

Abstract
It has been reported that DNA double‐strand breaks (DSB) can be induced by cytoplasm irradiation, and that both reactive free radicals and mitochondria are involved in DSB formation. However, the cellular antioxidative responses that are stimulated and the biological consequences of cytoplasmic irradiation remain unknown. Using the Single Particle Irradiation system to Cell (SPICE) proton microbeam facility at the National Institute of Radiological Sciences ([NIRS] Japan), the response of nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) antioxidative signaling to cytoplasmic irradiation was studied in normal human lung fibroblast WI‐38 cells. Cytoplasmic irradiation stimulated the localization of NRF2 to the nucleus and the expression of its target protein, heme oxygenase 1. Activation of NRF2 by tert‐butylhydroquinone mitigated the levels of DSB induced by cytoplasmic irradiation. Mitochondrial fragmentation was also promoted by cytoplasmic irradiation, and treatment with mitochondrial division inhibitor 1 (Mdivi‐1) suppressed cytoplasmic irradiation‐induced NRF2 activation and aggravated DSB formation. Furthermore, p53 contributed to the induction of mitochondrial fragmentation and activation of NRF2, although the expression of p53 was significantly downregulated by cytoplasmic irradiation. Finally, mitochondrial superoxide (MitoSOX) production was enhanced under cytoplasmic irradiation, and use of the MitoSOX scavenger mitoTEMPOL indicated that MitoSOX caused alterations in p53 expression, mitochondrial dynamics, and NRF2 activation. Overall, NRF2 antioxidative response is suggested to play a key role against genomic DNA damage under cytoplasmic irradiation. Additionally, the upstream regulators of NRF2 provide new clues on cytoplasmic irradiation‐induced biological processes and prevention of radiation risks.
Keywords: cytoplasm, double‐strand DNA break, mitochondria, NFE2L2 protein, radiation biology
1. INTRODUCTION
The nucleus has been recognized as the most critical target of ionizing irradiation to induce biological effects.1 However, the role of irradiated cytoplasm in the induction of biological effects remains largely unknown. Development of sophisticated microbeam facilities allowed carrying out cytoplasm‐targeted irradiation using precise doses.2 Oxidative DNA damage and DNA double‐strand breaks (DSB) in the nucleus were observed when only the cytoplasm was targeted, reflecting the indirect action of cytoplasmic irradiation on genomic DNA damage.3, 4 In addition, cytoplasmic irradiation led to a notable increase in micronucleus formation in non‐irradiated neighboring cells (ie, radiation‐induced bystander effects).5 Induced production of reactive free radicals, including reactive oxygen species (ROS) and nitric oxide (NO), was observed in mammalian cells exposed to irradiation.6 Reactive free radicals can directly damage DNA and are considered the key factor in cytoplasmic irradiation‐induced DNA oxidative damage and DSB,3, 4 also acting as signaling molecules of specific biological pathways to stimulate stress responses.7 As targets of cytoplasmic irradiation,8 mitochondria show altered dynamics, which is associated with mitochondrial malfunction and the occurrence of diseases,9 as observed in immortalized human small‐airway epithelial cells (SAE),10 accompanied by a reduction in respiratory chain functions and enhanced mitochondrial superoxide production. Another study showed that mitochondrial DNA largely depleted ρ0 Hela cells showing weaker p53 binding protein 1 (53BP1) foci formation after cytoplasmic irradiation than normal Hela cells.4 Moreover, ρ0 Hela cells could not produce bystander damage signal after cytoplasmic irradiation. These results indicated that mitochondria are not only a target but are also key mediators of DNA damage signaling under cytoplasmic irradiation. Nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) regulates the expression of proteins acting against oxidative damage11, 12 and NRF2‐mediated antioxidative pathways play important roles in maintaining the equilibrium of cellular oxidative status. Under normal conditions, cytoplasmic NRF2 is kept in association with a cluster of proteins that degrades it through the ubiquitin‐mediated pathway; under stress conditions, NRF2 dissociates from this protein complex and translocates from the cytoplasm to the nucleus where it binds DNA and initiates the transcription of its targets. Most NRF2 targets reported to date are proteins involved in decreasing the cellular oxidative level or anti‐inflammation, such as NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase‐1 (HO‐1), and sulfiredoxin 1 (SRXN1). Proteins involved in other physiological processes such as DNA damage repair and autophagy are also targets of NRF2; these include 53BP1, 8‐oxoguanine DNA glycosylase (OGG1), and sequestosome‐1 (SQSTM1).13, 14 Chemicals that stimulate NRF2 pathways can attenuate cell damage induced by various types of stresses, including ionizing radiation.15, 16
Because cytoplasmic irradiation resulted in nucleus DNA damage and bystander effects, the potential carcinogenic risk for cells when only the cytoplasm is irradiated has been surmised for the low‐dose environmental radiation emitted by radon.3 Therefore, understanding the biological processes that modulate cytoplasmic irradiation‐induced DNA damage will be valuable not only for radiation protection, but also for radiobiology considerations. In the present study, we found that the NRF2‐mediated antioxidative response was activated by cytoplasmic irradiation and alleviated the induced DSB levels. Additionally, p53, mitochondrial dynamics, and mitochondrial superoxide were associated with NRF2 activation.
2. MATERIALS AND METHODS
2.1. Cell culture
Normal human lung WI‐38 fibroblasts were obtained from RIKEN Bioresource Center and cultured in DMEM (Wako Pure Chemical Industries Ltd, Osaka, Japan) supplemented with 10% FBS plus 100 μg/mL streptomycin and 100 U/mL penicillin, under a humidified atmosphere with 5% CO2 at 37°C.
2.2. Reagents
The following primary antibodies were used: anti‐NRF2 (Abcam, Cambridge, UK), antiphosphorylated‐H2A.X (Merck KGaA, Darmstadt, Germany), anti‐HO‐1 (Proteintech, Rosemont, IL, USA), anti‐p53, anti‐RAD51 (both from Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti‐XRCC4 (BD Biosciences, Tokyo, Japan), and Alexa‐Fluor 594 conjugated anti‐COX IV (Cell Signaling Technology, Danvers, MA, USA). Alexa‐Fluor 488 conjugated goat antirabbit or mouse secondary antibodies and MitoSOX Red mitochondrial superoxide indicator (both from Thermo Fisher Scientific K.K., Tokyo, Japan), mitochondrial division inhibitor 1 (Mdivi‐1) and NRF2 inducer tert‐butylhydroquinone (tBHQ) (both from Merck KGaA), and pifithrin‐α and mitoTEMPOL (both from Abcam) were also used.
2.3. Cytoplasmic irradiation with microbeam
Cytoplasmic irradiation was carried out on the Single Particle Irradiation system to Cell (SPICE) proton microbeam facility at the National Institute of Radiological Sciences (NIRS) in Japan. Physical features of SPICE, methods for cell culturing, and cytoplasmic irradiation conditions were described previously.2 Briefly, SPICE provides a 3.4 MeV proton microbeam approximately 2 μm in size and with a precise number of protons. For WI‐38 cells, the absorbed doses of a single proton traversing the cell nucleus or the whole cell were estimated at 7.6 and 4.6 mGy, respectively.2 The bottom of the microbeam dish was 6‐μm thick polypropylene (PP) film (Chemplex Industries Inc., Palm City, FL, USA). Microbeam dishes were treated with 1 μg/mL fibronectin dissolved in PBS for 1 hour at 37°C to enhance attachment of cells to PP film. A cell suspension (1 mL; 15 000 cells/mL) was plated into the dish and cultured for 48 hours; 0.5 hours before irradiation, the cell culture medium was replaced with medium containing 1 μg/mL Hoechst 33342 (Dojindo Laboratories, Tokyo, Japan) for nuclei visualization and for determining the coordinates of each nucleus within the dish during irradiation. Just before irradiation, the medium was drained off the dish. To avoid dehydration, a 6‐μm‐thick PP film covered the cells during the irradiation process. The dish was set on the X‐Y motorized stage of the SPICE microscopic system2 to obtain fluorescence images of nuclei in a 1.64 × 1.64 mm2 area. All nuclei were considered elliptical in shape to calculate the center position of the nucleus, and the cytoplasm was targeted at two off‐centered positions, which were 14 μm apart from the nucleus center in the direction of the major axis. All irradiation procedures, including image capturing, X‐Y coordinates calculation, and irradiation itself were carried out within 10 minutes for each sample. After irradiation, cell culture media were added to the dish and the cells were incubated under routine culturing conditions for analysis.
2.4. Immunofluorescence staining
For immunostaining of NRF2, cells were fixed with −20°C methanol for 10 minutes, dried on a clean bench, and blocked with 5% goat serum plus 0.5% Triton‐X in PBS at 23°C for 2 hours. The primary antibody was then diluted in antibody buffer [1% BSA in PBS‐T (PBS with 0.1% Triton X‐100)] and added to cells. After overnight incubation at 4°C, cells were washed three times with PBS‐T for 5 minutes. The corresponding Alexa‐Fluor conjugated secondary antibody diluted in antibody buffer was then added, and cells were incubated for another 2 hours at 23°C in a dark box with gentle shaking. After counterstaining with 1 μg/mL Hoechst 33342 (Dojindo Laboratories) for 20 minutes, cells were washed and subjected to image capture on the off‐line SPICE microscope system. For immunostaining of other proteins, cells were fixed with 4% paraformaldehyde for 20 minutes and then treated with 0.5% Triton X‐100 at 23°C for 30 minutes. Remaining steps were as those used for immunostaining of NRF2. Fluorescence intensity was analyzed using NIH ImageJ.17 Quantitative analysis of the fluorescence intensity was as described below. The captured images were converted into 8‐bit grey scale format. Then the corresponding Hoechst 33342 staining or immunofluorescence staining pictures were used to set a threshold to determine the region of interest (ROI). Area sizes and mean fluorescence intensities of each ROI were measured by ImageJ. Further, the fluorescence intensity of each ROI was calculated and recorded. No less than 100 cells were analyzed for each sample. Fluorescence intensities of each ROI were then summed and divided by the number of ROI analyzed. Relative fluorescence intensity of each sample was represented as the value of normalizing the fluorescence intensity to that from the control sample. Representative raw data are provided in Tables [Link], [Link], [Link], [Link], [Link], [Link].
2.5. Mitochondrial morphology evaluation
Cytochrome c oxidase IV (COX IV), which is localized on the inner membrane of mitochondria, is involved in mitochondrial electron transport and is used as a mitochondrial marker. In the present study, COX IV immunofluorescence staining was used to evaluate whether cytoplasmic irradiation induced changes in mitochondrial morphology. Thus, after irradiation, cells were fixed and subjected to immunofluorescence staining of COX IV. After counterstaining with Hoechst 33342, images were captured on the SPICE off‐line microscope and NIH ImageJ was used to assess mitochondrial morphology. Mitochondrial dimensions were converted from pixels to actual size in μm, and the percentage of tubular mitochondria in the total mitochondrial mass was quantified to reflect mitochondrial fragmentation.
2.6. Measurement of mitochondrial superoxide production
To detect the level of mitochondrial superoxide production, 0.5 and 2 hours after cytoplasmic irradiation, cells were washed with a prewarmed buffer. A freshly prepared working solution containing 5 μmol/L MitoSOX Red was used to incubate the cells for 10 minutes at 37°C under a 5% CO2 atmosphere. After another wash with the prewarmed buffer, MitoSOX Red fluorescence was recorded by the off‐line SPICE microscope and its intensity was analyzed by NIH ImageJ.
2.7. Statistical analysis
At least 100 randomly selected cells were analyzed for each experimental sample. Data are presented as mean ± SD from at least three independent experiments. Statistically significant differences between treated and control groups were determined by ANOVA in IBM SPSS Statistics (International Business Machines Corporation, Somers, NY, USA) or by Student('s t test in SigmaPlot 12 (Systat Software Inc., San Jose, CA, USA). Symbols # and ## indicate significant differences at P < .05 and P < .01, respectively.
3. RESULTS
3.1. Cytoplasmic irradiation enhanced nucleus localization of NRF2
To confirm the precise cytoplasm‐targeted radiation to WI‐38 by SPICE‐NIRS microbeam facility, we first irradiated the cytoplasm or nucleus of WI‐38 cells with 200 protons. Four hours post‐radiation, immunofluorescence staining of γH2A.X, the biomarker of DNA DSB, was carried out. As shown in Figure 1A, nucleus‐targeted radiation showed sharp and focused γH2A.X foci in each WI‐38 nucleus. However, cytoplasmic radiation resulted in sporadic smaller γH2A.X foci in each WI‐38 nucleus, showing obvious contrast with that observed in radiation of the nucleus. Given the fact that the cytoplasm of WI‐38 cells can be successfully targeted while not hitting the nucleus with the SPICE‐NIRS microbeam facility, the following studies were carried out.
Figure 1.
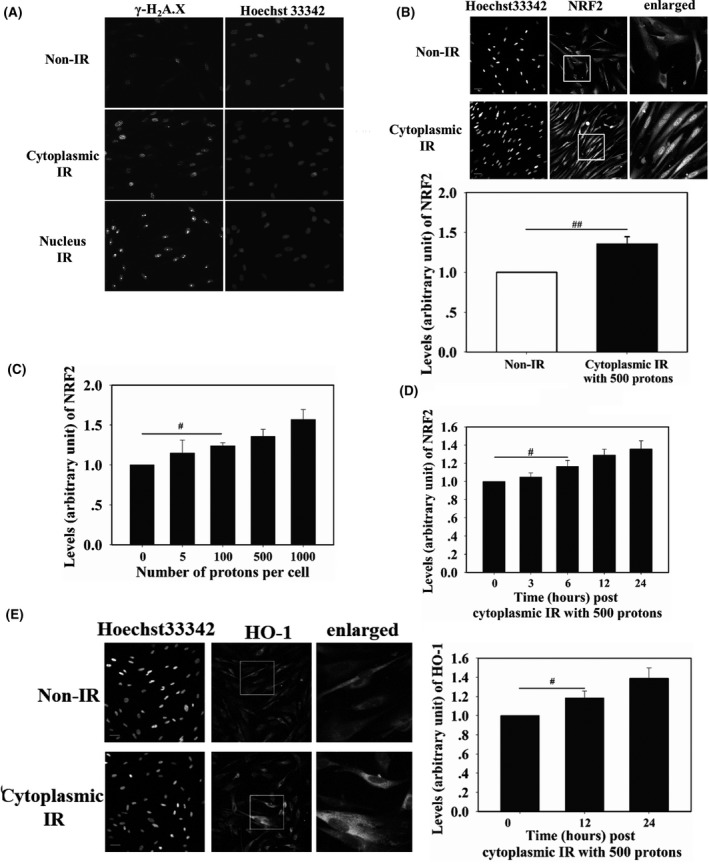
Precise cytoplasm and nucleus targeted irradiation of WI‐38 cells by Single Particle Irradiation system to Cell facility at the National Institute of Radiological Sciences (SPICE‐NIRS) proton microbeam. Each cell was irradiated by 200 protons. Four hours post‐radiation, γH2A.X immunofluorescence staining was carried out (A). The cytoplasm of WI‐38 cells was targeted by 500 protons (B) or by different amounts of protons (C) 24 h before nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) detection in the nucleus of targeted cells. At different time points post‐irradiation, nuclear NRF2 (D) and whole cell heme oxygenase‐1 (HO‐1) (E) were detected. Fluorescence intensity was normalized to that of non‐irradiated cells. Scale bar, 50 μm. IR, irradiation. # and ## indicate significant differences at P < .05 and P < .01, respectively
Translocation of NRF2 from the cytoplasm to the nucleus and upregulation of target proteins indicate NRF2 activation. To determine whether cytoplasmic irradiation activated NRF2, the cytoplasm of WI‐38 cells was irradiated with 500 protons and, 24 hours later, NRF2 was detected by immunostaining. As shown in Figure 1B, NRF2 fluorescence in the nucleus of irradiated cells was 30% higher than that in the nucleus of non‐irradiated cells. The accumulation of NRF2 in the nucleus was then measured in cells irradiated with five to 1000 protons, which are equivalent to 0.023 Gy to 4.6 Gy proton doses. As shown in Figure 1C, 24 hours after irradiation, levels of NRF2 in the nucleus increased with increasing numbers of protons hitting the cytoplasm. The 100‐proton irradiation resulted in 20% elevation of NRF2 in the nucleus, and when the dose reached 1000 protons, approximately 50% elevation was observed. Time‐dependent accumulation of NRF2 in the nucleus was also checked after irradiating 500 protons to the cytoplasm. As shown in Figure 1D, within 24 hours after irradiation, accumulation of NRF2 in the nucleus showed an increasing tendency. To confirm the activation of NRF2, expression of HO‐1 was detected at 12 and 24 hours after irradiating the 500 protons. It was found that HO‐1 expression at these time points increased by 18% and 39%, respectively (Figure 1E). These results indicated that cytoplasm‐targeted irradiation activated NRF2 antioxidative response.
3.2. Activation of NRF2 alleviated cytoplasmic radiation‐induced DSB
Cytoplasmic irradiation can lead to DNA damage in the nucleus, including DSB, which is considered the most hazardous to cells.4 We therefore investigated the effects of activated NRF2 on DSB formation after cytoplasmic irradiation. To activate NRF2, tBHQ (15 μmol/L final concentration) was added to WI‐38 cells 16 hours before irradiation. As shown in Figure 2A, tBHQ stimulated the accumulation of NRF2 in the nucleus. The cytoplasm of WI‐38 cells was then irradiated with 200 and 500 protons and, 4 hours after irradiation, the induced DSB levels were measured using immunofluorescence staining of γH2A.X,18 which is a marker for DSB. Cells irradiated with 200 and 500 protons showed DSB levels 24% and 53% higher than that shown by non‐irradiated cells, respectively. After the tBHQ treatment, the DSB levels of cells irradiated with 200 and 500 protons decreased to 115% and 136% of those observed in non‐irradiated cells (Figure 2B), indicating that activation of NRF2 protected cells from DNA damage induced by cytoplasmic irradiation. We also detected levels of RAD51 and XRCC4, which are involved in homologous recombination repair and non‐homologous end‐joining repair of DSB, respectively, in cells irradiated with 500 tBHQ‐treated or non‐treated protons (Figure 2C). Nucleus accumulation of RAD51 and XRCC4 increased in irradiated cells compared to that in control cells, and this increase was further enhanced in cells treated with tBHQ, indicating that NRF2 alleviated cytoplasmic irradiation‐induced DSB.
Figure 2.
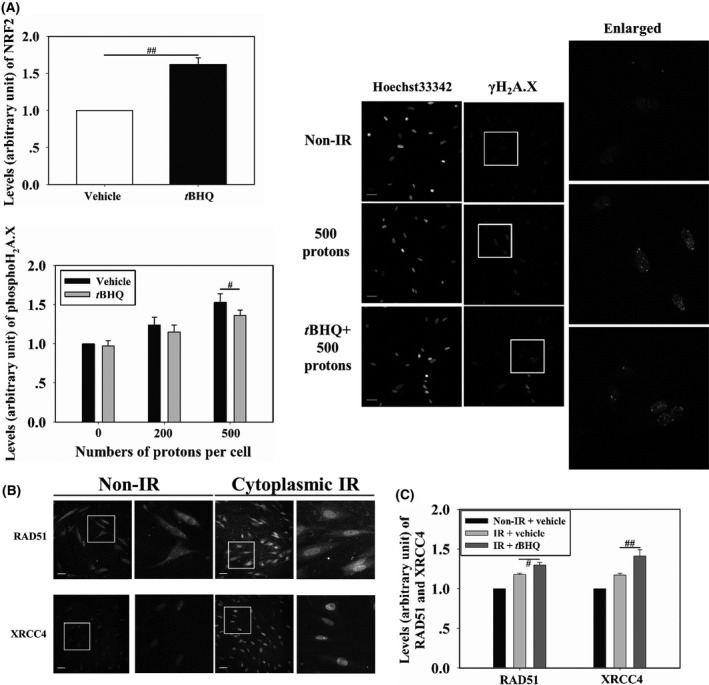
(A) Induction of nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) accumulation in the nucleus of WI‐38 cells pretreated with 15 μmol/L tert‐butylhydroquinone (tBHQ). Cells pretreated with tBHQ were cytoplasm targeted. γH2A.X (B) and RAD51 and XRCC4 levels (C) were detected. Fluorescence intensity was normalized to that of non‐irradiated cells. Scale bar, 50 μm. IR, irradiation. # and ## indicate significant differences at P < .05 and P < .01, respectively
3.3. Mitochondrial fragmentation promoted NRF2 translocation
At 3 and 6 hours after cytoplasmic irradiation by 500 protons, immunofluorescence visualization of COX IV was carried out to detect morphological changes in the mitochondria. As shown in Figure 3A, in the non‐irradiated cells, approximately 58% of the mitochondria showed tubular morphology. At 3 and 6 hours post‐irradiation, levels of tubular mitochondria decreased to 32% and 44%, respectively. Changes in mitochondria morphology were also detected at 6 hours post‐cytoplasmic irradiation by 200 protons, and tubular mitochondria corresponded to approximately 51%. These results indicated that cytoplasmic irradiation fragmented mitochondria. In addition, Mdivi‐1, a potent inhibitor of mitochondrial fragmentation,19 was added to the culture medium at the final concentration of 50 μmol/L from 30 minutes before cytoplasmic irradiation until 3 hours post‐irradiation. Mitochondrial fragmentation induced by cytoplasmic irradiation with 500 protons was reversed by the Mdivi‐1 treatment (Figure 3B). To test the relationship between mitochondrial fragmentation and NRF2 nucleus localization of cells subjected to Mdivi‐1 treatment, accumulation of NRF2 in the nucleus was measured at 6 and 12 hours after cytoplasmic irradiation with 500 protons. Accumulation of NRF2 was 12% lower in the nucleus of Mdivi‐1‐treated groups than in the vehicle‐treated group (Figure 3C). Furthermore, level of DSB 4 hours post‐500‐proton irradiation was aggravated by the Mdivi‐1 treatment, increasing by 20% (Figure 3D). These results showed that mitochondrial fragmentation induced by cytoplasmic irradiation contributed to the accumulation of NRF2 in the nucleus and decreased the residual DSB.
Figure 3.

WI‐38 cells were cytoplasm‐targeted by 500 protons. At different time points, mitochondrial morphology was observed (A). Cells were pretreated with 50 μmol/L mitochondrial division inhibitor 1 (Mdivi‐1) and then cytoplasm‐targeted. Effects of Mdivi‐1 treatment on cytoplasmic irradiation‐induced mitochondrial morphology changes (B), nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) localization to the nucleus (C), and γH2A.X formation (D) were detected. Scale bar, 20 μm. IR, irradiation. # and ## indicate significant differences at P < .05 and P < .01, respectively
3.4. p53 enhanced mitochondrial fragmentation and NRF2 translocation
Next, we examined how p53, an important DNA damage response protein, responded to cytoplasmic radiation in WI‐38 cells and whether p53 was involved in cytoplasmic radiation‐induced NRF2 activation. Expression of total p53 was downregulated 6 hours after cytoplasmic irradiation of WI‐38 cells (Figure 4A), as levels of p53 in cells irradiated with 500 and 200 protons were 47% and 66%, respectively, of those in non‐irradiated cells. We then checked the time‐course of p53 levels in WI‐38 cells with 500‐proton‐cytoplasmic radiation. A decreasing tendency in p53 levels within 6 hours post‐cytoplasmic irradiation was observed (Figure 4B). There was no change in p53 level 1 hour post‐cytoplasmic radiation. Three hours post‐radiation, the level of p53 decreased to approximately 80% of the level detected in non‐irradiated cells. Until the 6‐hour time point post‐radiation, p53 level continued to decrease to approximately 50% of that detected in non‐irradiated cells. We then examined whether p53 affected cytoplasmic irradiation‐induced nucleus localization of NRF2. Pifithrin‐α, which is an inhibitor of p53 transcription activity, was added to the culture medium at a final concentration of 20 μmol/L from 1 hour before irradiation until 6 hours post‐irradiation. After cytoplasmic irradiation with 500 protons, WI‐38 cells were incubated for 6 or 12 hours. Pifithrin‐α treatment decreased nucleus accumulation of NRF2 to 90% and 83% of the levels observed in non‐treated irradiated cells at 6 and 12 hours post‐irradiation, respectively, indicating that p53 transcription activity promoted nucleus localization of NRF2 in cytoplasm‐irradiated cells. Because mitochondrial fragmentation also stimulated accumulation of NRF2 in the nucleus, we tested the effect of p53 on mitochondrial fragmentation after irradiating the cytoplasm with 500 protons. Observation of mitochondrial morphology 3 hours after irradiation showed that under pifithrin‐α treatment, the fraction of tubular mitochondria was partially restored, from approximately 30% in non‐treated cells to 39% in treated cells, indicating that p53 transcription activity promoted cytoplasmic irradiation‐induced mitochondrial fragmentation (Figure 4D).
Figure 4.
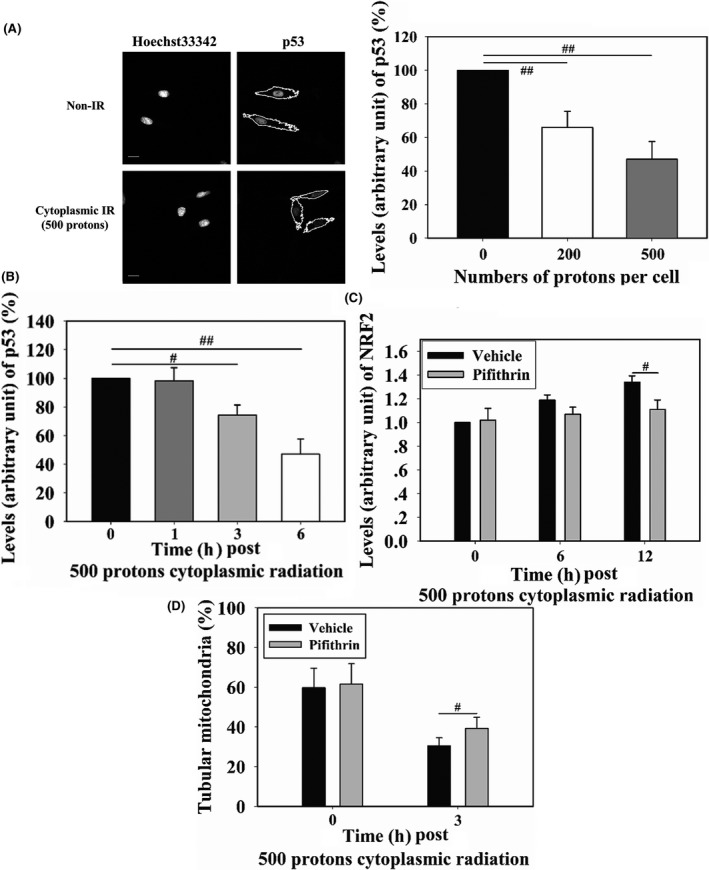
WI‐38 cells were cytoplasm targeted by 200 or 500 protons 6 h before measuring p53 levels (A). Fluorescence intensity was normalized to that of non‐irradiated cells. Cells were cytoplasm‐targeted by 500 protons and p53 levels were detected at different time points post‐irradiation (B). Effects of 20 μmol/L pifithrin‐α (p53 inhibitor) on cytoplasm irradiation‐induced nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) localization to the nucleus (C) and mitochondrial morphology changes (D) were detected. Scale bar, 20 μm. IR, irradiation. # and ## indicate significant differences at P < .05 and P < .01, respectively
Overall, these results showed that p53 transcription activity contributed to mitochondrial fragmentation and NRF2 nucleus localization, although its expression was decreased by cytoplasmic irradiation.
3.5. Mitochondrial superoxide stimulated NRF2 translocation
The mitochondrion is a defined target of cytoplasmic irradiation.8, 20 Because elevated levels of ROS in cells after ionizing radiation are common, and NRF2 is an oxidative stress responsive protein, we next investigated the relationship between mitochondrial superoxide production and nucleus accumulation of NRF2. At 0.5 and 2 hours after 500‐proton cytoplasmic irradiation, mitochondrial superoxide levels were detected in WI‐38 cells using the MitoSOX Red fluorescence probe. Mitochondrial superoxide levels were 15% and 48% higher in irradiated than in non‐irradiated cells at 0.5 and 2 hours post‐cytoplasmic irradiation, respectively. Treatment with mitoTEMPOL (10 μmol/L final concentration), a specific scavenger of mitochondrial superoxide, 30 minutes before irradiation inhibited the elevation of mitochondrial superoxide (Figure 5A). We next examined whether the increased levels of mitochondrial superoxide promoted nucleus accumulation of NRF2. Both the accumulation of NRF2 in the nucleus (12 hours after irradiation) and the upregulation of HO‐1 (24 hours after irradiation) in 500‐proton irradiated cells were inhibited by mitoTEMPOL treatment (Figure 5B), reflecting the role of mitochondria‐derived reactive species in the activation of NRF2 after cytoplasmic irradiation. We further checked the effects of mitoTEMPOL on p53 expression and mitochondrial fragmentation. Reduced downregulation of p53 was observed 6 hours after 500‐proton irradiation in the presence of mitoTEMPOL, restoring approximately 89% of the level observed in non‐irradiated cells. Non‐treated irradiated cells showed p53 expression levels approximately 50% that of non‐irradiated cells (Figure 5C). At 6 hours post 500‐proton cytoplasmic irradiation, level of tubular mitochondria in the presence of mitoTEMPOL was almost identical to that of non‐irradiated cells (Figure 5C). These results indicated that cytoplasmic irradiation‐induced elevation of mitochondrial superoxide played an important role in mediating NRF2 activation through p53 downregulation and mitochondrial fragmentation. We further checked the effect of mitoTEMPOL on cytoplasmic radiation‐induced γH2A.X levels. As shown in Figure 5D, in mitoTEMPOL pretreated cells, γH2A.X levels decreased to 87% and 79% compared to those in the mock treated and irradiated cells after 200 or 500 protons irradiation, respectively. Together with the above observations, cytoplasmic radiation‐induced mitochondrial superoxide elevation acted not only as the DNA‐damage agent, but also as a signaling factor to promote cellular self‐protection.
Figure 5.
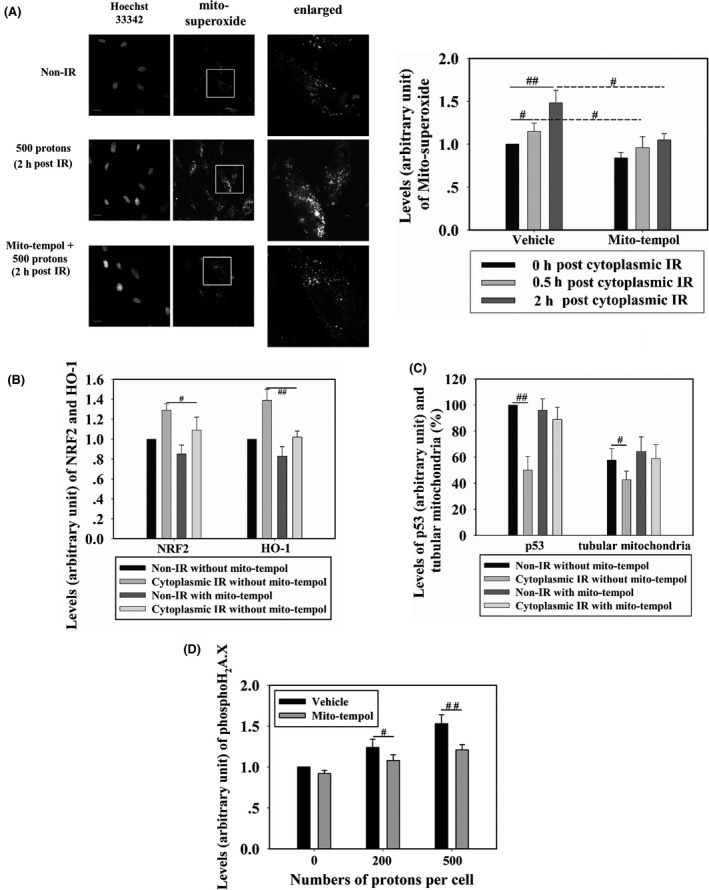
WI‐38 cells were cytoplasm‐targeted by 500 protons and, at different time points post‐irradiation, mitochondrial superoxide levels were detected in cells treated with 10 μmol/L mitoTEMPOL (scavenger of mitochondrial superoxide) or non‐treated (A). Fluorescence intensity was normalized to that of non‐irradiated cells. Effects of mitoTEMPOL treatment on the levels of nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) and heme oxygenase‐1 (HO‐1) (B) and on p53 expression and tubular mitochondria (C) were detected in cells targeted by 500 protons. Scale bar, 20 μm. (D) WI‐38 cells were treated with 10 μmol/L mitoTEMPOL or mock‐treated, then cytoplasm‐targeted by 200 or 500 protons. Four hours later, γH2A.X formation was detected. IR, irradiation. # and ## indicate significant differences at P < .05 and P < .01, respectively
4. DISCUSSION
A canonical theory in radiation biology is that genomic DNA is the main target for radiation‐induced genotoxicity. Genomic DNA damage can be caused directly by the radiation beam, or indirectly by the formation of reactive radical species. Within the past two decades, evidence from microbeam‐based studies showed that cytoplasm irradiation leads to DNA damage in the target cell,21 as well as in bystander cells.22 Elevated reactive radical species are considered one of the core reasons for such damage. Treatment with ROS scavenger DMSO largely decreased the mutation frequency of gene CD59 in AL cells irradiated by 4‐α particles in the cytoplasm. Moreover, reduction in intracellular glutathione levels led to a four‐ to fivefold increase in CD59 mutation frequency.3 Hong et al23 reported that in AL cells receiving 8‐α particle cytoplasmic irradiation, DMSO treatment could suppress the production of 8‐hydroxydeoxyguanosine to the level observed in non‐irradiated cells. This evidence led us to investigate the antioxidative response occurring in the cytoplasm of irradiated cells.
As a key regulator of oxidative stress in mammalian cells,24, 25 one of the functions of NRF2 is to transcriptionally activate the expression of proteins such as HO‐1, NQO1, and SRXN1, to reduce oxidative stress. In the present study, we first confirmed that the NRF2 antioxidative pathway could respond to cytoplasmic irradiation, which was reflected by increased nuclear localization of NRF2 and expression of its target protein HO‐1. Activation of NRF2 is known to protect cells against various external stresses, including ionizing radiation. Han et al26 showed that, in mice, theaflavin, a polyphenolic compound, could ameliorate irradiation‐induced hematopoietic stem cell injury through NRF2‐mediated decline of ROS levels and DNA damage. Fan et al27 reported that L‐carnitine preserved cardiac function in irradiated mice. To elucidate whether the stimulated activation of NRF2 could attenuate the level of DSB in the cytoplasm of irradiated cells, tBHQ, a selective NRF2 chemical inducer, was used in the present study. tBHQ activates NRF2 by interacting with thiol groups of cysteine molecules on the KEAP1 protein, which interferes with the ability of Keap1 to repress Nrf2.28 Wang et al29 reported that in tBHQ‐treated mouse heart, expression, transcription and nucleus accumulation of Nrf2 and its downstream target HO‐1 was significantly stimulated. In the present study, pretreatment with tBHQ significantly stimulated nucleus accumulation of NRF2 in WI‐38 cells, and decreased cytoplasmic radiation‐induced DSB levels, indicating the protective role of NRF2 toward irradiation. In addition, NRF2 is also a DNA damage repair regulator. In irradiated human colonic epithelial cells, Kim et al30 found that NRF2 activation increased DNA damage signaling by enhancing nucleus accumulation of BRCA1 and RAD51, and further identified that NRF2 could bind the promoter region of 53BP1 and induce its expression.30 Singh et al13 reported that NRF2 directly bound the promoter region of the DNA repair enzyme OGG1 and was correlated with estrogen‐induced oxidative DNA damage in breast cancer cells. In the present study, we found that RAD51 and XRCC4, two key proteins involved in DSB repair, showed enhanced nuclear accumulation in irradiated cells pretreated with tBHQ. This agreed with the results of previous studies showing that NRF2 protected cells against radiation‐induced damage,15, 16 and indicated that increased NRF2 activation might contribute to attenuate DSB levels in cytoplasm‐irradiated cells partially through stimulating DNA repair capability. The NRF2 antioxidative response has been reported to be activated by ROS and reactive nitrogen species (RNS), as both were increased in irradiated cells.12 Cytoplasm irradiation might influence the function of mitochondria, where ATP is synthesized, as electrons may leak from the mitochondrial respiration chain and react inappropriately with oxygen to form reactive free radicals.31 We therefore examined whether the mitochondria‐derived ROS was responsible for the activation of NRF2 in irradiated cells. In agreement with Zhang,8 an obvious elevation of mitochondrial superoxide was detected in our study. Furthermore, suppressing the production of mitochondrial superoxide with mitoTEMPOL resulted in the inhibition of NRF2 nuclear localization and in the upregulation of HO‐1, indicating that cytoplasmic irradiation‐induced mitochondrial superoxide production is a key stimulus for NRF2 activation. It was reported that in arsenite‐treated U937 cells, mitochondrial superoxide triggered two opposite pathways.32 One was the activation of NRF2 signaling, which enhanced glutathione biosynthesis to protect cells against arsenite toxicity. Our results also support this finding.
Mitochondrial superoxide production also affected p53 expression in irradiated cells. Because p53 is a DNA damage responsive protein, it is usually upregulated in irradiated cells.33 However, the opposite tendency (ie, p53 downregulation) was observed in cytoplasm‐irradiated WI‐38 cells. By suppressing mitochondrial superoxide in irradiated cells, mitoTEMPOL blocked the decreasing tendency of p53 expression under cytoplasmic irradiation. MDM2 is an E3 ubiquitin‐protein ligase that mediates ubiquitination of p53, leading to its degradation by the proteasome. You et al34 found that in nrf2‐deleted murine embryonic fibroblasts, Mdm2 expression was repressed, both at the protein and mRNA levels. A recent study provided direct evidence that NRF2 binds to the conserved binding site upstream of the MDM2/Mdm2 promoter in human MIA PaCa‐2 and mouse KPC cells.35 Using a promoter reporter assay, Todoric et al35 confirmed direct control of MDM2 expression by NRF2. In our study, activation of NRF2 was observed within 6 hours post‐cytoplasmic irradiation, and it might contribute to the downregulation of p53. However, a variety of post‐transcription modifications of p53 have been found to be associated with its stability.36 Thus, cytoplasmic irradiation might result in acetylation, methylation, or phosphorylation of p53 and its destabilization, which should be further investigated. We also observed that inhibition of p53 transcription activity by pifithrin‐α repressed NRF2 nucleus accumulation after cytoplasmic irradiation. Tung et al37 found that the expression of NRF2 was not suppressed in non‐small cell lung cancer cell lines harboring mutant p53. However, wild‐type p53 inhibited NRF2 expression by binding the NRF2 promoter and eliminating its recruitment of SP1. On the contrary, another report showed that in neuroblastoma with increased cellular nitric oxide level, p53 bound the promoter of peroxisome proliferator‐activated receptor‐γ coactivator‐1 α and stimulated its expression.38 This further resulted in the activation of NRF2‐mediated antioxidative responses. P21 is a transcription target of p53, and it can stabilize NRF2 by binding to KEAP1 and interfering with NRF2 ubiquitylation and degradation.39 Thus, the roles of p53 on NRF2 activation might be controlled by cellular and biological contexts.
Mitochondria are mobile organelles operating in dynamic networks that join each other by a process named fusion and fragment by a process named fission. There is emerging recognition that disordered mitochondrial dynamics contribute to the pathogenicity of complex diseases,9 and that extracellular stimuli affect mitochondrial dynamics.40 Mitochondrial fragmentation was observed in our research in cytoplasm‐irradiated cells, in line with the findings of Zhang et al.8 Furthermore, Wu et al10 found that the mitochondrial fragmentation inhibitor Mdivi‐1 could block cytoplasmic irradiation‐induced autophagy in SAE cells, and autophagy inhibition delayed the repair of cytoplasmic irradiation‐induced DSB.41 Overall, our results provided another mechanism for the protective role of mitochondrial fragmentation in decreasing cytoplasmic irradiation‐induced DSB level. Albeit scarcely reported, p53 greatly affects mitochondrial functions, and the current study showed that p53 affected mitochondrial dynamics. Li et al42 found that, in rat cardiac cells, p53 transcriptionally upregulated dynamin‐related protein‐1 (Drp1) and stimulated mitochondrial fragmentation, which initiated H2O2‐induced apoptosis. Yuan et al43 also found that in mouse podocytes, suppressed expression of p53 resulted in reduced transcription of Drp1. Furthermore, Qi et al44 reported that the acetylation of p53 was important for its binding to the DRP1 promoter and the upregulation of DRP1. Our results were consistent with the above findings that the transcription activity of p53 contributed to the expression of DRP1 and mitochondrial fragmentation. This was also in line with our observation that the inhibition of p53 or mitochondrial fragmentation resulted in the decrease of NRF2 nuclear localization. Moreover, Wang et al45 reported that, in cultured postnatal mouse cortical neurons, treatment with the DNA‐damaging agent camptothecin (CPT) resulted in elongated mitochondria, activated p53, and suppressed the expression of mitochondrial fission protein Drp1 and parkin. However, when p53 was deficient, CPT‐induced Drp1 and parkin downregulation were absent, suggesting that p53‐dependent declines in Drp1 and parkin contribute to alterations in mitochondrial morphology and lead to the death of mouse neurons under CPT regulation. Huntington's disease (HD) is associated with a variety of cellular dysfunctions including excessive mitochondrial fragmentation. Guo et al found that p53 bound DRP1 and mediated DRP1‐induced mitochondrial and neuronal damage. Silencing of p53 by siRNA reduced the number of cells with fragmented mitochondria and promoted cell viability.46 These studies indicated that p53 has different effects on stress‐induced mitochondrial fragmentation. We also observed that mitoTEMPOL treatment suppressed mitochondrial fragmentation, highlighting the importance of mitochondrial superoxide in disturbing the physiological equilibrium of cytoplasm‐irradiated cells.
In summary, by using a proton microbeam radiation facility, we identified that cytoplasmic irradiation stimulated the NRF2 antioxidative response for protection against cytoplasmic irradiation‐induced DNA damage. In addition, p53 transcription activity was found to be important for promoting mitochondrial fragmentation, which further led to the activation of the NRF2 antioxidative response. Elevation of mitochondrial superoxide after cytoplasmic irradiation was an elicitor for NRF2 activation and for changes in p53 level and mitochondrial morphology. Our results provided new evidence on the importance of oxidative stress responses in regulating nuclear DNA damage induced by cytoplasmic irradiation.
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank Mr Masakazu Oikawa and Ms Alisa Kobayashi from the National Institute of Radiological Sciences for their technical support on microbeam irradiation. This work was supported by NSFC program of China (11835014, 11575232, 31370842), International Partnership Program of Chinese Academy of Sciences (116134KYSB20160084), project from Hefei Center for Physical Science and Technology (2016FXCX005), the China Scholarship Council (201704910370), Japan Society for the Promotion of Science KAKENHI Grant‐in‐Aid for Challenging Exploratory Research (JP16K15586) and the International Open Laboratory program of National Institute of Radiological Sciences, Japan.
Wang J, Konishi T. Nuclear factor (erythroid‐derived 2)‐like 2 antioxidative response mitigates cytoplasmic radiation‐induced DNA double‐strand breaks. Cancer Sci. 2019;110:686–696. 10.1111/cas.13916
REFERENCES
- 1. Hall EJ, Giaccia AJ. Radiobiology for the Radiologist, 7th edn Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2012:1‐576. [Google Scholar]
- 2. Konishi T, Oikawa M, Suya N, et al. SPICE‐NIRS microbeam: a focused vertical system for proton irradiation of a single cell for radiobiological research. J Radiat Res. 2013;54:736‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu LJ, Randers‐Pehrson G, Xu A, et al. Targeted cytoplasmic irradiation with alpha particles induces mutations in mammalian cells. Proc Natl Acad Sci USA. 1999;96:4959‐4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tartier L, Gilchrist S, Burdak‐Rothkamm S, Folkard M, Prise KM. Cytoplasmic irradiation induces mitochondrial‐dependent 53BP1 protein relocalization in irradiated and bystander cells. Can Res. 2007;67:5872‐5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shao C, Folkard M, Michael BD, Prise KM. Targeted cytoplasmic irradiation induces bystander responses. Proc Natl Acad Sci USA. 2004;101:13495‐13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jella KK, Moriarty R, McClean B, Byrne HJ, Lyng FM. Reactive oxygen species and nitric oxide signaling in bystander cells. PLoS ONE. 2018;13:e0195371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Forman HJ. Redox signaling: an evolution from free radicals to aging. Free Radic Biol Med. 2016;97:398‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang B, Davidson MM, Zhou H, Wang C, Walker WF, Hei TK. Cytoplasmic irradiation results in mitochondrial dysfunction and DRP1‐dependent mitochondrial fission. Cancer Res. 2013;73:6700‐6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236‐2251. [DOI] [PubMed] [Google Scholar]
- 10. Wu J, Zhang Q, Wuu YR, Zou S, Hei TK. Cytoplasmic irradiation induces metabolic shift in human small airway epithelial cells via activation of Pim‐1 kinase. Radiat Res. 2017;187:441‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fan Z, Wirth AK, Chen D, et al. Nrf2‐Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6:e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh B, Chatterjee A, Ronghe AM, Bhat NK, Bhat HK. Antioxidant‐mediated up‐regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen‐induced breast cancer. BMC Cancer. 2013;13:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jain A, Lamark T, Sjottem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element‐driven gene transcription. J Biol Chem. 2010;285:22576‐22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen N, Wu L, Yuan H, Wang J. ROS/autophagy/Nrf2 pathway mediated low‐dose radiation induced radio‐resistance in human lung adenocarcinoma A549 cell. Int J Biol Sci. 2015;11:833‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen N, Zhang R, Konishi T, Wang J. Upregulation of NRF2 through autophagy/ERK 1/2 ameliorates ionizing radiation induced cell death of human osteosarcoma U‐2 OS. Mutat Res. 2017;813:10‐17. [DOI] [PubMed] [Google Scholar]
- 17. Rasband WS. ImageJ. Bethesda, MD: U.S. National Institutes of Health, 1997. ‐2016. [Google Scholar]
- 18. Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double‐strand breaks in vivo. J Cell Biol. 1999;146:905‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosdah AA, Holien JK, Delbridge LM, Dusting GJ, Lim SY. Mitochondrial fission ‐ a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect. 2016;4:e00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou H, Ivanov V, Lien Y‐C, Davidson M, Hei T. Mitochondrial function and NF‐κB mediated signaling in radiation‐induced bystander effects. Can Res. 2008;68:2233‐2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maeda M, Usami N, Kobayashi K, Cell XR. Low‐dose hypersensitivity in nucleus‐irradiated V79 cells studied with synchrotron X‐ray microbeam. Journal of Radiation Research. 2008;49:171‐180. [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi A, Tengku Ahmad TAF, Autsavapromporn N, et al. Enhanced DNA double‐strand break repair of microbeam targeted A549 lung carcinoma cells by adjacent WI38 normal lung fibroblast cells via bi‐directional signaling. Mutat Res. 2017;803–805:1‐8. [DOI] [PubMed] [Google Scholar]
- 23. Hong M, Xu A, Zhou H, et al. Mechanism of genotoxicity induced by targeted cytoplasmic irradiation. Br J Cancer. 2010;103:1263‐1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sekhar KR, Freeman ML. Nrf2 promotes survival following exposure to ionizing radiation. Free Radic Biol Med. 2015;88:268‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kitamura H, Motohashi H. NRF2 addiction in cancer cells. Cancer Sci. 2018;109:900‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han X, Zhang J, Xue X, et al. Theaflavin ameliorates ionizing radiation‐induced hematopoietic injury via the NRF2 pathway. Free Radic Biol Med. 2017;113:59‐70. [DOI] [PubMed] [Google Scholar]
- 27. Fan Z, Han Y, Ye Y, Liu C, Cai H. l‐carnitine preserves cardiac function by activating p38 MAPK/Nrf2 signalling in hearts exposed to irradiation. Eur J Pharmacol. 2017;804:7‐12. [DOI] [PubMed] [Google Scholar]
- 28. Li W, Kong A‐N. Molecular mechanisms of Nrf2‐mediated antioxidant response. Mol Carcinog. 2009;48:91‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang L‐F, Su S‐W, Wang L, et al. Tert‐butylhydroquinone ameliorates doxorubicin‐induced cardiotoxicity by activating Nrf2 and inducing the expression of its target genes. Am J Transl Res. 2015;7:1724‐1735. [PMC free article] [PubMed] [Google Scholar]
- 30. Kim SB, Pandita RK, Eskiocak U, et al. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc Natl Acad Sci USA. 2012;109:E2949‐E2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 2016;100:14‐31. [DOI] [PubMed] [Google Scholar]
- 32. Fiorani M, Guidarelli A, Capellacci V, Cerioni L, Crinelli R, Cantoni O. The dual role of mitochondrial superoxide in arsenite toxicity: signaling at the boundary between apoptotic commitment and cytoprotection. Toxicol Appl Pharmacol. 2018;345:26‐35. [DOI] [PubMed] [Google Scholar]
- 33. Fei P, El‐Deiry WS. P53 and radiation responses. Oncogene. 2003;22:5774‐5783. [DOI] [PubMed] [Google Scholar]
- 34. You A, Nam CW, Wakabayashi N, Yamamoto M, Kensler TW, Kwak MK. Transcription factor Nrf2 maintains the basal expression of Mdm2: an implication of the regulation of p53 signaling by Nrf2. Arch Biochem Biophys. 2011;507:356‐364. [DOI] [PubMed] [Google Scholar]
- 35. Todoric J, Antonucci L, Di Caro G, et al. Stress‐activated NRF2‐MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell. 2017;32:824‐839 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dai C, Gu W. p53 post‐translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tung MC, Lin PL, Wang YC, et al. Mutant p53 confers chemoresistance in non‐small cell lung cancer by upregulating Nrf2. Oncotarget. 2015;6:41692‐41705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G, Ciriolo MR. p53 orchestrates the PGC‐1α‐mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal. 2013;18:386‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen W, Sun Z, Wang X‐J, et al. Direct interaction between Nrf2 and p21Cip1/WAF1 upregulates the Nrf2‐mediated antioxidant response. Mol Cell. 2009;34:663‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062‐1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu J, Zhang B, Wuu YR, Davidson MM, Hei TK. Targeted cytoplasmic irradiation and autophagy. Mutat Res. 2017;806:88‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR‐30 regulates mitochondrial fission through targeting p53 and the dynamin‐related protein‐1 pathway. PLoS Genet. 2010;6:e1000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yuan Y, Zhang A, Qi J, et al. p53/Drp1‐dependent mitochondrial fission mediates aldosterone‐induced podocyte injury and mitochondrial dysfunction. American Journal of Physiology‐Renal Physiology. 2017;314:F798‐F808. [DOI] [PubMed] [Google Scholar]
- 44. Qi J, Wang F, Yang P, et al. Mitochondrial fission is required for angiotensin II‐induced cardiomyocyte apoptosis mediated by a Sirt1‐p53 signaling pathway. Frontiers in Pharmacology. 2018;9:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang DB, Garden GA, Kinoshita C, et al. Declines in Drp1 and parkin expression underlie DNA damage‐induced changes in mitochondrial length and neuronal death. J Neurosci. 2013;33:1357‐1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly‐Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington')s disease‐associated neurodegeneration. J Clin Invest. 2013;123:5371‐5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials