

Abstract
In the revised World Health Organization classification 2016, anaplastic pleomorphic xanthoastrocytoma (PXA) has been newly defined as a variant of the PXA entity. Furthermore, some anaplastic PXA were reported to have extremely poor prognosis which showed a type of pediatric glioblastoma (GBM) molecular profile. Recent integrated molecular classification for primary central nervous system tumors proposed some differences between histological and molecular features. Herein, in a genome‐wide molecular analysis, we show an extreme aggressive anaplastic PXA that resulted in a pediatric GBM molecular profile. A full implementation of the molecular approach is the key to predict prognosis and decide the treatment strategy for anaplastic PXA.
Keywords: anaplastic PXA, brain tumor, CpG island, DNA methylation, pediatric glioblastoma
Abbreviations
- Bev
bevacizumab
- CPT‐11
irinotecan
- GBM
glioblastoma
- GEO
Gene Enrichment Omnibus
- HGG
high‐grade glioma
- IDH
isocitrate dehydrogenase
- MI
mitotic index
- PXA
pleomorphic xanthoastrocytoma
- TMZ
temozolomide
- tSNE
t statistic‐based stochastic neighbor embedding
- WHO
World Health Organization
1. INTRODUCTION
Pleomorphic xanthoastrocytoma is a primary brain tumor categorized as WHO grade 2 with a predilection for the temporal lobe in pediatric and young adult patients.1 PXA have generally favorable prognoses with a 10‐year survival probability of over 70%.2 However, recent reports showed diversity of histological and clinical characteristics of PXA, and rapid recurrence and aggressive clinical behavior in subgroups of PXA. Anaplastic PXA is defined as a PXA variant categorized as WHO grade 3 with malignant pathological findings, such as increased mitosis and necrosis, according to the revised WHO classification of tumors of the central nervous system (CNS).1 In addition to the conventional pathological diagnostic strategy, genome‐wide gene aberration or DNA methylation analyses can profile the molecular characteristics of these tumors in detail. Some anaplastic PXA progress much faster than other anaplastic PXA.3 Through methylation profiling, Alexandrescu et al3 suggested the possibility that a subset of anaplastic PXA is closely related to epithelioid GBM which is an IDH‐wild‐type rare aggressive variant predominantly occurring in children and young adults. Recent integrated molecular analyses have shown that pediatric CNS tumors can be reclassified using genome‐wide DNA methylation profiling.4, 5 Herein, we conducted a genome‐wide DNA methylation analysis of a patient to identify anaplastic PXA with a set of PXA and pediatric GBM.
2. CASE REPORT
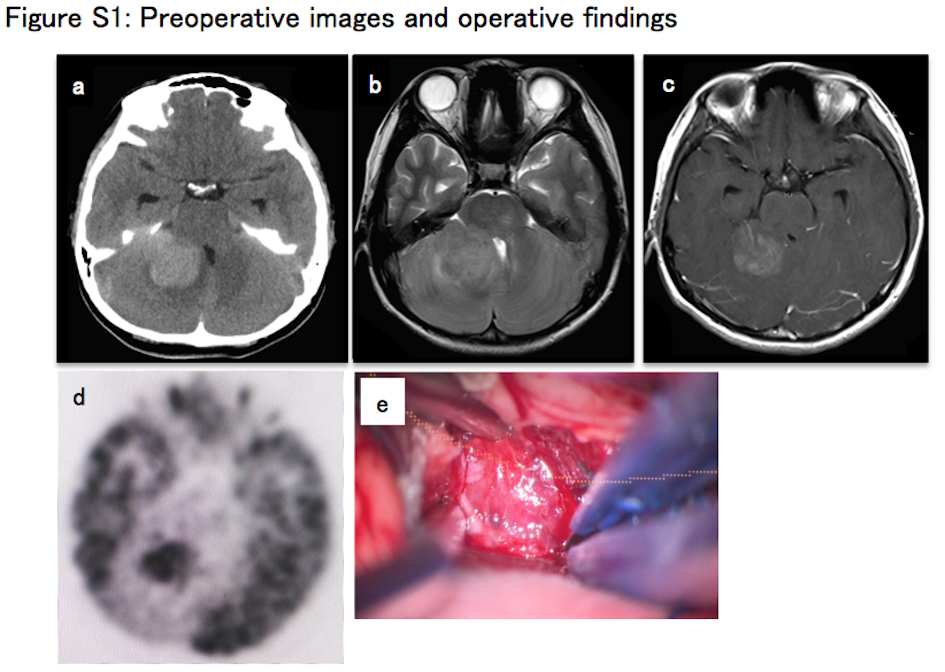
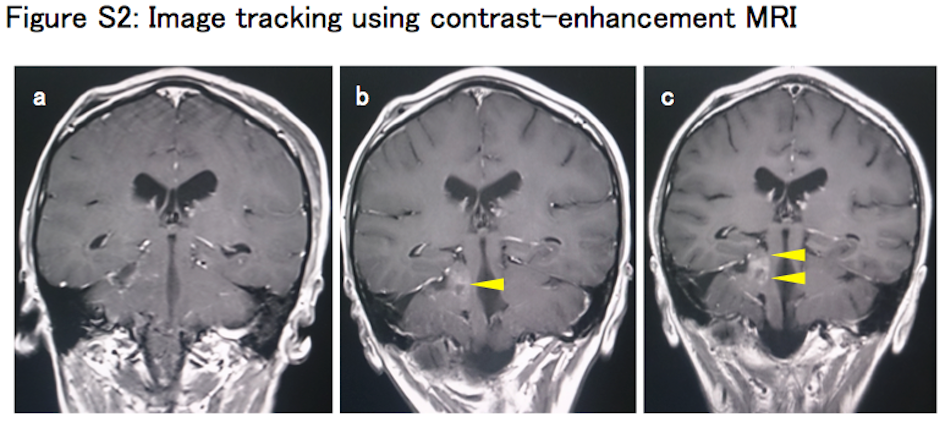
Detailed information of the patient(')s clinical course, pathological diagnosis, and analytic methods is described in Data S1. Briefly, a 14‐year‐old girl developed a brain tumor in the right cerebellar hemisphere. The tumor was surgically resected and histopathologically diagnosed as anaplastic PXA, WHO grade 3 (Data S1 [Case presentation], Figure S1A‐E). After surgical resection, combination therapy of chemotherapy with temozolomide (TMZ) (75 mg/m2; 42 days) and extended focal radiotherapy (54.0 Gy/30 fractions) was given. The patient was then subjected to TMZ maintenance therapy (150‐200 mg/m2), orally given for 5 days in every q28 days. Follow‐up MRI showed extension of the contrast‐enhanced mass lesion. High uptake of 11C‐methionine‐PET was identified and indicated disease progression just after primary adjuvant therapy (Figure S2A‐C). Three cycles of triple combination chemotherapy of Bev, irinotecan (CPT‐11), and TMZ (Bev 10 mg/kg per day on day 1 and day 15, CPT‐11 150 mg/m2 per day on day 1 and day 15, TMZ 200 mg/m2 per day on day 1 to day 5 of q28 days) were given. Despite treatment, the patient died of the disease on day 282 after initial diagnosis. The study was approved by the Institutional Review Board of each center and all participants gave informed consent.
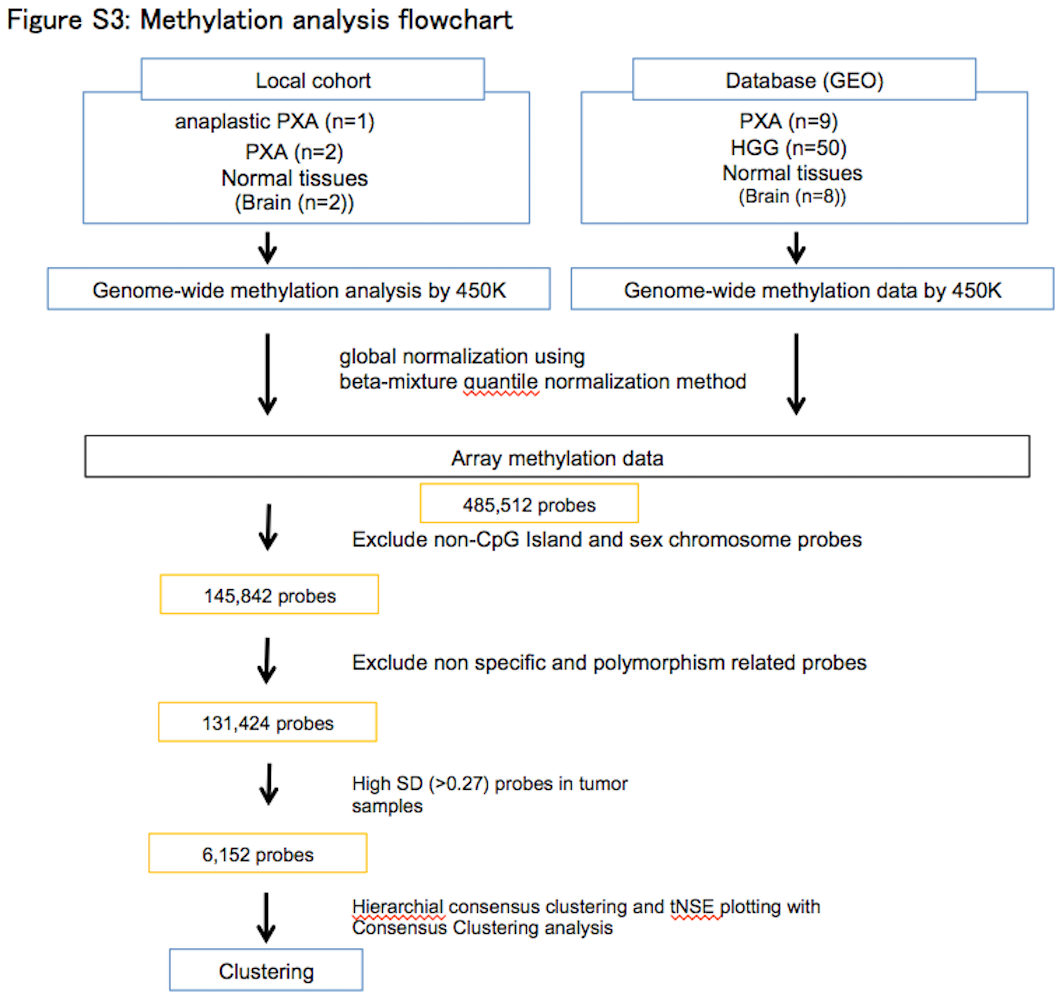
Surgical specimen of the patient (sample ID: anaplastic PXA01) was then extensively analyzed. Genomic DNA from the embedded specimen in Allprotect Tissue Reagent (Qiagen, Tokyo, Japan) was extracted using a DNeasy Blood & Tissue kit (Qiagen). DNA concentration was quantified using a fluorometer (Qubit 2.0; Thermo Scientific, Wilmington, DE, USA) and purity of the DNA was assessed using a spectrophotometer (NanoDrop 1000; Thermo Scientific) (Data S1 [DNA and RNA extraction, Annotating called variants]). Mutations in 93 genes were investigated through multiplex PCR analysis (Data S1 [Ion Torrent sequencing of multiplex PCR amplicons], Tables [Link], [Link]). This study was approved by the Institutional Review Boards of each center. Genomic DNA from the anaplastic PXA (anaplastic PXA01), two PXA (PXA10 and PXA11) were analyzed using an Infinium Human Methylation 450K BeadChip (Illumina, San Diego, CA, USA) (GPL13534) as described previously (Data S1 [Illumina Infinium 450K Methylation Array data generation]).6 DNA methylation and clinical data of nine pediatric PXA and 50 GBM generated by Sturm et al4 (GSE73801) were obtained from the GEO database for comparison (Table S3). We conducted a 450k methylation analysis for the anaplastic PXA (anaplastic PXA01), 11 PXA and 50 pediatric GBM through the pipeline (Figure S3, Data S1 [Statistical data processing for differential methylation]).
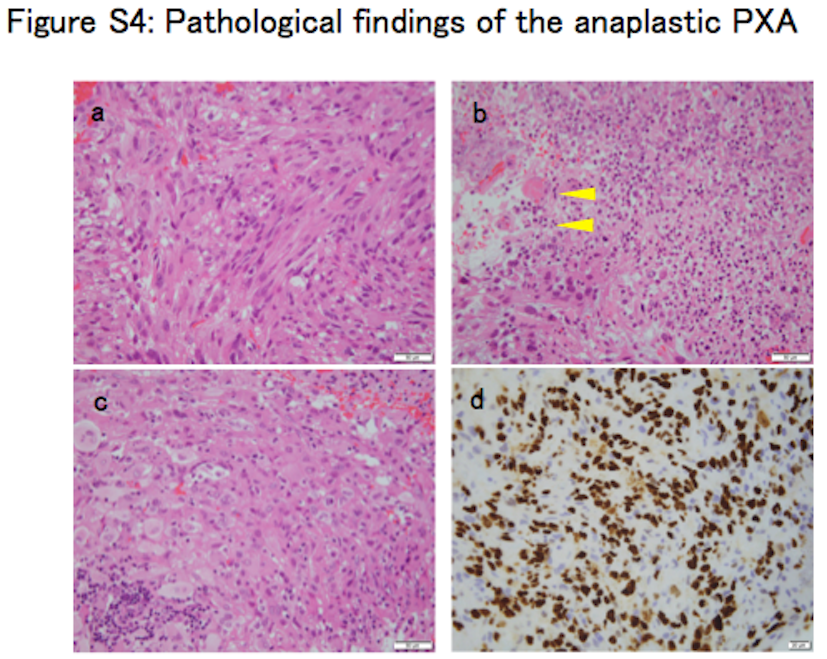
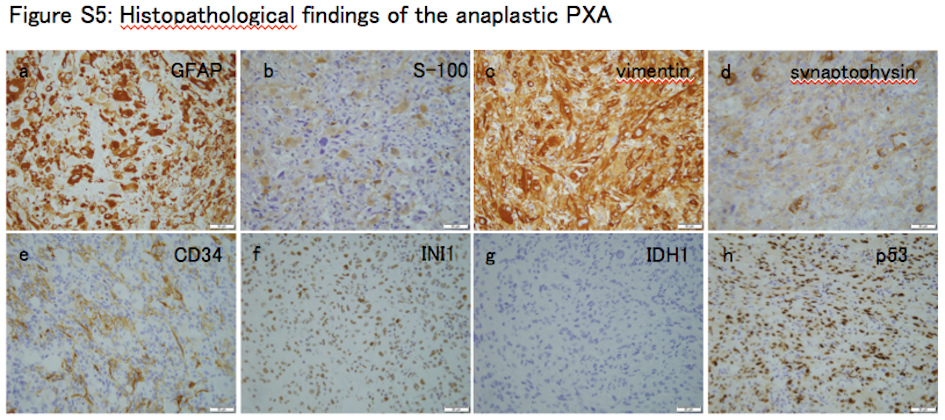
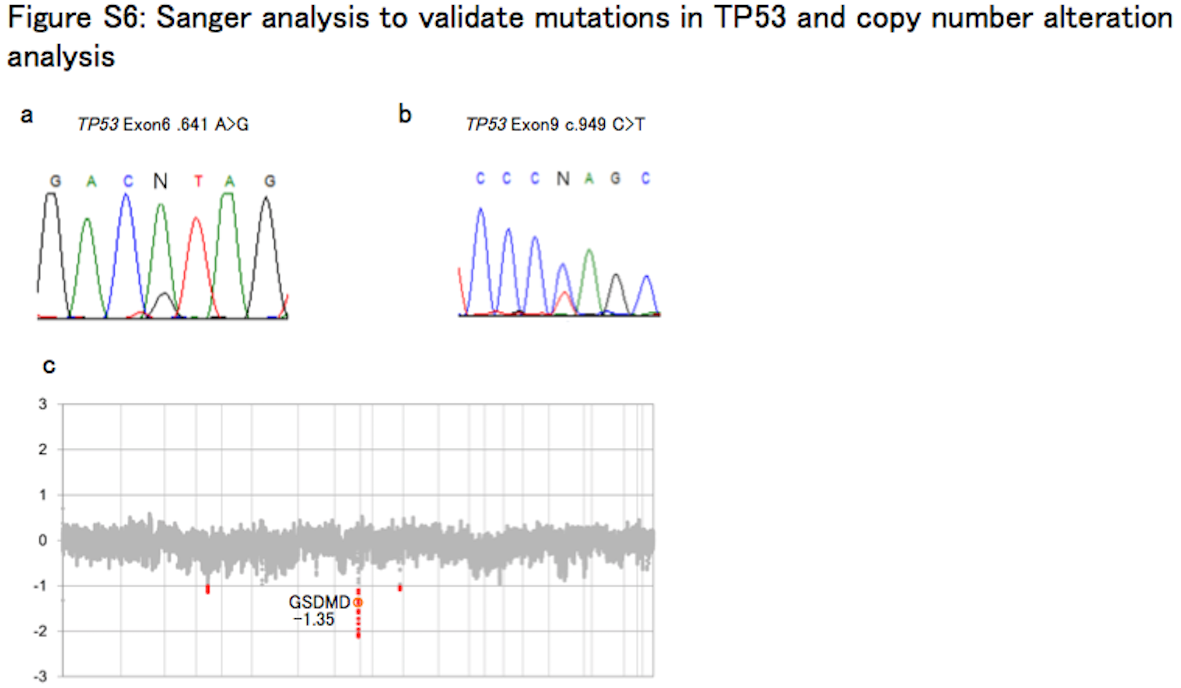
Tumor showed the characteristic histological features of anaplastic PXA upon H&E staining (Figure S4A‐C). There were pleomorphic spindle‐shaped cells with eosinophilic cytoplasm arranged in a bundle. Small cells with high nuclear cytoplasmic ratio were increased. Tumor cells also showed nuclear and cytoplasmic pleomorphism and xanthomatous change. Necrosis was often seen within the tumor, but microvascular proliferation and palisading necrosis were not observed. High mitotic activity was found with approximately 12 mitoses being observed per 10 high‐power fields. The Ki‐67 labeling index was determined to be 67.9%, which indicates high proliferative activity of the tumor. Immunohistochemical analysis showed positive staining for glial fibrillary acidic protein (GFAP), S‐100 protein, vimentin, INI1, CD34, synaptophysin, neurofilament protein and p53 (Figure S5A‐G). IDH1 R132H was negative (Figure S5H, Data S1 [Pathology]). Diagnosis of anaplastic PXA was thus confirmed after histopathological review. Two TP53 mutations (c.641A > G and c.949C > T) were identified by ion torrent targeted sequencing and were subsequently validated by Sanger sequencing (Figure S6A‐B, Table S4). There were no hotspot mutations in the IDH1 R132, IDH2 R172, TERT promoter lesions (C250 and C228), BRAF V600, H3F3A K27/G34, or FGFR1 N456/K656. Moreover, KIAA1549‐BRAF fusion was not detected by RT‐PCR (Data S1 [Pyrosequencing and Sanger sequencing]). A 16‐kb homozygous deletion at chromosome 8q24.3 involving the GSDMD gene was detected in the copy number analysis (Figure S6C, Data S1 [Generation of copy number profiles using 450K data]). DNA methylation analysis separated 62 cases of brain tumors, including the anaplastic PXA (anaplastic PXA01), 11 other PXA, and 50 pediatric GBM, into seven clusters; this was consistent with the previously described molecular classifiers, namely PXA and six HGG subgroups (MYC, RTK‐1, RTK‐2, K27M, G34R, and IDH) (Figure 1A).4, 5 Anaplastic PXA01 was clustered with HGG_MYC, whereas local cohort PXA (PXA10 and PXA11) were co‐clustered with the reference PXA. Methylation profile of the PXA‐type tumors was very similar to that of normal brain tissue, rather than that of the HGG subgroups. We also conducted a tSNE projection of the 450k methylation data. The analysis showed that anaplastic PXA01 was clustered into the HGG_MYC type, but not into the PXA nor the HGG_RTK‐1/‐2 types (Figure 1B). Based on the clustering and distributional projection analysis, we further focused on the tumors that belonged to the clusters PXA, HGG_MYC, or HGG_RTK‐1/‐2, and analyzed them for consensus clustering and unsupervised hierarchical clustering for the purpose of inner validation. In the consensus clustering, the tumor set was subdivided into three distinct subgroups, in which anaplastic PXA01 was included in the HGG_MYC type (Figure 1C‐D). We conducted additional analysis to generate top tables for a set of methylation probes (Table [Link], [Link]) and methylation volcano‐plotting (Figure S7A). It compared significant methylated CpG sites between a subgroup of PXA and that of HGG_RTK and HGG_MYC. Furthermore, we extracted four specific methylated probes referring gene CpG islands and generated a bee swarm boxplot with discovery anaplastic PXA (anaplastic PXA01) and PXA (PXA10 and PXA11) with validation analysis for methylation status for a CpG island site using pyrosequencing (Figure S7B‐C). A set of pyrosequencing assays to efficiently subclassify into PXA or HGG_RTK and HGG_MYC was developed. First, nine PXA and 20 HGG_RTK and HGG_MYC cases were investigated as a discovery set. Subsequently, two probes covering two genes, AUTS2 and PITX1, were identified. The probes that were highly methylated in HGG_RTK and HGG_MYC were also identified (IRX1 and TACSD2). A set of pyrosequencing assays were then designed to examine methylation statuses of the CpG sites targeted by those probes cg25714956 (AUTS2) and cg01294808 (IRX1) as well as their flanking CpG sites (primers and the dispensation orders are listed in Table S7). Methylation levels were measured using the CpG assay of PyroMark Q96, and mean methylation levels of all CpG sites included in each assay were used to represent the methylation status of each gene. The methylation levels at those CpG sites measured by pyrosequencing showed a certain concordance with that of the 450K array (Figure S7C). Our study suggested that anaplastic PXA may consist of a clinically and biologically heterogeneous group of tumors, and genome‐wide methylation profiling may help identify those.
Figure 1.
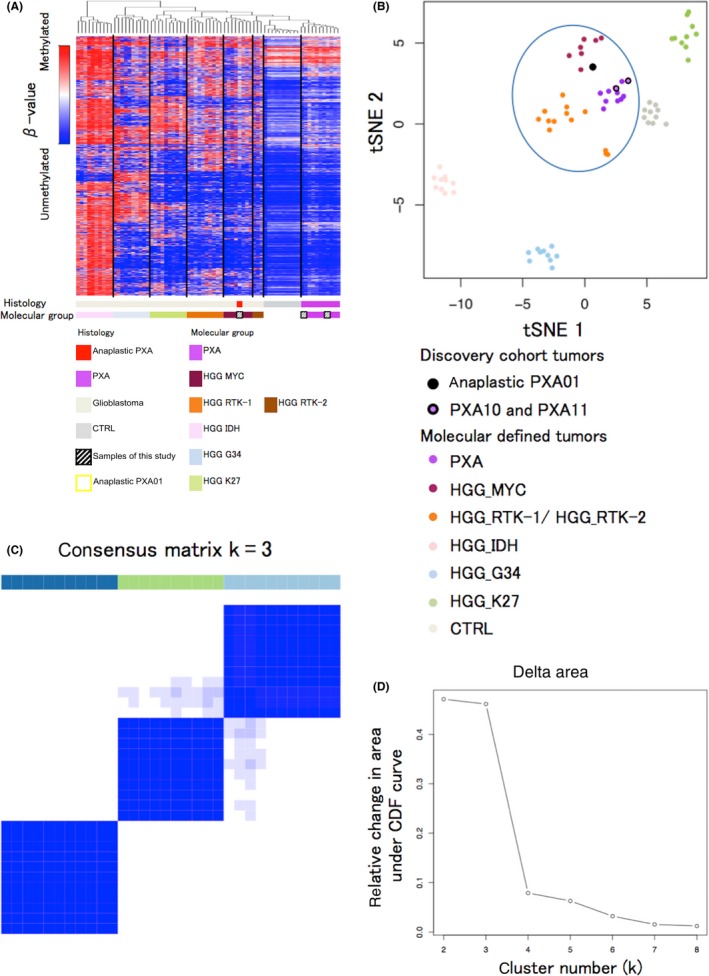
Genome‐wide methylation analysis. A, Unsupervised hierarchical clustering of DNA methylation and heatmap for the full tumor cohort based on the most variable 6152 methylated CpG island probes (standard deviation [SD] > 0.27) across the brain tumors methylation data sets. The probes are located within the CpG islands, whereas those on sex chromosomes are excluded. Anaplastic PXA01 was classified into the HGG_MYC type. B, A t statistic‐based stochastic neighbor‐embedding (tSNE) projection of the 450k methylation data. Anaplastic PXA01 and three clusters of GBM, ie, HGG_MYC, HGG_RTK (HGG_RTK‐1 and HGG_RTK‐2), and PXA clustered closely together (indicated by an oval). C, Heatmap for consensus matrix (k = 3) for tumors of anaplastic PXA01 and PXA with pediatric GBM of HGG_MYC and HGG_RTK (HGG_RTK‐1/‐2) types shows three distinct clusters, which is provided for the purpose of inner validation to confirm those relatively approximate classifiers in (B). These clusters are concordant with the unsupervised hierarchical clustering. D, Change in delta k for consensus matrix cumulative distribution function as k varies from 2 to 8, which showed the reliability of three distinct clusters. HGG, high‐grade glioma; PXA, pleomorphic xanthoastrocytoma
3. DISCUSSION
Pleomorphic xanthoastrocytoma has relatively good prognosis and is classified as WHO grade 2. In the revised WHO classification 2016, anaplastic PXA, WHO grade 3, has been newly defined as a variant of the PXA entity. The anaplastic feature is characterized by a MI of over five per 10 high‐power fields and the frequent presence of necrosis. Frequency of the hallmark BRAF V600E mutation is lower in anaplastic PXA than in PXA (47% vs 70%).7 Significance of the BRAF mutation for survival of anaplastic PXA is unknown.1 In our case (anaplastic PXA01), diagnosis of the tumor was histologically confirmed as an anaplastic PXA; however, early tumor regrowth was recognized by contrast MRI during the course of remission‐inducing radiochemotherapy on day 51 after the operation and the patient died 282 days after onset. Thus, the aggressive clinical course did not match what was expected from the histology. The BRAF V600E mutation was not detected, whereas non‐synonymous mutations in TP53 and a homozygous deletion at the 8q24.3 locus including GSDMD were detected. GSDMD was suggested as a tumor suppressor gene.8 Furthermore, genome‐wide 450k methylation analysis showed that this anaplastic PXA was clustered with the HGG_MYC type of pediatric GBM. It has been suggested that a subset of anaplastic PXA and epithelioid GBM share several genetic features including BRAF V600E mutations and may be closely related to each other, if not classified in the same entity.3 In contrast, methylation profiling showed that epithelioid GBM may be divided into PXA and GBM, suggesting that there may be distinct subgroups.9 Our case strongly suggests that DNA methylation‐based diagnosis can more precisely classify anaplastic PXA, as has recently been proposed.5 It is unlikely that histology alone is sufficient to diagnose anaplastic PXA, as pleomorphic gliomas with anaplastic features include both anaplastic PXA and epithelioid GBM. The BRAF V600E status alone is not sufficient as a diagnostic or prognostic biomarker either.10 Our study suggested that anaplastic PXA may consist of a clinically and biologically heterogeneous group of tumors, and genome‐wide methylation profiling may help identify those. Full implementation of the molecular approach is likely to be the key to predict prognosis and decide treatment strategy for PXA patients in the future.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
We are grateful to Dr David T. W. Jones for helping with statistical data processing in this project. The results published here include the data downloaded from the GEO database (GSE73801). This study was partially funded by KAKENHI Grants‐in‐Aid for Scientific Research, [18K16565 (T.N.)].
Nakamura T, Fukuoka K, Nakano Y, et al. Genome‐wide DNA methylation profiling shows molecular heterogeneity of anaplastic pleomorphic xanthoastrocytoma. Cancer Sci. 2019;110:828–832. 10.1111/cas.13903
REFERENCES
- 1. Louis D, Ohgaki H, Wiestler O, et al. WHO Classification of Tumours of the Central Nervous System, Revised. Lyon, France: IARC; 2016. [Google Scholar]
- 2. Marton E, Feletti A, Orvieto E, Longatti P. Malignant progression in pleomorphic xanthoastrocytoma: personal experience and review of the literature. J Neurol Sci. 2007;252:144‐153. [DOI] [PubMed] [Google Scholar]
- 3. Alexandrescu S, Korshunov A, Lai SH, et al. Epithelioid glioblastomas and anaplastic epithelioid pleomorphic xanthoastrocytomas—same entity or first cousins? Brain Pathol. 2016;26:215‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sturm D, Orr BA, Toprak UH, et al. New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell. 2016;164:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Capper D, Jones DT, Sill M, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakamura T, Yamashita S, Fukumura K, et al. Genome‐wide DNA methylation profiling identifies primary central nervous system lymphoma as a distinct entity different from systemic diffuse large B‐cell lymphoma. Acta Neuropathol. 2017;133:321‐324. [DOI] [PubMed] [Google Scholar]
- 7. Ida CM, Rodriguez FJ, Burger PC, et al. Pleomorphic xanthoastrocytoma: natural history and long‐term follow‐up. Brain Pathol. 2015;25:575‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saeki N, Usui T, Aoyagi K, et al. Distinctive expression and function of four GSDM family genes (GSDMA‐D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer. 2009;48:261‐271. [DOI] [PubMed] [Google Scholar]
- 9. Korshunov A, Chavez L, Sharma T, et al. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol. 2018;28:656‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones DT, Witt O, Pfister SM. BRAF V600E status alone is not sufficient as a prognostic biomarker in pediatric low‐grade glioma. J Clin Oncol. 2017;36:96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials