

Abstract
Hepatocellular carcinoma (HCC) is a common and aggressive malignant tumor with a poorly defined molecular mechanism. Cyclin‐dependent kinase 2 (CDK2) and Septin2 (SEPT2) are 2 known oncogenic molecules but the mechanism of functional interactions remains unclear. Here, we interestingly found that CDK2 and SEPT2 show very similar dynamic expression during the cell cycle. Both CDK2 and SEPT2 show the highest protein levels in the G2/M phase, resulting in CDK2 interacting with SEPT2 and stabilizing SEPT2 in HCC. In a panel of 8 pairs of fresh HCC tissues and corresponding adjacent tissues, both western blot and immunohistochemistry (IHC) assays demonstrate that CDK2 expression is highly correlated with SEPT2. HCC with high expression of both CDK2 and SEPT2 are more likely to relapse. This observation is further demonstrated by a large panel of 100 HCC patients. In this large panel, high expression of both CDK2 and SEPT2 significantly correlates with tumor differentiation and microvascular invasion, which is an independent prognostic factor in HCC patients. In summary, our results reveal a cooperative function between CDK2 and SEPT2. HCC with high expression of CDK2 and SEPT2 might be more aggressive and respond poorly to current therapy.
Keywords: CDK2, cell cycle, hepatocellular carcinoma, interaction, SEPT2
1. INTRODUCTION
Primary liver cancer is a common malignant tumor. China is a high‐risk area for liver cancer, with approximately 55% of the liver cancer patients worldwide living in China.1 Hepatocellular carcinoma (HCC) is the main type of liver cancer, accounting for around 90% of liver cancer cases. Approximately 700 000 people die of liver cancer globally each year, and it is on the rise.2 Despite some researchers and clinicians having made significant efforts, we still know very little about the underlying pathogenesis of HCC.3 It is worth noting that the infinite cell proliferation caused by the imbalance of cell cycle progression is the dominant hallmark for various tumor cells.4 Therefore, many scholars have explored this phenomenon, seeking to reveal the underlying mechanisms of the infinite proliferation of HCC cells.5, 6 These studies indicate that the imbalance of liver cancer cells at the G2/M checkpoint is an important reason for the promotion of HCC.
The smooth transition of cell cycle checkpoints relies on the precise regulation of the cell cycle by regulatory factors. Cyclin‐dependent kinases (CDKs) are essential to the cell cycle regulatory network, which is mainly regulated by cyclin and other proteins.7 CDK2, an important member of the CDK family that regulates the G1/S and G2/M checkpoints, mainly changes the activity of the substrate by directly phosphorylating the serine/threonine residues on the substrate, resulting in the promotion of cell cycle progression.8 Abnormally activated CDK2 can simultaneously cause abnormalities in G1/S and G2/M checkpoints and play an important role in tumor cell hyperproliferation.9 At present, the regulation mechanism of CDK2 on G1/S checkpoint is very clear. However, the specific regulation mechanism of the G2/M checkpoint has not been fully clarified.8 Therefore, exploring the mechanism of CDK2 on the G2/M checkpoint is of great significance for the diagnosis and treatment of HCC.
Septin2 is one of the conserved filamentous guanosine triphosphatases (GTPases), and has different effects in cell cycles. In interphase cells, SEPT2 are associated with actin stress fibers. However, in dividing cells, SEPT2 are associated with the contractile ring.10 In addition, SEPT2 plays an important role in the formation of spindles and the separation of sister chromatids by regulating the polymerization and separation of tubulin, thereby regulating G2/M phase transition and cell mitosis.11 Therefore, these findings guide us to continuously explore the mechanism of CDK2 and SEPT2 in promoting the G2/M phase transition, which promotes the development of liver cancer.
2. EXPERIMENTAL PROCEDURES
2.1. Western blot and immunoprecipitation analyses
Cells were lysed in RIPA lysis buffer (50 mmol/L Tris pH 7.4, 120 mmol/L NaCl, 1% Triton X‐100, 1% sodium deoxycholate, .1% SDS, Beyotime, Shanghai, China) supplemented with protease inhibitors (Complete Mini, Roche, Basel, Switzerland) and phosphatase inhibitors (PhosSTOP, Roche). Equal amounts of protein were analyzed by western blot assays with indicated antibodies.
For immunoprecipitations analysis, 1000 μg of total cell lysates was incubated with the 1 μg primary antibody or control rabbit IgG at 4°C overnight; 20 μL of Protein A+G agarose (Bioworld Technology, St. Louis Park, MN, USA) was then added for 2 hours at 4°C with rocking. The precipitates were washed 4 times with RIPA washing buffer (50 mmol/L Tris pH 7.4, 150 mmol/L NaCl, 1% NP‐40, .5% sodium deoxycholate, .1% SDS, Beyotime) and boiled for 15 minutes with SDS sample buffer.
Primary antibodies: CDK2 (ab32147; Abcam, Cambridge, UK), SEPT2 (ab88657, ab179436; Abcam), Flag (ab002‐100; Multi Sciences, HangZhou, China), HA (ab003‐100; Multi Sciences), Phospho‐Threonine (#9381; CST, Boston, MA, USA), phosphoserine (ab9332; Abcam), cyclin A (sc‐239; Santa Cruz, Dallas, TX, USA), cyclin B1 (sc‐245, Santa Cruz), PCNA (sc‐56, Santa Cruz), p44/42 MAPK(Erk1/2) (#9102; CST), Phospho‐p44/42 MAPK (Erk1/2) (#4370; CST) and GAPDH (60004‐1‐Ig; Proteintech, Rosemont, IL, USA).
2.2. Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde in PBS for 1 hour at room temperature and then permeabilized with 1% Triton X‐100 in PBS for 20 minutes. The cells were blocked with 1% BSA in PBS for 1 hour and incubated with primary antibodies overnight at 4°C. Secondary antibody incubation was performed using Alexa Fluor 568 donkey anti‐mouse, or Alexa Fluor 488 donkey anti‐rabbit antibodies (Invitrogen, Carlsbad, CA, USA) for 1 hour at room temperature. In all, 5 mg/mL DAPI was added to stain the nucleus. The slides were mounted and visualized using a Nikon Confocal Microscope (Nikon, Melville, NY, USA).
2.3. Preparation of stable expression cell lines
SMMC‐7721 cells were transfected with the plasmids and then screened by 1 μg/mL of puromycin (ApexBio, Houston, TX, USA), and stable expression cell lines were obtained.
2.4. Cell synchronization
For the double thymidine block, 1 × 106 cells were seeded on 10‐cm dishes; 2 mmol/L thymidine (Sigma, Milwaukee, WI, USA) was added for 15 hours. Cells were washed with PBS and released into growth medium for 9 hours. Cells were cultured in 2 mmol/L thymidine again for 15 hours, washed, and released into growth medium.
2.5. Cell proliferation assays and colony formation assays
Next, 2 × 103 7721 cells transfected with vectors, Flag‐SEPT2, Sh‐SEPT2 or HA‐CDK2, were seeded into a 96‐well plate and cell vitality was assessed by Cell Counting Kit‐8 (Biotechwell, Shanghai, China). The cells were incubated with CCK‐8 at 37°C for 1 hour and after 48 hours absorbance was recorded at 450 nm; 7721 cells transfected with vectors, Flag‐SEPT2, Sh‐SEPT2 or HA‐CDK2, were seeded in a 6‐well plate at a density of 500 cells/well and incubated at 37°C and 5% CO2 for 2 weeks. The cells were washed 3 times with PBS, 4% paraformaldehyde fixed for 1 hour and stained with .5% crystal violet for 30 minutes.
2.6. Flow cytometry
Cells were fixed with 70% ethanol at −20°C overnight and washed 3 times using cold PBS. The samples were stained with PI/RNase Staining Buffer (BD Pharmingen, Franklin Lakes, NJ, USA) for 15 minutes at room temperature according to the manufacturer's instructions. Stained cells were analyzed with a BD FACSCanto II Flow Cytometer. The results were analyzed by ModFit LT 3.1.
2.7. Xenograft mouse model
Nude mice were obtained from Nantong University Laboratory Animal Center. SMMC‐7721 cell suspension in DMEM (5 × 106/mouse) was subcutaneously injected into male nude mice. Mice were killed at 10 weeks after implantation, and tumor xenografts were collected.
2.8. Patients and tissue samples and immunohistochemistry analyses
Hepatocellular carcinoma tissues were obtained from 100 patients with HCC who underwent hepatic surgical resection without preoperative systemic chemotherapy in the Affiliated Hospital of Nantong University (Nantong, China). Tissue samples were immediately processed after surgical removal. For histological examination, all tumor and surrounding non‐tumor tissue portions were fixed in formalin and embedded in paraffin. Informed consent was obtained from all patients.
The paraffin‐embedded tissue samples were heated in a tissue drying oven at 60°C for 45 minutes. After deparaffinization in xylene, they were rehydrated in graded alcohol. Then, these tissue sections were incubated in sodium citrate buffer and heated for antigen retrieval. To block endogenous peroxidase activity, we used .3% hydrogen peroxide (H2O2) in methanol at room temperature for 10 minutes. These tissue sections were incubated overnight at 4°C with a primary antibody, and then treated with a secondary antibody at room temperature for 30 minutes. Furthermore, these sections were stained and counterstained with diaminobenzidine and hematoxylin. To obtain immunohistochemical results, 2 blinded investigators evaluated the clinical data.
The evaluation criteria of immunohistochemistry staining were as follows: staining intensity was scored as 0 (negative), 1 (weak), 2 (medium) and 3 (strong). Extent of staining was scored as 0 (0%), 1 (1%‐25%), 2 (26%‐50%), 3 (51%‐75%) and 4 (76%‐100%) according to the percentages of the positive staining areas in relation to the whole carcinoma area. The sum of the intensity and extent score was used as the final staining score (0‐7) for CDK2 and SEPT2. Tumors with a final staining score of ≥3 were considered positive.
2.9. Statistical analyses
Cumulative survival time was calculated using the Kaplan‐Meier method, and differences were compared by log‐rank test. Univariate and multivariate analyses were based on the Cox proportional hazards regression model. The χ2 test, Fisher's exact probability and Student's t test were used for comparison between groups. Statistical significance was defined by a P‐value <.05. Statistical analyses were performed using the Statistical Program for Social Sciences v20 software (SPSS Inc., Chicago, IL, USA).
3. RESULTS
3.1. Correlated dynamic expression of cyclin‐dependent kinase 2 and Septin2 during cell cycle
Studies have suggested that NEDD5 (SEPT2) is not only expressed in all brain tumors but also changes with the cell cycle progression, reaching a peak in the G2/M phase.12 However, It is well known that different CDK play distinct roles in different tumor cell cycles.13, 14 For the first time, we discovered through a serum starvation experiment that CDK2 was consistent with the trend of SEPT2 from the early G1 phase (Figure 1A). Subsequently, the expression of CDK2 and SEPT2 in different cell cycles was analyzed using the double thymidine synchronization method. We found that protein abundance of SEPT2 and CDK2 fluctuated during cell cycles in HCC cell lines, peaking in G2/M phase, followed by a sharp reduction in early G1 phase (Figure 1B).
Figure 1.
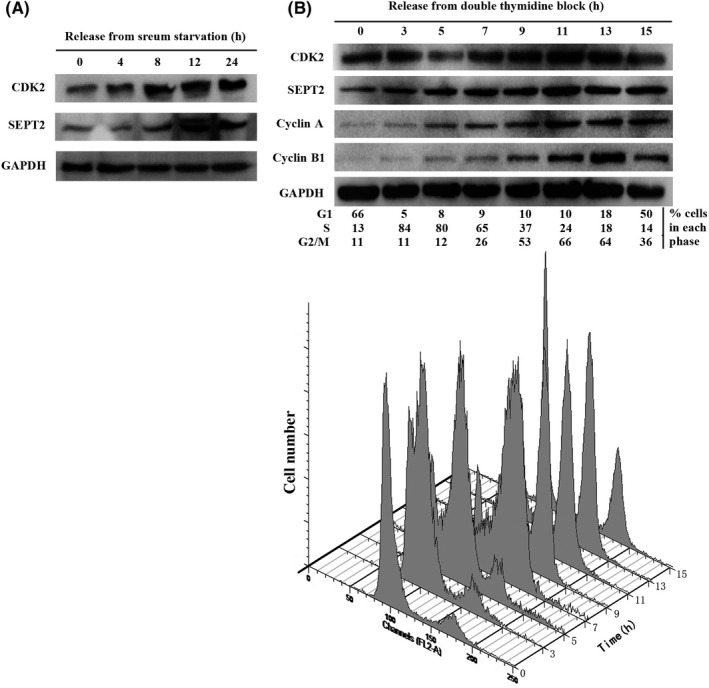
The protein abundance of Septin2 (SEPT2) and cyclin‐dependent kinase 2 (CDK2) during cell cycle progression. A, The protein abundance of SEPT2 and CDK2 released from serum starvation in SMMC‐7721 cells. SMMC‐7721 cells were serum‐starved for 72 h, and released with DMEM medium containing 10% FBS for the indicated period of time. The cells were harvested and analyzed for SEPT2 and CDK2 protein abundance by western blot. B, Western blot analysis of whole cell lysates derived from SMMC‐7721 cells synchronized in late G1/S phase by double thymidine followed by release back into the cell cycle. The cell‐cycle profiles in (B) were monitored by fluorescence‐activated cell sorting
3.2. Cyclin‐dependent kinase 2 activates Erk pathway by stabilizing Septin2
Cyclin‐dependent kinases play crucial roles in regulating the stability of cell cycle‐related proteins during cell cycle progression.15, 16 We constructed corresponding knockdown stable cell lines (Figure 2A). Knockdown CDK2 in SMMC‐7721 decreased the expression of SEPT2 (Figure 2B). In previous research, SEPT2 was shown to promote tumor development by activating the Erk signaling pathway in breast cancer and glioblastoma.17, 18 Therefore, we explored whether CDK2 activated the Erk signaling pathway by stabilizing SEPT2. Consistent with our conjecture, the results showed that the declined expression of SEPT2 after knockdown of CDK2 was accompanied by the decrease of p‐Erk protein expression without affecting the total Erk expression (Figure 2B). The above results indicate that CDK2 can stabilize SEPT2, thereby promoting the development of HCC by activating the Erk signaling pathway.
Figure 2.
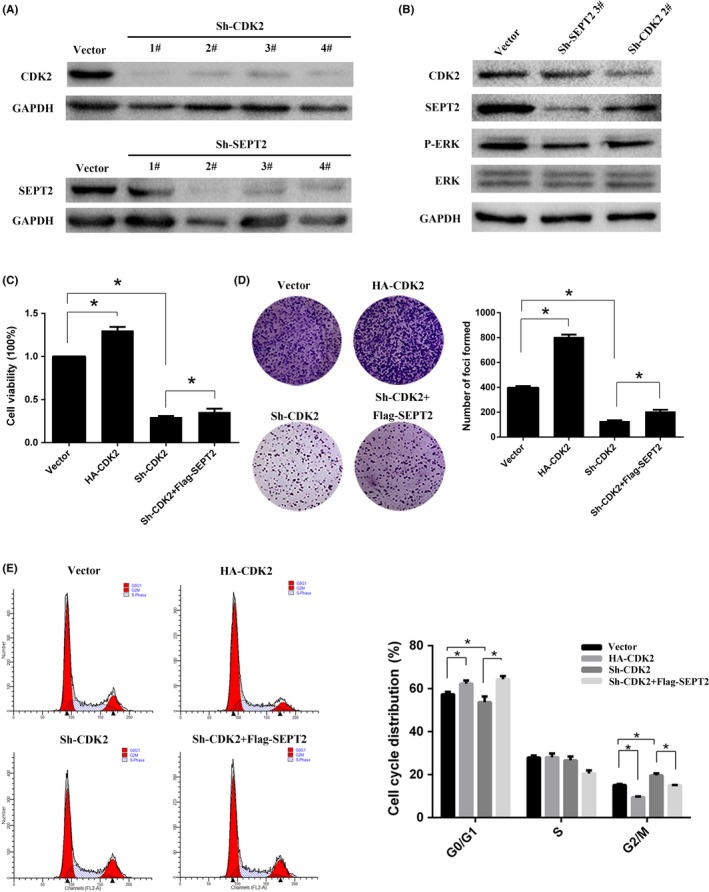
Cyclin‐dependent kinase 2 (CDK2) promotes hepatocellular carcinoma (HCC) cell proliferation by stabilizing Septin2 (SEPT2). A, SMMC‐7721 cells were transfected with 4 different Sh‐CDK2 or Sh‐SEPT2. The lysates were separated with SDS‐PAGE and blotted with the anti‐CDK2 or anti‐SEPT2 antibodies. B, The cell were transfected with Sh‐CDK2 or Sh‐SEPT2 and directly lysed followed by western blot analyses. C, SMMC‐7721 cells were transfected with vector, Flag‐SEPT2, Sh‐SEPT2 or HA‐CDK2 to examine the proliferation of SMMC‐7721 cells by CCK‐8 assay. D, The cells were transfected and seeded into 96‐well plates for 2 wk. The numbers of foci were captured and counted in the bar chart. E, Flow cytometry was performed to determine the cell cycle distribution of the cells that were transfected. The cell cycle distribution is displayed in the bar chart. All values are the mean + SD of 3 independent experiments. *P < .05
3.3. Cyclin‐dependent kinase 2 promotes hepatocellular carcinoma cell proliferation by regulating Septin2
Previous studies have shown that SEPT2 can promote tumor progression, enhancing the cell invasion ability.17, 19 According to our study, cell viability was decreased after CDK2 knockdown, and then restored after overexpressing SEPT2 (Figure 2C). Similarly, knockdown of CDK2 inhibited the ability of HCC cell clone formation, which was restored after overexpressing SEPT2 (Figure 2D). After CDK2 knockdown, the proportion of G2/M phase cells significantly increased, while the proportion of G0/G1 phase cells decreased, indicating that more cells were arrested at the G2/M transition point and could not be successfully induced to cell division. On the basis of the original overexpression of SEPT2, the number of cells arrested in the G2/M phase was reduced (Figure 2E). In summary, CDK2 can promote the proliferation of HCC cells by regulating SEPT2, resulting in promoting the G2/M phase transition of HCC cells.
3.4. Cyclin‐dependent kinase 2 interacts with Septin2 without phosphorylation
Early studies showed that CDK2, belonging to a CDK that phosphorylates downstream substrates, promotes chromatin instability and tumor development.20 In the present study, we demonstrated an endogenous interaction between CDK2 and SEPT2 (Figure 3A, top) and other exogenous interactions (Figure 3A, bottom). Further immunofluorescence experiments also revealed that CDK2 and SEPT2 are colocalization in the cytoplasm (Figure 3B). Previously, our experiments also demonstrated that the protein abundance of CDK2 and SEPT2 fluctuated with the progression of the cell cycle and peaked in the G2/M phase. Under the same conditions, we further found that in the G2/M phase, the interaction intensity between the CDK2 and SEPT2 was also greater than that of the G1/S phase (Figure 3C). These results revealed the interaction of CDK2 and SEPT2 in SMMC‐7721 cells. Some scholars have demonstrated that CDK has significant kinase function by recognizing the T/S‐P‐x‐K/R residue on the substrate.21, 22 Interestingly, when we analyzed the amino acid sequence of SEPT2 we found no such residues. In experiments, we also confirmed that even if CDK2 is overexpressed, there are no phosphorylated threonine and serine residues of SEPT2 (Figure 3D).
Figure 3.
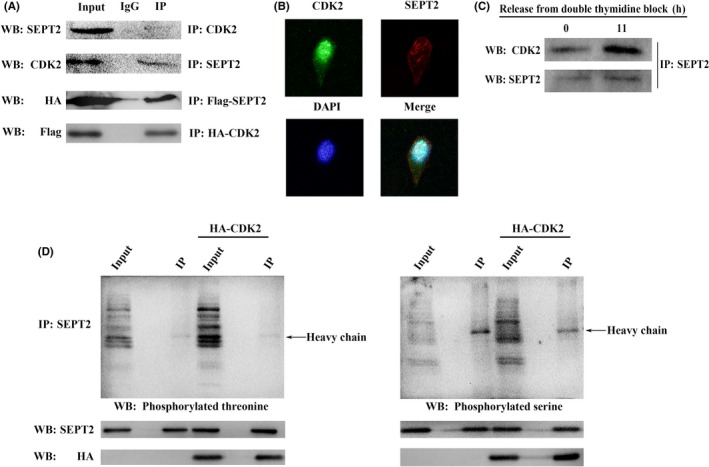
Cyclin‐dependent kinase 2 (CDK2) interacts with Septin2 (SEPT2) without phosphorylated threonine or serine. A, Lysates of SMMC‐7721 cells and HEK‐293T cells which co‐transfected with HA‐CDK2 and Flag‐SEPT2 were immunoprecipitated (IP) with anti‐CDK2, anti‐SEPT2, anti‐HA and anti‐flag antibodies or control IgG; western blot detected the immunoprecipitation with anti‐CDK2, anti‐SEPT2, anti‐HA and anti‐flag antibodies. B, Immunofluorescence analysis of CDK2 and SEPT2 in SMMC‐7721 cell using anti‐CDK2, and anti‐ SEPT2 antibodies. C, G1/S phase and G2/M phase of SMMC‐7721 cells were immunoprecipitated with anti‐ SEPT2 antibodies and then western blot analyses were performed with anti‐CDK2 and anti‐SEPT2 antibodies. D, SMMC‐7721 cells were transfected with HA‐CDK2 or not; total lysates were immunoprecipitated with anti‐SEPT2 antibodies and then western blot analyses were performed with anti‐SEPT2, anti‐HA, anti‐phosphoserine and anti‐phosphothreonine antibodies
3.5. Cyclin‐dependent kinase 2 promotes the tumorigenicity of HCC cells by regulating Septin2
Our data demonstrate that CDK2 could interact with SEPT2 and promote HCC cell proliferation. Next, we injected the corresponding stable cell line into nude mice and found that the tumorigenicity of HCC cells significantly decreased. CDK2 expression was very low as a result of CDK2 knockdown, and the tumor volume and quality were dramatically decreased. After re‐expressing SEPT2, the tumorigenicity increased, and the volume and quality also increased (Figure 4A‐C).
Figure 4.
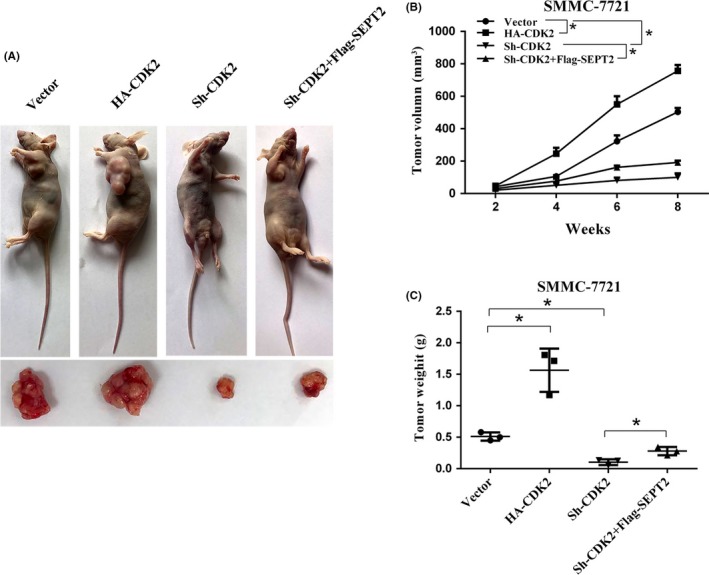
Cyclin‐dependent kinase 2 (CDK2) and Septin2 (SEPT2) promote tumor growth in vivo. A, Xenograft tumors were generated by subcutaneously injecting SMMC‐7721 cells overexpressing CDK2 or SEPT2, knocking down CDK2 or carrying a control vector. B, The growth of xenograft tumors was measured by volume. C, The xenograft tumor weight was recorded. All *P < .05
3.6. Cyclin‐dependent kinase 2 and Septin2 co‐express in hepatocellular carcinoma and promote recurrence
Next, we further explored protein abundance of CDK2 and SEPT2 in HCC tissues. We selected 8 pairs of fresh HCC tissues and corresponding adjacent tissues. It is worth noting that protein abundance of CDK2 and SEPT2 in tumor tissues was significantly higher than that of corresponding adjacent tissues (Figure 5A), which was consistent with IHC staining results (Figure 5B). We subsequently collected tumors and adjacent tissues from 100 patients with HCC, and statistically analyzed the pathological sections after IHC staining. Further analysis of these cases revealed that the time to recurrence of the CDK2/SEPT2 double positive group was significantly higher than that of others (Figure 5C).
Figure 5.
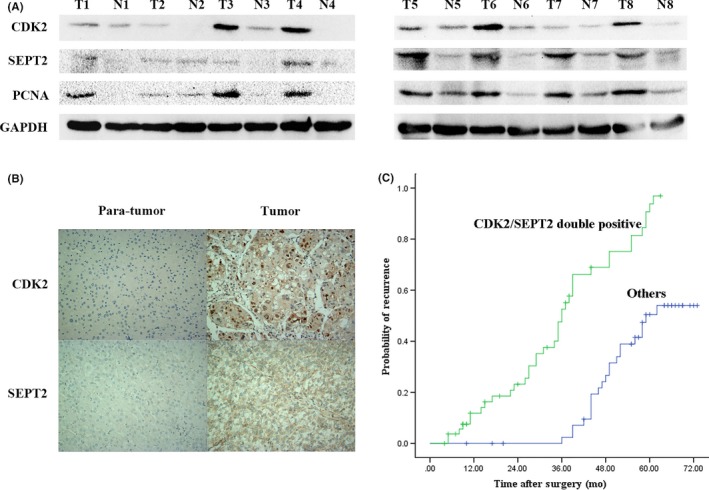
Correlation of cyclin‐dependent kinase 2 (CDK2) and Septin2 (SEPT2) expression in hepatocellular carcinoma (HCC) specimens. A, The representative expression of CDK2, SEPT2 and PCNA in 8 paired HCC tumor tissues (T) and adjacent normal tissues (N). B, Immunohistochemical analyses of CDK2 and SEPT2 in HCC tissues. C, Probability of recurrence of CDK2/SEPT2 double positive patients compared with CDK2 and/or SEPT2 negative patients (others)
3.7. High expression of cyclin‐dependent kinase 2 and Septin2 is an independent prognostic factor
We explored the correlation of CDK2/SEPT2 and clinical pathological parameters. The data, as shown as in Table 1, demonstrated that the expression of CDK2/SEPT2 was significantly associated with histological grade (P < .001) and microvascular invasion (P = .013). The results of univariate analysis and multivariate analysis, which are summarized in Table 2, showed that HBV‐DNA, tumor size, number of nodules, microvascular invasion and a patient being CDK2/SEPT2 double positive were prognostic factors of recurrence; among them, HBV‐DNA and a patient being CDK2/SEPT2 double positive were also considered to be independent prognostic factors for recurrence.
Table 1.
Associations between CDK2 and Sept2 expression with clinicopathological parameters
| Parameters | n | CDK2 and Sept2 expression | P | |
|---|---|---|---|---|
| Others | Double positive | |||
| Sex | 0.415 | |||
| Male | 82 | 36 | 46 | |
| Female | 18 | 9 | 9 | |
| Age, years | 0.106 | |||
| ≤50 | 48 | 18 | 30 | |
| >50 | 52 | 27 | 25 | |
| AFP(ng/ml) | 0.558 | |||
| <400 | 67 | 30 | 37 | |
| ≥400 | 33 | 15 | 18 | |
| Tumor size, cm | 0.182 | |||
| ≤5 | 45 | 23 | 22 | |
| >5 | 55 | 22 | 33 | |
| Tumor differentiation | <0.001 | |||
| II‐I | 71 | 40 | 31 | |
| III | 29 | 5 | 24 | |
| Microvascular invasion | 0.013 | |||
| Yes | 25 | 6 | 19 | |
| No | 75 | 39 | 36 | |
| Okuda stage | 0.094 | |||
| I | 93 | 44 | 49 | |
| II | 7 | 1 | 6 | |
| Child–Pugh score | 0.612 | |||
| A | 93 | 42 | 51 | |
| B | 7 | 3 | 4 | |
| Tumor number | 0.379 | |||
| Single | 82 | 38 | 44 | |
| Multiple | 18 | 7 | 11 | |
| HBV‐DNA, cps/m | 0.499 | |||
| ≤1000 | 70 | 31 | 39 | |
| >1000 | 30 | 14 | 16 | |
| Liver cirrhosis | 0.575 | |||
| Yes | 78 | 35 | 43 | |
| No Serum albumin, g/L 35.5(21.3‐50.6) Total bilirubin, μmol/L 18.4(6.6‐32.8) Prothrombin time, s 13.4(10.2‐15.6) | 22 | 10 | 12 | |
Table 2.
Univariate and multivariate analyses of factors associated with recurrence
| Variables | TTR | |
|---|---|---|
| HR (95%CI) | P | |
| Univariate analysis | ||
| Sex(male vs female) | 0.766 (0.363‐1.616) | 0.484 |
| Age, years (>50 vs ≤50) | 0.908 (0.544‐1.517) | 0.713 |
| HBV‐DNA, cps/ml (>1000 vs ≤1000) | 1.755 (1.034‐2.980) | 0.037 |
| Child‐Pugh score(B vs A) | 1.727 (0.688‐4.334) | 0.245 |
| Liver cirrhosis (yes vs no) | 1.389 (0.736‐2.622) | 0.311 |
| AFP, ng/ml (≥400 vs <400) | 1.180 (0.683‐2.039) | 0.552 |
| Serum albumin, g/L | 0.985 (0.933‐1.040) | 0.590 |
| Total bilirubin, μmol/L | 1.014 (0.973‐1.057) | 0.509 |
| Prothrombin time, s | 0.901 (0.703‐1.155) | 0.410 |
| Tumor differentiation(III vs II‐I) | 1.343 (0.709‐2.546) | 0.366 |
| Tumor size, cm (>5 vs ≤5) | 1.909 (1.118‐3.257) | 0.018 |
| Tumor number (multiple vs single) | 1.891 (1.016‐3.519) | 0.044 |
| Okuda stage (II vs I) | 1.442 (0.619‐3.355) | 0.396 |
| Microvascular invasion (yes vs no) | 2.116 (1.182‐3.788) | 0.012 |
| CDK2/Sept2 double positive (yes vs no) | 4.296 (2.480‐7.433) | <0.001 |
| Multivariate analysis | ||
| HBV‐DNA, cps/ml (>1000 vs ≤1000) | 2.130 (1.203‐3.773) | 0.010 |
| Tumor number (multiple vs single) | 2.143 (1.100‐4.173) | 0.025 |
| CDK2/Sept2 double positive (yes vs no) | 3.837 (2.181‐6.750) | <0.001 |
4. DISCUSSION
Clinical treatment of HCC is still based on traditional methods, such as surgical resection and chemotherapy. Although these methods can improve the prognosis of patients, the effects are unsatisfactory. In addition, multi‐kinase inhibitors (sorafenib and regorafenib) and immunology checkpoint inhibitors (nivolumab and lipilimumab) are available, and the latter can dramatically improve clinical outcomes. Therefore, it is of great significance to explore better adjuvant treatment strategies for improving the prognosis of patients with HCC.23, 24
Cell cycle disorder is a major feature of cancer cells, resulting in uncontrolled proliferation. CDK are a family of proteins involved in cell cycle regulation, which are frequently expressed or mutated in tumors. Hence, exploring the role of CDK as well as their related functional proteins in tumorigenesis is of great significance for the development of molecular inhibitors for tumor therapy.25 There have been many studies on CDK inhibitors, but most of them remain in the clinical trial stages.26 Exploring the mechanisms and targets is especially critical for developing new CDK inhibitors.
Previous studies have suggested that the Septin family is associated with a variety of human tumors.27 Studies have also analyzed the expression of the Septin gene in 35 human tumors, and found that septin 2, 8, 9 and 11 are significantly upregulated, while Septin4 and 10 are the opposite. In summary, SEPT2 may be a cancer‐promoting gene; however, knowledge of the mechanism is still limited.28 It is noteworthy that NEDD5 (SEPT2) is not only expressed in all brain tumors but also changes with the cell cycle progression.12 In this study, we found that the protein abundance of CDK2 and SEPT2 fluctuated with the cell cycle progression consistently, and all reached a peak in the G2/M phase. It has been confirmed that the CDK2/cyclin E complex can phosphorylate p27 in G1 to cause ubiquitin‐mediated degradation of p27, resulting in G1/S phase transition.29, 30 However, the mechanism by which CDK2 regulates G2/M phase conversion is not well understood. In this study, we attempted to clarify that CDK2 promotes G2/M detection point conversion by stabilizing SEPT2.
Our study demonstrated that CDK2 interacted with SEPT2 and activated the Erk signaling pathway, and the interaction between CDK2 and SEPT2 also peaked in G2/M phase. We also verified that the above mechanism was verified in vivo trials.
Finally, in fresh tumor tissues, western blot and IHC also demonstrated that CDK2 expression was highly correlated with SEPT2 and was significantly higher than that of paraneoplastic tissues. Data from 100 paraffin samples of HCC patients further supported the above results and revealed that high expression of CDK2 and SEPT2 was closely related to tumor differentiation and microvascular invasion, and could be an independent prognostic factor in HCC patient.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
Xu C, Zhang W, Zhang X, et al. Coupling function of cyclin‐dependent kinase 2 and Septin2 in the promotion of hepatocellular carcinoma. Cancer Sci. 2019;110:540–549. 10.1111/cas.13882
Chenzhou Xu and Wei Zhang are contributed equally to this work.
Contributor Information
Wenkai Ni, Email: niwenkai@sina.com.
Cuihua Lu, Email: lch670608@sina.com.
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69‐90. [DOI] [PubMed] [Google Scholar]
- 3. Finn RS. Development of molecularly targeted therapies in hepatocellular carcinoma: where do we go now? Clin Cancer Res. 2010;16:390‐397. [DOI] [PubMed] [Google Scholar]
- 4. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153‐166. [DOI] [PubMed] [Google Scholar]
- 5. Ehrlich SM, Liebl J, Ardelt MA, et al. Targeting cyclin dependent kinase 5 in hepatocellular carcinoma–A novel therapeutic approach. J Hepatol. 2015;63:102‐113. [DOI] [PubMed] [Google Scholar]
- 6. Pellegrino R, Calvisi DF, Ladu S, et al. Oncogenic and tumor suppressive roles of polo‐like kinases in human hepatocellular carcinoma. Hepatology. 2010;51:857‐868. [DOI] [PubMed] [Google Scholar]
- 7. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079‐3093. [DOI] [PubMed] [Google Scholar]
- 8. Chung JH, Bunz F. Cdk2 is required for p53‐independent G2/M checkpoint control. PLoS Genet. 2010;6:e1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paris J, Leplatois P, Nurse P. Study of the higher eukaryotic gene function CDK2 using fission yeast. J Cell Sci. 1994;107(Pt 3):615‐623. [DOI] [PubMed] [Google Scholar]
- 10. Joo E, Surka MC, Trimble WS. Mammalian SEPT2 is required for scaffolding nonmuscle myosin II and its kinases. Dev Cell. 2007;13:677‐690. [DOI] [PubMed] [Google Scholar]
- 11. Spiliotis ET, Kinoshita M, Nelson WJ. A mitotic septin scaffold required for mammalian chromosome congression and segregation. Science. 2005;307:1781‐1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sakai K, Kurimoto M, Tsugu A, Hubbard SL, Trimble WS, Rutka JT. Expression of Nedd5, a mammalian septin, in human brain tumors. J Neurooncol. 2002;57:169‐177. [DOI] [PubMed] [Google Scholar]
- 13. Liao P, Zeng SX, Zhou X, et al. Mutant p53 gains its function via c‐Myc activation upon CDK4 phosphorylation at serine 249 and consequent PIN1 binding. Mol Cell. 2017;68:1134‐1146.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dozier C, Mazzolini L, Cenac C, et al. CyclinD‐CDK4/6 complexes phosphorylate CDC25A and regulate its stability. Oncogene. 2017;36:3781‐3788. [DOI] [PubMed] [Google Scholar]
- 15. Malumbres M, Barbacid M. Mammalian cyclin‐dependent kinases. Trends Biochem Sci. 2005;30:630‐641. [DOI] [PubMed] [Google Scholar]
- 16. Hydbring P, Malumbres M, Sicinski P. Non‐canonical functions of cell cycle cyclins and cyclin‐dependent kinases. Nat Rev Mol Cell Biol. 2016;17:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang N, Liu L, Fan N, et al. The requirement of SEPT2 and SEPT7 for migration and invasion in human breast cancer via MEK/ERK activation. Oncotarget. 2016;7:61587‐61600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu D, Liu A, Wang X, et al. Repression of Septin9 and Septin2 suppresses tumor growth of human glioblastoma cells. Cell Death Dis. 2018;9:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu J, Zhang W, Tang H, et al. Septin 2 accelerates the progression of biliary tract cancer and is negatively regulated by mir‐140‐5p. Gene. 2016;589:20‐26. [DOI] [PubMed] [Google Scholar]
- 20. Takada M, Zhang W, Suzuki A, et al. FBW7 loss promotes chromosomal instability and tumorigenesis via cyclin E1/CDK2‐mediated phosphorylation of CENP‐A. Cancer Res. 2017;77:4881‐4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aregger M, Kaskar A, Varshney D, et al. CDK1‐cyclin B1 activates RNMT, coordinating mRNA cap methylation with G1 phase transcription. Mol Cell. 2016;61:734‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dinkel H, Chica C, Via A, et al. Phospho.ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res. 2010;39:D261‐D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893‐2917. [DOI] [PubMed] [Google Scholar]
- 24. Hlady RA, Robertson KD. A three‐pronged epigenetic approach to the treatment of hepatocellular carcinoma. Hepatology. 2018;68:1226‐1228. [DOI] [PubMed] [Google Scholar]
- 25. Cicenas J, Valius M. The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol. 2011;137:1409‐1418. [DOI] [PubMed] [Google Scholar]
- 26. Vijayaraghavan S, Moulder S, Keyomarsi K, Layman RM. Inhibiting CDK in cancer therapy: current evidence and future directions. Target Oncol. 2018;13:21‐38. [DOI] [PubMed] [Google Scholar]
- 27. Connolly D, Abdesselam I, Verdier‐Pinard P, Montagna C. Septin roles in tumorigenesis. Biol Chem. 2011;392:725‐738. [DOI] [PubMed] [Google Scholar]
- 28. Liu M, Shen S, Chen F, Yu W, Yu L. Linking the septin expression with carcinogenesis. Mol Biol Rep. 2010;37:3601‐3608. [DOI] [PubMed] [Google Scholar]
- 29. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev. 1999;13:1501‐1512. [DOI] [PubMed] [Google Scholar]
- 30. He G, Kuang J, Huang Z, et al. Upregulation of p27 and its inhibition of CDK2/cyclin E activity following DNA damage by a novel platinum agent are dependent on the expression of p21. Br J Cancer. 2006;95:1514‐1524. [DOI] [PMC free article] [PubMed] [Google Scholar]