

Abstract
Reliable biomarkers for renal cell carcinoma (RCC) have yet to be determined. Circulating tumor DNA (ctDNA) is an emerging resource to detect and monitor molecular characteristics of various tumors. The present study aims to clarify the clinical utility of ctDNA for RCC. Fifty‐three patients histologically diagnosed with clear cell RCC were enrolled. Targeted sequencing was carried out using plasma cell‐free DNA (cfDNA) and tumor DNA. We applied droplet digital PCR (ddPCR) to validate detected mutations. cfDNA fragment size was also evaluated using a microfluidics‐based platform and sequencing. Proportion of cfDNA fragments was defined as the ratio of small (50‐166 bp) to large (167‐250 bp) cfDNA fragments. Association of mutant allele frequency of ctDNA with clinical course was analyzed. Prognostic potential was evaluated using log‐rank test. A total of 38 mutations across 16 (30%) patients were identified from cfDNA, including mutations in TP53 (n = 6) and VHL (n = 5), and median mutant allele frequency of ctDNA was 10%. We designed specific ddPCR probes for 11 mutations and detected the same mutations in both cfDNA and tumor DNA. Positive ctDNA was significantly associated with a higher proportion of cfDNA fragments (P = .033), indicating RCC patients with ctDNA had shorter fragment sizes of cfDNA. Interestingly, the changes of mutant allele frequency in ctDNA concurrently correlated with clinical course. Positive ctDNA and fragmentation of cfDNA were significantly associated with poor cancer‐specific survival (P < .001, P = .011). In conclusion, our study shows the clinical utility of ctDNA status and cfDNA fragment size as biomarkers for prognosis and disease monitoring in RCC.
Keywords: cell‐free DNA, circulating tumor DNA, fragment size, next‐generation sequencing, renal cell carcinoma
Abbreviations
- BAM
binary alignment map
- ccRCC
clear cell renal cell carcinoma
- cfDNA
cell‐free DNA
- CSS
cancer‐specific survival
- ctDNA
circulating tumor DNA
- ddPCR
droplet digital PCR
- EBCall
empirical Bayesian mutation calling
- gDNA
genomic DNA
- MAF
mutant allele frequency
- NGS
next‐generation sequencing
- PCF
proportion of cell‐free DNA fragments
- PFS
progression‐free survival
- RCC
renal cell carcinoma
1. INTRODUCTION
Renal cell carcinoma is the seventh most common cancer and comprises 2.4% of all adult malignancies worldwide.1 The 5‐year overall survival is reported to be 74%, although the 30% of RCC patients who present with evidence of distant metastasis upon initial diagnosis have a poor prognosis of only 8% for 5‐year overall survival.2, 3 Currently, radiological examinations are commonly applied for the diagnosis of RCC and are subsequently confirmed by histopathological examinations. However, these approaches have several problems: radiological examinations are insufficient for qualitative characterization of the tumor, and histopathological examinations are invasive, unrepeatable, and thus not well suited for disease monitoring.
Blood‐based tests, also known as liquid biopsy, can offer a potential alternative measure that overcomes the problems posed by traditional methods. Liquid biopsies for circulating tumor cells or ctDNA constitute a promising and less invasive technique.4, 5, 6, 7, 8, 9, 10 ctDNA is circulating cfDNA derived from tumor cells. However, no satisfactory blood‐based markers for RCC currently exist, creating an urgent need for the identification of new molecular markers. cfDNA is released from both normal and tumor cells by different molecular processes, such as cell apoptosis, necrosis and secretion of gDNA fragments.11, 12 Generally, cfDNA fragment size falls within a range of multiples of 180 bp, consistent with the unit size of nucleosomes, similar to DNA from apoptotic cells.13 In addition to fragment size of cfDNA, mutation status in plasma cfDNA can be a universal marker for several malignancies,11, 14, 15, 16, 17, 18, 19, 20, 21, 22 yet there have been few reports regarding ctDNA analysis for RCC.23 In the present study, we investigated whether the cfDNA profile, such as mutation status and fragmentation, can be promising tools for monitoring as well as predicting prognosis in RCC patients. We showed that RCC patients with ctDNA had shorter fragment sizes of cfDNA. We also found that positive ctDNA and shorter fragment sizes of cfDNA were highly correlated with worse prognosis for RCC patients. Furthermore, we showed that changes of MAF in ctDNA correlated with the stages of disease progression in RCC patients. Collectively, these markers may lead to better alternative tools to track the clinical course of RCC patients.
2. MATERIALS AND METHODS
2.1. Study design
Between June 2015 and June 2017, a total of 53 patients with ccRCC were enrolled in this study. Two patients concurrently had colon cancer at the time of RCC diagnosis; the others had no sign of other active cancers within the study period. This study was approved by the Institutional Review Board of Osaka University Hospital (#13397‐2). All patients provided written informed consent for the collection and analysis of blood and tissue samples.
For all RCC patients, blood collections were carried out at pretreatment, post‐treatment, or both. In some patients, multiple blood collections were performed over time. All patients were pathologically diagnosed by surgical resection sample or needle biopsy. Histological diagnosis was determined on the basis of standard H&E‐stained sections. Two or more experienced senior pathologists assessed the pathological diagnosis according to the 7th American Joint Committee on Cancer TNM staging system (AJCC 2010 version). PFS was evaluated only in RCC patients who had yet to receive treatment, including surgical resection or systemic therapy, prior to the first day of blood collection, and irrespective of clinical metastasis status or whether surgical removal for primary RCC tumor was subsequently performed. PFS was calculated from the first day of blood sampling to the last follow‐up point or to the detection of a progressive event by computed tomography (CT) scan according to the RECIST 1.1 criteria.24 CSS was also evaluated from the first day of blood sampling (pretreatment) to the last follow‐up point or to the day of cancer death in RCC patients.
2.2. Preparation of genomic DNA from cancer tissue and germline DNA
In some patients with surgical resection, RCC tissues were frozen and preserved at −80°C. gDNA was isolated from RCC tissue using QIAamp DNA Mini kit (QIAGEN, Hilden, Germany) according to the manufacturer's protocol. Germline DNA was isolated from blood lymphocytes using QIAamp DNA Blood Mini kit (QIAGEN) according to the manufacturer's protocol.
2.3. Preparation of blood samples and cfDNA extraction from plasma
Whole blood (2.0‐7.0 mL) was collected directly into EDTA tubes. Within 3 hours of collection, all blood samples were centrifuged sequentially at 900 and 20 000 g for 10 minutes each, and supernatants were stored at −80°C as plasma. cfDNA was isolated from 1.0‐3.0 mL plasma samples using the QIAamp Circulating Nucleic Acid Kit (QIAGEN) according to the manufacturer's protocol.
2.4. Measurement of global concentration and fragment size of cfDNA
Global cfDNA concentration from 1 mL plasma was measured using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). cfDNA fragment size was measured using a microfluidics‐based platform, the Agilent 2100 Bioanalyzer with the High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA). Agilent 2100 Expert software (version B.02.08) offers a smear analysis with an integrator feature that allows precise measurement of the smear region. The software automatically determines the mean size for each defined smear region of plasma cfDNA.
2.5. Targeted sequencing
Targeted sequencing focused on 48 genes that have been previously identified as recurrently mutated and/or driver genes for ccRCC25, 26 (Table S1). Plasma cfDNA, gDNA from cancer tissue and germline (leukocyte) DNA samples were subjected to targeted capture sequencing. A sequence library was prepared using a combination of the KAPA Hyper Prep Kit (Kapa Biosystems, Wilmington, MA, USA) and the SureSelectXT Custom 1‐499 kb library (Agilent Technologies). Target capture and further library preparation processes were carried out according to the manufacturer's instructions for the Agilent SureSelectXT Target Enrichment System (Agilent Technologies) with minor modification. Amounts of input DNA were 10 ng for cfDNA and 50 ng for gDNA from tumor tissue and germline DNA. Post‐capture libraries were barcoded and pooled for sequencing. One hundred and twenty‐five bp paired‐end sequencing was carried out on an Illumina HiSeq2500 (Illumina, Inc., San Diego, CA, USA). Median sequencing output was 9.23, 0.40, and 0.40 Gb for plasma cfDNA, cancer DNA, and germline DNA, respectively.
2.6. Detection of somatic mutations using bioinformatics analysis
Renal cell carcinoma patients were defined to have positive ctDNA when they showed somatic mutations in plasma cfDNA. Paired‐end reads were aligned to the human reference genome (GRCh37) using the Burrows‐Wheeler Aligner (BWA)27 for plasma cfDNA, gDNA from cancer tissue, and matched germline DNA samples. Probable PCR duplications, for which paired‐end reads aligned to the same genomic position, were removed, and pileup files were generated as BAM files using SAMtools28 and our program developed in‐house. To find somatic point mutations (single nucleotide variations and short indels), the following cut‐off values were used for base selection: (i) a mapping quality score of at least 20; (ii) a base quality score of at least 15. Somatic mutations were selected using the following filtering conditions: (iii) total numbers of reads supporting each base were at least 50; (iv) numbers of reads supporting a mutation in cfDNA or gDNA were at least 4; (v) Fisher's exact P < .1; (vi) variant allele frequency of matched germline DNA was less than 0.01 and the variant allele number of matched germline DNA was under 2; (vii) mutations must be supported by both forward and reverse reads; (viii) known variants listed in the 1000 Genomes Project (Oct 2014 release) and NCBI dbSNP build 138 were excluded, although a variant that was also registered in the COSMIC database was included. As sequencing errors can occur in a sequence‐specific way, we screened for somatic mutations in each patient's cfDNA and cancer DNA by analyzing these reads against the corresponding pooled reads from all other patients’ germline DNA and cfDNA to discriminate true positives from false positives accurately using EBCall.29 Mutations with P ≥ .001 (EBCall) were excluded and mutations with .001 > P ≥ .0001 (EBCall) were also verified by ddPCR platform.
2.7. Droplet digital PCR platform
The ddPCR platform, Qx100 Droplet Digital PCR System (Bio‐Rad Laboratories, Hercules, CA, USA), was used to validate the mutations detected by NGS using existing or customized Droplet Digital PCR Assays (Bio‐Rad Laboratories) including primers and probes (FAM, mutant type; HEX, wild‐type; Table S2), and ddPCR Supermix for Probes (No dUTP; Bio‐Rad Laboratories) according to the manufacturer's instructions. For every experiment, we used gBlocks Gene Fragments 125‐500 bp (Integrated DNA Technologies, Coralville, IA, USA) that contained the relevant mutations and germline DNA from healthy controls as positive and negative controls, respectively, to determine the cut‐off value for allele calling. The initial amount of DNA used for ddPCR reaction was 12 ng plasma cfDNA and 80 ng gDNA from tumor samples and germline DNA. Thermal cycling conditions were as follows: 10 minutes incubation at 95°C followed by 40 cycles of 94°C for 30 seconds and 55°C for 1 minute, 1 cycle of 98°C for 10 minutes, and then 4°C hold. Droplet fluorescence was assessed in the droplet reader. Analysis of ddPCR data for allele calling and calculating absolute copy number was carried out using QuantaSoft software version 1.7.4 (Bio‐Rad Laboratories). Samples were designated positive for targeted mutations when they contained at least three droplets in the positive area of FAM signal. MAF was defined as the proportion of copies of mutant type relative to the sum of copies of mutant and wild‐type obtained by the ddPCR platform.
2.8. Analysis of fragment length from sequencing data
Fragment length was calculated from paired‐end alignment information according to the BAM format.30 Overlapping read pairs were treated as single observations. Fragment length information was extracted using Strand NGS 2.7 (Strand Life Sciences, Bangalore, India). To evaluate the differences in size distribution of plasma cfDNA in each RCC patient, the proportion of cfDNA fragments (PCF) was defined as the ratio of small cfDNA fragments (50‐166 bp) to large ones (167‐250 bp). For each of the mutations detected by NGS, fragment length was extracted using the integrative genomics viewer30 from BAM files of plasma cfDNA of between 50 and 250 bp and divided into two groups according to the presence of mutations.
2.9. Concurrent monitoring of clinical course
Concurrent monitoring of clinical course was evaluated using plasma cfDNA characteristics such as fragment size and presence of mutations. We developed specific ddPCR assays to monitor the disease status of six patients before and during therapeutic treatments. We also carried out ddPCR for gDNA from tumor tissue. In two patients (cases 47 and 50), gDNA from tumor thrombus was also evaluated by ddPCR and, then, in case 47, gDNA from tumor tissue upon pathological autopsy was evaluated.
2.10. Statistical analysis
Statistical analysis was done using JMP Pro 14.0.0 (SAS Institute Inc., Cary, NC, USA). Patient and cfDNA characteristics were presented as median + range, and data were compared using Wilcoxon test, correlation analysis. PFS rate and CSS were calculated using the Kaplan‐Meier method. Differences among the two groups were assessed by log‐rank test and were considered statistically significant when the P‐value was < .05.
3. RESULTS
3.1. Patient characteristics
Patient characteristics are summarized in Table S3. The RCC cohort consisted of 41 males and 12 females, and median age was 69 years (range 38‐90 years). In total, 53 RCC patients were histologically diagnosed with ccRCC and subsequently enrolled in this study (Figure 1). With regard to disease stage upon blood collection, 14 patients were classified as “pretreatment without metastasis”, 13 were classified as “pretreatment with metastasis”, and 26 were classified as “post‐treatment with metastasis or recurrence”. Median follow‐up duration was 15.6 months (range 0.2‐33.1 months). Global median plasma cfDNA concentration of RCC patients was 17.1 ng/mL (range 8.1‐219.0 ng/mL).
Figure 1.
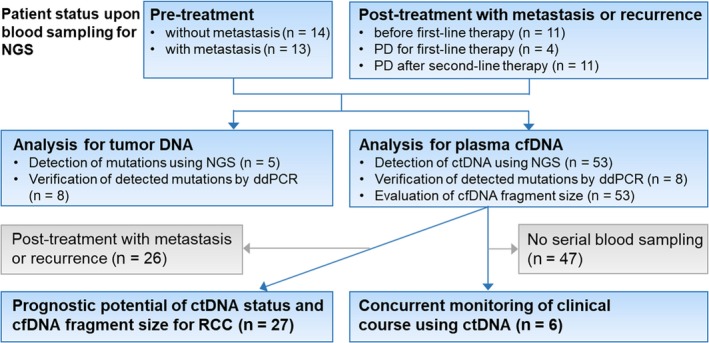
Study design and patient allocation. Blue squares indicate inclusion for analysis, and gray squares indicate exclusion. cfDNA, cell‐free DNA; ctDNA, circulating tumor DNA; ddPCR, droplet digital PCR; NGS, next‐generation sequencing; RCC, renal cell carcinoma
3.2. Somatic mutations detected by targeted sequencing of plasma cfDNA and gDNA from tumor tissue
We carried out targeted sequencing to analyze mutated genes in plasma cfDNA and gDNA from tumor tissue in RCC patients using the Illumina platform (Illumina, Inc.). In order to test clinical feasibility, we designed an original gene panel, focusing on 48 genes considered to be recurrent mutated genes and driver genes for ccRCC (Table S1). One hundred and six DNA libraries generated from 53 cfDNA and 53 matched germline DNA samples were pooled into two HiSeq2500 Flowcells. In five RCC patients, DNA libraries generated from gDNA from cancer tissue were pooled, making the total number of DNA libraries 111. Unique coverage depth was 204× on average (range 37‐955×) for cfDNA samples, 536× on average (range 496‐614×) for cancer DNA samples, and 358× on average (range 287‐580×) for germline DNA samples (excluding PCR duplications). In the five RCC patients whose plasma cfDNA and cancer tissue gDNA were sequenced, mutations were detected by targeted sequencing (Figure 2A). At least two mutations were detected in cancer tissue from all five patients, and the corresponding mutation in cfDNA was detected for two of these patients. In case 50, there was complete concordance for all five mutations between cfDNA sample and gDNA from cancer tissue. In contrast, in case 53, the same MTOR mutation was identified in cfDNA and gDNA from cancer tissue, yet a TSC1 mutation was detected in the cfDNA sample only. In 16 of 53 RCC patients, the total number of somatic mutations detected was 38 (median 2 mutations/patient, range 1‐7 mutations; Figure 2B). Among them, the most frequently mutated genes in the overall cohort included TP53 (n = 6), BAP1 (n = 5), VHL (n = 5), TSC1 (n = 4), and SETD2 (n = 3). Median MAF in ctDNA was 10.0% (range 1.2%‐54.5%). Of the 38 somatic mutations, there were 14 (36.8%) missense mutations, four (10.5%) nonsense mutations, 19 (50.0%) insertions/deletions, and one (2.6%) splicing site mutation.
Figure 2.

Somatic mutations detected by targeted sequencing of cell‐free DNA (cfDNA) and genomic DNA (gDNA) from tumor tissue. Mutated genes detected by targeted sequencing are shown in the left‐most column (arranged in descending order of the number of mutations). Numbers for each gene indicate the frequency of the mutant allele (%). MISSSENSE, missense mutation; NONSENSE, nonsense mutation; INDEL, insertion/deletion; SPLICING, splicing site mutation. A, Somatic mutations in plasma cfDNA and gDNA from cancer (n = 5). B, Somatic mutations of plasma cfDNA in 16 RCC patients with at least one mutation. Patient state at blood collection was classified as “Pretreatment without metastasis” (purple), “Pretreatment with metastasis” (green) and “Post‐treatment with metastasis or recurrence” (red). Right bar plot shows the number of samples
3.3. Verification of somatic mutations using the droplet digital PCR platform
To confirm the somatic mutations detected by NGS, we carried out ddPCR. Specific primers and probes were prepared for 12 of the somatic mutations detected by NGS (Table S2). In cfDNA samples, all 12 mutations were also detected by ddPCR (Table S4). Importantly, 11 of 12 mutations were verified by ddPCR in both cfDNA and gDNA from tumor tissue. In one sample (case 5), due to the lack of tumor tissue, we were not able to evaluate the mutation status in tumor DNA from this patient. Interestingly, in case 53, ddPCR enabled detection of the TSC1 mutation in both cfDNA and gDNA from tumor tissue, yet NGS showed the mutation only in the cfDNA sample.
3.4. Association between cfDNA fragment size and ctDNA status in RCC patients
Next, we examined the fragment size of plasma cfDNA using a microfluidics‐based platform. Median fragment size of plasma cfDNA that was sequenced was 168 bp (range 138‐181 bp). cfDNA fragment sizes in patients with metastasis showed no significant differences from those without metastasis (median value 165, 169.5 bp, respectively; P = .138). We also analyzed the distribution of cfDNA fragments according to the measured size by NGS (Figure 3A). The most prominent peak of cfDNA fragment distribution was 166 bp, consistent with previous reports.31, 32 In patients with ctDNA (Figure 3A, red line), short cfDNA fragments of between 130 and 150 bp were slightly more abundant compared to those in patients without ctDNA. To evaluate cfDNA fragmentation in each RCC patient, we defined PCF as the ratio of short fragments (50‐166 bp) to large ones (167‐250 bp) using NGS (Figure 3B). For example, case 50 had a higher PCF (1.61) than case 52, indicating that case 50 had more short fragments. Shorter fragment size of cfDNA using a microfluidics‐based platform tended to be associated with higher PCF (P = .085; Figure 3C) and showed weak correlation (R 2 = .105, P = .018; Figure 3D). Although patients with ctDNA showed no significant difference in cfDNA fragment size as measured by microfluidics‐based platform compared to those without ctDNA (P = .245; Figure 3E), positive ctDNA was significantly associated with higher PCF (P = .033; Figure 3F). These results suggest that RCC patients who had mutations in plasma cfDNA had significantly shorter cfDNA fragments than those without mutations.
Figure 3.
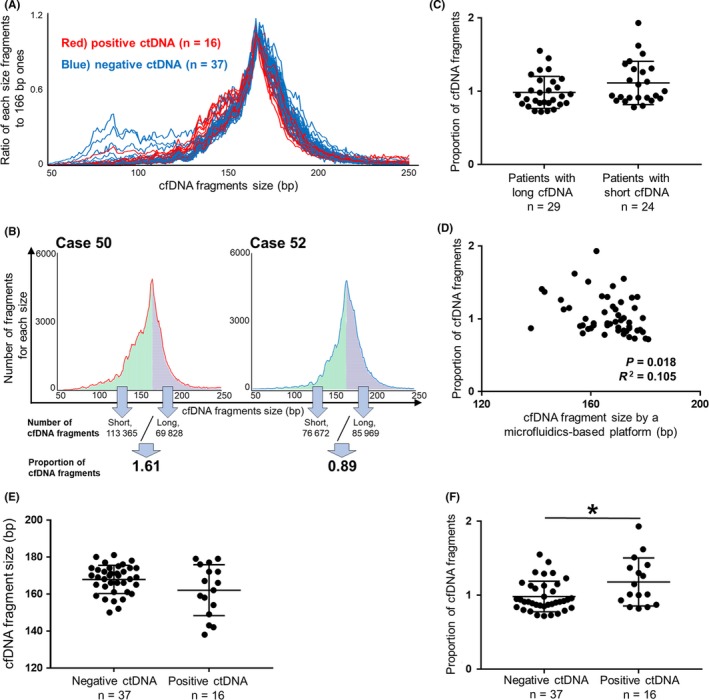
Renal cell carcinoma (RCC) patients with circulating tumor DNA (ctDNA) had shorter cell‐free DNA (cfDNA) fragments than those without ctDNA. A, Distributions of cfDNA fragments according to size were determined by targeted sequencing in 53 RCC patients. X‐axis shows cfDNA fragment size, and the Y‐axis shows the abundance of fragments of those specific sizes relative to the number of 166‐bp fragments. Red lines (n = 16) indicate the distributions of cfDNA fragments for patients with ctDNA, and blue lines (n = 37) for patients without ctDNA. B, Proportion of cfDNA fragments (PCF) was defined as the ratio of short cfDNA fragments (50‐166 bp; green) to long fragments (167‐250 bp; blue) as determined by next‐generation sequencing (NGS). Using a microfluidics‐based platform, average cfDNA fragment size in case 50 was classified as short (154 bp), whereas that in case 52 was classified as long (174 bp). C, PCF in patients with short cfDNA fragment size (≤166 bp, n = 24) as measured by a microfluidics‐based platform tended to be higher than in those with long cfDNA fragments (>166 bp, n = 29; P = .085). (Wilcoxon test). D, PCF was weakly correlated with cfDNA fragment size as determined by a microfluidics‐based platform (n = 53). (correlation analysis). E, Association between ctDNA status and cfDNA fragment size as determined by a microfluidics‐based platform (n = 53; P = .245). (Wilcoxon test). F, Association between ctDNA status and PCF (n = 53). *P < .05 (Wilcoxon test). Central line, mean; error bars, standard deviation
3.5. Fragment size of plasma cfDNA harboring mutant alleles tended to be short
To further investigate the relationship between cfDNA fragment size and ctDNA status, we compared cfDNA fragment sizes of mutation‐harboring alleles to those of wild‐type alleles. In case 10, mutations in plasma cfDNA were identified in the genes SETD2, BAP1 and NF2, and the cfDNA fragment sizes of mutant alleles were significantly shorter than those with wild‐type alleles for SETD2 (P = .012) and NF2 (P = .008; Figure 4A‐C). Likewise, in case 13, cfDNA fragment sizes of mutant alleles were significantly shorter than those with wild‐type alleles for TP53 (P < .001; Figure 4D,E). In case 50, cfDNA fragments harboring a TSC1 mutation were significantly shorter than those of the corresponding wild‐type allele (P < .001), with a similar trend for a FPGT mutation that did not reach significance (P = .095; Figure 4F‐J). In other cases, fragment sizes of cfDNA harboring mutant alleles tended to be short compared to those of the corresponding wild‐type alleles (Figure S1). This indirectly implies that fragment size of cfDNA derived from tumor is shorter on average than that from normal cells. Of course, further studies are needed to examine this phenomenon.
Figure 4.
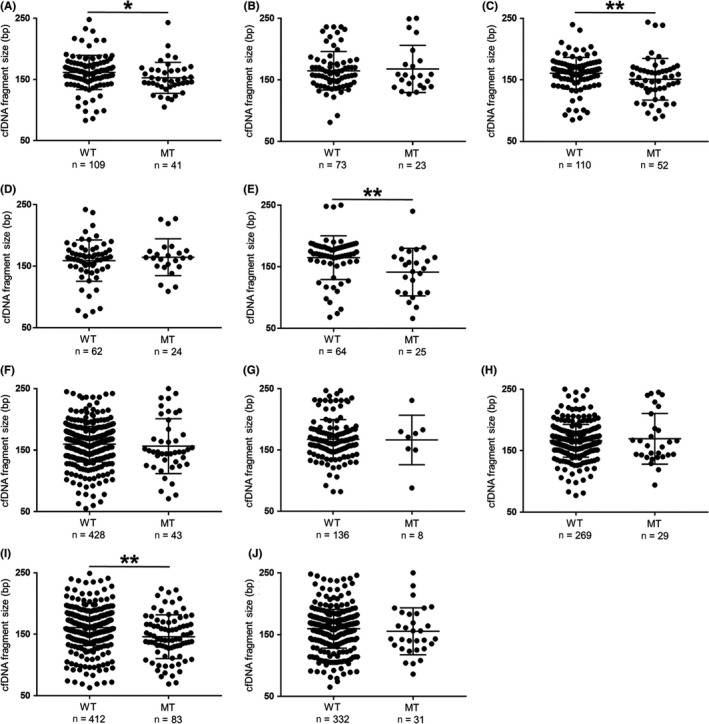
Cell‐free DNA (cfDNA) fragment sizes with mutations were often shorter than corresponding fragments of wild‐type alleles. For each mutation detected by next‐generation sequencing (NGS), cfDNA fragment sizes between 50 and 250 bp were extracted from binary alignment map files by integrative genomics viewer. MT, mutation; WT, wild‐type. *P < .05, **P < .01 (Wilcoxon test). Central line, mean; error bars, SD. A‐C, In case 10, SETD2 (A), BAP1 (B) and NF2 (C). D,E, In case 13, VHL (D) and TP53 (E). F‐J, In case 50, FPGT (F), VHL (G), BAP1 (H), TSC1 (I) and TP53 (J)
3.6. Concurrent monitoring of clinical course using ctDNA
We next evaluated whether the changes of plasma cfDNA characteristics such as fragment size and mutation status were consistent across the clinical course of RCC patients. In case 45 (pT3apN1M1 [lung metastasis], stage IV), VHL and TP53 mutations detected by NGS in plasma cfDNA were also examined using ddPCR in primary cancer tissue (Figure 5A). MAF of ctDNA in both genes decreased after surgical resection of primary tumor, yet re‐emerged coinciding with the appearance of brain metastasis. Cyber knife was performed for brain metastasis, after which the MAF of ctDNA decreased, only to rise again upon development of progressive disease. In case 53 (pT3bpN1M0, stage III), although the change in MAF of MTOR in plasma cfDNA similarly mirrored the clinical course of the disease, the change of MAF of TSC1 was different (Figure 5B). The TSC1 mutation in plasma cfDNA had been detected only before surgical resection of primary tumor and diminished after surgery. This finding is likely indicative of the heterogeneity of the ctDNA pool, and the dynamic shifts that occur in the prevalence of various alleles within that pool. Similarly, in case 48, a rare TP53 mutation that was detected in the primary site was not detected in plasma cfDNA before and between initial treatments, whereas a BAP1 mutation that existed abundantly in the primary site was only identified in plasma cfDNA before treatment (Figure S2A). Regarding cfDNA fragment size, some patients had dynamic changes depending on disease state, whereas others showed minimal differences. Overall, changes in the MAF of ctDNA generally mirrored the rise and fall of tumor burden throughout the clinical course of the disease (Figure S2).
Figure 5.
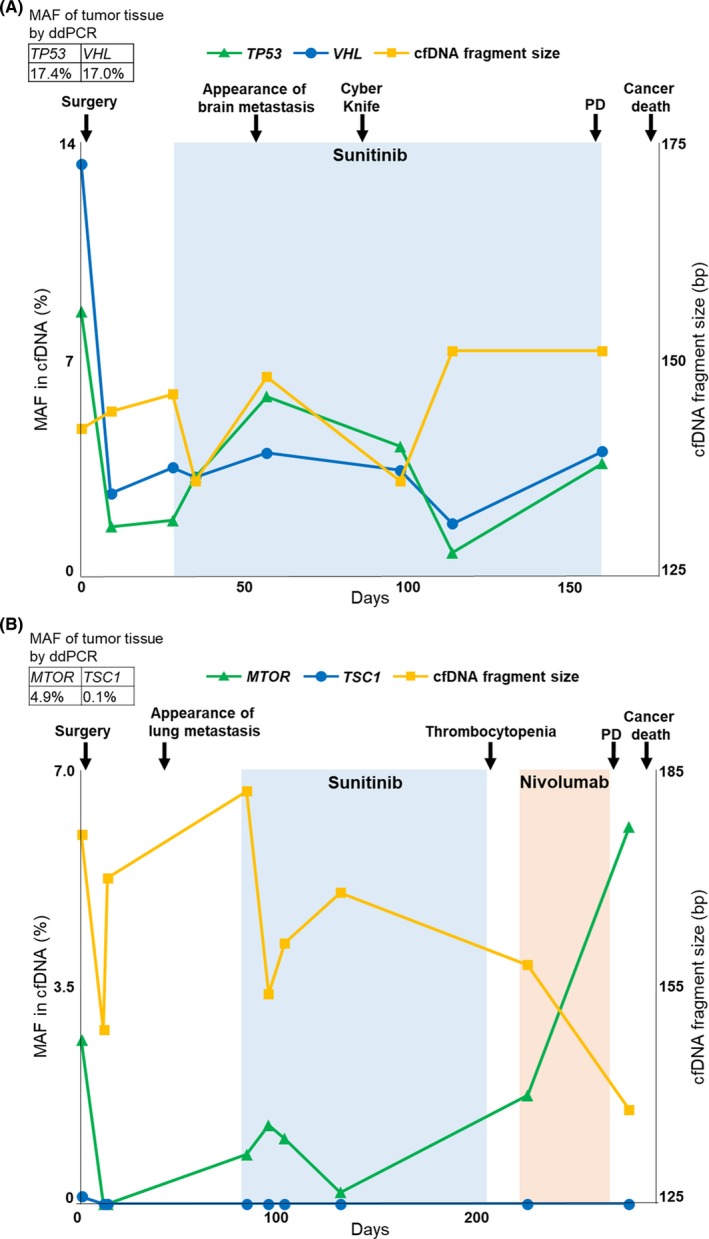
Clinical course monitoring in renal cell carcinoma (RCC) patients with circulating tumor DNA (ctDNA). Clinical course was analyzed using mutant allele frequency (MAF) of ctDNA by droplet digital PCR (ddPCR), and cell‐free DNA (cfDNA) fragment size by a microfluidics‐based platform in RCC patients with ctDNA. PD, progressive disease. A, In case 45, MAF of ctDNA was evaluated in TP53 and VHL. B, In case 53, MAF of ctDNA was evaluated in MTOR and TSC1
We examined the potential of cfDNA for both biomarker monitoring and predicting the treatment effect of any drugs (Figure S3). In terms of predicting drug response, patients with short fragment sizes of cfDNA showed significantly worse responsiveness (long vs short, P = .011; Figure S3A). Moreover, especially in the response for any tyrosine kinase inhibitors, positive ctDNA was significantly associated with weaker effect (positive vs negative, P = .049; FigureS3D), and short fragment sizes of cfDNA tended to be associated with worse outcome (long vs short, P = .090; Figure S3B).
3.7. Prognostic potential of ctDNA status and cfDNA fragment size for RCC
Finally, we evaluated whether ctDNA status and cfDNA fragment size correlated with prognosis of RCC patients. Using the Kaplan‐Meier method and log‐rank test, we found that ctDNA status was associated with PFS and CSS (positive vs negative, P = .061, P < .001, respectively; Figure 6A,B). We also found that cfDNA fragment size was significantly associated with PFS and CSS (long vs short, P = .004, P = .011, respectively; Figure 6C,D), although PCF showed no significant association (high vs low, P = .317, P = .127, respectively; Figure S4A,B). Moreover, in RCC patients with metastasis, positive ctDNA, short fragment size of cfDNA and high PCF were significantly associated with worse CSS (P = .010, P = .011 and P = .007, respectively; Figure 6E,F and Figure S4C), whereas patients without metastasis had no association between prognosis and these parameters of cfDNA and ctDNA (P = .190, P = .485 and P = .677, respectively). These data indicate that mutations and fragmentation of cfDNA could be used to identify more malignant tumors especially in patients with metastasis, warranting further study.
Figure 6.
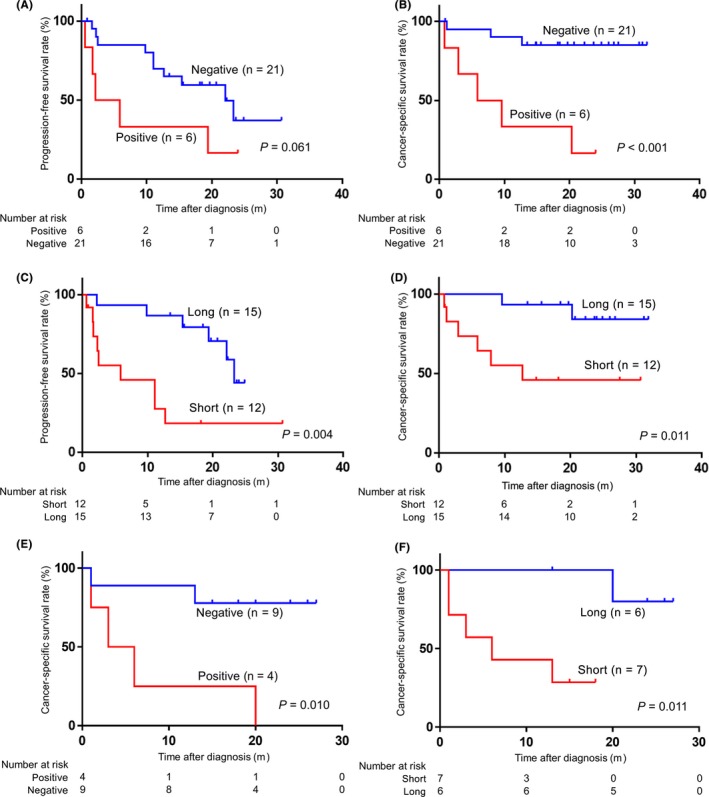
Positive circulating tumor DNA (ctDNA) and short fragment size of plasma cell‐free DNA (cfDNA) were associated with poor prognosis. (Kaplan‐Meier method and log‐rank test). A,B, Prognosis was analyzed in 27 renal cell carcinoma (RCC) patients whose cfDNA samples were sequenced at pretreatment state. Association of ctDNA status (positive vs negative) for progression‐free survival (PFS) (A) and cancer‐specific survival (CSS) (B). C,D, Association of cfDNA fragment size using a microfluidics‐based platform between ≤166 bp (the prominent peak of the distribution of cfDNA fragments according to size) (short) and >166 bp (long) for PFS (C) and CSS (D). E,F, Prognosis was analyzed in 13 RCC patients with metastasis whose cfDNA samples were sequenced at pretreatment state as in A‐D. Association of ctDNA status (positive vs negative) (E) and cfDNA fragment size (short vs long) (F) for CSS
4. DISCUSSION
Currently, there are no reliable biomarkers for RCC that are minimally invasive and facilitate diagnosis of early‐stage disease. Recently, blood‐based tests, also known as liquid biopsy, serve as potential alternative measures to radiological tests and tissue biopsies. In particular, ctDNA reflects disease status and holds advantages for the diagnosis, prognosis and monitoring of several cancers.16, 33, 34, 35 Recently, targeted analyses of specific cancer‐associated genes in plasma cfDNA obtained satisfactory clinical applicability for several cancers.36 cfDNA analysis can include not only somatic mutations but also fragment size, a metric that has received comparatively scant attention. Accordingly, we examined the potential of multiple cfDNA characteristics, such as somatic mutation and fragment size, as novel markers for RCC.
Regarding published mutation profiles from ccRCC tumor tissue, mutations in VHL (52.3%) is the most prominent, followed by PBRM1 (32.9%), SETD2 (11.5%) and BAP1 (10.1%), as well as TP53 (2.2%).25 In this study, TP53 mutations in cfDNA were most abundant, followed by VHL and BAP1, which is consistent with at least one previous report.23 The discrepancy between tumor DNA and cfDNA may be because TP53 mutations were induced by selection pressure from some drugs. A previous study for RCC reported that the mutation frequencies of TP53 and NF1 in cfDNA were higher after first‐line therapy than those before first‐line therapy.23 Another report for chronic lymphocytic leukemia showed the induction of TP53 mutation after treatment.37 To validate these findings, studies with much larger cohorts should be carried out.
Through the ctDNA analysis in the present study, we have shown several novel findings that may have utility in clinical settings. First, RCC patients with ctDNA had shorter cfDNA fragments. We have previously reported that RCC patients tended to have shorter fragment sizes of cfDNA compared to healthy controls.38 Moreover, we showed that cfDNA fragments harboring mutant alleles were often shorter than those with corresponding wild‐type alleles in RCC patients. Similarly, in lung cancer, melanoma and colorectal cancer, fragment sizes of cfDNA derived from tumor were short.39, 40 Recent research reported that higher nucleosome accessibility allowed endonuclease enzymes to cut gDNA within the nucleosome cores, contributing to a preponderance of shorter cfDNA.41 Further elucidation of the molecular mechanisms behind cfDNA fragmentation is needed. Second, some mutations could be detected in both cfDNA and gDNA from tumor tissue using the ddPCR platform. In RCC patients, the concordance rate of genomic alterations between plasma and tumor tissue using NGS was 8.6%, likely as a result of the heterogeneity of tumor tissue.42 Ability of the ddPCR platform to detect rare mutations might overcome the disparities between mutant allele detection in cfDNA and gDNA from tumor tissue in our study. These observations are of special importance as there is increasing interest toward integrating ctDNA applications into medical practice, and clinical practice will require precise, standardized methods to detect and characterize ctDNA. Third, regarding the potential of ctDNA as a clinical biomarker, shifts in MAF of ctDNA correlated with the clinical course of the disease, as has been reported for breast cancer.33 MAF of ctDNA could be superior to cfDNA fragment size for monitoring through analysis of serial sampling. Moreover, RCC patients with mutation‐harboring ctDNA and shorter cfDNA fragments showed significantly worse prognosis. Based on these findings, to our knowledge, this is the first report deciphering the utility of ctDNA for monitoring the clinical course of RCC. Interestingly, some mutations, such as TSC1 in case 53, were detected only before surgical resection, implying that individual somatic mutations have different roles during tumor evolution, some as drivers and some as passengers. Of course, further studies are needed to examine this phenomenon.
There are some apparent limitations in the present study. Our study was retrospective and had relatively short follow‐up duration. Sequencing depth for plasma cfDNA may also have been insufficient to detect rare mutations. Further investigations are needed to validate our results in larger numbers of patients by multi‐institutional studies.
In conclusion, our results imply that the mutational landscape and fragmentation of plasma cfDNA have promising prognostic potential in RCC patients. Change of MAF of ctDNA may be an auspicious monitoring marker for RCC. Given that plasma cfDNA is easily collected from peripheral blood, these newly discovered markers can be convenient and precise tools for understanding RCC.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported by a KAKENHI grant (16K20139, 18K16692). We thank Mutsumi Tuchiya for excellent technical support.
Yamamoto Y, Uemura M, Fujita M, et al. Clinical significance of the mutational landscape and fragmentation of circulating tumor DNA in renal cell carcinoma. Cancer Sci. 2019;110:617–628. 10.1111/cas.13906
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 2. Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev. 2008;34:193‐205. [DOI] [PubMed] [Google Scholar]
- 3. Choueiri TK, Motzer RJ. Systemic therapy for metastatic renal‐cell carcinoma. N Engl J Med. 2017;376:354‐366. [DOI] [PubMed] [Google Scholar]
- 4. Carter L, Rothwell DG, Mesquita B, et al. Molecular analysis of circulating tumor cells identifies distinct copy‐number profiles in patients with chemosensitive and chemorefractory small‐cell lung cancer. Nat Med. 2017;23:114‐119. [DOI] [PubMed] [Google Scholar]
- 5. Seitz AK, Thoene S, Bietenbeck A, et al. AR‐V7 in peripheral whole blood of patients with castration‐resistant prostate cancer: association with treatment‐specific outcome under abiraterone and enzalutamide. Eur Urol. 2017;72:828‐834. [DOI] [PubMed] [Google Scholar]
- 6. Alix‐Panabières C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6:479‐491. [DOI] [PubMed] [Google Scholar]
- 7. Rink M, Chun FK, Dahlem R, et al. Prognostic role and HER2 expression of circulating tumor cells in peripheral blood of patients prior to radical cystectomy: a prospective study. Eur Urol. 2012;61:810‐817. [DOI] [PubMed] [Google Scholar]
- 8. Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537:102‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sefrioui D, Blanchard F, Toure E, et al. Diagnostic value of CA19.9, circulating tumour DNA and circulating tumour cells in patients with solid pancreatic tumours. Br J Cancer. 2017;117:1017‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ignatiadis M, Dawson SJ. Circulating tumor cells and circulating tumor DNA for precision medicine: dream or reality? Ann Oncol. 2014;25:2304‐2313. [DOI] [PubMed] [Google Scholar]
- 11. Schwarzenbach H, Hoon DS, Pantel K. Cell‐free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426‐437. [DOI] [PubMed] [Google Scholar]
- 12. Choi JJ, Reich CF, Pisetsky DS. The role of macrophages in the in vitro generation of extracellular DNA from apoptotic and necrotic cells. Immunology. 2005;115:55‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659‐1665. [PubMed] [Google Scholar]
- 14. Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early‐stage lung cancer evolution. Nature. 2017;545:446‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamakawa T, Kukita Y, Kurokawa Y, et al. Monitoring gastric cancer progression with circulating tumour DNA. Br J Cancer. 2015;112:352‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wan JCM, Massie C, Garcia‐Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223‐238. [DOI] [PubMed] [Google Scholar]
- 17. Annala M, Vandekerkhove G, Khalaf D, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018;8:444‐457. [DOI] [PubMed] [Google Scholar]
- 18. Tabernero J, Lenz HJ, Siena S, et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 2015;16:937‐948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Christie EL, Fereday S, Doig K, Pattnaik S, Dawson SJ, Bowtell DDL. Reversion of BRCA1/2 germline mutations detected in circulating tumor DNA from patients with high‐grade serous ovarian cancer. J Clin Oncol. 2017;35:1274‐1280. [DOI] [PubMed] [Google Scholar]
- 20. Karachaliou N, Mayo‐de las Casas C, Queralt C, et al. Association of EGFR L858R mutation in circulating free DNA with survival in the EURTAC trial. JAMA Oncol. 2015;1:149‐157. [DOI] [PubMed] [Google Scholar]
- 21. Maia MC, Grivas P, Agarwal N, Pal SK. Circulating tumor DNA in bladder cancer: novel applications and future directions. Eur Urol. 2018;73:541‐542. [DOI] [PubMed] [Google Scholar]
- 22. Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pal SK, Sonpavde G, Agarwal N, et al. Evolution of circulating tumor DNA profile from first‐line to subsequent therapy in metastatic renal cell carcinoma. Eur Urol. 2017;72:557‐564. [DOI] [PubMed] [Google Scholar]
- 24. Sohaib A. RECIST rules. Cancer Imaging. 2012;12:345‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Network CGAR. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear‐cell renal cell carcinoma. Nat Genet. 2013;45:860‐867. [DOI] [PubMed] [Google Scholar]
- 27. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shiraishi Y, Sato Y, Chiba K, et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jiang P, Chan CW, Chan KC, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci USA. 2015;112:E1317‐E1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome‐wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2:61ra91. [DOI] [PubMed] [Google Scholar]
- 33. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199‐1209. [DOI] [PubMed] [Google Scholar]
- 34. Nakano Y, Kitago M, Matsuda S, et al. KRAS mutations in cell‐free DNA from preoperative and postoperative sera as a pancreatic cancer marker: a retrospective study. Br J Cancer. 2018;118:662‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD‐1 inhibition in metastatic gastric cancer. Nat Med. 2018;24:1449‐1458. [DOI] [PubMed] [Google Scholar]
- 36. Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science. 2018;359:926‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malcikova J, Stano‐Kozubik K, Tichy B, et al. Detailed analysis of therapy‐driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia. 2015;29:877‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamamoto Y, Uemura M, Nakano K, et al. Increased level and fragmentation of plasma circulating cell‐free DNA are diagnostic and prognostic markers for renal cell carcinoma. Oncotarget. 2018;9:20467‐20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Underhill HR, Kitzman JO, Hellwig S, et al. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12:e1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mouliere F, El Messaoudi S, Gongora C, et al. Circulating cell‐free DNA from colorectal cancer patients may reveal high KRAS or BRAF mutation load. Transl Oncol. 2013;6:319‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun K, Jiang P, Wong AIC, et al. Size‐tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc Natl Acad Sci USA. 2018;115:E5106‐E5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hahn AW, Gill DM, Maughan B, et al. Correlation of genomic alterations assessed by next‐generation sequencing (NGS) of tumor tissue DNA and circulating tumor DNA (ctDNA) in metastatic renal cell carcinoma (mRCC): potential clinical implications. Oncotarget. 2017;8:33614‐33620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials