

Abstract
Rho GTPase Rac1 is a central regulator of F‐actin organization and signal transduction to control plasma membrane dynamics and cell proliferation. Dysregulated Rac1 activity is often observed in various cancers including breast cancer and is suggested to be critical for malignancy. Here, we showed that the ubiquitin E3 ligase complex Cullin‐3 (CUL3)/KCTD10 is essential for epidermal growth factor (EGF)‐induced/human epidermal growth factor receptor 2 (HER2)‐dependent Rac1 activation in HER2‐positive breast cancer cells. EGF‐induced dorsal membrane ruffle formation and cell proliferation that depends on both Rac1 and HER2 were suppressed in CUL3‐ or KCTD10‐depleted cells. Mechanistically, CUL3/KCTD10 ubiquitinated RhoB for degradation, another Rho GTPase that inhibits Rac1 activation at the plasma membrane by suppressing endosome‐to‐plasma membrane traffic of Rac1. In HER2‐positive breast cancers, high expression of Rac1 mRNA significantly correlated with poor prognosis of the patients. This study shows that this novel molecular axis (CUL3/KCTD10/RhoB) positively regulates the activity of Rac1 in HER2‐positive breast cancers, and our findings may lead to new treatment options for HER2‐ and Rac1‐positive breast cancers.
Keywords: Cullin‐3, HER2, membrane ruffle, Rac1, RhoB
Abbreviations
- BafA1
bafilomycin A1
- BC
breast cancer
- BTBP
Bric‐a‐brac/Tramtrack/Broad complex (BTB) domain‐containing proteins
- CA
constitutive‐active
- CUL3
Cullin‐3
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐mesenchymal transition
- ER
estrogen receptor
- GAP
GTPase activating protein
- GEF
GTP exchange factor
- HER2
human epidermal growth factor receptor 2
- METABRIC
Molecular Taxonomy of Breast Cancer International Consortium
- PR
progesterone receptor
- SEM
scanning electron microscopy
- SrGAP3
Slit‐Robo GAP3
- TNF
tumor necrosis factor
1. INTRODUCTION
Cullin‐3, a cullin family protein, is a scaffold protein that forms a RING ubiquitin E3 ligase complex. Its adaptor proteins, BTBP, bridge CUL3 and substrate proteins leading to their ubiquitination.1, 2 CUL3/BTBP/substrates axes are essential for development and regulate a variety of cellular functions (e.g. cell cycle progression, membrane trafficking, transcription, signaling in stress response, and cytoskeletal reorganization). Dysfunction of CUL3 has been implicated in the development of human diseases such as hypertension and cancer.3, 4 CUL3 also regulates endothelial functions (e.g. cell proliferation and barrier function) and angiogenesis by formation of complexes with multiple BTBP.5, 6, 7, 8, 9 Recently, we found that CUL3 or KCTD10, one of the BTBP, is essential for endothelial barrier functions in HUVEC.7 Loss of CUL3 or KCTD10 induces strong actin polymerization and cell contraction, inhibiting the proper generation of endothelial barriers.7 We also found that RhoB, an endosomal Rho GTPase, suppresses endothelial barrier formation in HUVEC.7 An inhibitory role of RhoB in endothelial barrier formation was also implicated in TNF‐simulated HUVEC and vascular endothelial cells in Crohn('s disease.10 Mechanistically, we found that RhoB was constitutively degraded in lysosomes, and the degradation process was mediated through K63 polyubiquitination at lysine 162 and 181 of RhoB by CUL3/KCTD10.7
Rac1, another Rho GTPase, is a central regulator of F‐actin organization at the plasma membrane. Rac1 is translocated from endosomes to cell borders during endothelial barrier formation and stabilizes endothelial barriers in HUVEC.10 Interestingly, the translocation of Rac1 was inhibited by high expression of RhoB, resulting in impaired barrier restoration.10 Given our finding of the constitutive degradation of RhoB by CUL3/KCTD10,7 CUL3/KCTD10 may be critical for Rac1 trafficking and activation through RhoB degradation.
In addition to the physiological roles of Rac1 in endothelial cells, the oncogenic roles of Rac1 in various cancers have been recognized.11 Elevated expression or hyperactivation of Rac1 is frequently observed in human cancers, correlating with their aggressiveness and poor prognosis.12 NSC23766, a selective inhibitor of Rac1 activation, has been developed and was found to show anti‐cancer effects in lymphoma.13 In BC, both upregulation of Rac1 GEF and downregulation of Rac1 GAP have been reported. Overexpression of P‐Rex1, a Rac1 GEF, played a critical role in metastasis of luminal‐type BC.14, 15 Low expression of SrGAP3, a Rac1/Cdc42 GAP, was observed in invasive ductal breast carcinomas.16, 17 Downregulation of SrGAP3 enhanced anchorage‐independent cell growth in a Rac1‐dependent way in human mammary epithelial cells.17 Activation of Rac1 contributed to trastuzumab (an inhibitory antibody to Human epidermal growth factor receptor 2 [HER2]) resistance which poses a serious problem during chemotherapy for HER2‐positive BC.18, 19
In the present study, we found that among the subtypes of human BC, high expression of Rac1 is correlated with poor prognosis, specifically of HER2‐positive BC patients. We investigated the molecular mechanism underlying Rac1 activation in HER2‐positive BC cells. Our results suggest that Rac1 activation requires downregulation of RhoB that is mediated by the CUL3/KCTD10 E3 complex.
2. MATERIALS AND METHODS
2.1. Cell culture
SKBR‐3 cells and HEK293T cells were maintained at 37°C with 5% CO2 in DMEM (Wako, Tokyo, Japan) supplemented with 10% FBS, 20 units/mL penicillin and 100 μg/mL streptomycin. MCF‐7 cells were maintained at 37°C with 5% CO2 in EMEM (Wako) supplemented with 10% FBS, 20 units/mL penicillin and 100 μg/mL streptomycin. MDA‐MB‐231 cells and MDA‐MB‐453 cells were maintained at 37°C without CO2 in Leiboviz's L‐15 medium (Wako) supplemented with 10% FBS, 20 units/mL penicillin and 100 μg/mL streptomycin. Other Materials and Methods are described in Doc S1.
3. RESULTS
3.1. In HER2‐positive BC, high expression of Rac1 mRNA significantly correlates with poor prognosis
We first examined whether the expression level of Rac1 correlates with the prognosis of human BC, the most common cancer in women, using the METABRIC database.20 METABRIC database contains transcriptome information of over 2000 clinically annotated primary BC, of which long‐term clinical outcomes are available.20 Expression level of Rac1 mRNA did not significantly correlate with the prognosis of human BC (Figure 1A; P = .091, High: n = 1636, Low: n = 268). Human BC can be classified by gene expression profiles into five subtypes: luminal‐A (ER+, PR+, Ki‐67low, HER2−), luminal‐B (ER+, PR+, Ki‐67high, HER2+/−), HER2‐positive (ER−, PR−, HER2+), basal (ER−, PR−, HER2−, cytokeratin5/6+, EGFR+), and claudin‐low (ER−, PR−, HER2−, claudinlow) types.20, 21, 22 We found that high mRNA expression of Rac1 significantly correlated with poor prognosis of HER2‐positive BC (Figure 1B top left panel: P = .0012, High: n = 49, Low: n = 171). The other three types (basal, claudin‐low, or luminal‐B type) did not show significant correlation between the expression levels of Rac1 mRNA and their prognosis (Figure 1B top middle panel: P = .15, High: n = 97, Low: n = 102; Figure 1B top right panel: P = .052, High: n = 110, Low: n = 89; Figure 1B bottom right panel: P = .17, High: n = 70, Low: n = 391, respectively). In luminal‐A‐type BC, low mRNA expression of Rac1 significantly correlated with poor prognosis (Figure 1B bottom left panel: P = .0046, High: n = 492, Low: n = 187).
Figure 1.
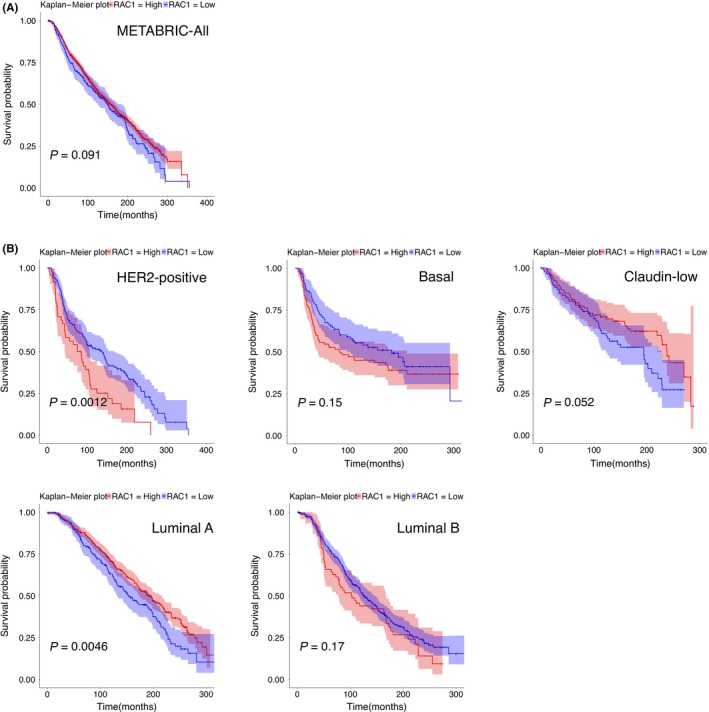
Survival analysis with Rac1 expression profiles in human breast cancer (BC) subtype. Kaplan‐Meier plot with Rac1 expression profiles in Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) cohort using overall survival status. Shading along the curve shows 95% confidential interval. A, Analysis with all samples shows that Rac1 expression was not correlated with prognosis (High: n = 1636, Low: n = 268). B, In human epidermal growth factor receptor 2 (HER2)‐positive subtype, high expression of Rac1 correlated with poor prognosis (High: n = 49, Low: n = 171). In other subtypes, luminal A subtype (High: n = 492, Low: n = 187), luminal B subtype (High: n = 70, Low: n = 391), basal subtype (High: n = 97, Low: n = 102), and claudin‐low subtype (High: n = 110, Low: n = 89), Rac1 expression was not positively correlated with poor prognosis
3.2. Cullin‐3/KCTD10 ubiquitin E3 ligase complex is essential for Rac1 activation through RhoB degradation in HER2‐positive BC SKBR‐3 cells
Because high mRNA expression of Rac1 significantly correlated with poor prognosis of HER2‐positive BC, we sought to understand how Rac1 is activated in this BC subtype. We envisaged that the outcome may lead to new treatment options for HER2‐ and Rac1‐double‐positive BC. Given the previous findings in HUVEC that RhoB was constitutively degraded by CUL3/KCTD107 and that overexpression of RhoB suppressed the activation of Rac1,10 we assumed that the activation of Rac1 in HER‐2‐positive BC cells would also require the function of CUL3/KCTD10. SKBR‐3 cells (HER‐2‐positive BC cells) were treated with siRNA oligos designed for CUL3 or KCTD10, and knockdown efficiency was confirmed by western blot (Figure 2A). We noticed that RhoB expression increased in cells depleted of CUL3 or KCTD10, suggesting that CUL3 and KCTD10 regulate the degradation of RhoB in SKBR‐3 cells (Figure 2A), as in HUVEC.7 Expression of FLAG‐tagged siRNA‐resistant CUL3 or siRNA‐resistant non‐tagged KCTD10 reduced RhoB expression in CUL3 or KCTD10 knockdown cells, respectively, excluding the off‐target effect of siRNA (Figure 2B). RhoB expression was also increased by treatment with MLN‐4924, an inhibitor of cullin family proteins that functions by suppressing the neddylation of cullin, and BafA1, an inhibitor of vacuolar‐type H+‐ATPase (Figure 2C). MG‐132, a proteasome inhibitor, slightly increased RhoB expression (Figure 2C). Other proteasome inhibitors, lactacystin and epoxomicin, also slightly increased RhoB expression (Figure S1). Thus, RhoB may be degraded mainly through the lysosomal pathway after ubiquitination by CUL3/KCTD10 in SKBR‐3 cells as in HUVEC.7 Expression of other Rho family small GTPases, such as RhoA, RhoC, Rac1/2/3, or Cdc42, was not affected by knockdown of CUL3 or KCTD10 (Figure 2A). The same results were obtained in another HER2‐positive BC cell line, MDA‐MB‐453 cells (Figure S2A). By knockdown of CUL3 or KCTD10, protein expression of RhoB was increased and expression of other Rho family small GTPases (RhoA and RhoC) was not affected in MDA‐MB‐453 cells (Figure S2A). We also examined the effects of CUL3 or KCTD10 knockdown on RhoB expression in other subtypes of BC cell lines, MCF‐7 cells (luminal type) and MDA‐MB‐231 cells (basal type) (Figure S2B,C). In contrast to HER2‐positive BC cells, CUL3‐ or KCTD10‐knockdown did not affect protein expression of RhoB in both MCF‐7 cells and MDA‐MB‐231 cells (Figure S2B,C). These results suggest that the CUL3/KCTD10/RhoB axis functions specifically in HER2‐positive BC cells.
Figure 2.
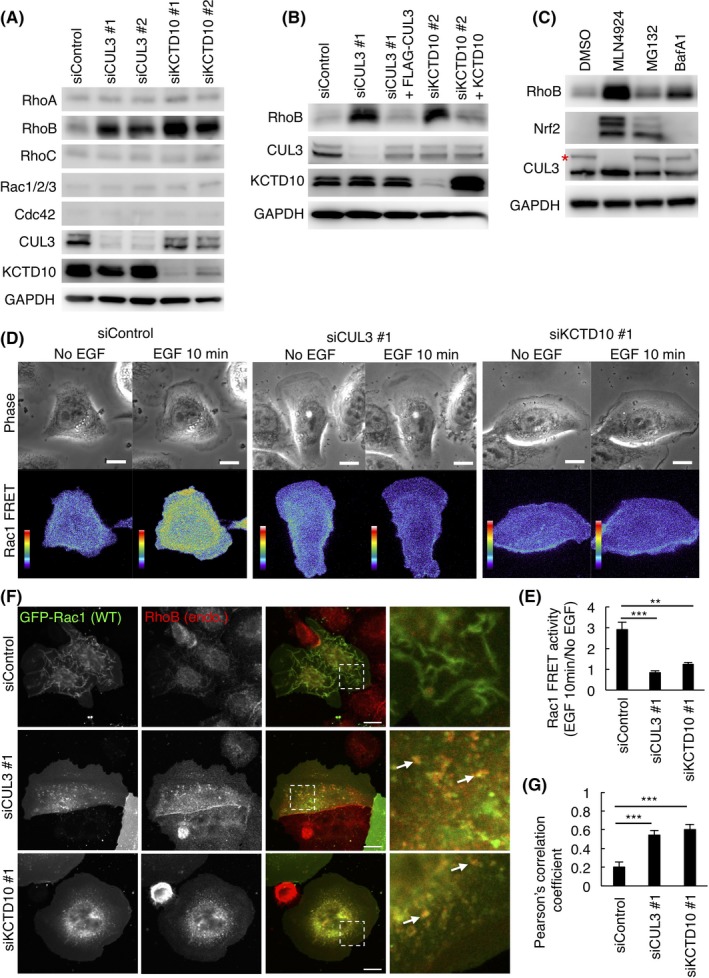
Cullin‐3 (CUL3) and KCTD10 regulate protein expression of RhoB and activation of Rac1 in SKBR‐3 cells. A, Western blots of SKBR‐3 cell lysates, 72 h post‐transfection with siRNAs. B, Rescue experiments of CUL3 or KCTD10 knockdown. Western blots of SKBR‐3 cell lysates infected with siRNA‐resistant‐FLAG‐CUL3‐, or siRNA‐resistant non‐tagged KCTD10‐carrying lentivirus. C, Western blots of cell lysates of SKBR‐3 cells treated with 1 μM MLN4924 for 24 h, 10 μM MG‐132 for 6 h or 0.5 μM bafilomycin A1 (BafA1) for 24 h. Asterisk indicates neddylated CUL3. Nrf2 was a positive control to examine the effect of MG‐132. D, Serum‐starved SKBR‐3 cells expressing RaichuEV‐Rac1, a FRET biosensor for Rac1 activity, were stimulated with epidermal growth factor (EGF). Phase‐contrast image and fluorescence images of FRET and cyan fluorescent protein (CFP) of SKBR‐3 cells treated with control siRNA, CUL3 siRNA #1 or KCTD10 siRNA #1 were acquired every 15 s. Ratio images, showing Rac1 FRET efficiency, were created by the image arithmetic of the FRET/CFP ratio using MetaMorph software. Rac1 FRET efficiency showing Rac1 activity levels before and 10 min after EGF stimulation are shown as pseudo‐color spectral images. Scale bars, 10 μm. See also Videos S1, S2 and S3. E, Quantitation of (D). Intracellular signal intensity of FRET/CFP ratio at 10 min after EGF stimulation was normalized to that of quiescent cells (No EGF). Data are mean ± SEM (n = 5 cells). **P < .01. ***P < .001. F, Confocal images of SKBR‐3 cells treated with control siRNA, CUL3 siRNA #1 or KCTD10 siRNA #1 for 72 h. Forty‐eight hours after transfection of the GFP‐Rac1 vector, cells were stimulated with EGF for 15 min, fixed, permeabilized, and stained for RhoB. Magnifications of the dashed squared areas are shown in the right images. Representative colocalized GFP‐Rac1 and RhoB are indicated by arrows. Bars, 10 μm. G, Quantitation of (F). Colocalization of GFP‐Rac1 with RhoB was determined using Pearson')s correlation coefficient. Twenty cells from three independent experiments were analyzed. Data are mean ± SEM. ***P < .001
We next biochemically examined the detection of the active Rac1 in SKBR‐3 cells by pull‐down with PAK‐PBD affinity beads, which can bind to Rac1‐GTP. As shown in Figure S3, Rac1‐GTP was slightly detected in control cells by the addition of EGF. This result suggested that EGF‐induced Rac1 activation may occur transiently and/or locally in SKBR‐3 cells and, thus, it might be difficult to detect Rac1‐GTP from total cell lysates by the pull‐down assay. Alternatively, we then visualized the activation of Rac1 in SKBR‐3 cells using a Rac1‐FRET biosensor, RaichuEV‐Rac1.23, 24 In control cells, only a basal level of FRET signal that indicates the presence of Rac1‐GTP was observed without EGF. Ten minutes after EGF stimulation, the FRET signal became prominent at membrane ruffles at the dorsal cell surface (Figure 2D,E; Video S1), suggesting that EGF‐dependent activation of Rac1 occurred transiently and locally in SKBR‐3 cells. In contrast, cells depleted of CUL3 or KCTD10 did not show an increase in FRET signals after EGF stimulation (Figure 2D,E, Videos S2 and S3). These results suggest that Rac1 could not be activated with EGF in cells depleted of CUL3 or KCTD10.
It has been suggested that overexpression of RhoB inhibited Rac1 activation at the plasma membrane by suppressing endosome‐to‐plasma membrane traffic of Rac1 in HUVEC.10 Therefore, we examined subcellular localization of Rac1 in SKBR‐3 cells. In control cells, GFP‐Rac1 mostly localized in the plasma membrane of dorsal cell surface ruffles at 15 minutes after EGF stimulation (Figure 2F). In contrast, in cells depleted of CUL3 or KCTD10, plasma membrane localization of GFP‐Rac1 was not observed; instead, it was found to be associated with intracellular vesicles. We observed partial colocalization of GFP‐Rac1 with endogenous RhoB (Figure 2F,G; arrows). These results suggest that CUL3 and KCTD10 are required for translocation of Rac1 from endosomes to the plasma membrane in SKBR‐3 cells.
3.3. Cullin‐3/KCTD10 regulates plasma membrane dynamics of SKBR‐3 cells
Given that knockdown of CUL3 or KCTD10 in SKBR‐3 cells mostly abolished the activation of Rac1, as indicated by FRET analysis (Figure 2C), we reasoned that cellular events regulated by Rac1 would also be affected by knockdown of CUL3 or KCTD10. Because SKBR‐3 cells formed dorsal membrane ruffles in response to EGF (Figure 2D) and the formation of dorsal membrane ruffles requires Rac1 activation,25, 26 we examined whether knockdown of CUL3 or KCTD10 affects EGF‐induced membrane ruffle formation at the dorsal cell surface.
We first examined the formation of membrane ruffles by phase‐contrast live‐cell imaging. In control cells, a number of membrane ruffles, which can be detected as phase‐dark objects in phase‐contrast microscopy, originated from the cell periphery after EGF stimulation and moved along the dorsal plasma membrane (Figure 3A, arrowheads; Video S4). In contrast, in cells depleted of CUL3 or KCTD10, fewer membrane ruffles were formed by EGF stimulation (Figure 3A; Videos S5 and S6). Interestingly, instead of generating dorsal membrane ruffles, cells formed lamellipodia, sheet‐like thin membrane protrusions at the periphery of cells (Figure 3A, arrows). The cell area of CUL3‐ or KCTD10‐knockdown cells was approximately twice as large as that of the control cells after EGF stimulation (Figure 3B).
Figure 3.
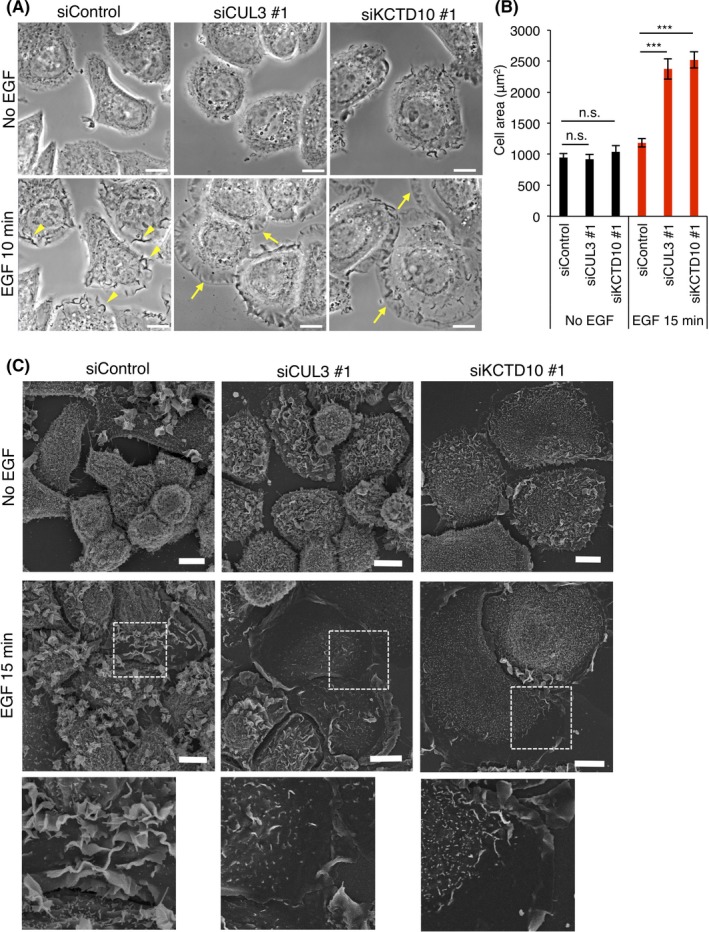
Plasma membrane dynamics of cullin‐3 (CUL3)‐ or KCTD10‐depleted SKBR‐3 cells. A, Phase‐contrast images, before EGF stimulation (No EGF) and 10 min after EGF stimulation, of SKBR‐3 cells treated with the indicated siRNAs. Bars, 10 μm. Representative dorsal membrane ruffles and lamellipodia are indicated by yellow arrowheads and arrows, respectively. See also Videos S4, S5 and S6. B, Areas of SKBR‐3 cells treated with the indicated siRNAs, with or without epidermal growth factor (EGF) stimulation. Fifty cells from three independent experiments were analyzed. Data are mean ± SEM. ***P < .001. n.s., not significant. C, Scanning electron micrographs of SKBR‐3 cells depleted of CUL3 or KCTD10 with or without EGF stimulation. Magnifications of dashed squared areas are shown in the lower panels. Bars, 10 μm
We next examined the fine architecture of the plasma membrane by SEM imaging.27 Without EGF stimulation, we did not find a significant difference among control cells and cells depleted of CUL3 or KCTD10; they had microvillus‐like protrusions and small membrane ruffles on the cell surface (Figure 3C). After EGF stimulation, control cells generated a number of dorsal membrane ruffles (Figure 3C). In contrast, cells depleted of CUL3 or KCTD10 did not generate dorsal membrane ruffles (Figure 3C). These cells became flat with a smooth cell surface, consistent with the generation of lamellipodia, as indicated by phase‐contrast microscopy (Figure 3A).
3.4. Aberrant F‐actin organization by CUL3‐ or KCTD10‐depletion in SKBR‐3 cells is Rac1‐dependent
As membrane ruffle formation was accompanied by actin polymerization beneath the dorsal plasma membrane,28 we expected that F‐actin organization would be different among EGF‐stimulated control, CUL3‐, or KCTD10‐knockdown cells. Cells were stimulated with EGF for 15 minutes, fixed, stained with fluorophore‐labeled phalloidin that binds to F‐actin, and imaged at the bottom and top by confocal microscopy (Figure 4A). In control cells, a number of F‐actin‐rich structures (Figure 4B, yellow arrowheads) were observed at the top section, corresponding to membrane ruffles of the dorsal cell surface. In contrast, such cell surface structures were hardly observed in CUL3‐ or KCTD10‐knockdown cells (Figure 4B).
Figure 4.
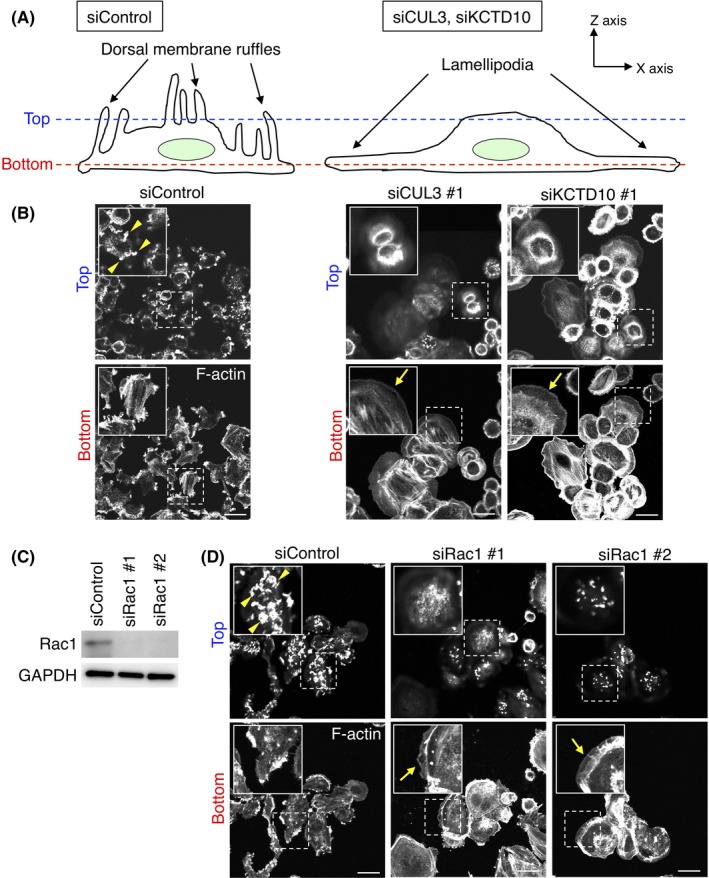
Visualization of F‐actin in cullin‐3 (CUL3)‐, KCTD10‐ or Rac1‐depleted SKBR‐3 cells. A, Schematic diagrams of control and CUL3‐ or KCTD10‐depleted cells. Confocal images in (B, D) were obtained at the top or bottom sections of cells. B, F‐actin staining by phalloidin in CUL3‐ or KCTD10‐depleted SKBR‐3 cells 15 min after epidermal growth factor (EGF) stimulation. Confocal images at the top and bottom sections of cells are shown. Inset: magnifications of the area indicated by a dashed square. Representative F‐actin‐positive dorsal membrane ruffles and lamellipodia are indicated by yellow arrowheads and arrows, respectively. Bars, 20 μm. C, Western blots of SKBR‐3 cell lysates, 72 h post‐transfection with siRNAs. D, F‐actin staining by phalloidin in Rac1‐depleted SKBR‐3 cells 15 min after EGF stimulation. Confocal images at the top and bottom sections of cells are shown. Inset: magnifications of the area indicated by a dashed square. Representative F‐actin‐positive dorsal membrane ruffles and lamellipodia are indicated by yellow arrowheads and arrows, respectively. Bars, 20 μm
At the bottom section of control cells, accumulation of F‐actin was observed at the tip of the extensions of the plasma membrane (Figure 4B). In contrast, at the bottom section of CUL3‐ or KCTD10‐knockdown cells, continuous cortical actin was observed along the edge of the plasma membrane (Figure 4B, yellow arrows). Fan‐shaped protrusions were often observed, indicating the formation of lamellipodia in these cells. In summary, these results showed that CUL3‐ or KCTD10‐knockdown SKBR‐3 cells had aberrant F‐actin organization after EGF stimulation.
As Rac1 is required for the formation not only of membrane ruffle but also of lamellipodia in some cell lines,29, 30 we examined Rac1‐dependency of the formation of membrane ruffle and lamellipodia in SKBR‐3 cells (Figure 4C,D). SKBR‐3 cells were treated with siRNA oligos designed for Rac1, and knockdown efficiency was confirmed by western blot (Figure 4C). In Rac1‐depleted cells, fewer dorsal membrane ruffles were observed at the top section compared to control cells (Figure 4D, yellow arrowheads), and lamellipodia formation was still observed by EGF stimulation (Figure 4D, yellow arrows), as seen in CUL3‐ or KCTD10‐knockdown cells (Figure 4B). We also found that enforced expression of constitutive‐active Rac1, GFP‐Rac1 (CA), in CUL3‐ or KCTD10‐knockdown cells restored dorsal membrane ruffle formation at the top section (Figure S4A,B, yellow arrowheads) compared to CUL3‐ or KCTD10‐knockdown cells that did not express GFP‐Rac1 (CA) exogenously (Figure S4A,B, yellow arrows). Overexpression of GFP‐Rac1 (CA) in CUL3‐ or KCTD10‐knockdown cells did not induce lamellipodia formation at the bottom section (Figure S4A,B). Together, these results suggest that Rac1 is essential for the formation not of lamellipodia but of dorsal membrane ruffle by EGF stimulation in SKBR‐3 cells, and that aberrant membrane dynamics by CUL3‐ or KCTD10‐knockdown is due to inability to activate Rac1 by EGF stimulation.
3.5. Cullin‐3/KCTD10 regulates F‐actin organization in a RhoB‐dependent manner
As shown (Figure 2A), we noticed that RhoB expression drastically increased in SKBR‐3 cells depleted of CUL3 or KCTD10. As RhoB inhibited Rac1 activation in HUVEC,10 the increased expression of RhoB by knockdown of CUL3 or KCTD10 in SKBR‐3 cells may contribute to the inactivation of Rac1, which results in the aberrant F‐actin organization. To test this, we further depleted RhoB in CUL3‐ or KCTD10‐depleted cells (Figure 5A). As indicated by F‐actin staining, RhoB knockdown restored EGF‐induced membrane ruffle formation at the dorsal surface of CUL3‐ or KCTD10‐depleted cells (Figure 5B; yellow arrowheads). RhoB knockdown also restored the accumulation of F‐actin at the tip of the lateral extensions of the cell periphery (Figure 5B; yellow arrows), as was seen in control cells. The cell area of CUL3‐ or KCTD10‐depleted cells was reduced by RhoB knockdown compared to that of control cells (Figure 5C). In addition, enforced expression of RhoB exogenously in SKBR‐3 cells inhibited the dorsal membrane ruffle formation (Figure 5D; yellow arrowheads). In contrast, SKBR‐3 cells that did not overexpress RhoB normally formed membrane ruffles at the dorsal cell surface (Figure 5D; yellow arrows). These results suggest that aberrant F‐actin organization caused by CUL3 or KCTD10 knockdown is at least, in part, due to the accumulation of RhoB.
Figure 5.
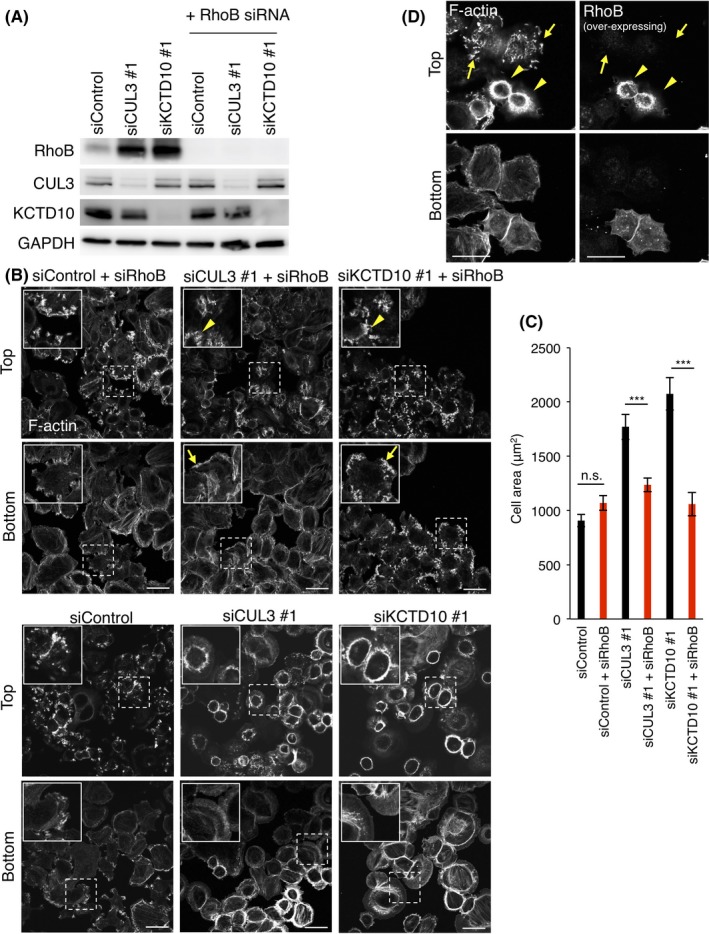
Accumulation of RhoB by cullin‐3 (CUL3) or KCTD10 knockdown inhibits dorsal membrane ruffle formation. A, Western blots of SKBR‐3 cells lysates 72 h post‐transfection of the indicated siRNAs. B, F‐actin staining by phalloidin in SKBR‐3 cells treated with the indicated siRNAs. Confocal images 15 min after epidermal growth factor (EGF) stimulation at the top and bottom sections of cells are shown. Inset: magnifications of the area indicated by a dashed square. By RhoB knockdown, dorsal membrane ruffle formation (yellow arrowheads) and the accumulation of F‐actin at the tip of membrane extensions of the cell periphery (yellow arrows) were restored at the top and bottom sections, respectively, of CUL3 or KCTD10‐depleted cells. Bars, 20 μm. Note that aberrant F‐actin morphology was observed in almost all SKBR‐3 cells treated with siRNAs targeting CUL3 or KCTD10, indicating that siRNAs were successfully transfected in almost all SKBR‐3 cells. Therefore, RhoB siRNA was also transfected in almost all CUL3‐ or KCTD10‐knockdown cells. See also the blot for RhoB protein in Figure 5A. C, Areas of SKBR‐3 cells treated with the indicated siRNAs with EGF stimulation for 15 min. Fifty cells from three independent experiments were analyzed. Data are mean ± SEM. ***P < .001. n.s., not significant. D, F‐actin staining by phalloidin in RhoB‐overexpressing SKBR‐3 cells 15 min after EGF stimulation. Confocal images at the top and bottom sections of cells are shown. Note that exogenously expressing RhoB was visualized using a rabbit anti‐RhoB polyclonal antibody (sc‐180; Santa Cruz Biotechnology, Dallas, TX, USA), which can recognize not endogenous but overexpressing RhoB in immunostaining. Representative cells overexpressing RhoB exogenously are indicated by yellow arrowheads, and non‐transfected cells are indicated by yellow arrows. Bars, 20 μm
3.6. Human epidermal growth factor receptor 2 signaling is required for generation of dorsal membrane ruffles and Rac1 activation
We examined whether HER2 signaling contributes to membrane ruffle formation in SKBR‐3 cells. HER2 is transactivated by EGF‐activated EGFR.31, 32 In control cells, after EGF stimulation, HER2 colocalized well with F‐actin, indicating the presence of HER2 at the dorsal membrane ruffles (Figure 6A). In contrast, as is the case with cells depleted of CUL3 or KCTD10 (Figure 4), cells depleted of HER2 did not show F‐actin concentration at the dorsal side of the plasma membrane (Figure 6A). Instead, as indicated by F‐actin staining, flat lamellipodia extended approximately all over the cell periphery (Figure 6A). The same results were essentially obtained by treating the cells with Herceptin (Figure 6B), an inhibitory antibody that targets the extracellular domain of HER2, or with lapatinib or gefitinib (Figure S5A,B), HER2 or EGFR specific tyrosine kinase inhibitors, respectively. These results suggest that the kinase activity of HER2 is required for dorsal membrane ruffle formation in response to EGF stimulation. We also monitored EGF‐induced generation of Rac1‐GTP in HER2‐knockdown or Herceptin‐treated SKBR‐3 cells using a Rac1‐FRET biosensor (Figure 6C,D). FRET signals by EGF stimulation were attenuated by knockdown of HER2 or treatment with Herceptin, compared to control cells (Figure 6C,D, Videos S7‐S10). These results suggest that HER2 is essential for EGF‐induced Rac1 activation in SKBR‐3 cells.
Figure 6.
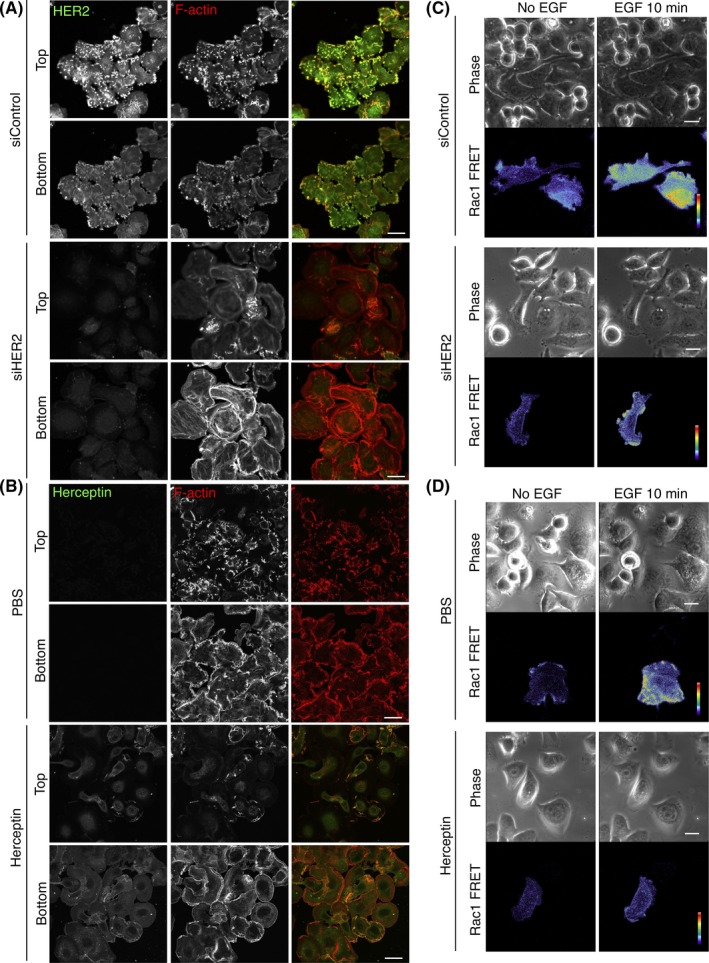
Human epidermal growth factor receptor 2 (HER2) is essential for dorsal membrane ruffle formation and Rac1 activation. A, HER2 and F‐actin were stained by Herceptin as a primary antibody and phalloidin, respectively, in SKBR‐3 cells treated with the indicated siRNAs. Confocal images 15 min after epidermal growth factor (EGF) stimulation at the top and bottom sections of cells are shown. Bars, 20 μm. B, F‐actin staining by phalloidin in SKBR‐3 cells treated with Herceptin (1 mg/mL, for 24 h). Confocal images 15 min after EGF stimulation at the top and bottom sections of cells are shown. Herceptin was visualized with Alexa488‐conjugated anti‐human IgG antibody. Bars, 20 μm. C,D, Serum‐starved SKBR‐3 cells expressing RaichuEV‐Rac1, a FRET biosensor for Rac1 activity, were stimulated with EGF. Phase‐contrast images and fluorescence images of FRET and cyan fluorescent protein (CFP) of SKBR‐3 cells treated with control siRNA (C, upper panels), HER2 siRNA (C, lower panels), PBS (D, upper panels) or Herceptin (D, lower panels) were acquired every 15 s. Ratio images, showing Rac1 FRET efficiency, were created by image arithmetic of the FRET/CFP ratio using MetaMorph software. Rac1 FRET efficiency showing Rac1 activity levels before and 10 min after EGF stimulation are shown as pseudo‐color spectral images. Scale bars, 10 μm. See also Videos S7, S8, S9 and S10
We then examined the relationship between HER2 signaling and CUL3/KCTD10‐mediated RhoB degradation pathway. As shown (Figure S6A), knockdown of HER2 or lapatinib treatment did not affect the protein levels of CUL3, KCTD10, or RhoB in SKBR‐3 cells (Figure S6A). EGF stimulation did not affect the expression levels of these proteins (Figure S6A). Intracellular localization of RhoB was not affected by HER2 knockdown (Figure S6B). Both EGFR and HER2 localized on the plasma membrane in CUL3 or KCTD10‐depleted cells, as seen in control cells (Figure S6C). As HER2 depletion did not affect the amount of RhoB, the results suggest that HER2 signaling has a distinct site of action from CUL3/KCTD10.
3.7. Cullin‐3/KCTD10 is required for cell proliferation of SKBR‐3 cells
Finally, as Rac1 activation is also known to regulate cell growth11 and we found that Rac1 activation requires constitutive degradation of RhoB by CUL3/KCTD10 ubiquitin E3 complex, we hypothesized that CUL3 and KCTD10 regulate cell proliferation of SKBR‐3 cells. As expected, cell proliferation of SKBR‐3 cells in the presence of EGF was drastically suppressed by CUL3‐ or KCTD10‐knockdown, as seen in Rac1 or HER2 knockdown cells (Figure 7A). Together with Rac1‐FRET imaging shown in Figure 6C,D, HER2 signaling may function in the translocation step of Rac1‐loaded intracellular vesicles to the plasma membrane and/or the activation step of Rac1 at the plasma membrane for the generation of dorsal membrane ruffles and signal transduction for cell proliferation (Figure 7B).
Figure 7.
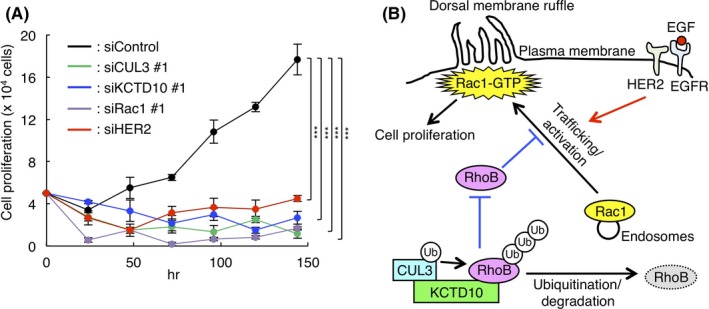
Cullin‐3 (CUL3) and KCTD10 regulate cell proliferation in SKBR‐3 cells. A, SKBR‐3 cells were treated with indicated siRNA for CUL3, KCTD10, human epidermal growth factor receptor 2 (HER2) or Rac1 for 48 h. Trypsinized cells (total 0.5 × 105 cells) were then replated and treated with the same siRNA 24 h after replating in the presence of epidermal growth factor (EGF; 100 ng/mL). Cell number was counted every 24 h after replating. Data are mean ± SEM from three independent experiments. ***P < .001. B, Scheme of the present study. CUL3/KCTD10 E3 complex constitutively ubiquitinates RhoB leading to its degradation. Low level of RhoB enables activation of Rac1 by stimulation through the EGF/EGFR/HER2 signaling pathway in HER2‐positive breast cancer cells. In CUL3 or KCTD10 knockdown cells, accumulated RhoB impairs Rac1 trafficking from endosomes to the plasma membrane, resulting in the inhibition of Rac1‐dependent dorsal membrane ruffle formation and cell proliferation
4. DISCUSSION
Human epidermal growth factor receptor 2 is a member of the ErbB receptor tyrosine kinase family. Although HER2 has no specific ligands, HER2 forms heterodimers with other ErbB receptors (EGFR, HER3, and HER4) and contributes to various cellular signaling pathways.33, 34, 35, 36, 37 HER2‐positive BC progress more aggressively than other subtypes of BC, and gene amplification and overexpression of HER2 in BC are correlated with poor prognosis.38 Mechanistically, overexpression of HER2 induces the activation of signaling pathways for cell proliferation (e.g. MAPK pathway and PI3K/Akt pathway) and upregulates gene expressions that are critical for EMT.35, 36, 37, 39, 40 Recently, overexpression of HER2 was shown to deform the plasma membrane in a kinase activity‐independent way, leading to disruption of normal epithelial morphology,41 which argues an interesting possibility that the deformation contributes to the EMT of BC with high expression of HER2. In the present study, we found that HER2 is essential for the generation of dorsal membrane ruffles in SKBR‐3 cells (Figure 6). Dorsal membrane ruffle is a form of plasma membrane, frequently found in a motile cell surface.42 Therefore, in addition to cell proliferation and EMT, HER2 may contribute to cell movement, which is relevant to metastatic potentials of BC. Of note, HER2 homodimers and HER2‐containing clusters are preferentially found in membrane ruffles in SKBR‐3 cells,43, 44 suggesting a positive feedback loop between HER2 and HER2 signaling on the membrane ruffles.
Although HER2‐targeted therapy, such as that with trastuzumab (an inhibitory humanized monoclonal antibody to HER2), is the first‐line and effective therapy towards HER2‐positive BC, development of trastuzumab resistance has been recognized. The majority of patients who showed an initial response to trastuzumab subsequently acquired resistance within 1 year.45, 46, 47 Approximately half of the patients treated with chemotherapy combined with trastuzumab acquired resistance to trastuzumab within approximately 7 months.48 Ex vivo study using SKBR‐3 cells suggested that Rac1 activation could be one of the molecular mechanisms underlying the development of trastuzumab resistance. Overexpression of CA Rac1 attenuated the efficacy of trastuzumab in SKBR‐3 cells.18 In contrast, treatment of SKBR‐3 cells with NSC23766, a selective inhibitor of Rac1 activation, reduced trastuzumab resistance.18, 19 In the present study, we found a novel CUL3‐mediated molecular mechanism to regulate Rac1 activation. As Rac1 is also known to regulate cell proliferation through activation of mammalian target of rapamycin (mTOR) and p38/MAPK pathways,49 and knockdown of CUL3 or KCTD10 suppressed cell proliferation in SKBR‐3 cells (Figure 7A), inhibition of the complex formation of CUL3/KCTD10 will be a new strategy to suppress Rac1 activation and may lead to new treatments for HER‐2 positive BC. Interestingly, mRNA expression of CUL3 and KCTD10 was not correlated with prognosis of HER2‐positive BC (Figure S7; P = .14, High: n = 39, Low: n = 181; P = .091, High: n = 51, Low: n = 169, respectively). Together with Rac1 transcriptome data in HER2‐positive BC (Figure 1B), CUL3/KCTD10 could constitutively activate Rac1 in HER2‐positive BC in the response of growth factors, and inhibition of CUL3/KCTD10 function might be more effective in Rac1high/HER2‐positive BC than in Rac1low/HER2‐positive BC.
It has been extensively studied as to how Rac1 is activated and regulates actin dynamics at the plasma membrane. Recently, the steady‐state distribution of Rac1 was analyzed in endothelial cells and the results suggested that Rac1 was mainly localized on intracellular vesicles.10 With various stimuli, Rac1‐loaded vesicles immediately translocated to the plasma membrane so that Rac1 functions at the plasma membrane.50 RhoB appears to be a negative regulator of this translocation. Overexpression of RhoB in endothelial cells suppressed the translocation of Rac1‐loaded vesicles to the plasma membrane, resulting in impaired barrier reformation.10 In the present study, we found that the protein level of RhoB was increased and that Rac1 remained intracellularly after EGF stimulation in SKBR‐3 cells depleted of CUL3 or KCTD10. Further knockdown of RhoB in these cells restored dorsal membrane ruffle formation, indicating that RhoB negatively regulates the translocation of Rac1 in BC cells. Together with the results in endothelial cells,10 these results suggest the general role of RhoB in the suppression of Rac1 translocation and activation. Further investigations to elucidate the molecular mechanisms as to how RhoB regulates Rac1 trafficking/activation are needed. In addition to manipulation of the protein levels of RhoB through the CUL3/KCTD10 complex, the manipulation of RhoB activity by RhoB GEF51 may be considered to cure HER2‐positive BC.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
We thank Ms Ayako Fujisaki, Ms Mami Chosei, Ms Kaori Kubo, Ms Misaki Hyodo, Drs Tomohisa Sakaue, Shinji Fukuda (Ehime University, Toon, Japan) for technical assistance, Drs Igor Kovačević (Sanquin Research, Amsterdam, The Netherlands), Peter L Hordijk (VU University Medical Center, Amsterdam, The Netherlands) for comments on this work. This work was supported by JSPS KAKENHI Grant Number JP17K16510 to AM and JSPS KAKENHI Grant Numbers JP16K19038, JP18K15244, SGH Cancer Research Grant 2017 to MM, JSPS KAKENHI Grant Numbers JP18K06831 to NA and JSPS KAKENHI Grant Numbers JP16H046980 to SH.
Murakami A, Maekawa M, Kawai K, et al. Cullin‐3/KCTD10 E3 complex is essential for Rac1 activation through RhoB degradation in human epidermal growth factor receptor 2‐positive breast cancer cells. Cancer Sci. 2019;110:650–661. 10.1111/cas.13899
Murakami and Maekawa contributed equally to this work.
Contributor Information
Masashi Maekawa, Email: masashim@m.ehime-u.ac.jp.
Shigeki Higashiyama, Email: shigeki@m.ehime-u.ac.jp.
REFERENCES
- 1. Petroski MD, Deshaies RJ. Function and regulation of cullin‐RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9‐20. [DOI] [PubMed] [Google Scholar]
- 2. Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3‐RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 2013;32:2307‐2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng J, Guo J, Wang Z, et al. Functional analysis of Cullin 3 E3 ligases in tumorigenesis. Biochem Biophys Acta. 2018;1869:11‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sakaue T, Sakakibara I, Uesugi T, et al. The CUL3‐SPOP‐DAXX axis is a novel regulator of VEGFR2 expression in vascular endothelial cells. Sci Rep. 2017;7:42845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maekawa M, Tanigawa K, Sakaue T, et al. Cullin‐3 and its adaptor protein ANKFY1 determine the surface level of integrin beta1 in endothelial cells. Biol Open. 2017;6:1707‐1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kovacevic I, Sakaue T, Majolee J, et al. The Cullin‐3‐Rbx1‐KCTD10 complex controls endothelial barrier function via K63 ubiquitination of RhoB. J Cell Biol. 2018;217:1015‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ohnuki H, Inoue H, Takemori N, et al. BAZF, a novel component of cullin3‐based E3 ligase complex, mediates VEGFR and Notch cross‐signaling in angiogenesis. Blood. 2012;119:2688‐2698. [DOI] [PubMed] [Google Scholar]
- 9. Sakaue T, Fujisaki A, Nakayama H, et al. Neddylated Cullin 3 is required for vascular endothelial‐cadherin‐mediated endothelial barrier function. Cancer Sci. 2017;108:208‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marcos‐Ramiro B, Garcia‐Weber D, Barroso S, et al. RhoB controls endothelial barrier recovery by inhibiting Rac1 trafficking to the cell border. J Cell Biol. 2016;213:385‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kazanietz MG, Caloca MJ. The Rac GTPase in cancer: from old concepts to new paradigms. Can Res. 2017;77:5445‐5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Olson MF. Rho GTPases, their post‐translational modifications, disease‐associated mutations and pharmacological inhibitors. Small GTPases. 2018;9:203‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thomas EK, Cancelas JA, Chae HD, et al. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR‐ABL‐induced myeloproliferative disease. Cancer Cell. 2007;12:467‐478. [DOI] [PubMed] [Google Scholar]
- 14. Sosa MS, Lopez‐Haber C, Yang C, et al. Identification of the Rac‐GEF P‐Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell. 2010;40:877‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lopez‐Haber C, Barrio‐Real L, Casado‐Medrano V, Kazanietz MG. Heregulin/ErbB3 signaling enhances CXCR4‐driven Rac1 activation and breast cancer cell motility via hypoxia‐inducible factor 1alpha. Mol Cell Biol. 2016;36:2011‐2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Endris V, Wogatzky B, Leimer U, et al. The novel Rho‐GTPase activating gene MEGAP/srGAP3 has a putative role in severe mental retardation. Proc Natl Acad Sci USA. 2002;99:11754‐11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lahoz A, Hall A. A tumor suppressor role for srGAP3 in mammary epithelial cells. Oncogene. 2013;32:4854‐4860. [DOI] [PubMed] [Google Scholar]
- 18. Dokmanovic M, Hirsch DS, Shen Y, Wu WJ. Rac1 contributes to trastuzumab resistance of breast cancer cells: Rac1 as a potential therapeutic target for the treatment of trastuzumab‐resistant breast cancer. Mol Cancer Ther. 2009;8:1557‐1569. [DOI] [PubMed] [Google Scholar]
- 19. Zhao Y, Wang Z, Jiang Y, Yang C. Inactivation of Rac1 reduces Trastuzumab resistance in PTEN deficient and insulin‐like growth factor I receptor overexpressing human breast cancer SKBR3 cells. Cancer Lett. 2011;313:54‐63. [DOI] [PubMed] [Google Scholar]
- 20. Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pereira B, Chin SF, Rueda OM, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komatsu N, Aoki K, Yamada M, et al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22:4647‐4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ikeda Y, Kawai K, Ikawa A, Kawamoto K, Egami Y, Araki N. Rac1 switching at the right time and location is essential for Fcgamma receptor‐mediated phagosome formation. J Cell Sci. 2017;130:2530‐2540. [DOI] [PubMed] [Google Scholar]
- 25. Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP‐binding protein rac regulates growth factor‐induced membrane ruffling. Cell. 1992;70:401‐410. [DOI] [PubMed] [Google Scholar]
- 26. Koivusalo M, Welch C, Hayashi H, et al. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol. 2010;188:547‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maekawa M, Terasaka S, Mochizuki Y, et al. Sequential breakdown of 3‐phosphorylated phosphoinositides is essential for the completion of macropinocytosis. Proc Natl Acad Sci USA. 2014;111:E978‐E987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol. 2008;9:639‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kurokawa K, Itoh RE, Yoshizaki H, Nakamura YO, Matsuda M. Coactivation of Rac1 and Cdc42 at lamellipodia and membrane ruffles induced by epidermal growth factor. Mol Biol Cell. 2004;15:1003‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kato T, Kawai K, Egami Y, Kakehi Y, Araki N. Rac1‐dependent lamellipodial motility in prostate cancer PC‐3 cells revealed by optogenetic control of Rac1 activity. PLoS One. 2014;9:e97749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kokai Y, Myers JN, Wada T, et al. Synergistic interaction of p185c‐neu and the EGF receptor leads to transformation of rodent fibroblasts. Cell. 1989;58:287‐292. [DOI] [PubMed] [Google Scholar]
- 32. Stern DF, Kamps MP. EGF‐stimulated tyrosine phosphorylation of p185neu: a potential model for receptor interactions. EMBO J. 1988;7:995‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER‐2/neu proto‐oncogene in human breast and ovarian cancer. Science. 1989;244:707‐712. [DOI] [PubMed] [Google Scholar]
- 34. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127‐137. [DOI] [PubMed] [Google Scholar]
- 35. Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469‐6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neve RM, Sutterluty H, Pullen N, et al. Effects of oncogenic ErbB2 on G1 cell cycle regulators in breast tumour cells. Oncogene. 2000;19:1647‐1656. [DOI] [PubMed] [Google Scholar]
- 37. Lenferink AE, Busse D, Flanagan WM, Yakes FM, Arteaga CL. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Can Res. 2001;61:6583‐6591. [PubMed] [Google Scholar]
- 38. Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology. 2001;61(Suppl 2):1‐13. [DOI] [PubMed] [Google Scholar]
- 39. Johnson E, Seachrist DD, DeLeon‐Rodriguez CM, et al. HER2/ErbB2‐induced breast cancer cell migration and invasion require p120 catenin activation of Rac1 and Cdc42. J Biol Chem. 2010;285:29491‐29501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Giordano A, Gao H, Anfossi S, et al. Epithelial‐mesenchymal transition and stem cell markers in patients with HER2‐positive metastatic breast cancer. Mol Cancer Ther. 2012;11:2526‐2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chung I, Reichelt M, Shao L, et al. High cell‐surface density of HER2 deforms cell membranes. Nat Commun. 2016;7:12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Borm B, Requardt RP, Herzog V, Kirfel G. Membrane ruffles in cell migration: indicators of inefficient lamellipodia adhesion and compartments of actin filament reorganization. Exp Cell Res. 2005;302:83‐95. [DOI] [PubMed] [Google Scholar]
- 43. Peckys DB, Korf U, de Jonge N. Local variations of HER2 dimerization in breast cancer cells discovered by correlative fluorescence and liquid electron microscopy. Sci Adv. 2015;1:e1500165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hommelgaard AM, Lerdrup M, van Deurs B. Association with membrane protrusions makes ErbB2 an internalization‐resistant receptor. Mol Biol Cell. 2004;15:1557‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Esteva FJ, Valero V, Booser D, et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER‐2‐overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:1800‐1808. [DOI] [PubMed] [Google Scholar]
- 46. Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2‐targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269‐280. [DOI] [PubMed] [Google Scholar]
- 47. Dokmanovic M, Shen Y, Bonacci TM, Hirsch DS, Wu WJ. Trastuzumab regulates IGFBP‐2 and IGFBP‐3 to mediate growth inhibition: implications for the development of predictive biomarkers for trastuzumab resistance. Mol Cancer Ther. 2011;10:917‐928. [DOI] [PubMed] [Google Scholar]
- 48. Slamon DJ, Leyland‐Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783‐792. [DOI] [PubMed] [Google Scholar]
- 49. Bid HK, Roberts RD, Manchanda PK, Houghton PJ. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther. 2013;12:1925‐1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bouchet J, Del Rio‐Iniguez I, Lasserre R, et al. Rac1‐Rab11‐FIP3 regulatory hub coordinates vesicle traffic with actin remodeling and T‐cell activation. EMBO J. 2016;35:1160‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kamon H, Kawabe T, Kitamura H, et al. TRIF‐GEFH1‐RhoB pathway is involved in MHCII expression on dendritic cells that is critical for CD4 T‐cell activation. EMBO J. 2006;25:4108‐4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials