

Abstract
The present state of therapy for colorectal cancer (CRC) is far from satisfactory, highlighting the need for new targets for this disease. We identified a new CRC‐specific molecule, TMEM180, a predicted 11‐pass transmembrane protein that apparently functions as a cation symporter. We developed an anti‐TMEM180 mAb and then succeeded in humanizing the mAb. Immunohistochemistry (IHC) in CRC with the mAb showed a similar positivity rate as compared with anti‐epidermal growth factor receptor mAb, and IHC with anti‐TMEM180 mAb did not show staining in major organs used in this study. Immune electron microscopy clearly indicated that TMEM180 was present on the tumor exosome. The TMEM180 promoter region contains 10 hypoxia‐responsive element consensus sequences; accordingly, SW480 cells upregulated TMEM180 under low‐oxygen conditions. Anti‐TMEM180 mAb has in vitro antibody‐dependent cell‐mediated cytotoxicity and complement‐dependent cytotoxicity activity, and SW480 CRC xenografts were eradicated by the mAb. These data indicate that TMEM180 may be a new CRC marker and that a mAb against this protein could be used as antibody‐based therapy against CRC.
Keywords: antibody drug, antibody therapy, diagnosis by tumor marker and biomarker, gastrointestinal tract, tumor antigen
1. INTRODUCTION
Colorectal cancer (CRC) is the third leading cause of cancer‐related mortality in the world.1 Despite recent progress in chemotherapeutic options, including mAb therapeutics, the prognosis for metastatic CRC is still very poor. Of the mAb therapies for CRC, mAbs against vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) are clinically available.2, 3 Although EGFR is expressed in several normal tissues, the mAb can preferentially accumulate in CRC tumor tissues as a result of the enhanced permeability and retention effect of tumor tissue.4, 5 However, because EGFR is expressed at a high level in normal skin tissue, skin toxicity is common, and cessation of mAb treatment is sometimes inevitable even if the therapy is effective in patients.6, 7 In the case of anti‐VEGF mAb, some patients receiving the mAb have life‐threatening side‐effects including serious bleeding and gastrointestinal perforation.8
In this context, we need to find a new CRC‐specific molecule and develop mAb against the molecule in order to produce an effective antibody therapy in the treatment of metastatic CRC with minimal side‐effects.
To find a CRC‐specific molecule, comprehensive expression analysis is usually carried out between CRC cells and their normal counterpart, mucoepithelial cells. However, in reality, obtaining pure live normal mucoepithelial cells is more difficult than obtaining CRC cells even if a laser micro‐dissection method is used. We have reported that many mucoepithelial cells are exfoliated in stool, some of which contain intact mRNA.9 Based on the results, we previously developed a method for obtaining almost pure normal mucoepithelial cells from lavage solution following colonoscopies of healthy examinees. We then identified several new CRC‐specific molecules after comprehensive expression analyses between the pure mucoepithelial cells and the CRC cell lines.10 TMEM180 was one of them and is a predicted 11‐pass transmembrane protein (UniProtKB, http://www.uniprot.org/uniprot/). In the present study, we developed a mAb against TMEM180 and evaluated the potential of using its mAb for CRC therapy.
2. MATERIALS AND METHODS
2.1. Collection of human samples
Human exfoliated colonocytes in a saline mucosal wash fluid were obtained from two healthy donors at the time of their colonoscopy examination. Cell purification and processing methods were conducted as previously described.10
The experimental protocols and procedures were approved by the institutional review board of the National Cancer Center, Japan. All methods were carried out in accordance with the relevant guidelines and regulations.
2.2. Cells and cell culture
Human colon cancer cell lines of SW480, LoVo, DLD‐1, HT‐29, HCT116, and Colo320 and hematopoietic cell lines of K562, Ramos, RL and Raji were purchased from ATCC. TMEM180 gene knockdown SW480 cells were established using shRNA vector (MISSION TRC clones TRC0000243137; Sigma‐Aldrich, St Louis, MO, USA) according to our previously reported protocol.11 For overexpression of the TMEM180 gene, plasmid pCMV‐TMEM180 (OriGene, Rockville, MD, USA) was transfected into DLD‐1 cells (DLD‐1‐OE) using Lipofectamine LTX (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol.
Growth and survival of the cells were evaluated using the WST‐8 assay (CCK‐8; Dojindo, Tokyo, Japan). Reaction signals were evaluated by measuring the absorbance at 450 nm and using a microplate reader (SpectraMax Paradigm; Molecular Devices, San Jose, CA, USA).
To evaluate the cellular response to hypoxia, DLD‐1 and SW480 cells were cultured in DMEM (Wako Pure Chemical Industries, Osaka, Japan) supplemented with 10% FBS (Life Technologies, Carlsbad, CA, USA) and a 1% penicillin‐streptomycin‐amphotericin B suspension (Wako Pure Chemical Industries) under normoxic (37°C, 5% CO2, 21% O2) or hypoxic (37°C, 5% CO2, 1% O2) conditions. BIONIX2 hypoxic cell culture kit (Sugiyama‐Gen Co., Ltd., Tokyo, Japan) was also used to generate hypoxic conditions according to the manufacturer's instructions.
2.3. DNA microarray
DNA microarray analysis was conducted using 10 μg total RNA from colon cancer cells or exfoliated colonocytes and the Affymetrix GeneChip Human Genome U133 plus 2.0 arrays; the analysis was conducted according to standard Affymetrix protocols.12 In the microarray analysis, 91 genes encoding membrane proteins, which were highly expressed in CRC cells but not in the colonocytes from healthy donors, were selected from 38 500 genes contained in the chip array.
2.4. Real‐time quantitative RT‐PCR
RNA extraction and qRT‐PCR were conducted as previously described. 13 PCR consisted of 10 μL TaqMan Fast Universal PCR Master Mix (Thermo Fisher Scientific), 1 μL TaqMan primer/probe mixture (Thermo Fisher Scientific) and 9 μL template cDNA diluted to a total volume of 20 μL. Real‐time PCR was carried out in an Applied Biosystems 7500 Fast System (Thermo Fisher Scientific). Relative quantification of the total RNA in each sample was conducted using the comparative Ct (threshold cycle) method. Relative expression of each gene was normalized against expression of GAPDH or ACTB.
2.5. In situ hybridization
In situ hybridization was conducted as previously described.10
Deparaffinized sections of human tissue (Genostaff Co., Ltd., Tokyo, Japan) were fixed with 4% paraformaldehyde (PFA; Wako Pure Chemical Industries). After treatment with 7 μg/mL proteinase K (Roche, Basel, Switzerland), the sections were refixed with 4% PFA and acetylated with 0.25% acetic anhydride. After the tissue was dehydrated, hybridization was done with digoxigenin‐labeled RNA probes (475‐bp fragment of TMEM180, GeneBank accession number NM_024789, nucleotide positions 1314‐1789). After the tissue was washed, RNase treatment was conducted. The sections were then rewashed and treated with 0.5% blocking reagent (Roche), followed by treatment with 20% heat‐inactivated sheep serum (Sigma‐Aldrich). After incubation with an alkaline phosphatase (AP)‐conjugated anti‐DIG antibody (Roche) for 2 hours, the sections were visualized using a nitro‐blue tetrazolium and 5‐bromo‐4‐chloro‐3’‐indolyphosphate (NBT/BCIP) solution (Roche).
2.6. Antibodies
Hybridoma cells producing anti‐TMEM180 monoclonal antibodies (mAbs) were established using myeloma cells (p3x63) and lymph node cells from a rat with immunizing recombinant human extradomain (355‐400 aa, UniProtKB entry number Q14CX5) of TMEM180‐ or TMEM180‐positive tumor exosomes. The TMEM180‐positive tumor exosomes were purified from the culture supernatant of DLD‐1‐OE cells using hydroxyapatite and gel filtration (Superdex 200 pg; GE Healthcare, Tokyo, Japan). Initially, the anti‐TMEM180 mAb (rat IgM) was obtained using recombinant human extradomain of TMEM180. Expression level of TMEM180 in the exosome was confirmed by ELISA with the mAb. Finally, the anti‐TMEM180 mAb (rat IgM, clone 669) was obtained using TMEM180‐positive tumor exosomes. Heavy‐chain variable and the kappa light‐chain variable‐region cDNAs were humanized and cloned into the vector for human IgG1 expression. The vectors were transfected into CHO cells (Riken BioResource Center, Ibaraki, Japan). A stable clone (humanized IgG, clone 669) was isolated. The procedure was conducted according to standard protocols.14
2.7. Exosome isolation, immunogold electron microscopy and ELISA
DLD‐1 cells (6.8 × 106) were plated on a 15‐cm dish and grown overnight. FBS‐supplemented medium was depleted by washing twice with PBS. The cells were then incubated with serum‐free culture medium at 30 mL/dish for 24 hours. After collection and filtration through a 0.22‐μm filter, the exosome‐containing supernatant was obtained. This supernatant was preserved at 4°C with a protease inhibitor (Wako Pure Chemical Industries) in each experiment.
For immunogold electron microscopy, isolated exosomes were adsorbed on nickel grids with supported films. Anti‐TMEM180 mAb, anti‐CD9 (Ancell Corporation, Bayport, MN, USA) or CD63 mAb (Ancell Corporation) was added to the grids, which were then incubated at room temperature. After the grids were washed with 1% BSA/PBS, they were incubated at room temperature with gold‐conjugated goat anti‐rat/mouse IgG polyclonal antibody (British BioCell International Ltd., Crumlin, UK) and washed with 1% BSA/PBS. After glutaraldehyde fixation, negative staining was carried out using a 2% solution of phosphotungstic acid (pH 7.0). The samples were then dried and visualized with transmission electron microscopy (JEM‐1400Plus; JEOL Ltd., Tokyo, Japan). Images were acquired with a CCD camera (VELETA; Olympus Soft Imaging Solutions GmbH, Münster, Germany).
For sandwich ELISA, anti‐TMEM 180 mAb or anti‐CD9 mAb (Ancell Corporation) as a capture antibody was coated onto 96‐well plates. Isolated exosomes from DLD‐1 or K562 cells were added to each plate and incubated for 1 hour at room temperature. After washing, the plates were incubated with HRP‐labeled anti‐TMEM180 mAb as a detection antibody for 1 hour at room temperature. After washing, absorbance at 450 nm was measured by Spectra Max paradigm (Molecular Devices, San Jose, CA, USA).
2.8. Immunohistochemistry
Human specimens were purchased from BioChain Institute, Inc (Newark, CA, USA). Immunohistochemistry (IHC) was carried out as previously described.15 Anti‐TMEM180 antibody or anti‐EGFR mAb (cetuximab, Merck, Darmstadt, Germany) was labeled with HRP using Peroxidase Labeling Kit‐SH (Dojindo). Tissues were then incubated with the HRP‐conjugated anti‐TMEM180 mAb or anti‐EGFR mAb for 1 hour at room temperature. After the tissue was washed with PBS, the reaction was visualized using diaminobenzidine (DAB) (Dako, Santa Clara, CA, USA), and the slides were counterstained with hematoxylin.
For cell staining, cultured cells were fixed with 4% PFA/PBS. After permeabilization with 0.1% Triton X‐100/PBS and blocking with 5% skimmed milk, the cells were stained with R‐phycoerythrin‐labeled anti‐TMEM180 using R‐Phycoerythrin Labeling Kit‐SH (Dojindo) as the primary antibody. The cells were then incubated with goat anti‐mouse IgG antibody conjugated to Alexa Fluor 488 (1:200; Thermo Fisher Scientific) as a secondary antibody and DAPI solution for nuclear staining. Images were acquired using a BZ‐9000 digital high‐definition microscopic system (Keyence Co., Osaka, Japan). Fluorescence intensity of TMEM180 from at least 1500 DAPI‐positive cells was analyzed using ImageJ (NIH). Representative results were from at least two independent experiments.
2.9. Western blotting
Cells were lysed with buffer containing 20 mmol/L Tris‐HCl (pH 7.5), 150 mmol/L NaCl, 0.1% SDS, 1% Triton X‐100 and cOmplete Protease Inhibitor Cocktail (Roche). The lysate was boiled at 95°C for 5 minutes for denaturing and inactivation of the endogenous proteases. The samples were separated using SDS‐PAGE (4%‐15% Mini‐PROTEAN TGX Precast Gels; Bio‐Rad, Hercules, CA, USA) and transferred onto PVDF membranes (Bio‐Rad). The membranes were blocked with TBS containing 5% skimmed milk and 0.1% Tween 20 (Sigma‐Aldrich). The blots were then incubated with anti‐hypoxia‐inducible factor 1‐alpha (anti‐HIF‐1α) (1:500; GeneTex, Inc., Irvine, CA, USA) or β‐actin (1:1000; Epitomics, Inc., Burlingame, CA, USA) antibodies as the primary antibody at room temperature for 2 hours or at 4°C overnight. The membranes were washed three times with TBS containing 0.1% Tween 20 (TBS‐T) and then incubated with HRP‐conjugated anti‐mouse (1:1000; R&D Systems, Minneapolis, MN, USA) or anti‐rabbit IgG (1:1000; Cell Signaling Technology, Inc., Danvers, MA, USA) as the secondary antibody. Can Get Signal solution (Toyobo, Osaka, Japan) was used to reduce background noise. After the membranes were washed with TBS‐T, proteins were visualized using ECL prime (GE Healthcare). The images were acquired using a ChemiDoc imager (Bio‐Rad).
2.10. Flow cytometry
Flow cytometry was conducted as previously described method.10 In vitro cultured cells were stained with anti‐TMEM180 mAbs (clone 669) as a first antibody and an Alexa Fluor 488/647‐conjugated anti‐mouse/human‐Ig polyclonal antibody (Thermo Fisher Scientific) as a secondary antibody. Stained cells were analyzed using a Guava easyCyte 10HT (Merck Millipore Co., Burlington, MA, USA) or Aria flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Dead cells, which were stained using propidium iodide (PI) (Thermo Fisher Scientific), were excluded from the analysis. The data were analyzed using the FlowJo program (Tree Star, Inc., San Carlos, CA, USA).
2.11. Antibody‐dependent cell‐mediated cytotoxicity and complement‐dependent cytotoxicity assay
51Cr release was conducted to evaluate antibody‐dependent cell‐mediated cytotoxicity (ADCC) and complement‐dependent cytotoxicity (CDC) activities. Here, 5 × 106 DLD‐1, SW480 or K562 cells were labelled for 2 hours at 37°C with 3.7 MBq of 51Cr (100 μCi).
For ADCC evaluation, cells were placed in 96‐well plates at 1 × 105 or 1 × 104 cells/well. The cells were exposed to anti‐TMEM180 mAb or anti‐EGFR mAb at 40‐fold the number of natural killer (NK) cells (Chemicals Evaluation and Research Institute Japan, Tokyo, Japan) as an effector target ratio and used as effector cells.
For CDC evaluation, cells were placed in 96‐well plates at 5 × 104 cells/well. The cells were exposed to anti‐TMEM180 mAb or anti‐EGFR mAb with complement human sera (Chemicals Evaluation and Research Institute Japan).
The cells were then incubated at 37°C, 5% CO2 for 4 hours. Cytotoxicity (%) = ([test sample release − background release]/[maximum release − background release]) × 100. All samples were run in triplicate.
2.12. Animal model and antitumor activity
Female BALB/c nude mice (5 weeks old) were purchased from Charles River Laboratories Japan Inc (Kanagawa, Japan).
To evaluate the antitumor activity of anti‐TMEM180 mAb, mice were inoculated s.c. in the flank with 5 × 106 DLD‐1 or SW480 cells. Length (L) and width (W) of tumor masses were measured every 4 days, and tumor volume was calculated using (L × W2)/2. When the mean tumor volume reached approximately 100‐200 mm3, the mice were randomly divided into groups that consisted of five mice each. PBS, anti‐TMEM180 mAb or anti‐EGFR mAb cetuximab was given on day 0 by i.p. injection. We selected an i.p. injection because it has been well used in similar experiments of antibody therapeutics.16, 17, 18, 19 Moreover, as it was reported in other references,20, 21 we confirmed that there was no clear difference in the biodistribution and tumor accumulation between i.p. injection and i.v. injection of the mAb (data not shown).
All animal procedures were carried out in compliance with the Guidelines for the Care and Use of Experimental Animals that were established by the Committee for Animal Experimentation of the National Cancer Center. These guidelines meet the ethical standards required by law and comply with the guidelines for the use of experimental animals in Japan.
2.13. Database analysis
Number of hypoxia response element consensus sequences (ACGTG or GCGTG)22, 23 at 3000 bp upstream (NC_000010.11, location: 102457895‐102460895) from exon 1 of the TMEM180 gene was analyzed. The sequence was obtained from NCBI gene databases (https://www.ncbi.nlm.nih.gov/gene/). TMEM180 gene expression in normal tissues and cancer tissues was analyzed using the microarray gene expression dataset from RefEx (http://refex.dbcls.jp/)24 and Oncomine (http://www.oncomine.org/),25 respectively. Comparison of TMEM180 gene expression between 286 colorectal adenocarcinoma patient samples and 41 normal tissue samples using The Cancer Genome Atlas (TCGA) cancer genomics data was carried out using UALCAN (http://ualcan.path.uab.edu/index.html).26
2.14. Generation of TMEM180 KO mice
TMEM180 gene KO mice were generated using CRISPR/Cas9‐mediated genome editing technology.27 Briefly, guide RNAs (gRNAs) at exon 2 of mouse TMEM180 genome immediately downstream of the ATG start codon were designed using CRISPRdirect.28 Two gRNAs [gS01(5′‐AGCTGTGGTGTACGGCTCGTTGG‐3′), gS03(5′‐TGTGTTCCTGCTGTACTACGTGG‐3′)] were selected in an in vitro digestion assay system29 using the target TMEM180 PCR product. These two gRNAs were inserted into the BbsI restriction site in the px330 plasmid and constructed as a px330‐TMEM180 genome editing vector.
Pronuclear stage ova of C57BL/6J (Clea Japan, Tokyo, Japan) were prepared by in vitro fertilization and injection of 1.67 ng/μL px330‐TMEM180 DNA vector into the pronucleus according to standard protocols.30 The injected ova were transferred into the oviduct of the pseudopregnant Jcl:MCH (Clea Japan) female on that day.
Two viable mice from gS01 and 14 viable mice from gS03 were obtained independently by cesarean section. Genotyping of mouse tail DNA by PCR [forward (5′‐CTAATAGGCAACCGCAGAGC‐3′), reverse (5′‐CTAAACAGCACAGTCTGCCC‐3′)] and purified PCR products were sequenced using the forward primer. Results of the sequence analysis are shown in Table S1.
2.15. Statistical analysis
Significant differences between the groups were determined using Student's t test for ELISA data, ANOVA to compare tumor size and body weight, and chi‐squared test. All analyses were carried out using SPSS software version 20 (IBM, Armonk, NY, USA).
3. RESULTS
3.1. Identification and characterization of TMEM180 as a new CRC marker
We previously developed a method to obtain almost pure normal mucoepithelial cells from the lavage solution following colonoscopies of healthy examinees using immune beads with anti‐EpCAM mAb.10 Moreover, the previous study reported a comprehensive DNA microarray analysis comparing CRC cell lines (SW480, LoVo, DLD‐1, HT‐29 and HCT116) and normal mucoepithelial cells.10 We reanalyzed these microarray data and found that TMEM180 was highly expressed in five CRC cell lines but not in the normal colonocytes obtained from lavage of two healthy volunteers (Figure 1A). In quantitative RT‐PCR (qPCR) analysis, carried out as a first validation, TMEM180 was expressed at high levels in CRC tissue samples (Figure 1B). In situ hybridization (ISH) was then conducted for the final validation (Figure 1C,D and Figure S1). All of these methods indicated that TMEM180 was a new CRC marker (Figure 1).
Figure 1.

Identification of TMEM180 as a colorectal cancer‐specific marker. A, TMEM180 gene expression in five colorectal cancer cell lines and two colonocyte samples from healthy donors was evaluated using DNA microarray analysis. B, TMEM180 gene expression in five clinical samples was evaluated using quantitative RT‐PCR. Relative quantification as tumor‐to‐normal tissue ratio. C,D, In situ hybridization of TMEM180 in the clinical samples. Arrows indicate cancer (D)
3.2. First production of anti‐TMEM180 antibody with a recombinant TMEM180 peptide
To evaluate the potential of TMEM180 as a new target for the diagnosis and/or therapy of CRC, we attempted to produce a mAb. However, the TMEM180 gene is predicted to encode an 11‐pass transmembrane protein with limited extracellular domain expression. First, we immunized rats with a recombinant extracellular domain of the TMEM180 protein and obtained an IgM mAb. During the evaluation process, we unexpectedly noticed that the TMEM180 protein was present in the culture supernatant of SW480 and DLD‐1 CRC cells. We collected the supernatant and subjected it to hydroxyapatite and gel filtration chromatography. We found that a high concentration of the TMEM180 protein existed in the void fraction (Figure S2A).
As a result of the detection of TMEM180 in the culture supernatant, we hypothesized that TMEM180 coexisted with exosomes or other extracellular microparticles. To test whether TMEM180 exists on tumor‐derived exosomes, we conducted immune electron microscopy for the enriched exosomes with CD9 or CD63 mAbs and the TMEM180 mAb. The images clearly indicated that TMEM180 was present on the tumor exosome, similar to CD9 and CD63 (Figure S2B). In the sandwich ELISA, both TMEM180‐positive and CD9‐positive exosomes were detected (Figure S2C). We concluded that the tumor exosomes released from DLD‐1 cells contain TMEM180.
3.3. Second production of anti‐TMEM180 antibody with TMEM180‐positive exosomes and validation of a CRC‐specific target
We judged that purifying the full‐length TMEM180 protein while maintaining the native structure necessary for producing a useful mAb recognizable for the live cell would be the most difficult step. Moreover, we thought that several mAbs with a higher affinity could be obtained by antigens with native structure. Therefore, we immunized rats again with TMEM180‐positive tumor exosomes purified from the supernatant of DLD‐1 cells. The newly obtained anti‐TMEM180 mAb (IgM, clone 669) was able to recognize most CRC cells, but not hematopoietic cells (Figure S3A,B).
We then converted the rat IgM to humanized IgG1 in order to evaluate the potential for therapeutic application of the mAb. First, we examined the expression of TMEM180 in CRC and normal major tissues (colon, skin, liver, brain, kidney, lung and heart) by IHC. Cetuximab (an anti‐EGFR mAb) was used as a reference as it is currently widely used for CRC patients. We found that nine of 37 CRC tissues (24.3%) were TMEM180‐strong positive and 43.2% (16/37) were TMEM180‐weak positive. Regarding EGFR, seven of 37 CRC tissues (18.9%) were alternate EGFR‐strong positive and 35.1% (13/37) were EGFR‐weak positive. However, there was no significant difference in the expression level between TMEM180 and EGFR (Figure 2A,B). IHC with the mAb did not show staining in major organs including the brain, heart, lung, liver, kidney, colon and skin (Figure 2C). In contrast, IHC with cetuximab showed strongly positive staining in the skin, moderately positive staining in the liver and colon and weakly positive staining in the brain, kidney and lung (Figure 2C).
Figure 2.
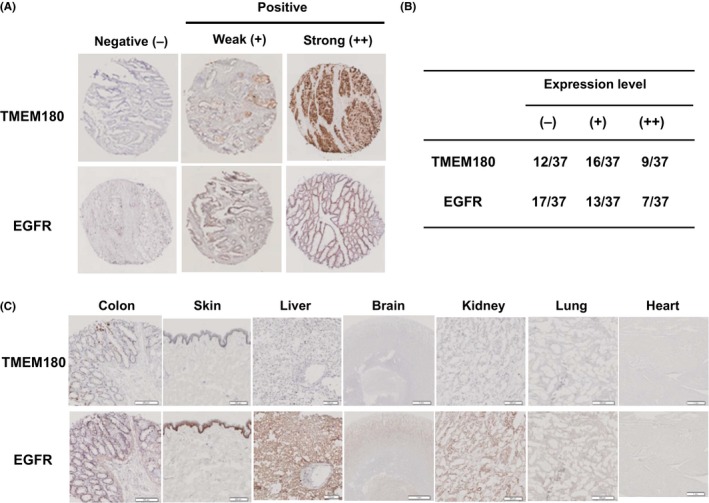
TMEM180 expression in colorectal cancer (CRC) and normal tissues, A, Representative immunostaining patterns of TMEM180 and epidermal growth factor receptor (EGFR) in CRC tissues were classified as negative (−), a weak positive (+) and strong positive (++). B, Number of cases with each expression level (−), (+) or (++) of TMEM180 or EGFR in CRC tissues (total number 37) is shown. C, Immunohistochemistry showing the expression of TMEM180 or EGFR in normal tissues (colon, brain, heart, lung, liver, kidney and skin). Scale bar, 100 μm
According to a database, the expression of TMEM180 at the mRNA level is very low not only in normal organs but also in their respective cancer tissues (Figure S4A,B). However, clinical samples used in these datasets would contain various stromal cells, such as fibroblasts or blood cells. Therefore, the expression level of TMEM180 was underestimated in the regular methods used in the database. By contrast, our unique comprehensive expression analysis using pure CRC cells and normal colonocytes without stroma cells could increase the expression level of TMEM180 to enable detection stably as a new CRC marker.
3.4. Diagram and functional role of TMEM180
TMEM180 is classified in the major facilitator superfamily and appears to function as a cation symporter (Figure 3A). To address the functional role of TMEM180, we established two TMEM180 gene knockdown (KD) SW480 cell lines: KD1 cells with the lowest expression of TMEM180 protein, and KD2 cells with intermediate TMEM180 protein expression (Figure 3B). We investigated the uptake of glutamine and arginine in the cation symport mechanism because the utilization of both amino acids was strongly increased in tumor cells.31, 32 We found that SW480‐WT cells were capable of growing in a serum‐free medium with glutamine and arginine. By contrast, both KD1 and KD2 could not grow in the same medium (Figure 3C). These findings suggest TMEM180 has an important role in the uptake or metabolism of glutamine and arginine in tumor growth and proliferation.
Figure 3.

Diagram and functional role of TMEM180. A, Diagram of TMEM180 as a cation symporter is shown. B, Flow cytometric analysis of the SW480 wild‐type (WT), gene knockdown clone no. 1 (KD1) or gene knockdown clone no. 2 (KD2) cells. Difference in the TMEM180 expression level in SW480‐WT, ‐KD1 and ‐KD2 cells is shown. C, Cell growth of SW480‐WT, ‐KD1 or ‐KD2 cells in (i) glutamine (Gln)‐positive (+), arginine (Arg) (+) and Serum (+); (ii) Gln (+), Arg (+) and Serum‐negative (−); (iii) Gln (+), Arg (−) and Serum (−); or (iv) Gln (−), Arg (+) and Serum (−) Each growth activity was evaluated by WST‐8
3.5. Anti‐TMEM180 mAb is highly effective in the targeted inhibition of CRC tumors
Anti‐TMEM180 mAb showed little to no direct cytotoxicity in DLD‐1 and SW480 cells in vitro (Figure S5A). We next evaluated ADCC and CDC in DLD‐1 and SW480 cells. Cetuximab was also used in this study as a reference. Anti‐TMEM180 mAb appeared to have ADCC and CDC in both cell lines (Figure S5B,C).
Because of in vitro ADCC activity of both mAbs, we evaluated the in vivo antitumor activity of the anti‐TMEM180 mAb and cetuximab in mice bearing DLD‐1 or SW480 xenografts. Preliminary experiments showed that cetuximab exerted statistically significant antitumor effects in both tumor xenografts. In the case of anti‐TMEM180 mAb, the antitumor effect was significant in DLD‐1 xenografts. Strikingly, all SW480 xenografts were completely eradicated by the anti‐TMEM180 mAb. We then confirmed these results using a dosage of 25 or 12.5 mg/kg given twice a week for a total of eight injections for both mAbs in mice bearing DLD‐1 or SW480 xenografts, respectively. In mice with the DLD‐1 xenografts, both anti‐TMEM180 mAb and cetuximab showed a significant antitumor effect compared to the control treatment (Figure 4A). In mice with the SW480 xenografts, cetuximab showed a significant antitumor effect at 25 mg/kg but not at 12.5 mg/kg. In contrast, anti‐TMEM180 mAb elicited a striking antitumor effect, eradicating the tumor at both 25 and 12.5 mg/kg (Figure 4A). There was no significant difference in the treatment‐related body weight loss among the five groups (Figure 4B).
Figure 4.
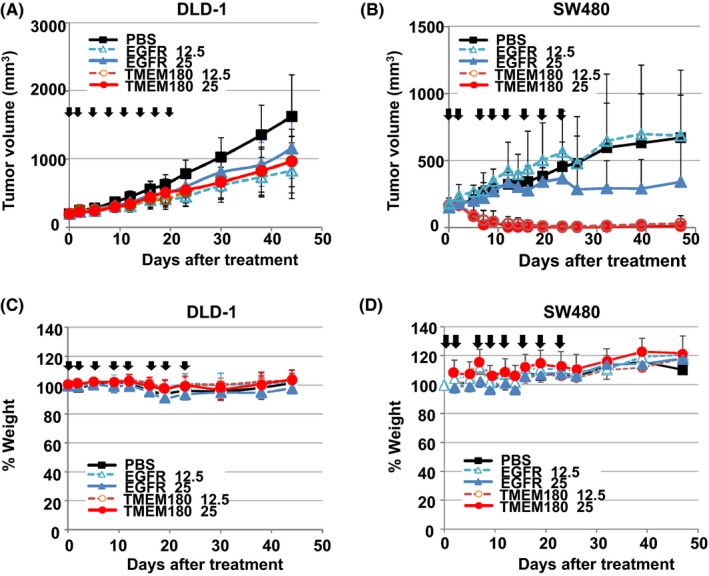
Antitumor effect of the anti‐TMEM180 antibody against colorectal cancer tumors. A,B, Antitumor activity of the anti‐TMEM180 mAb against DLD‐1 (A) and SW480 (B) xenograft models. When the tumor volume reached approximately 200 mm3, each treatment started (day 0). Anti‐TMEM180 mAb (TMEM180), anti‐epidermal growth factor receptor (EGFR) mAb cetuximab (EGFR) or saline (PBS) as a control was given at a dose of 12.5 or 25 mg/kg to each group of mice (n = 5) by i.p. injection twice a week from days 0 to 22. Arrows indicate the days of dosage, and the curves show the effect of the treatment on tumor size. P < .001 (SW480 xenograft, PBS compared with TMEM180 12.5 mg/kg or 25 mg/kg; EGFR 12.5 mg/kg compared with TMEM180 25 mg/kg), P < .01 (DLD‐1 xenograft, PBS compared with EGFR 25 mg/kg, TMEM180 12.5 mg/kg or 25 mg/kg; SW480 xenograft, EGFR 25 mg/kg compared with TMEM180 12.5 mg/kg). P < .05 (DLD‐1 xenograft, PBS compared with EGFR 12.5 mg/kg). Bar, SD. C,D, Percent change in body weight in the same mice with the same treatments as in A and B. Bar, SD
3.6. Biological characteristics of TMEM180 expression in CRC cells and tissues
Using a public database to analyze the promoter region of the TMEM180 gene, we found that there were 10 hypoxia response element consensus sequences22, 23 in the gene (Figure 5A). Moreover, HIF‐1α expression was elevated in both DLD‐1 and SW480 cells in hypoxic conditions (Figure 5B). We therefore evaluated the change in TMEM180 expression in response to the same treatment. Hypoxia significantly increased TMEM180 mRNA expression in SW480 cells and also tended to increase expression in DLD‐1 cells (Figure 5C). Protein expression level of TMEM180 was significantly increased in both DLD‐1 and SW480 cells after hypoxia treatment (Figure 5D,E). Therefore, TMEM180 expression in CRC cells is regulated by hypoxia.
Figure 5.
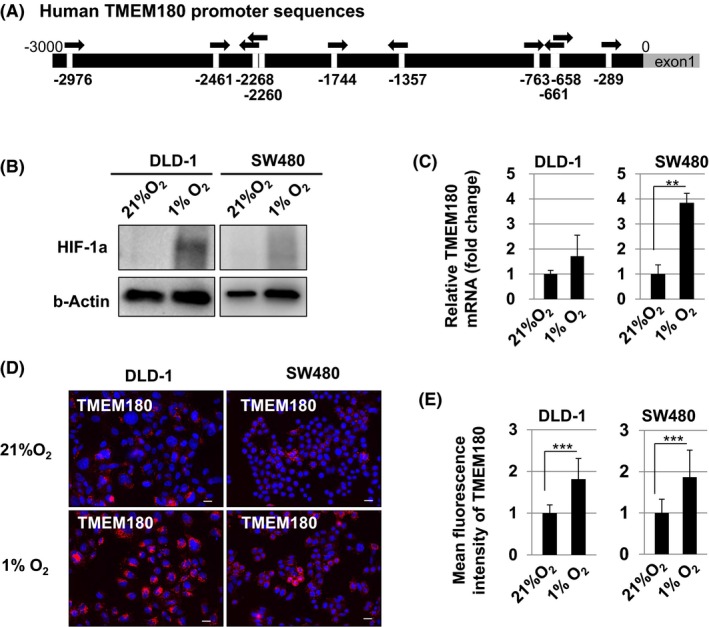
TMEM180 expression is controlled by hypoxia in colorectal cancer (CRC) cells. A, Schema of the promoter in the TMEM180 gene. Ten hypoxia response element consensus sequences (HRE) were found in the promoter sequence. HRE are indicated by arrows. B, Western blotting of hypoxia‐inducible factor 1‐alpha (HIF‐1α). Difference in the HIF‐1α protein expression level in the CRC cell lines DLD‐1 and SW480 under normoxia (21% O2) and hypoxia (1% O2) is shown. Expression of β‐actin was used as a control. C, Quantitative RT‐PCR of TMEM180. Difference in the TMEM180 mRNA expression level in DLD‐1 and SW480 cells under normoxia (21% O2) and hypoxia (1% O2) is shown. **P < .01. Bar, SD. D,E, Immunostaining of TMEM180. Change in the TMEM180 expression pattern in DLD‐1 and SW480 cells under normoxia (21% O2) and hypoxia (1% O2) is shown (D). Scale bar, 10 μm. Data were quantified and are shown in (E). ***P < .001. Bar, SD
4. DISCUSSION
In the present study, we identified a new CRC‐specific molecule, TMEM180, by taking advantage of our original method,9, 10 and succeeded in establishing a mAb against TMEM180 followed by humanizing it. The expression profile of TMEM180 differed from the expression profile of EGFR, and TMEM180 mAb staining was almost absent in normal tissues, unlike staining with the EGFR mAb. We also found that TMEM180 was expressed at levels as high as the levels of other existing markers such as EGFR and CD44 in cancers and at extremely low levels compared to the levels of other molecules in normal tissues (Figure S6). We, therefore, think that the tumor specificity of TMEM180 is substantially higher than that of other molecules.
More importantly, anti‐TMEM180 mAb had in vitro ADCC and CDC activity and significant in vivo antitumor activity in mice bearing TMEM180‐positive CRC cell xenografts. Anti‐TMEM180 mAb treatment suppressed DLD‐1 tumor growth equally to cetuximab treatment. Surprisingly, SW480 tumors were completely eradicated by anti‐TMEM180 mAb treatment but not by cetuximab treatment. This remarkable antitumor effect is now under evaluation.
In further experiments, we found that the TMEM180 gene had 10 hypoxia response element consensus sequences, which may be HIF‐1α binding sites controlled by hypoxia.22, 23, 33 Recent studies have indicated that not only hypoxia but also chemotherapy induces HIF‐1α expression followed by T‐cell anergy and increased dissemination of cancer cells.34 HIF‐1α is currently considered a key factor of cancer stem cell function. In cancer tissues, unlike in normal tissues, angiogenesis and hypoxia associated with the necrosis caused by cancer‐induced blood coagulation are repeated alternately during clinical progression.35, 36, 37 However, HIF‐1α is upregulated in many types of cancer besides CRC. Therefore, another transcription factor specific in CRC might regulate TMEM180 expression in conjunction with HIF‐1α. Further molecular and translational studies are needed to elucidate the detailed biological role of TMEM180 and the antitumor mechanism of its targeting therapy.
Safety of a drug should also be considered for therapeutic use in clinical practice. According to the database, TMEM180 mRNA is hardly expressed in any normal tissues (Figure S4A). Also, the expression of TMEM180 is extremely low in all organs compared to the expression of other known CRC markers. In the present study, IHC also showed that there was no TMEM180 expression in all major organs studied. Finally, we succeeded in obtaining a TMEM180 gene KO mouse that does not show embryonic, neonatal, and postnatal lethality (Table S1). According to these data, we expect that unpredictable adverse effects are unlikely to occur in a clinical trial of anti‐TMEM180 mAb.
Our mAb is now being processed according to Good Manufacturer Practice principles for testing in a first‐in‐human clinical trial. We think that there are several concerns to be clarified before entering a clinical trial. We must investigate further antitumor activity using other CRC cell lines and PDX tumors. We will make progress on the functional analysis of TMEM180 including the involvement in glutamine and arginine metabolism as a cation transporter after obtaining an F2 generation of the KO mice. We hope that a future clinical trial will verify the significance of the mAb for the treatment of CRC.
CONFLICTS OF INTEREST
Yasuhiro Matsumura is co‐founder, shareholder and Board Member of RIN Institute Inc., the company that owns the anti‐TMEM180 antibody. Masahiro Yasunaga is shareholder of RIN Institute Inc. Shinji Saijou, Shingo Hanaoka, Takahiro Anzai and Ryo Tsumura declare no conflict of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported in part by a research and development grant by the New Energy and Industrial Technology Development Organization (NEDO), National Cancer Center Research and Development Fund (26‐A‐14, 26‐A‐12, 29‐A‐9, 29‐S‐1), and Project for Cancer Research and Therapeutic Evolution from the Japan Agency for Medical Research and Development (AMED) (17cm0106415 h0002).
Yasunaga M, Saijou S, Hanaoka S, Anzai T, Tsumura R, Matsumura Y. Significant antitumor effect of an antibody against TMEM180, a new colorectal cancer‐specific molecule. Cancer Sci. 2019;110:761–770. 10.1111/cas.13907
REFERENCES
- 1. World Health Organisation . The World Health Organization’s Mortality Database. 2002. http://www.ciesin.org/IC/who/MortalityDatabase.html. Accessed December 10, 2016.
- 2. Damelin M, Zhong W, Myers J, Sapra P. Evolving strategies for target selection for antibody‐drug conjugates. Pharm Res. 2015;32:3494‐3507. [DOI] [PubMed] [Google Scholar]
- 3. Mack F, Ritchie M, Sapra P. The next generation of antibody drug conjugates. Semin Oncol. 2014;41:637‐652. [DOI] [PubMed] [Google Scholar]
- 4. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Can Res. 1986;46:6387‐6392. [PubMed] [Google Scholar]
- 5. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441‐1454. [DOI] [PubMed] [Google Scholar]
- 6. Lyon RP, Bovee TD, Doronina SO, et al. Reducing hydrophobicity of homogeneous antibody‐drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33:733‐735. [DOI] [PubMed] [Google Scholar]
- 7. Bosman FT. Prognostic value of pathological characteristics of colorectal cancer. Eur J Cancer. 1995;31A:1216‐1221. [DOI] [PubMed] [Google Scholar]
- 8. Arora N, Gupta A, Singh PP. Biological agents in gastrointestinal cancers: adverse effects and their management. J Gastrointest Oncol. 2017;8:485‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamao T, Matsumura Y, Shimada Y, et al. Abnormal expression of CD44 variants in the exfoliated cells in the feces of patients with colorectal cancer. Gastroenterology. 1998;114:1196‐1205. [DOI] [PubMed] [Google Scholar]
- 10. Yasunaga M, Matsumura Y. Role of SLC6A6 in promoting the survival and multidrug resistance of colorectal cancer. Sci Rep. 2014;4:4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saito Y, Hashimoto Y, Kuroda J, et al. The inhibition of pancreatic cancer invasion‐metastasis cascade in both cellular signal and blood coagulation cascade of tissue factor by its neutralisation antibody. Eur J Cancer. 2011;47:2230‐2239. [DOI] [PubMed] [Google Scholar]
- 12. Yasunaga M, Tada S, Torikai‐Nishikawa S, et al. Induction and monitoring of definitive and visceral endoderm differentiation of mouse ES cells. Nat Biotechnol. 2005;23(12):1542‐1550. [DOI] [PubMed] [Google Scholar]
- 13. Koga Y, Yasunaga M, Moriya Y, et al. Detection of colorectal cancer cells from feces using quantitative real‐time RT‐PCR for colorectal cancer diagnosis. Cancer Sci. 2008;99:1977‐1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuramochi T, Igawa T, Tsunoda H, Hattori K. Humanization and simultaneous optimization of monoclonal antibody. Methods Mol Biol. 2014;1060:123‐137. [DOI] [PubMed] [Google Scholar]
- 15. Obonai T, Fuchigami H, Furuya F, Kozuka N, Yasunaga M, Matsumura Y. Tumour imaging by the detection of fibrin clots in tumour stroma using an anti‐fibrin Fab fragment. Sci Rep. 2016;6:23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luo FR, Yang Z, Dong H, et al. Correlation of pharmacokinetics with the antitumor activity of Cetuximab in nude mice bearing the GEO human colon carcinoma xenograft. Cancer Chemother Pharmacol. 2005;56:455‐464. [DOI] [PubMed] [Google Scholar]
- 17. Goldenberg DM, Rossi EA, Stein R, et al. Properties and structure‐function relationships of veltuzumab (hA20), a humanized anti‐CD20 monoclonal antibody. Blood. 2009;113:1062‐1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wild R, Fager K, Flefleh C, et al. Cetuximab preclinical antitumor activity (monotherapy and combination based) is not predicted by relative total or activated epidermal growth factor receptor tumor expression levels. Mol Cancer Ther. 2006;5:104‐113. [DOI] [PubMed] [Google Scholar]
- 19. Chen Q, Weng Z, Lu Y, et al. An experimental analysis of the molecular effects of Trastuzumab (Herceptin) and Fulvestrant (Falsodex), as single agents or in combination, on human HR+/HER2+ breast cancer cell lines and mouse tumor xenografts. PLoS ONE. 2017;12:e0168960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koppe MJ, Soede AC, Pels W, et al. Experimental radioimmunotherapy of small peritoneal metastases of colorectal origin. Int J Cancer. 2003;106:965‐972. [DOI] [PubMed] [Google Scholar]
- 21. Lee CM, Tannock IF. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer. 2010;10:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ke Q, Costa M. Hypoxia‐inducible factor‐1 (HIF‐1). Mol Pharmacol. 2006;70:1469‐1480. [DOI] [PubMed] [Google Scholar]
- 23. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature. 1999;399:271‐275. [DOI] [PubMed] [Google Scholar]
- 24. Ono H, Ogasawara O, Okubo K, Bono H. RefEx, a reference gene expression dataset as a web tool for the functional analysis of genes. Sci Data. 2017;4:170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: a cancer microarray database and integrated data‐mining platform. Neoplasia. 2004;6:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naito Y, Hino K, Bono H, Ui‐Tei K. CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off‐target sites. Bioinformatics. 2015;31:1120‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gordon JW, Scangos GA, Plotkin DJ, Barbosa JA, Ruddle FH. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci USA. 1980;77:7380‐7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palm W, Thompson CB. Nutrient acquisition strategies of mammalian cells. Nature. 2017;546:234‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Can Res. 2015;75:1782‐1788. [DOI] [PubMed] [Google Scholar]
- 33. Chiu DK, Tse AP, Xu IM, et al. Hypoxia inducible factor HIF‐1 promotes myeloid‐derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu H, Chen I, Shimoda LA, et al. Chemotherapy‐induced Ca(2+) release stimulates breast cancer stem cell enrichment. Cell Rep. 2017;18:1946‐1957. [DOI] [PubMed] [Google Scholar]
- 35. Hisada Y, Yasunaga M, Hanaoka S, et al. Discovery of an uncovered region in fibrin clots and its clinical significance. Sci Rep. 2013;3:2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yasunaga M, Manabe S, Matsumura Y. New concept of cytotoxic immunoconjugate therapy targeting cancer‐induced fibrin clots. Cancer Sci. 2011;102:1396‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Semenza GL. Hypoxia‐inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials