

Abstract
Molecular mechanisms involved in the relapse of T‐cell acute lymphoblastic leukemia (T‐ALL) are not fully understood, although activating NOTCH1 signaling due to NOTCH1/FBXW7 alterations is a major oncogenic driver. To unravel the relevance of NOTCH1/FBXW7 mutations associated with relapse, we performed whole–exome sequencing in 30 pediatric T‐ALL cases, among which 11 diagnosis‐relapse paired cases were further investigated to track the clonal evolution of relapse using amplicon–based deep sequencing. NOTCH1/FBXW7 alterations were detected in 73.3% (diagnosis) and 72.7% (relapse) of cases. Single nucleotide variations in the heterodimerization domain were the most frequent (40.0%) at diagnosis, whereas proline, glutamic acid, serine, threonine–rich (PEST) domain alterations were the most frequent at relapse (54.5%). Comparison between non–relapsed and relapsed cases at diagnosis showed a predominance of PEST alterations in relapsed cases (P = .045), although we failed to validate this in the TARGET cohort. Based on the clonal analysis of diagnosis‐relapse samples, we identified NOTCH1 “switching” characterized by different NOTCH1 mutations in a major clone between diagnosis and relapse samples in 2 out of 11 diagnosis‐relapse paired cases analyzed. We found another NOTCH1 “switching” case in a previously reported Berlin‐Frankfurt‐Münster cohort (n = 13), indicating NOTCH1 importance in both the development and progression of T‐ALL. Despite the limitations of having a small sample size and a non–minimal residual disease–based protocol, our results suggest that the presence of NOTCH1 mutations might contribute to the disease relapse of T‐ALL.
Keywords: NOTCH1, pediatric leukemia, relapse, T‐cell acute lymphoblastic leukemia, whole–exome sequencing
1. INTRODUCTION
Although pediatric cases of T‐cell acute lymphoblastic leukemia (T‐ALL) have a cure rate exceeding 80% because of intensified chemotherapies and appropriate prognostic classifications, the outcome of T‐ALL patients with primary resistant or relapsed leukemia remains extremely poor.1, 2, 3 Recent insights into the biology of the disease have uncovered the genomic landscape of T‐ALL at diagnosis and defined T‐ALL subgroups.4, 5, 6, 7 However, the molecular basis of refractory/relapsed T‐ALL is largely unknown, except for the involvement of NT5C2 mutations in relapsed T‐ALL.8, 9 In addition, the underlying clonal evolution leading to relapse and treatment resistance has been poorly studied in pediatric T‐ALL. In contrast, NOTCH1 and/or FBXW7 alterations, leading to constitutive activation of NOTCH1 signaling, are among the most common changes detected in T‐ALL patients.6, 7, 10 It is well known that alterations in the negative regulatory region (NRR), such as single nucleotide variations in the heterodimerization (HD) domain (HD‐SNV) and small in–frame insertions or deletions in the HD domain (Indel), lead to constitutive activation of NOTCH1 without ligand binding.10 NOTCH1 transcriptional activation is terminated by the proteasomal degradation of the NOTCH1 intracellular domain (NICD), which is induced by the proline, glutamic acid, serine, threonine–rich (PEST) domain recognition of the FBXW7‐SCF ubiquitin ligase complex.11 Alterations in the PEST domain of NOTCH1 and FBXW7 result in impaired degradation (ID) of NICD, leading to aberrantly prolonged NOTCH1 signaling.11, 12 Thus, there are 2 patterns of activation of NOTCH1 signaling: ligand–independent activation (LIA) of NOTCH110, 13, 14, 15, 16 and ID of NOTCH1.10, 11, 12, 17
Although activated NOTCH1 signaling constitutes the most predominant oncogenic event involved in the pathogenesis of T‐ALL, it is widely reported that T‐ALL patients with NOTCH1 mutations have a favorable early therapeutic response18, 19 or outcome.20, 21, 22 However, some groups have reported no effect of NOTCH1 mutations in the outcome of T‐ALL.18, 23, 24 Thus, the prognostic relevance of NOTCH1 and/or FBXW7 alterations is still controversial and may be protocol–specific. Moreover, the role of NOTCH1 and/or FBXW7 mutations in T‐ALL relapse is unclear. Despite recent advances in genome–wide analyses of diagnostic T‐ALL, little is known about the involvement of NOTCH1 in the relapse and progression of T‐ALL.25, 26 To investigate relapse–related genes in the progression of T‐ALL from diagnosis to relapse, especially the impact of NOTCH1 and/or FBXW7 alterations, we performed genetic analysis of 30 pediatric T‐ALL cases using whole–exome sequencing (WXS) and amplicon–based deep sequencing. Among these 30 cases, 12 cases were relapsed, and 11 diagnosis‐relapse paired cases available were further investigated to track the clonal evolution of the relapses.
2. MATERIALS AND METHODS
2.1. Patients and materials
Thirty pediatric T‐ALL patients were enrolled in this study (Table S1); results for 24 cases were reported previously.6 Analyzed samples were mainly offered from the Tokyo Children’s Cancer Study Group (TCCSG) and the Japan Association of Childhood Leukemia Study (JACLS). All patients received Berlin‐ Frankfurt‐ Münster (BFM)–based chemotherapy. No minimal residual disease (MRD)–based risk stratification was performed. Written, informed consent was attained according to protocols approved by the Human Genome, Gene Analysis Research Ethics Committee of the University of Tokyo and other participating institutes. Peripheral blood, bone marrow blood and lymph node samples were collected from T‐ALL patients. Among 12 relapsed cases, relapse samples were available in 11 cases.
2.2. Whole–exome sequencing
Whole–exome sequencing of diagnosis/relapse tumor and matched normal specimens was performed as previously described.27 Whole–exome capture was accomplished using SureSelect Human All Exon Kit V3 or V5 (Agilent Technology, Wilmington, DE) and was subjected to sequencing using HiSeq 2000 (Illumina, San Diego, CA) according to the manufacturer's protocol. Raw sequence data were processed using our in–house pipelines (Genomon 2.3.0, https://github.com/Genomon-Project/GenomonPipeline). Sequence reads with a mapping quality score <25, base quality score <30, or 5 or more mismatched bases were excluded. Relevant somatic mutations were filtered by excluding variants: (i) with incomplete open reading frame information; (ii) listed in the 1000 Genomes Project (May 2011 release), NCBI SNP database (dbSNP) build 131, National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) 5400, the Human Genome Variation Database (HGVD; October 2013 release) or our in–house SNP database; (iii) represented only in unidirectional reads; (iv) occurring in repetitive genomic regions; (v) having a variant allele frequency (VAF) <.1; (vi) represented in <5 reads in tumor samples; (vii) occurring in non–paired normal samples with VAF >.0025; and (viii) occurring in paired normal samples with VAF ≥.1. Stringent criteria were used for calling mutations, requiring a P‐value for EBCall28 <10−4 and a Fisher's P‐value <10−2. Finally, mapping errors were removed by visual inspection using the Integrative Genomics Viewer browser.29
2.3. Validation and detection of variant allele frequency for mutations detected by whole–exome sequencing using amplicon–based deep sequencing
To validate the somatic mutations detected via WXS and to obtain more accurate VAF, we conducted amplicon–based deep sequencing at ≥1500× coverage (an average coverage of 7942×) using paired or trio DNA samples with MiSeq (Illumina). Because mutations in the 3′ untranslated region (UTR) of NOTCH1 could not be detected using WXS given the predetermined bait design, primers were specifically designed (Table S2). Target sequences were amplified using primers (Table S3) tagged with NotI cleavage sites (AAGCGGCCGC), and the PCR products were ligated and fragmented for deep sequencing as described previously.27
2.4. Analysis of T‐cell receptor rearrangements
T‐cell receptor (TCR) rearrangements were determined at the TCR γ locus using T Cell Receptor Gamma Gene Rearrangement Assay 2.0 for ABI Fluorescence Detection (Invivoscribe, San Diego, CA, USA, #1‐207‐101) according to the manufacturer's protocol.
2.5. Clonal analysis
Intrasample subpopulations were estimated using a hierarchical Beta Binomial emission model implemented in PyClone30 and the variational Bayesian beta mixture model in SciClone31 as described previously.32, 33 Mutations in indels, for which VAFs were poorly estimated, were not used for the analysis. We constructed branch–based phylogenetic trees using a bootstrap resampling technique implemented in ClonEvol.34 In each estimated clone, we defined minor clones and major clones as VAF <.15 and VAF >.15, respectively. To categorize the types of clonal evolution, we used a cutoff of VAF = .01.
2.6. Statistical analysis
Statistical analyses were performed using R v3.4.0 software with the R packages Survival and maftools. Statistical significance was assessed using Student's 2–tailed t test, and resulting values were considered statistically significant at P < .05.
3. RESULTS
3.1. Mutation detection at diagnosis and relapse of T‐cell acute lymphoblastic leukemia using whole–exome sequencing
We performed WXS on 30 cases of matched T‐ALL and normal (samples in complete remission phase) pairs, including 11 trios comprising samples at relapse, diagnosis, and normal (Table S1). One T‐ALL case (TALL023) developed Langerhans cell histiocytosis (LCH) after achieving complete remission. In this case, the LCH cells are not only a derivative of the same ancestral clone but should be a branched clone from the one that already gave rise to T‐ALL, because the LCH cells have the same TCR rearrangements (Figure S1D,E).35, 36 WXS was performed at an average coverage of 107.8× (range: 66.2‐201.9) (Figure S2). All predicted SNVs were validated by amplicon–based deep sequencing (an average coverage of 7942×). Across the coding regions of 30 cases (total 41 samples: 30 samples at diagnosis and 11 samples at relapse), we detected 777 somatic mutations and structural variants in 515 genes (Tables [Link], [Link]). Each T‐ALL sample at diagnosis harbored a mean of 16.9 somatic mutations (range: 3‐29), of which the mean number of mutations without synonymous SNV was 14.4 per sample (range: 2‐24). The mutation rate of relapse samples was significantly higher than that of samples at diagnosis, with a mean of 24.6 mutations per sample (range: 9‐40, P = .017; Figure S3) and a mean of 20.5 mutations without synonymous SNV per sample (range: 9‐31; P = .014; Figure 1A). No difference was observed between non–relapsed and relapsed cases in the mutation rate of diagnostic T‐ALL samples (Figure 1A and Figure S3). Because of the small sample number of cases analyzed, we could not find different features of immunophenotype and molecular subgroups between non–relapse and relapse samples (Table 1 and Figure S4). These subgroups were defined in our previous study6 based on expression data of whole–transcriptome sequencing. Consistent with previous reports, mutations in NOTCH1 (66.7%), FBXW7 (20.0%), DNM2 (20.0%) and PHF6 (20.0%) were common in T‐ALL samples at diagnosis (Figure 1B and Figure S5A). Among these, NOTCH1 (72.7%) mutations were also frequently observed in relapsed T‐ALL, but FBXW7, DNM2 and PHF6 mutations were less frequent. As previously reported, NT5C2 mutations (27.3%) were predominantly detected in relapse samples (Figure 1C and Figure S5B). An overrepresentation of C>T and C>A transitions was observed in samples at both diagnosis and relapse (Figure S6).
Figure 1.
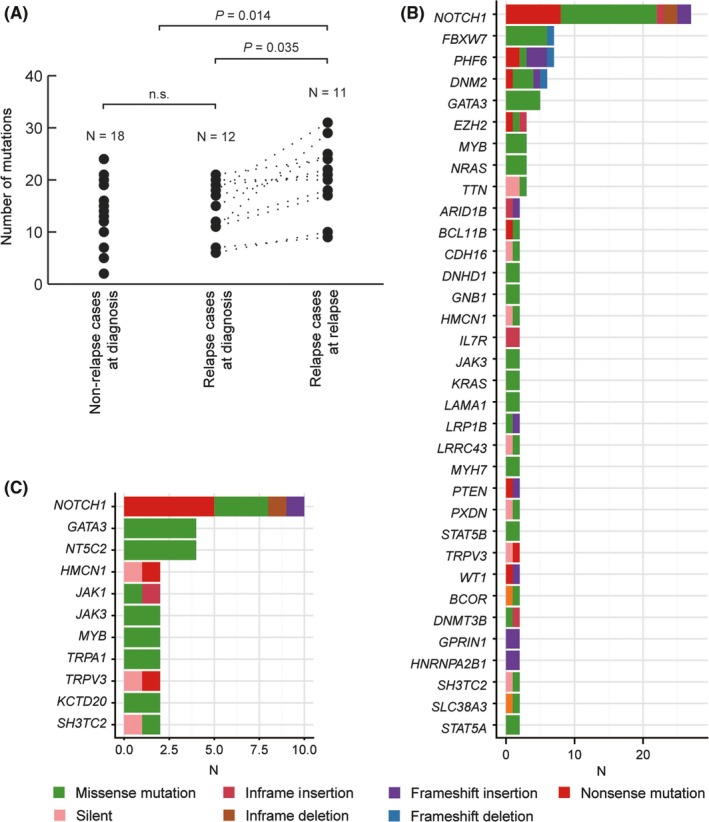
Whole–exome sequencing (WXS) data of 11 cases of relapse‐diagnosis‐remission trios and 19 matched T‐cell acute lymphoblastic leukemia (T‐ALL) remission pairs. A, The number of mutations detected by WXS without synonymous mutations. More mutations were observed in relapse samples (P = .014; Student's t test). Genes recurrently mutated in (B) 30 T‐ALL samples at diagnosis and (C) 11 relapse samples. The most common mutations were detected in NOTCH1 in both diagnosis (66.7%) and relapse (36.6%) samples. NT5C2 (27.3%) mutations were present only in relapse samples. n.s., not significant
Table 1.
Clinical characteristics of relapse and non–relapse cases at diagnosis
| Characteristics | Non–relapse cases (N = 18) | Relapse cases (N = 12) | Fisher's exact test |
|---|---|---|---|
| N (%) | N (%) | P‐value | |
| Sex | n.s. | ||
| Male | 13 (72.2) | 10 (90.9) | |
| Female | 5 (27.8) | 1 (9.1) | |
| Not available | 0 | 1 | |
| Age at diagnosis | n.s. | ||
| 1 to 9 y | 11 (61.1) | 5 (41.7) | |
| ≥10 y | 7 (38.9) | 7 (58.3) | |
| Not available | 0 | 0 | |
| WBC counts at diagnosis | n.s. | ||
| <100 000/μL | 12 (66.7) | 4 (33.3) | |
| ≥100 000/μL | 6 (33.3) | 8 (66.7) | |
| Not available | 0 | 0 | |
| Prednisolone response | n.s. | ||
| Good (day 8 WBC <1000/μL) | 9 (52.9) | 8 (80.0) | |
| Poor (day 8 WBC ≥1000/μL) | 8 (47.1) | 2 (20.0) | |
| Not available | 1 | 2 | |
| CNS involvement | n.s. | ||
| CNS‐1, CNS‐2 | 17 (94.4) | 10 (90.9) | |
| CNS‐3 | 1 (5.6) | 1 (9.1) | |
| Not available | 0 | 1 | |
| Mediastinal mass | n.s. | ||
| Yes | 7 (38.9) | 7 (58.3) | |
| No | 11 (61.1) | 5 (41.7) | |
| Not available | 0 | 0 | |
| Previously analyzed expression clustera | |||
| TAL1‐RA | 5 (33.3) | 4 (44.4) | |
| TAL1‐RB | 2 (13.3) | 0 (0) | |
| TLX | 3 (20.0) | 1 (11.1) | |
| ETP | 4 (26.7) | 2 (22.2) | |
| SPI1‐fusion | 1 (6.7) | 2 (22.2) | |
| Follow–up duration days (median) | 507‐5427 (3069) | 209‐3211 (750) | |
CNS, central nervous system; ETP, early T‐cell precursor; WBC, white blood cell.
These 5 clusters were identified using whole–transcriptome sequencing based on expression profiles in our previous study.6 ETP, immature cluster showing similar expression profiles to early T‐cell precursor; SPI1‐fusion; SPI1‐highly expressed cluster with SPI1 fusion; TAL1‐RA, TAL1‐highly expressed cluster with low gene mutation rate; TAL1‐RB, TAL1‐highly expressed cluster with high gene mutation rate; TLX, TLX1/TLX3‐highly expressed cluster.
3.2. Germline mutations in predisposition genes detected in whole–exome sequencing
We identified 3 missense germline variants, FAP (NM_004460: K533X), IDH1 (NM_005896: F355V) and RET (NM_020630: M848V), in 2 specimens (Table S5). FAP, encoding a tumor suppressor involved in cell proliferation and survival, was truncated at exon 18. The IDH1 variant was reported as one of the cancer predisposition tumor suppressor genes.37 The detected IDH1 variant was predicted as a high risk factor for developing cancers according to the mutation assessor (http://mutationassessor.org/v1/), although no germline and somatic mutations have been reported at the same position. The mutation in RET was located in its kinase domain, which is consistent with previous reports.37 Importantly, neither family history of cancer nor underlying disease was observed in these cases.
3.3. Types of NOTCH1 and FBXW7 mutations observed in T‐cell acute lymphoblastic leukemia
We detected mutations leading to NOTCH1 signaling activation (NOTCH1 and/or FBXW7 alterations) in 22 out of 30 (73.3%) and 8 out of 11 (72.7%) samples at diagnosis and relapse, respectively (Figure 2A and Table [Link], [Link]). Both LIA and ID mutation types were detected in 11 out of 30 (36.7%) and 3 out of 11 (27.3%) samples at diagnosis and relapse, respectively (Figure 2A,B and Figure [Link], [Link]). These mutations were mutually exclusive within each LIA and ID pattern, except for 1 sample that harbored mutations with low allele frequencies less than 15% (Figure 2A). In our cohort, HD‐SNVs were the most frequent (40.0%; 12 out of 30 samples), followed by PEST alterations (33.3%; 10 out of 30 samples). In–frame internal duplication in exon 28, known as juxta‐membrane expansion (Dup), intragenic NOTCH1 deletion (Del‐N) and SNV at 3′UTR (UTR) were each detected in 1 sample (Table 2). The frequency of PEST alterations in relapsed cases of diagnosis samples (7 out of 12; 58.3%) was significantly higher than that in non–relapsed cases (3 out of 18; 16.7%) (P = .045; Figure 2C and Table 2). We also analyzed the WXS data of 200 T‐ALL cases generated by Therapeutically Applicable Research to Generate Effective Treatment (TARGET)7; however, no predominance of PEST alterations was observed in relapsed cases (Table S6).
Figure 2.
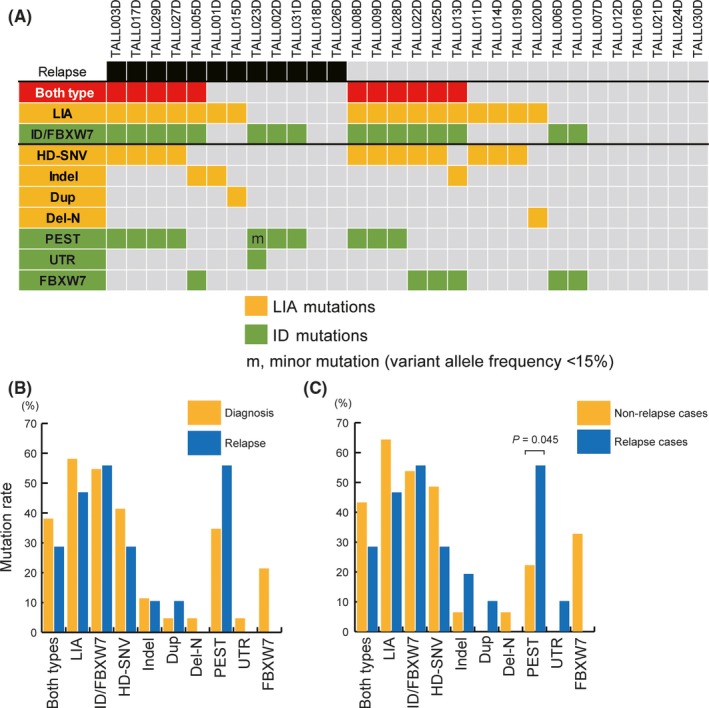
Characteristics of NOTCH1 and FBXW7 alterations leading to NOTCH1 signaling activation. A, An overview of NOTCH1 and FBXW7 mutations in 30 T‐cell acute lymphoblastic leukemia (T‐ALL) samples at diagnosis. Detected mutation types (HD‐SNV, Indel, Dup, Del‐N, PEST, UTR and FBXW7) and patterns (LIA and ID) are colored. Each T‐ALL case did not possess mutations of multiple types within the same patterns (LIA or ID) except minor mutation. B, Frequencies of NOTCH1 or FBXW7 mutations detected at diagnosis and relapse. C, Frequencies of NOTCH1 or FBXW7 mutations detected at diagnosis in non–relapsed and relapsed cases. Mutations in the PEST domain were detected significantly more frequently in relapsed cases than in non–relapsed cases (P = .045; Fisher's exact test). Del‐N, intragenic NOTCH1 deletion; Dup, in–frame internal duplication in exon 28; HD‐SNV, single nucleotide variations in the heterodimerization domain; ID, impaired degradation; Indel, small in–frame insertions or deletions in the HD domain; LIA, ligand–independent activation; m, minor mutation (VAF <15%); PEST, proline, glutamic acid, serine, and threonine; UTR, SNV in 3′ untranslated region
Table 2.
Frequencies of NOTCH1 or FBXW7 alterations detected at diagnosis of T‐ALL
| Non–relapsed cases (%) n = 18 | Relapsed cases (%) n = 12 | P‐value | References | |
|---|---|---|---|---|
| LIA, ligand–independent activation. | ||||
| NOTCH1 HD domain non–synonymous mutation (HD‐SNV) | 8 (44.4) | 4 (33.3)a | .71 | 10 |
| NOTCH1 HD domain non–frameshift indel (Indel) | 1 (5.6) | 2 (16.7)c | .55 | 10 |
| NOTCH1 internal duplication (Dup) | 0 (0) | 1 (8.3)c | .4 | 13 |
| Deletion in extracellular domains of NOTCH1 (Del‐N) | 1 (5.6) | 0 (0) | — | 14, 15 |
| NOTCH1 fusion (Fusion) | 0 (0) | 0 (0) | — | 16 |
| Impaired degradation: ID | ||||
| NOTCH1 PEST domain truncation (PEST) | 3 (16.7)b | 7 (58.3) | .045 | 10 |
| NOTCH1 3′UTR region mutation (UTR) | 0 (0) | 1 (8.3)a | — | 17 |
| FBXW7 mutation (FBXW7) | 5 (27.8) | 1 (8.3)c | .35 | 11, 12 |
All cases had PEST as well.
All cases had HD‐SNV as well.
No cases had PEST additionally.
3.4. Clonal analysis of samples at diagnosis and relapse identified mutational “switching” of NOTCH1
Based on the results of amplicon–based deep sequencing of all mutations detected by WXS, we evaluated the clonal evolution of T‐ALL relapses using accurate VAFs of each detected mutation. The clonal analysis of diagnosis and relapse samples identified 3 different types of clonal evolution at relapse in T‐ALL samples: acquired new mutations (VAF >.01) at relapse which were not detected or very low level (VAF <.01) at diagnosis without losing any mutations (VAF >.01) (type 1; Figure 3A), acquired new mutations (VAF >.01) and lost some mutations (VAF <.01) at relapse (type 2; Figure 3B); and no mutations were acquired (VAF >.01) or lost (VAF <.01) at relapse (type 3; Figure 3C).
Figure 3.
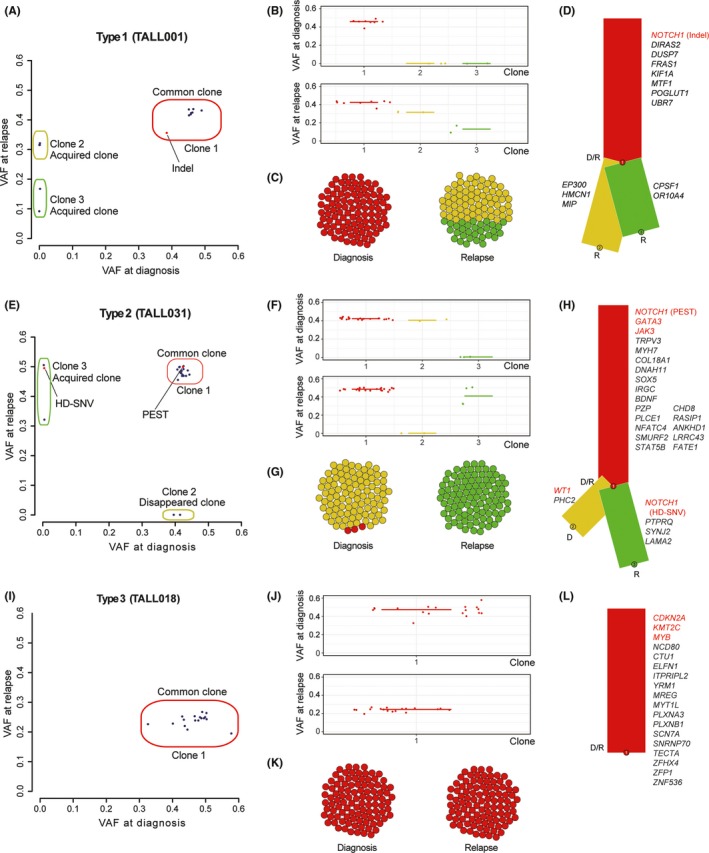
Depictions of clonal evolution from diagnosis to relapse in each relapse type. A‐D, Clonal analysis in the representative case (TALL001) of relapse type 1, which acquired additional mutations at relapse in addition to all detected mutations at diagnosis. E‐H, Clonal analysis in the representative case (TALL031) of relapse type 2. Several mutations presented in the major clone at diagnosis were lost, and additional mutations were acquired at relapse. I‐L, Clonal analysis of relapse type 3. Same gene mutations were observed at both diagnosis and relapse in type 3 samples. A,E,I, Observed VAF of each mutation are shown in diagonal plots. Red dots indicate NOTCH1. B,F,J, VAF of detected mutations in each clone are indicated. C,G,K, Schematic description of population in each clone at diagnosis and relapse is shown. D,H,L, Schematic description of clonality in each clone at diagnosis (D) and relapse (R) is shown in phylogenetic trees. Detected mutations in each clone are listed. VAF, variant allele frequency
Type 1 clonal evolution at relapse was observed in 3 samples (TALL001, TALL015 and TALL026). These samples acquired additional mutations later in the process of leukemogenesis at relapse. All mutations detected at diagnosis were also detected in relapse samples (Figure 3A‐D). NOTCH1 and/or FBXW7 alterations were detected in the common major clone (VAF >.15, both diagnosis and relapse) in TALL001 and TALL015, and none of these 3 type 1 cases acquired additional NOTCH1 and/or FBXW7 alterations at relapse. Unlike the previous report,26 all cases of type 1 clonal evolution relapsed after 12 months of diagnosis (Figure S9A).
In type 2 clonal evolution, several mutations detected in the major clone at diagnosis were lost and additional new mutations were acquired at relapse (Figure 3E‐H). Type 2 clonal evolution was observed in 7 cases (TALL002, TALL003, TALL005, TALL017, TALL023, TALL029 and TALL031), of which PEST alterations were identified in 6 cases at relapse. Three cases also acquired HD‐SNVs and GATA3 mutations at the same time. In TALL031, PEST alteration was detected in the common major clone both at diagnosis and relapse. The other major clone (Clone 2) at diagnosis disappeared and the new major clone (Clone 3) with the acquired NOTCH1 mutation (HD‐SNV) expanded at relapse (Figure 3E). In contrast, in TALL029, both the HD‐SNV and PEST alterations were detected at diagnosis; of these, the HD‐SNV disappeared and only the PEST alteration was identified with other acquired mutations at relapse.
It should be noted that we identified NOTCH1 “switching” in 2 cases (TALL017 and TALL023) with type 2 clonal evolution, showing different NOTCH1 mutations at diagnosis and relapse. In these cases, the dominant clone at diagnosis with NOTCH1 mutations had disappeared at relapse with an expansion of a new dominant clone with a different NOTCH1 mutation derived from the same ancestor clone (Figure 4 and Figure S1). For example, TALL017 harbored 3 NOTCH1 mutations at diagnosis, PEST alteration in Clone 1 (common major clone, VAF >.15 both at diagnosis and at relapse), and 2 independent HD‐SNVs in Clone 2 (VAF >.15 at diagnosis) and Clone 3 (VAF <.15 at diagnosis), respectively (Figure 4A). The treatment of T‐ALL at diagnosis might eradicate 1 of the HD‐SNVs present in Clone 2. The other HD‐SNV in Clone 3 expanded as a “switched” major clone (VAF >.15) at relapse (Figure 4A‐D). In addition to NOTCH1 expansion, NT5C2 mutation was also acquired in this “switched” clone, and seemed to have a selective advantage for relapse (Figure 4D). Intriguingly, TALL023 was the other case of NOTCH1 “switching” that developed LCH from a common clone in diagnostic T‐ALL (Figure 4E‐H and Figure S1). In the T‐ALL sample, 2 NOTCH1 mutations (both with ID pattern) were identified in the UTR (Clone 2, VAF = .19) and the other in the PEST domain (Clone 4, VAF = .07). In the LCH sample, only the PEST alteration was observed, suggesting that the mutation in the UTR disappeared after the initial T‐ALL treatment. Moreover, the clone with PEST alteration (Clone 4) expanded as a “switched” major clone (VAF >.15) at LCH from a minor subclone (VAF <.15) in the T‐ALL sample at diagnosis (Figure 4E‐H). In this case, the acquisition and expansion of BRAF alteration, important for the pathogenesis of LCH, were observed at LCH in addition to NOTCH1 “switching.” By contrast, the GATA3 mutation involved in T‐ALL pathogenesis was eradicated. Using multiplex PCR for the detection of TCR rearrangement between diagnosis and relapse samples of these “switching” cases, we confirmed that diagnosis‐relapse paired samples had the same TCR rearrangement pattern (Figure S1). Intriguingly, we detected multiple waves in TALL017 that might indicate the existence of multiple clones harboring different TCR rearrangements within the tumor. This is consistent with the ClonEvol analysis result that TALL017 had 2 major subclones at relapse originated from the same ancestor clone (Figure 4A‐D). We further investigated previously reported paired diagnosis‐relapse T‐ALL cases and found another NOTCH1 “switching” case (T‐ALL‐H‐S00285) out of 13 paired cases.26 In this case, 2 NOTCH1 mutations (both HD‐SNV) were detected at diagnosis. One in minor subclones had disappeared (VAF = from .12 to .0003) and the other HD‐SNV in major clones was replaced (VAF = from .33 to .04) by newly expanded HD‐SNV at relapse (VAF = .26), which was not detected at diagnosis (VAF = .0008).
Figure 4.
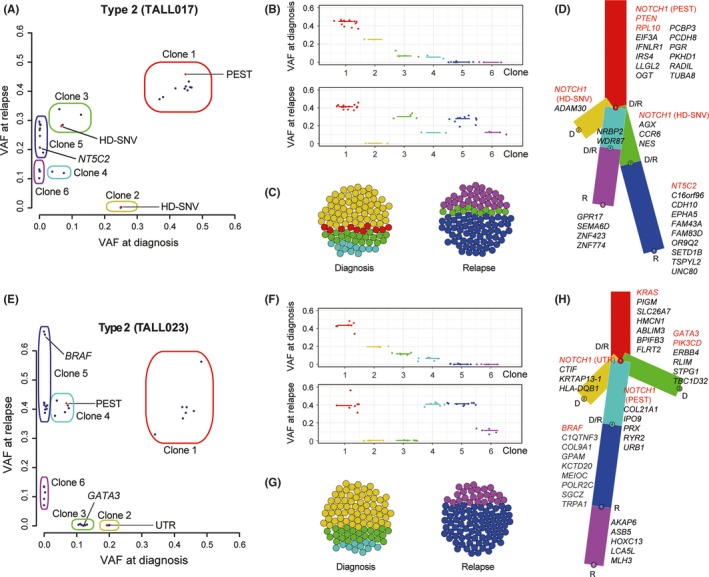
Schematic description of clonal evolution in the cases of NOTCH1 “switching.” Clonal analysis in TALL017 (A‐D) and TALL023 (E‐H) is shown; representative cases of NOTCH1 “switching” detecting different NOTCH1 mutations between T‐ALL diagnosis and relapse. A,E, Observed VAF of each mutation are shown in diagonal plots. Red dots indicate NOTCH1. Each clone was estimated based on the results of PyClone and SciClone. B,F, VAF of detected mutations in each clone are indicated. D,H, Schematic description of clonality in each clone at diagnosis (D) and relapse (R) is shown in phylogenetic trees. Detected mutations in each clone are listed. VAF, variant allele frequency
Type 3 clonal evolution at relapse was observed in 1 case (TALL018). In this case, the same gene mutations were observed in both diagnosis and relapse samples (Figure 3I‐L). Insufficient treatment of T‐ALL at diagnosis was considered to induce expansion of the same clone in this type of relapse. In this case, relapse occurred within 10 months of diagnosis. TALL018 was treated without MRD–based protocol. This case was assumed to remain MRD positive after remission induction. The mean VAF of all validated mutations in the corresponding normal sample was above 10−3 level at the end of the induction (mean VAF = .0026; Figure S9B).
4. DISCUSSION
Based on the results of clonal analysis of samples at diagnosis and relapse, we identified NOTCH1 “switching” characterized by different NOTCH1 mutations in a major clone between diagnosis and relapse samples. This NOTCH1 “switching” has been previously reported using WAVE chromatograms.38 In chronic lymphocytic leukemia (CLL), ultra–deep next–generation sequencing demonstrated similar changes of TP53 mutations in relapse and re–relapse samples.39 Dysfunction of TP53 is considered the major cause of genomic instability in CLL cells, leading to resistance to treatment.40, 41 Taken together, “switching” of NOTCH1 mutations indicates that NOTCH1 plays an important role in both leukemogenesis and progression. According to previous studies,10, 42 increased luciferase activity by HD‐SNV and PEST domain mutations seemed to be different depending on the position of mutations. These HD‐SNV and PEST domain mutations were recognized as a weak tumor initiator even when they existed in cis, because these mutations failed to efficiently initiate T‐ALL in mice. In contrast, they were reported to accelerate T‐ALL with other driver mutations such as KRAS (detected in “switched” TALL023 sample), and they showed addiction to NOTCH1 signaling. Thus, we estimate that functional advantage in replacement of NOTCH1 mutations (HD‐SNV to HD‐SNV in TALL017 and UTR to PEST in TALL023, respectively) in “switched” T‐ALL cases itself was not big, but the existence of NOTCH1 mutations in the “switched” clone might be important for selection advantage in relapse as the secondary event. Furthermore, we identified the acquisition of NT5C2 and BRAF mutations in cases of “switched” clones, respectively. NT5C2 mutation involves in clonal evolution during disease progression and relapse with induction of resistance to 6‐mercaptopurine.8, 9, 43 Mutations in MAPK pathway, including BRAF, were frequently observed in LCH.44 Acquisition of these mutations in “switched” clones might be associated with selective advantage during tumor clonal evolution.
All 3 cases representing NOTCH1 “switching” or expansion at relapse contained the PEST alteration in the major clone. In TALL023, the case of NOTCH1 “switching,” the PEST alteration at relapse was enriched from minor populations (VAF = from .07 to .42), and the UTR–specific mutation was eradicated (VAF = from .19 to 0). This suggests that NOTCH1 pathway activation via PEST alterations might offer a selection advantage in this case. In CLL, NOTCH1 mutations occur almost exclusively in PEST and are associated with poor prognosis.45 Furthermore, PEST alterations were predominantly detected in diagnosis samples of relapsed cases rather than in non–relapsed cases, although this enrichment of PEST alterations in relapse cases was not confirmed in TARGET data. These results imply that PEST alterations are involved in the progression and early relapse of T‐ALL. However, further research is warranted to decide the impact of PEST alterations in T‐ALL.
The limitations of this study include its small sample size, treatment without MRD–based protocol, and its retrospective design in a single facility, leading to a selection bias. Nonetheless, mutations detected in this study using WXS of 30 T‐ALL cases are consistent with previous reports.6, 7, 46, 47 Recent improvement in survival might be largely due to MRD–based risk stratification. Thus, further analysis is required to confirm the relevance of PEST mutations in the progression to relapse using large cohorts with or without MRD–based protocol.
In conclusion, we described that “switching” of NOTCH1 mutations demonstrates the importance of NOTCH1 signaling activation not only in the development but also in the progression of T‐ALL. In addition, mutations in the PEST domain of NOTCH1 may be involved in T‐ALL relapse in pediatric cases. Our results might be helpful for developing a new anti–NOTCH1 therapeutic strategy for refractory or relapsed T‐ALL patients, although further studies are warranted.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
S.K. and J.T. wrote the manuscript; S.K., M.S., M.A., K.K., T.I., A.M. and Y.H. collected and analyzed data; S.K., M.S. and K.Y. performed experiments; Y.S. and S.M. developed bioinformatics pipelines; M.K., A.O., S.O. and J.T. gave conceptual advice; J.T. designed the study. All authors read and approved the final manuscript.
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank the TCGA Consortium and all its members for making their invaluable data publicly available. The results published here are in whole or part based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatment (TARGET) initiative managed by the NCI. Information about TARGET can be found at http://ocg.cancer.gov/programs/target. The authors are also grateful to T. Isobe, M. Kato, M. Matsumura, N. Hoshino, K. Yin and F. Saito for their excellent technical assistance. The authors also wish to express our appreciation to K. Chiba, H. Tanaka, S. Tsuji and J. Mitsui (The University of Tokyo) for next–generation sequencing.
Kimura S, Seki M, Yoshida K, et al. NOTCH1 pathway activating mutations and clonal evolution in pediatric T‐cell acute lymphoblastic leukemia. Cancer Sci. 2019;110:784–794. 10.1111/cas.13859
REFERENCES
- 1. Winter SS, Dunsmore KP, Devidas M, et al. Safe integration of nelarabine into intensive chemotherapy in newly diagnosed T‐cell acute lymphoblastic leukemia: Children's Oncology Group Study AALL0434. Pediatr Blood Cancer. 2015;62(7):1176‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pui CH. Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood. 2004;104(9):2690‐2696. [DOI] [PubMed] [Google Scholar]
- 3. Nguyen K, Devidas M, Cheng S‐C, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children's Oncology Group study. Leukemia. 2008;22(12):2142‐2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1(1):75‐87. [DOI] [PubMed] [Google Scholar]
- 5. Bongiovanni D, Saccomani V, Piovan E. Aberrant signaling pathways in T‐cell acute lymphoblastic leukemia. Int J Mol Sci. 2017;18(9):1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seki M, Kimura S, Isobe T, et al. Recurrent SPI1 (PU.1) fusions in high‐risk pediatric T cell acute lymphoblastic leukemia. Nat Genet. 2017;22:569. [DOI] [PubMed] [Google Scholar]
- 7. Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T‐lineage acute lymphoblastic leukemia. Nat Genet. 2017;373:1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meyer JA, Wang J, Hogan LE, et al. Relapse‐specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45(3):290‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tzoneva G, Perez‐Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19(3):368‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269‐271. [DOI] [PubMed] [Google Scholar]
- 11. Thompson BJ, Buonamici S, Sulis ML, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204(8):1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma‐secretase inhibitors. J Exp Med. 2007;204(8):1813‐1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sulis ML, Williams O, Palomero T, et al. NOTCH1 extracellular juxtamembrane expansion mutations in T‐ALL. Blood. 2008;112(3):733‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ashworth TD, Pear WS, Chiang MY, et al. Deletion‐based mechanisms of Notch1 activation in T‐ALL: key roles for RAG recombinase and a conserved internal translational start site in Notch1. Blood. 2010;116(25):5455‐5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haydu JE, De Keersmaecker K, Duff MK, et al. An activating intragenic deletion in NOTCH1 in human T‐ALL. Blood. 2012;119(22):5211‐5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ellisen LW, Bird J, West DC, et al. TAN‐1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66(4):649‐661. [DOI] [PubMed] [Google Scholar]
- 17. Puente XS, Beà S, Valdés‐Mas R, et al. Non‐coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519‐524. [DOI] [PubMed] [Google Scholar]
- 18. Clappier E, Collette S, Grardel N, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T‐cell acute lymphoblastic leukemia (T‐ALL) treated on EORTC trials 58881 and 58951. Leukemia. 2010;24(12):2023‐2031. [DOI] [PubMed] [Google Scholar]
- 19. Zuurbier L, Homminga I, Calvert V, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T‐cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. 2010;24(12):2014‐2022. [DOI] [PubMed] [Google Scholar]
- 20. Park M‐J, Taki T, Oda M, et al. FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non‐Hodgkin lymphoma. Br J Haematol. 2009;145(2):198‐206. [DOI] [PubMed] [Google Scholar]
- 21. Kox C, Zimmermann M, Stanulla M, et al. The favorable effect of activating NOTCH1 receptor mutations on long‐term outcome in T‐ALL patients treated on the ALL–BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. 2010;24(12):2005‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jenkinson S, Koo K, Mansour MR, et al. Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T‐cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia. 2012;27(1):41‐47. [DOI] [PubMed] [Google Scholar]
- 23. van Grotel M, Meijerink JPP, Beverloo HB, et al. The outcome of molecular‐cytogenetic subgroups in pediatric T‐cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica. 2006;91(9):1212‐1221. [PubMed] [Google Scholar]
- 24. Ma J, Wu M. The indicative effect of Notch1 expression for the prognosis of T‐cell acute lymphocytic leukemia: a systematic review. Mol Biol Rep. 2012;39(5):6095‐6100. [DOI] [PubMed] [Google Scholar]
- 25. Knoechel B, Roderick JE, Williamson KE, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46(4):364‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kunz JB, Rausch T, Bandapalli OR, et al. Pediatric T‐cell lymphoblastic leukemia evolves into relapse by clonal selection, acquisition of mutations and promoter hypomethylation. Haematologica. 2015;100(11):1442‐1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64‐69. [DOI] [PubMed] [Google Scholar]
- 28. Shiraishi Y, Sato Y, Chiba K, et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41(7):e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roth A, Khattra J, Yap D, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11(4):396‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miller CA, White BS, Dees ND, et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol. 2014;10(8):e1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Makishima H, Yoshizato T, Yoshida K, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2016;49(2):204‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Isobe T, Seki M, Yoshida K, et al. Integrated molecular characterization of the lethal pediatric cancer pancreatoblastoma. Cancer Res. 2017;78:865‐876. [DOI] [PubMed] [Google Scholar]
- 34. Dang HX, White BS, Foltz SM, et al. ClonEvol: clonal ordering and visualization in cancer sequencing. Ann Oncol. 2017;28(12):3076‐3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kato M, Seki M, Yoshida K, et al. Genomic analysis of clonal origin of Langerhans cell histiocytosis following acute lymphoblastic leukaemia. Br J Haematol. 2015;175(1):169‐172. [DOI] [PubMed] [Google Scholar]
- 36. Yokokawa Y, Taki T, Chinen Y, et al. Unique clonal relationship between T‐cell acute lymphoblastic leukemia and subsequent Langerhans cell histiocytosis with TCR rearrangement and NOTCH1 mutation. Genes Chromosom Cancer. 2015;54(7):409‐417. [DOI] [PubMed] [Google Scholar]
- 37. Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336‐2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mansour MR, Duke V, Foroni L, et al. Notch‐1 mutations are secondary events in some patients with T‐cell acute lymphoblastic leukemia. Clin Cancer Res. 2007;13(23):6964‐6969. [DOI] [PubMed] [Google Scholar]
- 39. Malcikova J, Stano‐Kozubik K, Tichy B, et al. Detailed analysis of therapy‐driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia. 2014;29(4):877‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T. p53 dysfunction in B‐cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood. 2001;98(3):814‐822. [DOI] [PubMed] [Google Scholar]
- 41. Ouillette P, Fossum S, Parkin B, et al. Aggressive chronic lymphocytic leukemia with elevated genomic complexity is associated with multiple gene defects in the response to DNA double‐strand breaks. Clin Cancer Res. 2010;16(3):835‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiang MY, Xu L, Shestova O, et al. Leukemia‐associated NOTCH1 alleles are weak tumor initiators but accelerate K‐ras‐initiated leukemia. J Clin Invest. 2008;118(9):3181‐3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tzoneva G, Dieck CL, Oshima K, et al. Clonal evolution mechanisms in NT5C2 mutant‐relapsed acute lymphoblastic leukaemia. Nature. 2018;553(7689):511‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Allen CE, Parsons DW. Biological and clinical significance of somatic mutations in Langerhans cell histiocytosis and related histiocytic neoplastic disorders. Hematology Am Soc Hematol Educ Program. 2015;2015(1):559‐564. [DOI] [PubMed] [Google Scholar]
- 45. Strefford JC. The genomic landscape of chronic lymphocytic leukaemia: biological and clinical implications. Br J Haematol. 2014;169(1):14‐31. [DOI] [PubMed] [Google Scholar]
- 46. De Keersmaecker K, Atak ZK, Li N, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T‐cell acute lymphoblastic leukemia. Nat Genet. 2012;45(2):186‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kalender Atak Z, Gianfelici V, Hulselmans G, et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T‐cell acute lymphoblastic leukemia. PLoS Genet. 2013;9(12):e1003997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials