sEPSC in LHGABA-innervated CA3GABA neurons in WT mice (Fig 6j-k), which led to an elevated AMPAR/NMDAR ratio (Supplemental Fig S6a). Such learning-induced changes were not observed in NS-V mice (Fig 6j-k). Training slightly increased the expression of the GABRA2 in LHGABA neurons (Supplemental Fig S6b-d). We did not observe obvious changes in firing activities of CA3-innervating LHGABA neurons (labeled with retrograde beads) in response to MWM training (Supplemental Fig S6e-h), although a small increase in the mIPSCs amplitude was observed (Fig S6e). This suggests that learning-induced CA3 plasticity in WT mice is not directly driven by a possible suppression of LHGABA neurons. Rather, LHGABA neurons may play a permissive role in learning-induced CA3 plasticity.
To directly address the role of LHGABA in learning-induced plasticity in NS-V mice in vivo, we injected AAV-FLEX-hM4Di or AAV-FLEX-mCherry into the LH of the NS-V mice (Fig 6l). Upon recovery, both groups of mice were trained in the MWM tests after CNO administration in each training session, followed by the recording of AMPAR- and NMDAR-dependent sEPSC. Such chemogenetic suppression of LHGABA neural activity in NS-V mice largely rescued training-induced increase in AMPAR-dependent sEPSC (Fig 6m-n, Supplemental Fig S6i). Chemogenetic suppression of LHGABA did not affect AMPAR-dependent sEPSCs in naive untrained NS-V mice (Fig 6o), which suggests that the learning experience and the normal LHGABA neural activity are both required for CA3 plasticity. These results demonstrate that LHGABA hyperexcitability contributes to the impaired synaptic plasticity in NS-V mice in vivo. Collectively, the data obtained from the animal models delineated a circuitry pathway through which NCORs regulate neurocognition (Fig 6p).
Genetic variants of the NCORs complex in human neurocognitive disorders
Estimates from exome sequencing data on over 60,000 apparently healthy individuals suggest that the observed numbers of loss-of-function variants for NCOR1 (n=8), NCOR2 (n=6) and HDAC3 (n=3) are significantly smaller than that for the expected numbers (NCOR1, n=87; NCOR2, n=65; HDAC3, n=20), with the probability of loss-of-function intolerance (pLI) scores of 1, 1, and 0.91, respectively, strongly suggesting that NCOR1, NCOR2, or HDAC3 are intolerant to loss-of-function variants in humans (a pLI score > 0.9 is considered to reflect intolerance35).
Importantly, we identified subjects carrying deleterious mutations in NCOR1, NCOR2 or HDAC3 with neurocognitive disorders from the DECIPHER database36, including one individual with a pathogenic deletion, one individual with a pathogenic single nucleotide variant (SNV), and two individuals with likely pathogenic SNVs (Fig 7a). For NCOR1, a 152 kb heterozygous de novo deletion was found in patient #251746, a boy with severe global developmental delay and no language expression. He developed refractory epilepsy before the age of 4 years, severe mental retardation, poor motor coordination, stereotyped hand and head movements (jactatio capitis), and auto-aggressive attitude (Fig 7a, b). The deletion removes the last 25 exons of NCOR1, which is predicted to result in depletion of the truncated allele through nonsense-mediated decay. The deletion also removes three other genes, ADORA2B, TTC19, and ZSWIM7. However, heterozygous loss-of-function variants in any of these three genes are tolerant in healthy individuals from the Exome Aggregation Consortium database (ExAC), with the pLI score of 0.09 and 0 for ZSWIM7 and ADORA2B, respectively. Defects in TTC19 cause mitochondrial complex III deficiency, nuclear type 2 (MIM: 615157), an autosomal recessive disorder that cannot explain the phenotypes in this patient with a heterozygous deletion. No other known pathogenic deletion was found in this patient. Therefore, we consider that NCOR1 is the likely contributing gene in patient #251746. A de novo heterozygous c.2182+2T>G variant of NCOR1 was identified in another 8-year old boy with global developmental delay, moderate learning difficulties, autistic spectrum disorder, epilepsy, absences and drop attacks, tip-toe walker, and double incontinence (Fig 7a, b). This SNV affects the canonical splice donor site of NCOR1 and has not been observed in the control cohort in the ExAC35. It is therefore classified as a likely pathogenic variant.
Figure 7. Genetic variants involving NCORs/HDAC3 in human with neurocognitive disorders.

. (a) Patients carrying copy number variants (CNV) or single nucleotide variants (SNV) in NCOR1, NCOR2, or HDAC3. Genomic coordinates are shown in hg19. DDD_SNV, single nucleotide variants retrieved from Deciphering Developmental Disorders (DDD) website (United Kingdom); kb, kilobase. (b-d) Schematic representations for the deletions and point mutations affecting NCOR1, NCOR2, or HDAC3, respectively, observed in patients with neurodevelopmental disorders. The locations of deletions are depicted in red, and the point mutations in pink. (e) Western blot of HEK-293 cells transfected with plasmids expressing wild-type (WT) HDAC3 with or without mutant L266S. The experiment was repeated independently once with similar results. The blot images have been cropped. (f) Fluorescence-based HDAC enzyme assay after anti-HDAC3 immunoprecipitates from cell lysates overexpressing the indicated HDAC3 proteins. Box plots center line, median; box limits, upper and lower quartiles; whiskers, minimal and maximum values. Data were analyzed by two-tailed unpaired t test. n=4 biological independent samples for each group. (g) Western blot of HEK-293 cells transfected with plasmids expressing WT HDAC3, WT NCOR1, with or without the NCOR1 deletion mutant (Del). The experiment was repeated independently once with similar results. Data were analyzed by two-tailed unpaired t test. n=3 biological independent samples for each group. The blot images have been cropped. (h) Chromatin immunoprecipitation (ChIP) with anti-HDAC3 antibodies followed by qPCR using primers targeting promoters of the indicated genes ARNTL and CDKN1A. RPLP0 serves as a negative control. Data is expressed as mean ± S.E.M. For detailed statistics results, see Supplementary Table 1. * P ≤ 0.05 is set as significance.
Heterozygous de novo missense variants in NCOR2 [c.6887G>A (p.R2296Q)] and HDAC3 [c.797T>C (p.L266S)] were observed in two patients (Fig 7c, d). The NCOR2 variant was found in a 17-year old girl with dentinogenesis imperfecta, obesity, severe learning difficulties, and coordination problems. The HDAC3 variant was found in a 12-year old girl who displayed learning difficulties, focus of nodular heterotopia on brain MRI, subcutaneous scalp arteriovenous malformation, and joint hypermobility. These two variants have not been detected in over 60,000 control individuals and are predicted to be deleterious to protein function by both the SIFT and the PolyPhen2 prediction programs. Selected patients are not known to carry additional pathogenic variants that can complicate their clinical presentations.
We performed cellular assays to further characterize the human genetic variants. Cells co-overexpressing a wild-type (WT) and the mutant HDAC3 (L266S) at 1:1 molar ratio showed dramatically reduced HDAC3 enzyme activity compared to the control cells overexpressing two copies of WT HDAC3 (Fig 7e, f). This is consistent with the HDAC3 crystal structure showing that L266 forms one wall of the catalytic tunnel and is likely important for the substrate to access the active site37. Overexpression of WT HDAC3 itself did not increase the total HDAC3 deacetylase activity, in keeping with the previous finding that the endogenous NCORs is a limiting factor for HDAC3 enzyme activity18. L266S overexpression reduced the enzyme activity below the baseline, suggesting a dominant negative effect. The de novo deletion variant in NCOR1 in patient #251746 results in a C-terminal deleted NCOR1 protein (Del) with amino acids 1-666. Cells co-overexpressing NCOR1 WT and Del at 1:1 molar ratio showed significantly reduced chromatin occupancy of HDAC3 as compared to the control cells overexpressing two copies of WT NCOR1 (Fig 7g, h). These results suggest that the human genetic variants could cause loss-of-function for the NCORs/HDAC3 complex in the heterozygous scenario. The causative role of these mutations in cognitive behaviors needs to be further addressed in vivo using new genetic mouse models. In summary, NCOR1, NCOR2, and HDAC3 are promising Mendelian disease genes whose loss-of-function in humans could contribute to intellectual disability, neurodevelopmental defects, or multiple congenital anomalies.
DISCUSSION
Our results delineated a molecular and circuitry pathway through which NCORs regulate neurocognition (Fig 6p). NCORs loss-of-function causes downregulation of the GABAA receptor expression in LHGABA neurons, leading to hyperexcitability of these inhibitory neurons. Such an E/I imbalance in the hypothalamus inhibits LTP formation in the hippocampus, likely through monosynaptic LHGABA > CA3GABA projections, which underlies memory deficits caused by NCORs loss-of-function. This model is supported by the following observations. (1) Depletion of NCORs specifically in GABAergic neurons in NS-V mice recapitulated memory deficit observed in the NS-DADm mice and impaired LTP formation in the hippocampal CA3 region. (2) Electrophysiology in NS-V mice identified hyperexcitability in LHGABA neurons. (3) LHGABA neurons send monosynaptic projects to CA3GABA neurons. (4) Transcriptomic profiling identified GABAA receptor subunits as the molecular targets of NCORs in the hypothalamus, and downregulation of GABRA2 in LHGABA neurons was further confirmed by immunostaining and ribosomal profiling. (5) Targeted activation of the LHGABA > CA3GABA projection is sufficient to cause memory deficit. (6) Learning behaviors facilitate synaptic plasticity in LHGABA-innervated CA3GABA neurons in an NCORs-dependent manner. (7) Targeted manipulation of LHGABA neurons or the LHGABA > CA3GABA projection rescued LTP in hippocampal CA3 and cognitive deficits in NS-V mice. (8) Targeted manipulation of CA3GABA neurons rescued cognitive deficits in NS-V mice. (9) Positive modulators of GABAA receptor administered in the LH rescued cognitive deficits in NS-V mice. Collectively, these findings delineate a molecular and circuitry mechanism through which NCORs regulate synaptic plasticity and cognitive function.
Our study established a pivotal role of LHGABA neurons in memory and hippocampal synaptic plasticity. LH glutamatergic neurons expressing orexin were previously implicated in regulating memory formation38,39. LHGABA neurons were less studied in the context of learning and memory. In addition to the direct monosynaptic LHGABA to CA3GABA projection, it is possible that LHGABA neurons could affect hippocampal synaptic plasticity indirectly through projections to other brain regions. LHGABA neurons have been shown to regulate feeding40. However, we did not observe abnormal eating behaviors in NS-V mice or NS-DADm mice. It is possible that loss of NCORs in other brain regions compensates for orexigenic effects of LHGABA activation. It is also possible that the alteration of LHGABA excitability in NS-V or NS-DADm mice is not sufficient to change consummatory behaviors, unlike high-frequency optogenetic stimulation or genetic ablation of the LHGABA neurons.
It remains an open question whether LHGABA hyperexcitability can fully explain the seemingly hypoexcitability of CA3GABA neurons in the NS-V mice. The frequency of IPSCs/EPSCs generally reflects presynaptic inputs, while the amplitude is often attributable to postsynaptic mechanisms. However, synapses are complex and this general principle may not be applicable in all situations. CA3GABA neurons likely receive multiple inputs that are integrated in a complex manner that is yet to be fully defined. It is possible that the dysfunction of NCORs in other regions of the brain can also contribute to the cognitive deficits.
We found that several human patients with genetic variants involving NCORs or HDAC3 show growth abnormalities in multiple systems. This is consistent with the previous findings that NCORs and HDAC3 are essential for development41,42. However, growth abnormalities were not observed in NS-DADm mice or NS-V mice, except slightly lower body weight in both mouse lines compared to their littermate control. This suggests that the abnormal growth in various systems in human patients is attributable to HDAC3-independent functions of NCORs in cells that are distinct from GABAergic neurons. This further suggests that the function of NCORs in neurocognition can be separated from developmental regulation. Our previous study has demonstrated normal development and survival of NS-DADm mice despite lacking HDAC3 enzymatic acitivity19. Therefore, the developmental function of HDAC3 does not require its enzymatic activity, in line with its known deacetylase-independent function10. Our circuit mapping study revealed intact LHGABA > CA3GABA projections in NS-V mice, demonstrating normal anatomy and development of GABAergic neurons in those regions. The memory deficit in NS-DADm mice and NS-V mice can be rescued by small molecule modulators or chemogenetic manipulation in adult mice, indicating that the cognitive dysfunction is not caused by an irreversible anomaly originated from early development. One caveat is that the rescue experiments in the current study were performed acutely, so the long-term reversibility of cognitive deficits remains an open question.
Several previous studies have nicely demonstrated that HDAC3 can play different or opposite roles in neurocognition. Depletion of HDAC3 in hippocampus CA1 regions facilitated the formation of context memory22, while depletion of HDAC3 in forebrain excitatory neurons caused memory deficits in multiple neurobehavioral tests43. Transcriptomic changes were also context-dependent. Our RNA-seq showed that expression of Fos, Nr4a1, and Nr4a2 were not changed in the hypothalamus in NS-DADm mice, while Fos and Nr4a2 gene expression were upregulated in hippocampal CA1 after depletion of HDAC3 in CA1 using AAV-Cre injection22. In contrast, Fos and Nr4a1 were downregulated after depletion of HDAC3 in forebrain excitatory neurons using the Camk2a-Cre transgenic line43. These findings highlight the heterogeneity of gene expression regulation in the central nervous system.
The NCOR complex is one of several corepressor complexes that interact with MeCP244, and thus could partially contribute to MeCP2 function. MeCP2 mutations that either abolish or reinforce interactions with NCORs cause Rett-like phenotype, as nicely demonstrated in mouse models7,8. These data suggest that the NCOR/HDAC3 complex is required for normal function of MeCP2, although they do not exclude possibilities that these mutations could also affect other aspects of the MeCP2 function such as DNA binding45. Another recent study clearly showed that MeCP2 is required for maximum HDAC3 genomic recruitment in the hippocampus43. Comparison of our NS-DADm or NS-V mice with MeCP2-KO mice or MeCP2loxP/loxP/Vgat-Cre mice revealed some overlapping, but yet distinct phenotypes. Both MeCP2-KO mice and NS-DADm exhibit less anxiety, reduced social interaction, and explicit memory deficit46. But unlike MeCP2-KO mice, NS-DADm mice exhibit normal brain weight, normal hindlimb activity, regular breathing, and normal survival for at least a year. Also contrary to the MeCP2-KO mice, NS-DADm mice show hyperactivity, superior locomotor coordination, and better cerebellar learning than control. Neurocognitive functions of both MeCP2 and NCORs can be largely attributed to their function in GABAergic neurons. However, MeCP2loxP/loxP/Vgat-Cre mice display lower locomotor activity without memory deficit47, while NS-V mice show a robust deficit in explicit memory without changes in total locomotor activity. At the molecular level, transcriptome and cistrome analysis of MeCP2 and NCORs revealed some, but limited, similarities in the hypothalamus. Expression of GABRA2 is downregulated in both NS-DADm, NS-V, and MeCP2-KO hypothalamus48,49. However, MeCP2 target genes BDNF and GAD1/2 remain unaltered in the NS-DADm hypothalamus. Although both NCORs and MeCP2 are known as transcription repressors, loss-of-function of either NCORs or MeCP2 causes more genes downregulated than upregulated in the hypothalamus. The molecular basis of such regulation remains elusive in the hypothalamus and further studies are required to delineate the relationship between NCORs and MeCP2 in various brain regions. As far as neuronal activity is concerned, both MeCP2-KO mice and NS-V mice showed neuronal hyperexcitability at certain brain regions50. These findings support excitation/inhibition imbalance as an important factor contributing to cognitive dysfunction and pinpoint the inhibitory GABA signaling as a possible therapeutic target. We attempted treatment in the adult NS-DADm and NS-V mice with small molecule modulators or chemogenetic manipulation and successfully reversed the cognitive and memory deficits, which provided strategic insights for the treatment of NCORs-related neurocognitive disorders in human patients.
NCORs are fundamental players in the action of many endocrine factors. Hypothalamus is the central nexus of neuroendocrine regulation that governs many basic biological processes through hormones. Our finding thus established a hypothalamic-hippocampal communication that potentially links endocrine signals with synaptic plasticity through NCORs-mediated regulation of GABA signaling.
METHODS
Animals
NS-DADm mice were generated from crossing N-DADm and S-DADm mice19. HDAC3loxP/loxP mice were described before51. Vgat-Cre mice24 and Rosa26-tdTomato26 mice were obtained from JAX. The NCOR1/2loxP/loxP mice were provided by PHENOMIN, Institut Clinique de la Souris (ICS), CNRS, INSERM, University of Strasbourg, France (http://www.phenomin.fr/). NCOR1loxP/loxP/NCOR2loxP/loxP/Vgat-Cre (NS-V) mice were generated through crossbreeding. tdTomatoloxP/loxP/NCOR1loxP/loxP/NCOR2loxP/loxP/Vgat-Cre mice (tdTomato-labeled NS-V mice) were further generated for circuit mapping and electrophysiology studies. All mice were C57BL/6 genetic background. Male mice at the age of 2-6 months were used for all experiments except otherwise noted. Female mice were also used for some of the tests and no sexual dimorphism was observed for the phenotypes of NS-DADm mice or NS-V mice. For NS-DADm mice, wild-type littermates were used as the control and referred to as WT. For the initial characterization of NS-V mice, NCOR1loxP/loxP/NCOR2loxP/loxP mice were used as the control and referred to as WT. For electrophysiology studies, ribosomal profiling, circuit mapping, and chemogenetic experiments involving NS-V mice, Vgat-Cre mice or tdTomato/Vgat-Cre mice served as control and referred to as WT. All tests were repeated at least 2 times. 5 mice were housed in each cage in a 12-12 light-dark (7am - 7pm) facility with free access to water and food. After surgery, mice were housed singly in the same facility. All animals were grouped according to their sex, age, and genotype. Mice with the same sex, age, and genotypes were randomized into different surgery groups or treatment groups. All the animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at Baylor College of Medicine.
Behavioral tests:
All the behavior tests were done during 12pm-6pm in a dim light environment unless specifically mentioned. Mice were given a 2-weeks interval to recover from the first behavioral test to have a next behavioral test. Mice were not subjected to more than four behavioral tests.
Novel Object Recognition Test:
For Novel Object Recognition test (NOR), the objects used in the Novel Object Recognition test were built from Legos and were provided by the Neurobehavioral Core at Baylor. We used a small (22cm x 44 cm) arena to facilitate object exploration and reduce the time needed to habituate mice to the arena. The arena was surrounded on three sides by a white screen to limit spatial information and prevent spatial biases. During the training session, two identical objects were placed at both right and left side of the arena. During the first days, mice were put into the center of the arena and allowed to explore freely for 5 min. After that, mice were returned to their home-cages. 24 hours later, one of the objects was replaced by a novel object with different color and shape, and mice were allowed to freely explore the whole arena for 5 min. Animal behavior during the training and test session was tracked by a top camera and analyzed by ANY-maze software (Stoelting Co.). Discrimination index was calculated as (Exploration time on the novel object - Exploration time on the familiar object) / (Exploration time on the novel object + Exploration time on the familiar object). Sniffing, touching (>1s), and staring the objects were judged as exploration behaviors52.
Elevated Plus Maze:
For Elevated Plus Maze test, we used a plus-shaped platform that was elevated to 40 cm above the floor. Two opposite arms of the maze were walled (15cm high), whereas the other two arms were open with a 5mm high ridge to prevent falling. Each arm was 8cm wide, 25 cm long. The test lasted for 10 min and was started by placing a mouse in the center part of the maze facing one of the two open arms under a bright environment. An overhead camera and the ANY-maze software program were used to track the position of the mouse in the elevated plus maze. The number of visits to the open arms and the time spent on the open arms was used as measures for anxiety53.
Open-Field Arena Test:
Open-Field Arena Test was performed using the Versamax animal activity monitor equipped with infra-red photo beams as horizontal X-Y sensors and/or Z sensors. Mice were placed in the center of the open-field arena (40cm x 40cm x 30cm) and allowed to explore for 60-min. The locomotor activity and location of the mice were scored automatically by VersaMax software. The percentage of time spent in the center area measures anxiety levels.
Light-Dark Test:
The Light-Dark Test was performed in a box (42cm x 20cm x 25 cm) that contained a dark area (1/3) and light area (2/3) connected by a small opening to allow mice to move from one area to the other. The test lasted for 10 min and started by placing a mouse in the bright area. The activity and location of the mouse was scored automatically by VersaMax software. The number of transitions between dark and light zones and the time spent in the light and dark area was the index for anxiety54.
Morris Water Maze:
Morris Water Maze test (MWM) was performed as described55 with modification. During the training session, a transparent rescue platform was submerged under the painted water (0.5cm-1cm) and was placed in a fixed position of the pool. On the first day training, mice were first let stand on the platform for 10s. After that, mice were gently placed into the water facing the wall of the pool and are allowed to freely explore the whole maze for 1min. Mice were then guided to the rescue platform if they did not find it. Mice were allowed to take a rest on the platform for 10s, and then re-trained from a different start position with the same procedure. After four training trials, they were dried by a paper towel and returned to home cages. Twenty-four hours later, mice were trained again following the same procedure without the initial habituation session. Mice were trained for five consecutive days. At the end of the 4th trial on day 5, mice were returned to home cages for a rest. One hour later, mice were put into the water maze for 1 min, where the platform had been removed. Mouse behaviors were videotaped and analyzed by the Noldus EthoVision XT. The Morris Water Maze was virtually divided into 4 quadrants. The rescue platform located in the target quadrant. Escape latency was defined as time spent before finding the platform. Escape latency during the 5-day training sessions served as an independent measurement of spatial learning and memory.
Rotarod Test:
Rotarod test was performed using an accelerating rotarod (UGO Basile)56 with minor modification. Mice were placed on a rotating drum, which was accelerated from 4 to 40 rpm over a 5 min period. Time spent walking on top of the rod before falling off the rod or hanging on and riding completely around the rod was recorded. The rod was only 20 cm above the platform, so mice were not injured falling off the rod. Mice were given 2 trials per day for 4 consecutive days, with a maximum time of 5 min per trial and a 30-60 min inter-trial rest interval.
Social Interaction Test:
Social Interaction Test was performed in a 3-chamber (Crawley) apparatus (60.8 × 40.5 × 23 cm) that was comprised of three chambers (left, center, right) of equal size with 10 × 5 cm openings between the chambers with minor modification57. Mice were given two consecutive 10 min tests: the first measured baseline activity in the apparatus and the second test measured sociability of the test mouse given a choice of exploring a chamber containing another mouse under a mesh pencil cup or a chamber containing a novel object under a mesh pencil cup. A camera and the ANY-maze Behavioral Tracking software program (Stoelting Co.) were used to track the position of the mouse in the 3-chambered box, while the user scores the approaches to the object or partner mouse using a wireless keyboard. Partner mice (sex-, age- and weight-matched) were purchased from the Center for Comparative Medicine (CCM) at Baylor College of Medicine and habituated to the mesh pencil cups in the apparatus for 1 hour per day for 2 days prior to the day of testing. Partner mice were used up to 3 times, with one test per day.
Virus, stereotaxic injection, DREADD, and Optogenetics
AAV.CAG.FLEX.Rpl10a-GFP-Flag vector was constructed based on the AAV.CAG.FLEX.GFP plasmid (Addgene 28304, from Dr. Edward Boyden), and was pseudotyped with AAV9 serotype during virus production. AAV5.FLEX.hM4Di-mCherry, and AAV5.FLEX.mCherry were was obtained from Dr. Bryan Roth58 through the Vector Core at the University of Northern Carolina at Chapel Hill. AAV5.FLEX.hM3Dq-mCherry was obtained from Addgene (44361-AAV-5)59. AAV2.EF1a.FLEX.hChR2(H134R)-EYFP and AAV2.EF1a.FLEX.eNpHR3.0-EYFP were from Dr. Karl Deisseroth at Stanford University through the Vector Core at the University of Northern Carolina. AAV8.TR.eGFP and AAV8.hSyn.GFP.Cre were provided by the Vector Core at University of Northern Carolina at Chapel Hill. Ad.iN.WGA-GFP was provided by Dr. Martin Meyer at the University of Michigan27. AAV.EF1α.FLEX.synaptophysin-EGFP.WPRE.hGHpA, serotype DJ/8, was from Dr. Ben Arenkiel at BCM for synaptophysin tracing. All virus was tittered at around 10E13 GC/ml. Mice were anesthetized with ketamine and xylazine (100mg/kg:10mg/kg) and the head were fixed on the surgery platform by a stereotaxic system (Stoelting Co). For virus injection, a blur hole was drilled on each side of the skull (posterior 1.06 mm and lateral 1.2 mm to the bregma; depth: 5.48 mm for LH; and posterior 1.98mm and lateral 2.0mm to the bregma, depth: 2.25mm for CA3), and 150nl of each virus/side/mouse was injected. For i.c.v. injection, a mouse was implanted with a guide cannula (posterior 0.8, lateral 1.6 to the bregma; depth 2.2mm for i.c.v.). One or two weeks later after the surgery, diazepam (0.5µg/mouse/µl) or SL651498 (1µg/2µl/mouse) was injected through the cannula in the freely moving mouse 2hr. prior to the tests. DREADD (Designer Receptor Exclusively Activated by Designer Drugs) uses engineered G-protein coupled receptors (GPCRs) to activate or silence specific neurons in response to clozapine-N-oxide (CNO)29. Mice were i.p. injected with 3 mg/kg CNO shortly before each session of neurobehavioral tests. For optogenetics, two photo-fibers (Thorlabs, CFML12U-20) were implanted above the CA3 regions (posterior 2.18, lateral 2.2mm to the bregma, depth 2.5 mm for CA3) after the virus injection. Mice were singly housed after the surgeries and were allowed to recover for one or two weeks prior to any test. For examining the mIPSCs and mEPSCs at CA3-innervating LHGABA neurons, the green Retrobeads™ IX (Lumafluor Inc.) were injected into the CA3 (posterior 2.18, lateral 2.2mm to the bregma, depth 2.5 mm for CA3)60. Mice were allowed to recover in their home cages for one week before any training and test. For optogenetic activation in CA3GABA neurons, 10-ms 473-nM blue light pulses with 3s on and 2s off were delivered from a 473-nm blue solid state laser (MBL-III-473, Changchun New Industries Optoelectronics Technology Co., Ltd., China). The intensity power at the fiber tip was about 10-15 mW. The stimulation frequency and duration varied for the different experiments (see figure legends). The optogenetic inhibition of LHGABA neurons was achieved using a continuously 10-ms 598-nm yellow light (MGL-F-515, Changchun New Industries Optoelectronics Technology Co., Ltd., China). The power at the fiber tip was 7 mW. A fiber-optic rotary joint (FRJ_1×1_FC-FC, Doric Lenses) was used to avoid winding of the fiber-optic cable in freely behaving animals. Accurate injection or implantation was validated by either immunostaining or dye infusion. Mis-injected mice were excluded during the final data analysis. Mice that did not recover to their pre-surgery body weight were also excluded.
DNA constructs, Immunoprecipitation, HDAC assay, and western blot
Human full-length cDNA for NCOR1 was obtained from transOMIC and was sub-cloned into the pcDNA3.1-Zeo+ plasmid with C-terminal Flag-tag using standard PCR method for both WT and the 1-666 truncation. pcDNA3-based Flag-tagged HDAC3 cDNA construct was described before10 and the L266S point mutation was introduced using standard PCR method. All plasmids were validated by Sanger sequencing. HEK293 cells were co-transfected with plasmids for HDAC3 WT: L266S at 1:1 molar ratio, with the control cells transfected with double amount of plasmids for WT (equivalent with WT: WT at 1:1 ratio). Cells were lysed in buffer containing 1% NP40 and immunoprecipitated with anti-HDAC3 antibodies, followed by a fluorescence-based HDAC enzyme assay (Active Motif). For testing NCOR1 deletion (Del) mutants, HEK293 cells were co-transfected with plasmids for HDAC3 WT: NCOR1 WT: NCOR1 Del at 2:1:1 molar ratio, with 2:2:0 serving as the control condition. Empty pcDNA3 plasmid serves as a baseline control (Vector).
For HDAC assay, brain tissues were lysed in RIPA lysis buffer containing 0.1% SDS, 1% NP40, 0.5% sodium deoxycholate, and phosphatase/ protease inhibitors. Hypothalamus from 6 mice were pooled as one sample, whereas the cortex or hippocampus from each mouse was treated as one sample. The equal amount of total protein from each sample was subjected to immunoprecipitation with HDAC3 antibodies (Abcam 7030) or HDAC1 antibodies (Abcam ab7028) followed by protein A agarose beads (Invitrogen Cat#15918014). After washing with lysis buffer, the beads were dried using an insulin syringe and mixed with the working solution containing a fluorescence-tagged acetylated peptide from the HDAC assay kit (Active Motif Cat#56200). The reaction was allowed to last for 40 min before quenching with the developing solution containing HDAC inhibitors, followed by fluorescence measurement in a plate-reader. For western blot, immunoprecipitates and total tissue lysates were resolved by Tris-glycine SDS-PAGE, transferred to PVDF membranes, and blotted with antibodies against NCOR1 (lab-made)10, TBLR1 (IMGENEX, IMG591), and HDAC3 (Abcam 7030).
Immunostaining, in-situ hybridization, and anterograde tracing
Mice were anesthetized for transcardiac perfusion with cold PBS and 4% paraformaldehyde. Overnight-fixed brains were immersed in 30% sucrose, and embedded in the O.C.T. compound and frozen in isopentane. Coronal brain sections (10 µm) were prepared on the Leica CM1850 cryostat slicer. The coronal sections corresponding to Bregma: −2.8 mm to −0.3 mm were collected. Anti-GABRA2 antibodies (Cat#224104, Synaptic Systems, Geottingen, Germany. 1:50), anti-HDAC3 (SC-11417, Santa Cruz, 1:100), anti-glutamate (#G6642, Sigma-Aldrich, 1:100), anti-GFP (#2956, Cell Signaling. 1:75), Alexa Fluor 488 goat anti rabbit IgG(H+L) (A1034, Life Tech. 1:1000), Alexa Fluor 647 goat anti-rabbit IgG(H+L) (A32733, Life Tech, 1:1000), Alexa Fluor 546 goat anti-rabbit IgG(H+L) (A11010, Life Tech, 1:1000), and Alexa Fluor 488 goat anti-rabbit IgG(H+L) (A1034, Life Tech. 1:1000), and Alexa Fluor 488 goat anti-guinea pig IgG (H+L) (A11073, Life Tech. 1:1000) were diluted in PBS blocking buffer separately before use. Brain sections were incubated with 1st antibodies at 4 °C overnight. After that, sections were washed for 3 times in PBS at room temperature and furtherly incubated with 2nd antibodies tagged with fluorescence group for 1 hour at room temperature. These sections were then washed 3 times in PBS at room temperature, counterstained with Hoechst, and sealed with the coverslip. For in-situ hybridization, adult male Vgat-Cre mice and NS-V mice were sent to the RNA In Situ Hybridization Core (Baylor College of Medicine). For detection of GABAergic neurons in the brain, a pre-proved probe of Gad1 was used (GENEPAINT ID: EG1070, genepaint.org). For determining the knock-out of NCORs in NS-V mice, we designed a probe of NCOR1 with the sequence below. Immunofluorescence of brain sections is viewed and captured with the Zeiss Axio imager.M2m microscope and processed by ImageJ software.
(GCTAGGAGTGAGCATGAGATTTCTGAAATTATTGATGGTCTTTCTGAACAGGAGAATAATGAGAAGCAAATGCGTCAGCTTTCTGTGATTCCACCTATGATGTTTGATGCAGAACAAAGAAGGGTCAAATTCATCAATATGAATGGGCTGATGGAGGATCCAATGAAGGTTTATAAAGACAGACAGTTTATGAATGTTTGGACTGACCATGAAAAGGAGATCTTTAAGGACAAGTTTATCCAGCATCCAAAAAACTTTGGACTAATTGCATCCTATTTGGAAAGGAAGAGTGTTCCTGATTGTGTTTTATATTACTATTTAACCAAGAAAAATGAGAATTATAAGGCCCTCGTGAGAAGGAATTATGGAAAACGCAGAGGCAGAAATCAGCAGATTGCCCGTCCCTCACAAGAAGAAAAAGTAGAAGAAAAGGAAGAGGATAAAGCAGAAAAAACAGAGAAAAAGGAAGAAGAAAAGAAGGATGATGAAGAAAAAGATGATAAGGAAGACTCTAAAGAAACAACCAAGGAAAAGGACAGAACGGAAGCCACAGCAGAAGAACCTGAAGAAAGAGAG).
Electrophysiology
Whole-cell patch clamp recordings were performed on tdTomato-labeled GABAergic neurons from the hippocampus and lateral hypothalamus (LH) of NS-V and WT control mice. Mice of 6-12 weeks old were deeply anesthetized with isoflurane and transcardially perfused with a modified ice-cold sucrose-based cutting solution (pH 7.3) containing 10 mM NaCl, 25 mM NaHCO3, 195 mM Sucrose, 5 mM Glucose, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM Na-Pyruvate, 0.5 mM CaCl2, and 7 mM MgCl2, bubbled continuously with 95% O2 and 5% CO261. The mice were then decapitated, and the entire brain was removed and immediately submerged in the cutting solution. Slices (250 µm) were cut with a Microm HM 650V vibratome (Thermo Scientific). Three brain slices containing the hippocampus and LH were obtained for each animal, and recordings were made at levels throughout this brain region. The slices were recovered for 1 h at 34°C and then maintained at room temperature in artificial cerebrospinal fluid (aCSF, pH 7.3) containing 126 mM NaCl, 2.5 mM KCl, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 11.1 mM glucose, and 21.4 mM NaHCO3 saturated with 95% O2 and 5% CO2 before recording. Slices were transferred to a recording chamber and allowed to equilibrate for at least 10 min before recording. The slices were perfused at 34°C in oxygenated aCSF at a flow rate of 1.8-2 ml/min. tdTomato-labeled neurons in the LH and hippocampus (CA3, CA1 and DG regions) were visualized using epifluorescence and IR-DIC imaging on an upright microscope (Eclipse FN-1, Nikon) equipped with a moveable stage (MP-285, Sutter Instrument). Patch pipettes with resistances of 3-5 MΩ were filled with intracellular solution (pH 7.3) containing 128 mM K-Gluconate, 10 mM KCl, 10 mM HEPES, 0.1 mM EGTA, 2 mM MgCl2, 0.05 mM (Na)2GTP, and 0.05 mM (Mg)ATP. Recordings were made using a MultiClamp 700B amplifier (Axon Instrument), sampled using Digidata 1440A and analyzed offline with pClamp 10.3 software (Axon Instruments). Series resistance was monitored during the recording, and the values were generally <10 MΩ and were not compensated. The liquid junction potential was +12.5 mV, and was corrected after the experiment. Data were excluded if the series resistance increased dramatically during the experiment or without overshoot for the action potential. Currents were amplified, filtered at 1 kHz, and digitized at 20 kHz. The current clamp was engaged to test neural firing frequency and resting membrane potential (RM). The values for RM and firing frequency were averaged within 2-min bin.
For the miniature excitatory postsynaptic current (mEPSC) recordings, the internal recording solution contained 125 mM CsCH3SO3, 10 mM CsCl, 5 mM NaCl, 2 mM MgCl2, 1 mM EGTA, 10 mM HEPES, 5 mM (Mg)ATP, and 0.3 mM (Na)2GTP (pH 7.3 with NaOH)62. The mEPSCs were recorded in whole-cell voltage-clamp mode, by holding the membrane potential at Vh = −60 mV in the presence of 1μM TTX, 50 μM bicuculline. The miniature inhibitory postsynaptic current (mIPSC) recordings were recorded in whole-cell voltage-clamp mode by holding the membrane potential at Vh = −70 mV. The CsCl-based pipette solution contains 140mM CsCl, 10mM HEPES, 5mM MgCl2, 1mM BAPTA, 5mM (Mg)ATP, and 0.3mM (Na)2GTP (pH 7.30 adjusted with NaOH; 295 mOsm kg−1). The mIPSCs were recorded in the presence of 1μM TTX, 30 μM D-AP5, and 30 μM CNQX63. Frequency and peak amplitude were measured using the Mini Analysis program (Synaptosoft Inc.).
The E/I ratio was measured in a separate experiment. We used another general pipette solution containing 55 mM CsCH3SO3, 70 mM CsCl, 5 mM NaCl, 2 mM MgCl2, 1 mM EGTA, 10 mM HEPES, 5 mM (Mg)ATP, and 0.3 mM (Na)2GTP (pH 7.3 with NaOH). 1uM TTX was added in the bath to block the sodium current. A total post-synaptic current baseline was recorded at the holding voltage Vh = −70mV. mIPSC current amplitude was calculated by subtracting the average current amplitude in the presence of 50 μM bicuculline from that recorded in its absence. mEPSC current amplitude was then calculated by subtracting the background current amplitude (recorded in the presence of 50 μM bicuculline, 50 μM D-AP5, and 30 μM CNQX) from that recorded in the presence of 50 μM bicuculline only62–64.
For measuring AMPAR and NMDAR sEPSC currents, CA3GABA neurons were recorded in whole-cell voltage-clamp mode by holding the membrane potential at Vh = −60 mV. The pipette solution contained 125 mM CsCH3SO3, 10 mM CsCl, 5 mM NaCl, 2 mM MgCl2, 1 mM EGTA, 10 mM HEPES, 5 mM (Mg)ATP, and 0.3 mM (Na)2GTP (pH 7.3 with NaOH). A baseline of average sEPSC total current amplitude was recorded and analyzed. NMDAR sEPSC amplitude was calculated by subtracting the average sEPSC total current amplitude in the presence of 50 μM D-AP5 from that recorded in its absence. AMPAR sEPSC amplitude was then calculated by subtracting the background current amplitude (recorded in the presence of 50 μM D-AP5 and 30 μM CNQX) from that recorded in the presence of D-AP5 only62.
For recording LTP, whole-cell EPSPs were recorded in CA3GABA neurons (labeled by both tdTomato and WGA-GFP) in standard aCSF containing 1mM (MgCl2) and 2.5mM (CaCl2) at 34°C. Series resistance was fully compensated, and the pipette capacitance was ~70% compensated. Both resting membrane potential and input resistance of the recorded CA3 neurons were monitored over experimental time. Pipettes were filled with solution (in mM): 135 K-gluconate, 5 KCl, 10 Hepes, 5 phosphocreatine, 2 (Mg)ATP, 0.4 (Na)2GTP and 0.07 CaCl2 (pH 7.2). Bipolar stimulating electrodes were put in the dentate gyrus (DG). LTP was induced by high-frequency stimulation (HFS) paradigms65,66. The HFS protocol consisted of 3 trains of 100 stimuli at 100 Hz delivered every 10 s66. The inhibitory DREADD expressed in LHGABA neurons was validated by bath perfuse 10 μM CNO under current clamp. Resting membrane potential of LHGABA neurons hyperpolarized or depolarized more than 2 mV was defined as inhibition or activation.
Some recordings were done using the perforated patch. Briefly, the patch pipette solution for gramicidin perforated patch recording contained (in mM): KCl 150 and HEPES 10, pH 7.2 adjusted with Tris-OH. The gramicidin was first dissolved in DMSO in a stock solution of 60 mg/mL and then diluted to a final concentration of 30 μg/mL. The gramicidin free pipette solution was back filed into the pipette first and gramicidin containing pipette solution was then added into the pipette67. After 10 to 15 min of cell-attached formation, series resistance (Rs) decreased and stabilized at around 10 to 50 MΩ. Resting membrane was then recorded in the current clamp model.
Channelrhodopsin-2–assisted circuit mapping (CRACM)
To determine the LH project to CA3 neural circuits, AAV2.EF1α.FLEX.hChR2(H134R)-EYFP and Ad-iN/GFP-WGA were co-injected into the LH region of Vgat-Cre mice. Evoked IPSCs of the mCherry and GFP positive CA3 neurons (holding potential at −70mV) in brain slices were recorded in response to 473 nM blue light to validate the functional neural circuits from LHGABA neurons to CA3GABA neurons68. TTX (1 μ m) and 4-AP (100 μM) were added to the aCSF in order to confirm the response is monosynaptic responses69.
In vivo Optogenetic Behavior assays
For reducing the interference from the optic cord, mice injected with AAV.FLEX.ChR2-EYFP, AAV.FLEX.eNpHR3.0-EYFP, or AAV.FLEX.EYFP were acclimated to attached with the optic cord in the open arena with two identical objects with the light on for consecutive 3 days. On each day the mice were trained and acclimated to the environment for 5 min. During the training session, mice were chronically stimulated with trains of blue light (5-7 mW, 15 ms pulses, 20 Hz, 3 s ON and 2 s OFF) or yellow light (5-7 mW, 10 ms pulses, 20 Hz, 6000 pulse sequence). 24h later after the 3rd training, mice were re-exposed in a similar environment and the same light stimulus with one familiar object and one novel object. The animal behavior was recorded and analyzed by an experienced technician.
Ribosomal profiling, RT-qPCR, ChIP-qPCR, RNA-seq, and ChIP-seq
AAV9.FLEX.Rpl10a-Flag was stereotaxically injected into the LH of NS-V mice and Vgat-Cre WT mice. Mice were decapitated 3 weeks later, and hypothalamus tissues were harvested. Tissues from 6 mice were pooled and treated as one biological sample for the subsequent procedure. Tissues were homogenized in lysis buffer containing 100 µg/ml cycloheximide (CHX), 40 U/ml RNase inhibitor (Ambion) with a Downs homogenizer on ice. 1/9 sample volume of 10% NP-40 and 1% Saponin were added and mixed, followed by centrifugation at 20,000 ×g in 4 oC. An aliquot of total RNA was saved, while the rest were gently mixed with anti-FLAG M2 affinity gel (Sigma-Aldrich) for 30 min in the cold room with end-over-end. After washing the gel beads three times with washing buffer (20 mM HEPES-KOH [pH 7.4], 5 mM MgCl2, 350 mM KCl, 1% NP-40, 0.5 mM DTT, and 100 ug/ml CHX), we recovered enriched mRNA with phenol-chloroform extraction and ethanol precipitation with glycogen. The RT-qPCR analysis was performed using the reverse transcription kit (Life Tech) and SYBR green master mixture (Life Tech) to determine the fold enrichment.
RNA-seq was performed using total RNA extracted from the hypothalamus in NS-DADm mice and their littermate WT control mice (n = 3). ChIP and ChIP-seq were described previously10. Briefly, brain tissues were grounded in liquid N2 and cross-linked in 1% formaldehyde for 20 min at room temperature. Whole cell extracts were sonicated followed by immunoprecipitation with antibodies for HDAC3 (Abcam, ab7030) and Protein A Sepharose CL-4B (GE, 17-0780-01). After the immunoprecipitation, beads were washed by ChIP dilution buffer (50 mM HEPES [pH 7.5], 155 mM NaCl, 1.1% TritonX-100, 0.11% NaDeoxycholate, 1mM EDTA) for 3 times followed once wash by ChIP wash buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 0.5 % NaDeoxycholate, 250 mM LiCl, and 0.5 % NP-40). Sepharose beads were further washed one by TE buffer (Promega, E260A). DNA were enriched with phenol/chloroform extraction and dissolved in 20 µl TE buffer followed the RNA degradation by RNase (Roche, 11119915001) for 30min at 37°C. Four HDAC3 ChIP reactions were performed with each reaction containing samples from 3 mice. The ChIP reactions were then pooled to be sequenced as one sample, together with the pooled total input DNA as the control. The precipitated DNA was then pooled and amplified according to the guide of Illumina, followed by 50 bp single-end deep sequencing on Illumina Genome Analyzer IIx. For ChIP-qPCR analysis, primers were as follows: GABRA2 forward GATTAAGAAGGTTCCAGGGAATAGG, reverse GGTGGCTTGGCCAGGATT, distance to TSS is around 160 bp; GABRA4 forward CCAACCGACCCATCAACTGT, reverse TGATGCCCCAGCGTAGGT, distance to TSS is around 1.5 kb; GABRD forward CTGCCTGGAGCACAAACGA, reverse GCACTACCTGCAAAAGGGAATC, distance to TSS is around 270 bp. ChIP-qPCR was performed on 4 independent ChIP reactions with each reaction containing samples from 3 mice. A non-binding region near the gene Arbp served as the negative control (forward primer GAGGTGGCTTTGAACCAGAG; reverse primer TCTTTGTCTCTGTCTCGGAAAA).
Analysis of Behavioral Trials
For all behavioral studies, including those results from circuitry-targeted chemogenetic and optogenetic experiments, Student’s two-tail unpaired t-test, one-way ANOVA, two-way ANOVA, and repeated ANOVA followed by Tukey or LSD post hoc analysis were used to analyze data when it is appropriate. All intracranial injected mice were analyzed postmortem, and mis-injected mice were excluded during final data analysis. Mice that did not recover to their pre-surgery body weight were also excluded. Sample sizes were based on literature and were not determined by statistical methods. All data meet the assumptions of the specific statistical tests. Data collection and analysis were not completely blind to the conditions of the experiments. Behavioral experiments were done by experimentalists who know the drug treatment information but do not know the genotype or surgery information. During the early stage of analysis such as counting the time duration from video clips, experimentalists were blind to both genotype and treatment information. The statistical analysis and data plotting was then done by experimentalists who knew both genotype and treatment information. All data were individually plotted (Prism 7, GraphPad). The exact value of all N (number of animals), F, T, and P are reported in the figure legends and Supplemental Table 1. P<0.05 is set as significance.
Analysis of ex-vivo Electrophysiological Data
Multiple single neurons were recorded from 2-4 mice in each group and were pooled for statistical analysis. We performed recording in pairs of mice, with one mouse from each group per day. We used two-way ANOVA to compare between two groups. The genotype or treatment serves as one variable, and the recording day served as another independent variable to take into account the animal-to-animal or day-to-day variability. We also analyzed the data using the linear mixed-effects (LME) models with the packages “lme4” and “lmerTest” in the statistical package R version 3.5.1. The comparison between the experimental groups was made using the fixed effects while the possible correlation due to the mouse was modeled using the random effects70,71. The results were generally in line with the ANOVA analysis and were summarized in Supplemental Table 1 and Supplemental Table 2.
Analysis of Immunostaining, in-situ Hybridization, and Anterograde Tracing
For evaluating the expression of GABRA2LH in NS-DADm mice and its WT littermates, coronal brain sections including the lateral hypothalamus were used. The averaged optical density was measured by ImageJ software. The background was manually measured in the area near the analyzed GABARA2+ cells. The final results were calculated as O.D. = O.D.GABRA2 – O.D.background. Student’s two-tail t-test was used to analyze all tests when it is appropriate. P<0.05 is set as significance.
For evaluating the AAV-mediated HDAC3 depletion efficiency in targeted regions, the averaged optical density in the CA3 region and the LH region was measured by ImageJ software. The background optical density was based on the area nearby and was subtracted from the targeted regions. The final results were calculated as the percentage of the optical density of the control wild-type mice.
For evaluating the expression of NCOR1 in GABAergic neuron, GABAergic neurons were manually identified according to the expression of GAD1 and the optical density of NCOR1 in the GABAergic neurons was measured by ImageJ software. The final results were calculated as O.D. = O.D.GABAergic – O.D.background. Student’s two-tail t-test was used to analyze all tests when it is appropriate. P<0.05 is set as significance.
For identifying the projection of LH, Ad-iN/GFP-WGA was injected into one side of the lateral hypothalamus of tdTomatolox/lox/Vgat-Cre. The number of GFP+ or tdTomato+ cells in CA3 region was characterized. The number of co-localized GFP+ and tdTomato+ cells were also characterized. The number of GFP+ or Glutamate+ cells in CA3 was characterized. The number of co-localized GFP+ and Glutamate+ cells was also characterized. Student’s two-tail t-test was used to analyze all tests when it is appropriate. P<0.05 is set as significance. For further confirming the projection of LHGABA neurons, AAV.EF1α.FLEX.synaptophysin::EGFP.WPRE.hGHpA was injected into one side of the lateral hypothalamus of Vgat-Cre mice.
Analysis of ChIP-seq and RNA-seq
ChIP-seq reads were aligned to the mus musculus genome (mm10) using Bowtie2 V2.1.0 with following parameters: -D 20 -R 3 -N 0 -L 20 -i S,1,0.50. The average mappability across all the samples was ~96%. SAMtools V0.1.19 and BEDtools v2.17.0 were used to convert Sequence Alignment/Map (SAM) files to Binary Alignment Map (BAM) and Browser Extensible Data (BED) files respectively. HDAC3 ChIP samples were ratio normalized to their respective inputs and converted to bigwig format using the bamCompare (with --normalizeTo1x) module from deepTools. Normalized bigwig files were visualized using Integrative Genome Browser (IGV). HDAC3 peaks were called using used SICER v1.1 with a window size of 200nt, a gap of 300nt and a false discovery rate (FDR) of ≤ 0.05. Called peaks were annotated and plotted using ChIPseeker. For RNA-seq, raw sequence reads from each biological replicate of total RNA were first aligned to the UCSC mm10 genome with Tophat v2.1.0 using default parameters72. Then, HTSeq was used to obtain read counts from the aligned reads73. Finally, DESeq2 (version 1.8.2) was used to normalized the read counts and perform differential gene analysis74. Significantly differentially expressed genes were identified based on 5% false discovery rate (FDR) threshold.
Patient data
Data from the Exome Aggregation Consortium (ExAC) were used to estimate the intolerance of heterozygous loss-of-function variants in the apparently healthy human population. Data regarding the observed and the expected numbers of loss-of-function variants and the probability of being loss-of-function intolerant (pLI) for each gene were obtained from http://exac.broadinstitute.org/ and calculated based on the previous description35. In brief, the higher the pLI score (ranging from 0 to 1) is, the more likely that the gene is intolerant to heterozygous loss-of-function variants in humans. A pLI score higher than 0.9 is defined as being loss-of-function intolerant.
Patient data were obtained from the DECIPHER database (https://decipher.sanger.ac.uk/). For copy number variants, only small deletions affecting NCOR1, NCOR2 or HDAC3 were selected for analysis; large deletions that encompass additional disease genes for which heterozygous deletions are known or can potentially be associated with Mendelian disorders are excluded. Potential deleterious effects of the variants on protein functions were assessed by both the SIFT and the PolyPhen2 prediction programs75,76. The patients selected do not have additional known pathogenic deletions. For single nucleotide variants, only de novo changes are included. Single nucleotide variants that have been observed in apparently healthy individuals from the ExAC database are excluded. The patients selected are not known to carry additional pathogenic variants that could complicate their clinical presentations. Pathogenicities (pathogenic or likely pathogenic significance) of the variants from the patients are classified according to the guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology77. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC).
Statistical Analysis
Data analysis was performed using SPSS (V.21.0, IBM), or packages “lme4” and “lmerTest” (statistical package R version 3.5.1). For statistical significance, two-tail unpaired t test, or one-way repeated ANOVA was used for experiments with two groups. Experiments with more than two groups were tested by one-way ANOVA, two-way ANOVA, or two-way repeated ANOVA followed by post hoc Tukey or LSD for multiple comparisons. We aslo analysis the ex-vivo electrophysiological data using linear mixed-effects models, due to the limited mouse number. RNA-seq data were analyzed by DESeq2 (version 1.8.2) with the significant threshold of 5% FDR. All experiments were performed at least twice using independent biological samples or distinct cohorts of mice, except the RNA-seq and ChIP-seq studies that were performed once. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications78–83. All data met the assumptions of the statistical tests used. All data were tested for normality to determine the usage of parametric or nonparametric tests. Under the condition of nonequivalent variances, non-parametric equivalent of the independent t-test or nonparametric ANOVA was used to test the significance. Experiments were not completely blinded to experimental conditions. The behavior test experimenter was blinded to the genotype information, but not blinded to the pharmacological agent. Animals were excluded and euthanized before behavior tests if they showed distress, infection, bleeding, or anorexia due to surgery. Animals were excluded after behavior tests if they showed mis-injection at postmortem examination. All data were expressed as mean ± SEM.
Reporting Summary and Key reagents list
The detailed information of research design is available in the Life Sciences Reporting Summary linked to this article. A list of key reagents were summarized in the Supplementary Table S3.
Data Availability
The data that support the findings of this study are available from the corresponding author upon request. RNA-seq and ChIP-seq data are available in GEO (GSE92452).
Supplementary Material
ACKNOWLEDGEMENT
We thank Dr. Mitchell Lazar at University of Pennsylvania (UPenn) for the NS-DADm and HDAC3 loxP mice, Dr. Martin Myers Jr. at University of Michigan for the WGA adenovirus, Dr. Bryan Roth at University of North Carolina for the hM4Di AAV viral vector, Dr. Ben Arenkiel at Baylor College of Medicine (BCM) for the Syn-GFP AAV viral vector, Dr. Zhiping Pang at Rutgers and Dr. Long-Jun Wu at Mayo Clinic for critical reading of the manuscript, Dr. Tim O’Brien at UPenn for technical consultancy in behavior tests, Dr. Pingwen Xu and Justin Qian at BCM for technical assistance. We thank the BCM Neurobehavioral Core for providing space and training for behavioral tests (U54HD083092), BCM Genomic and RNA Profiling Core (Texas Medical Center Digestive Disease Center P30DK56338) and Texas A&M AgriLife Research Genomics and Bioinformatics Service for nucleotide sequencing, BCM Gene Vector core (Diabetes Research Center P30DK076938) for viral vector production, and BCM RNA In Situ Hybridization Core (U54HD083092, P30DK056338, U42OD011174) for assistance with histology studies. This study makes use of data generated by the DECIPHER community. A full list of centers that contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via e-mail from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome. Those who carried out the original analysis and collection of the data bear no responsibility for the further analysis or interpretation of it by the recipient or its registered users. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome and the Department of Health, and the Wellcome Sanger Institute (grant number WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome or the Department of Health. The study has UK Research Ethics Committee (REC) approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The DDD research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. The authors’ laboratories were supported by Taishan Scholarship (X.H.), American Diabetes Association (ADA1-17-PDF-138) (Y.H.), US Department of Agriculture (USDA) Cris6250-51000-059-04S (Y.X.), National Institutes of Health R01DK101379, R01DK117281, P01DK113954, R01DK115761 (Y.X.), and American Heart Association (AHA30970064), R01DK111436, R21CA215591, and R01ES027544 (Z.S.).
Footnotes
COMPETING FINANCIAL INTERESTS STATEMENT
The authors disclose no competing financial conflict of interest.
ACCESSION CODES
RNA-seq and ChIP-seq data are available in GEO (accession code GSE92452).
REFERENCES
- 1.Rudenko A & Tsai L-H Epigenetic regulation in memory and cognitive disorders. Neuroscience 264, 51–63 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Lonard DM & O’Malley BW Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat. Rev. Endocrinol. 8, 598–604 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mottis A, Mouchiroud L & Auwerx J Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 27, 819–835 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perissi V, Jepsen K, Glass CK & Rosenfeld MG Deconstructing repression: evolving models of co-repressor action. Nat. Rev. Genet. 11, 109–123 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Guenther MG et al. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 14, 1048–1057 (2000). [PMC free article] [PubMed] [Google Scholar]
- 6.Wen YD et al. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. U. S. A. 97, 7202–7207 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J et al. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 19, 4342–4350 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guenther MG, Barak O & Lazar MA The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 21, 6091–6101 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Z et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 18, 934–942 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Z et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol. Cell 52, 769–782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong S et al. Dissociation of muscle insulin sensitivity from exercise endurance in mice by HDAC3 depletion. Nat. Med. 23, 223–234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kokura K et al. The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 276, 34115–34121 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Amir RE et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Lyst MJ et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 16, 898–902 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebert DH et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 499, 341–345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoon H-G et al. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 22, 1336–1346 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. https://www.omim.org/entry/608628.
- 18.Ishizuka T & Lazar MA The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. Mol. Cell. Biol. 23, 5122–5131 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You S-H et al. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 20, 182–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischle W et al. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 9, 45–57 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Lahm A et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U. S. A. 104, 17335–17340 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McQuown SC et al. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. Off. J. Soc. Neurosci. 31, 764–774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lenègre A, Chermat R, Avril I, Stéru L & Porsolt RD Specificity of piracetam’s anti-amnesic activity in three models of amnesia in the mouse. Pharmacol. Biochem. Behav. 29, 625–629 (1988). [DOI] [PubMed] [Google Scholar]
- 24.Vong L et al. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnütgen F et al. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol. 21, 562–565 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Louis GW, Leinninger GM, Rhodes CJ & Myers MG Direct innervation and modulation of orexin neurons by lateral hypothalamic LepRb neurons. J. Neurosci. Off. J. Soc. Neurosci. 30, 11278–11287 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herman AM et al. A cholinergic basal forebrain feeding circuit modulates appetite suppression. Nature 538, 253–256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth BL DREADDs for Neuroscientists. Neuron 89, 683–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez JL et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357, 503–507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griebel G et al. SL651498: an anxioselective compound with functional selectivity for alpha2- and alpha3-containing gamma-aminobutyric acid(A) (GABA(A)) receptors. J. Pharmacol. Exp. Ther. 298, 753–768 (2001). [PubMed] [Google Scholar]
- 32.Laezza F & Dingledine R Voltage-controlled plasticity at GluR2-deficient synapses onto hippocampal interneurons. J. Neurophysiol. 92, 3575–3581 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Pelkey KA, Topolnik L, Yuan X-Q, Lacaille J-C & McBain CJ State-dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron 60, 980–987 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galván EJ et al. Synapse-specific compartmentalization of signaling cascades for LTP induction in CA3 interneurons. Neuroscience 290, 332–345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Firth HV et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 84, 524–533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watson PJ, Fairall L, Santos GM & Schwabe JWR Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 481, 335–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L et al. Hypocretin/orexin neurons contribute to hippocampus-dependent social memory and synaptic plasticity in mice. J. Neurosci. Off. J. Soc. Neurosci. 33, 5275–5284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selbach O et al. Orexins/hypocretins control bistability of hippocampal long-term synaptic plasticity through co-activation of multiple kinases. Acta Physiol. Oxf. Engl. 198, 277–285 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Jennings JH, Rizzi G, Stamatakis AM, Ung RL & Stuber GD The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science 341, 1517–1521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jepsen K et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 102, 753–763 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Norwood J, Franklin JM, Sharma D & D’Mello SR Histone deacetylase 3 is necessary for proper brain development. J. Biol. Chem. 289, 34569–34582 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nott A et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat. Neurosci. 19, 1497–1505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guy J, Cheval H, Selfridge J & Bird A The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Heckman LD, Chahrour MH & Zoghbi HY Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. eLife 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W & Pozzo-Miller L Beyond Widespread Mecp2 Deletions to Model Rett Syndrome: Conditional Spatio-Temporal Knockout, Single-Point Mutations and Transgenic Rescue Mice. Autism-Open Access 2012, 5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chao H-T et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chahrour M et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L et al. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. U. S. A. 112, 5509–5514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dani VS et al. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U. S. A. 102, 12560–12565 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 51.Mullican SE et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 25, 2480–2488 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou W, Kavelaars A & Heijnen CJ Metformin Prevents Cisplatin-Induced Cognitive Impairment and Brain Damage in Mice. PloS One 11, e0151890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polter A et al. Forkhead box, class O transcription factors in brain: regulation and behavioral manifestation. Biol. Psychiatry 65, 150–159 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khalil R & Fendt M Increased anxiety but normal fear and safety learning in orexin-deficient mice. Behav. Brain Res. 320, 210–218 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Vorhees CV & Williams MT Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 1, 848–858 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou W et al. The effects of glycogen synthase kinase-3beta in serotonin neurons. PloS One 7, e43262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou W, Chen L, Yang S, Li F & Li X Behavioral stress-induced activation of FoxO3a in the cerebral cortex of mice. Biol. Psychiatry 71, 583–592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Armbruster BN, Li X, Pausch MH, Herlitze S & Roth BL Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U. S. A. 104, 5163–5168 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krashes MJ et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 121, 1424–1428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grippo RM, Purohit AM, Zhang Q, Zweifel LS & Güler AD Direct Midbrain Dopamine Input to the Suprachiasmatic Nucleus Accelerates Circadian Entrainment. Curr. Biol. CB 27, 2465–2475.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ren H et al. FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 149, 1314–1326 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu T et al. Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron 73, 511–522 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fenselau H et al. A rapidly acting glutamatergic ARC→PVH satiety circuit postsynaptically regulated by α-MSH. Nat. Neurosci. 20, 42–51 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith KR et al. Cadherin-10 Maintains Excitatory/Inhibitory Ratio through Interactions with Synaptic Proteins. J. Neurosci. Off. J. Soc. Neurosci. 37, 11127–11139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yeckel MF, Kapur A & Johnston D Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat. Neurosci. 2, 625–633 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Urban NN, Henze DA, Lewis DA & Barrionuevo G Properties of LTP induction in the CA3 region of the primate hippocampus. Learn. Mem. Cold Spring Harb. N 3, 86–95 (1996). [DOI] [PubMed] [Google Scholar]
- 67.Khalilov I, Minlebaev M, Mukhtarov M, Juzekaeva E & Khazipov R Postsynaptic GABA(B) Receptors Contribute to the Termination of Giant Depolarizing Potentials in CA3 Neonatal Rat Hippocampus. Front. Cell. Neurosci. 11, 179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petreanu L, Huber D, Sobczyk A & Svoboda K Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat. Neurosci. 10, 663–668 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Adhikari A et al. Basomedial amygdala mediates top-down control of anxiety and fear. Nature 527, 179–185 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.R: a language and environment for statistical computing. Available at: https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing. (Accessed: 3rd September 2018)
- 71.Fitting Linear Mixed-Effects Models Using lme4 | Bates | Journal of Statistical Software. doi: 10.18637/jss.v067.i01 [DOI] [Google Scholar]
- 72.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinforma. Oxf. Engl. 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anders S & Huber W Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ng PC & Henikoff S SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adzhubei IA et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Richards S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 17, 405–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Faraco G et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat. Neurosci. 21, 240–249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y & Sabatini BL Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(−/−) mice. Nat. Neurosci. 19, 716–724 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Witton J et al. Hippocampal circuit dysfunction in the Tc1 mouse model of Down syndrome. Nat. Neurosci. 18, 1291–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu P et al. Estrogen receptor-α in medial amygdala neurons regulates body weight. J. Clin. Invest. 125, 2861–2876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perusini JN et al. Optogenetic stimulation of dentate gyrus engrams restores memory in Alzheimer’s disease mice. Hippocampus 27, 1110–1122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang W et al. Chemogenetic Activation of Prefrontal Cortex Rescues Synaptic and Behavioral Deficits in a Mouse Model of 16p11.2 Deletion Syndrome. J. Neurosci. Off. J. Soc. Neurosci. 38, 5939–5948 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. RNA-seq and ChIP-seq data are available in GEO (GSE92452).