

SUMMARY
Activated T cells differentiate into functional subsets with distinct metabolic programs. Glutaminase (GLS) converts glutamine to glutamate to support the tricarboxylic acid cycle and redox and epigenetic reactions. Here, we identify a key role for GLS in T cell activation and specification. Though GLS deficiency diminished initial T cell activation and proliferation and impaired differentiation of Th17 cells, loss of GLS also increased Tbet to promote differentiation and effector function of CD4 Th1 and CD8 CTL cells. This was associated with altered chromatin accessibility and gene expression, including decreased PIK3IP1 in Th1 cells that sensitized to IL-2-mediated mTORC1 signaling. In vivo, GLS null T cells failed to drive Th17-inflammatory diseases, and Th1 cells had initially elevated function but exhausted over time. Transient GLS inhibition, however, led to increased Th1 and CTL T cell numbers. Glutamine metabolism thus has distinct roles to promote Th17 but constrain Th1 and CTL effector cell differentiation.
Graphical Abstract
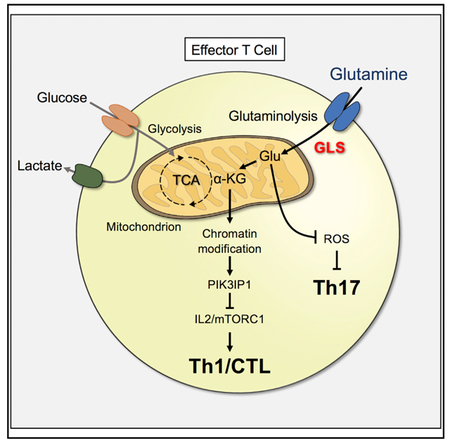
In Brief
Glutamine metabolism, and its effects on chromatin, promotes Th17 but constrains Th1 and CTL effector cell differentiation.
INTRODUCTION
Stimulated T cells exit quiescence to proliferate and develop effector functions that are essential for immunity. To support the metabolic demands of an immune response, antigen receptor signals and co-stimulation activate the phosphatidtyl-inosi-tide-3 kinase (PI3K)/Akt/mTORC1 signaling pathway to induce Myc and metabolic flux through glycolysis and mitochondrial oxidative phosphorylation (Ho et al., 2015; Sena et al., 2013). Elevated glycolysis and mitochondrial production of reactive oxygen species also promote T cell calcium signaling (Ho et al., 2015; Sena et al., 2013) and specific effector functions, including transcription and translation of interferon-γ (IFNγ) (Chang et al., 2013; Jacobs et al., 2008; Peng et al., 2016) in activated Th1 CD4 T cells and CD8 T cells. Importantly, each T cell subset utilizes and requires a distinct metabolic program (Michalek et al., 2011; Nakaya et al., 2014). If activated T cells fail to induce appropriate metabolic pathways, effector function and ability to induce inflammatory disease In vivo are impaired (Macintyre et al., 2014; Yin et al., 2015). How specific metabolic programs establish and promote the function of T cell subsets remains poorly understood but may allow selective modulation of immunity in inflammation and cancer.
In addition to increased use of glucose in activated effector T cells, glutamine uptake and glutaminolysis are also upregulated (Carr et al., 2010; Wang et al., 2011). Glutamine is a conditionally essential amino acid in rapidly proliferating cells (Curthoys and Watford, 1995) and a potential target in cancer treatment (DeBerardinis and Cheng, 2010; Cheng et al., 2011; Wise and Thompson, 2010). Activated T cells upregulate amino acid transporters (Sinclair et al., 2013) and enzymes that metabolize glutamine (Nakaya et al., 2014; Wang et al., 2011). Glutamine is initially hydrolyzed via the enzyme glutaminase (GLS) (Wang et al., 2010) to produce glutamate, which is used in protein synthesis and generation of glutathione to regulate reactive oxygen species (ROS) and exchanged to promote cystine uptake (Siska et al., 2016). Glutamate is further metabolized to α-ketoglutarate (α-KG), which provides anaplerotic support of the tricarboxylic acid cycle (TCA) cycle in growing cells (Yuan et al., 2014) and is a substrate for histone and DNA demethylases that regulate chromatin accessibility (Nakajima and Kunimoto, 2014).
In addition to effector T cell activation, glutamine metabolism is implicated in the establishment of specific CD4 T cell subsets. Glutamine deprivation or deletion of ASCT2 was shown to promote Foxp3 expression, the transcription factor of regulatory T cells (Treg) (Klysz et al., 2015; Nakaya et al., 2014). Inhibition or silencing of the glutamic-oxaloacetate transaminase 1 (GOT1) enzyme that mediates conversion of glutamate to α-KG led to a shift in differentiation of Th17 to Treg via methylation of the Foxp3 locus (Xu et al., 2017). Further, direct treatment of T cells with α-KG altered gene expression and chromatin methylation, in part through the CCCTC-binding factor (CTCF) (Chisolm et al., 2017).
Conversion of glutamine to glutamate by GLS may play a critical role in T cell function and fate. Although under investigation as a target to inhibit the metabolism of cancer cells (Cerione and Richard, 2010; DeBerardinis et al., 2008), the role of GLS in T cells has been unclear. Here, we explore whether inhibition of glutaminolysis at the level of GLS alters T cell effector function and show distinct T cell dependencies on this metabolic pathway.
RESULTS
GLS and Glutaminolysis Contribute to T Cell Metabolism upon Activation
Activated T cells have significant metabolic requirements to support proliferation and differentiation. To determine the relative roles of glucose and glutamine in these processes, intracellular metabolites were measured following activation of CD4 T cells. In addition to increased pyruvate and lactate, glutamate and α-KG levels increased, suggesting elevated glutamine metabolism (Figure 1A). Intracellular glutamate is primarily generated from glutamine by GLS or from α-KG and aspartate by GOT1 and GOT2 and is converted to α-KG by glutamate dehydrogenase 1 (GLUD1), which are each expressed in CD4 and CD8 T cells (Figure S1A). The increased levels of both α-KG and glutamate, and high relative ratio of glutamine to glutamate (Figure S1B), suggested GLS as a key source of glutamate and α-KG. To determine the relative roles of glutaminolysis and glycolysis as fuels for mitochondrial metabolism, we measured oxygen consumption of stimulated T cells treated with mitochondrial pyruvate carrier (UK5099) or GLS (CB839) inhibitors. While neither UK5099 nor CB839 was sufficient to reduce T cell respiration alone, the combination led to a significant decrease in oxygen consumption (Figure 1B).
Figure 1. Activated T Cells Rely on Both Glucose and Glutamine to Sustain Cell Metabolism.

(A) Metabolites extracted for mass spectrometry and presented as fold change from naive in T cells stimulated for 16 hr (S) or naive (N) conditions (one-way ANOVA).
(B) Oxygen consumption rate (OCR) assayed from naive CD4 cells from wild-type (WT) mice stimulated for 3 days on αCD3/CD28, injected with drug described (top). OCR at time point 200 min (bottom, one-way ANOVA).
(C and D) Abundance of metabolites (left, unpaired t test) and fractional labeling (right, one-way ANOVA) of stimulated CD4+ T cells in the presence of CB839 and 13C-glucose for glutaminolytic intermediates (C) and TCA intermediates (D).
Also see Figure S1.
To directly determine how inhibition of GLS affects glucose metabolism, CD4 T cells were stimulated in uniformly labeled 13C-glucose with or without CB839, and glucose-derived carbons were traced. As expected, inhibition of GLS led to increased intracellular glutamine and decreased glutamate (Figure 1C). Aspartate levels also decreased significantly. Glucose-derived 13C was increased in both glutamate and aspartate, indicating a greater fraction of glucose contribution to synthesis of these amino acids. Serine and alanine overall abundance also decreased while glycine was unchanged, and each showed a decreased portion derived from glucose (Figure S1C). Overall levels of TCA intermediates were also reduced, yet with increased fractional labeling from glucose-derived 13C (Figure 1D). Glycolytic intermediates were more abundant upon GLS inhibition, suggesting elevated glycolysis (Figure S1D). However, lactate levels and 13C labeling were unchanged, and pyruvate levels decreased (Figure S1E). Anabolic pathways were also affected, and total levels of the nucleotide precursor N-carbamoyl-aspartate decreased (Figure S1F).
CD4 T Cell Subsets Have Distinct Programs of Glutamine Metabolism
Distinct cytokines led activated T cells to induce specific metabolic programs. Th1, Th17, and Treg cells (Gerriets et al., 2015) were examined to assess whether CD4 T cell subsets had different patterns and reliance on glutamine metabolism. T cells activated and differentiated into each subset showed increased glutamate and α-KG levels relative to naive T cells. This was most pronounced in Th17 cells (Figure 2A), which also had the highest relative ratio of glutamate to glutamine (Figure S2A). To test the role of glutamine and GLS in Th1, Th17, and Treg T cell subsets, CD4 T cells were differentiated with or without glutamine or with GLS inhibitor. Both Th1 and Th17 required glutamine, as glutamine-deficient media markedly reduced Th1 production of IFNγ and Th17 production of IL-17, yet GLS inhibition decreased cytokine production and proliferation only from Th17 cells and appeared to increase Th1 cytokine secretion (Figure 2B). Glutamine deficiency reduced proliferation at days 3 and 5 in both Th1 and Th17 cells. GLS inhibition impaired proliferation of both Th1 and Th17 cells after 3 days in culture (Figures 2C and 2D). Importantly, CB839-treated Th1 cells partially recovered proliferation by day 5. Glutamine deprivation also induced Treg under Th1 and Th17 skew conditions, yet GLS inhibition did not (Figure 2E).
Figure 2. Th1 and Th17 Cells Differ in Their Use of Glutaminolysis, and GLS Deficiency Is Distinct from Glutamine Deficiency.

(A) Metabolite fold change from naive in wild-type CD4+ cells maintained in IL-7 (N) or differentiated for 5 days into Th1 (1), Th17 (17), or Treg (R) cells (one-way ANOVA).
(B) Cytokine production from Th1 (top) and Th17 (bottom) differentiated T cells in the presence of glutamine (left), absence of glutamine (middle), or presence of GLS1-inhibitor CB839 (right).
(C) Proliferation of cell trace violet (CTV)-labeled T cells stimulated and differentiated in Th1 or Th17 conditions with (black lines) or without (red lines) glutamine after 3 and 5 days of culture.
(D) Same as in (C), but with vehicle (black lines) or CB839 (green lines).
(E) Foxp3 expression in CD4 T cells activated in Th1- or Th17-skewing conditions in glutamine deficient (red, left) conditions or in the presence of CB839 (green, right).
(F) Heatmap (left) and principle component analysis (right) of metabolites from Th1 and Th17 cells with or without CB839.
Several enzymes contribute to regulation of glutaminolysis in T cells. Th17 cells had greater expression of GLS than Th1 at protein and RNA levels (Figures S2B and S2C). Th1 and Th17 cells expressed low levels of Gls2 mRNA and expressed similar levels of other glutamine and anaplerotic metabolic enzymes (Figures S2C–S2E). Th1 and Th17 cells had distinct metabolic profiles, and intracellular metabolites shifted in both Th1 and Th17 cells upon GLS inhibition, including alanine, aspartate, and glutamate metabolism pathways (Figure 2F; Table S1). Nutrient uptake and secretion also differed between Th1 and Th17 cells and were modified by GLS inhibition. Glutamine uptake and glutamate, pyruvate, and lactate secretion were higher in Th17 but reduced upon GLS inhibition (Figure S2F). GLS may contribute to cellular redox regulation through generation of glutamate for glutathione synthesis, and ROS increased in both Th1 and Th17 cells when treated with CB839 (Figure S2G).
GLS Deficiency Has Little Effect on Resting T Cells but Modulates Activation
A GLSfl/fl model was generated and crossed to CD4-Cre to specifically delete GLS late in T cell thymic development to test the role of GLS in T cells. Although GLSfl/flCD4-Cre+ T cells efficiently deleted Gls compared to control GLSfl/fl T cells (Figure 3A), lymphocyte frequencies and numbers were unaltered (Figure 3B). Treg cells have been previously shown to be increased by ASCT2 or GOT1 deficiency (Nakaya et al., 2014; Xu et al., 2017) but were unchanged with GLS deficiency. Resting GLSfl/flCD4-Cre+ CD4 T cells also had normal cell size and phenotype (Figure 3C).
Figure 3. GLS Is Dispensable for T Cell Homeostasis but Constrains Development of a Th1-like Phenotype.

(A) Immunoblot (left) and genomic DNA (right) in isolated Pan T cells (CD4+ and CD8+) from GLSfl/fl CD4-Cre+ (GLS KO) and littermate wild-type controls (WT).
(B) Cell counts (left) and percent of total splenocytes (right) from WT and GLS KO animals. No significance versus wild-type, one-way ANOVA.
(C and D) Flow cytometry analysis of T cell activation markers and cell size of CD4+ T cells (C) freshly isolated from WT and GLS KO T spleens or (D) activation markers and proliferation of WT and GLS KO CD4+ T cells activated on αCD3/CD28 over 48 hr.
(E) Flow cytometry analysis of CD44 in CB839- or vehicle-treated T cells activated on αCD3/CD28 at day 5.
(F–K) Naive CD4+ T cells activated without cytokines over 3 days, split with IL-2, then stimulated to measure cytokines on day 5. (F) Cytokine production of wild-type and GLS KO T cells. (G) Average percent total IFNγ+ producers (left), percent double positive IFNγ+IL2+ producers (middle), and the median fluorescence intensity (MFI) (right) of all IFNγ+ cells in (F) (unpaired t test). (H) Tbet protein expression in WT, GLS KO, and isotype control T cells. Representative of n = 2 experiments. (I–K) Same as in (F–H), except with GLS-inhibitor CB839 and vehicle.
(L and M) CD8+ T cells from WT or GLS KO animals activated on αCD3/CD28 + IL2 for 5 days. (L) Expression of CD8+ granzyme B protein at day 5 (left) and average of granzyme B MFI signal (right) (Student’s t test, n = 3 replicates/group). (M) Tbet protein expression in WT, GLS KO, and isotype control (representative of n = 2 experiments).
Also see Figure S3.
GLS deficiency did, however, impact T cell activation. Measurement of lactate secretion showed that acute GLS inhibition did not impair immediate events in T cell activation to rapidly induce glycolysis (Figure S3A). However, in vitro-stimulated GLS-deficient T cells failed to efficiently undergo blastogenesis and increase in cell size in the first 2 days (Figure S3B). GLS-deficient CD4 T cells had reduced induction of CD25 and CD44 and downregulation of CD62L (Figure 3D) at 48 hr. In addition, in vitro accumulation of viable stimulated T cells was reduced by GLS deficiency (Figure S3C). By day 5 of stimulation in IL2 (Th0 conditions), however, GLS-deficient CD4 T cells had adapted, and activation markers were similar to control (Figure 3E).
Delayed activation-marker expression and proliferation of GLS-deficient T cells suggested impaired function and differentiation. Surprisingly, a greater frequency of GLSfl/flCD4-Cre+ T cells produced IFNγ after 5 days in Th0 conditions than did control T cells (Figures 3F and 3G). In addition, GLS-deficient cells that expressed IFNγ did so to a higher level than IFNγ-producing control T cells. IFNγ expression is regulated in part by the transcription factor, Tbet, and Tbet levels were elevated in activated GLSfl/flCD4-Cre+ Th0 T cells (Figure 3H). Similarly, inhibition of GLS with CB839 also led to greater expression of IFNγ and Tbet (Figures 3I–3K).
The ability of T cells to adapt to GLS deficiency and display enhanced function in vitro suggested that In vivo responses may be altered. Control and GLSfl/flCD4-Cre+ mice were immunized with 2W peptide to measure proliferation and IFNγ secretion. At 8 days after immunization, 2W peptide (EAWGALANWAVDSA)-major histocompatibility complex II (2W-MHC) tetramer binding CD4 T cells proliferated similarly regardless of GLS expression (Figures S3D and S3E). At day 15, IFNγ levels, however, were increased in GLS-deficient 2W-MHC tetramer binding T cells (Figure S3F). In contrast, proliferation to weaker homeostatic cues was reduced for GLSfl/flCD4-Cre+ T cells in both spleen and lymph node compared to wild-type T cells 5 days after transfer to recipient RAG1−/−mice (Figure S3G).
Because cytotoxic CD8 T cells are also driven by Tbet (Knox et al., 2014), the dependence of CD8 T cells on GLS was assessed. Similar to CD4 cells, in vitro-stimulated GLSfl/flCD4-Cre+ CD8 T cells survived and accumulated less efficiently than control T cells (Figure S3H). Importantly, GLSfl/flCD4-Cre+ CD8 T cells had increased expression of the effector protein granzyme B (Figure 3L) and Tbet (Figure 3M). Acute inhibition of GLS with CB839 led to increased granzyme B and perforin after 5 days stimulation (Figures S3I and S3J). In addition to increased effector proteins, CB839-treated CD8 T cells expressed increased levels of Tbet and Eomes and markers of proliferation (Figures S3K–S3M). However, GLS inhibition also increased the portion of CD8 T cells that expressed the inhibitory receptors Lag3 and PD-1 (Figure S3O). GLS deficiency thus can impair initial activation and proliferation of CD4 and CD8 cells while promoting Th1-like and cytotoxic T lymphocyte (CTL) effector programs that may ultimately sensitize to inhibition.
GLS Plays Differential Roles in CD4 T Cell Effector Subsets
Given the differences in glutamine metabolism between Th1 and Th17 cells and spontaneous Th1-like differentiation with IL2 in Th0 conditions, we next tested whether GLS deficiency affected T cell subset specification and function. Control and GLSfl/flCD4-Cre+ CD4 T cells were differentiated in vitro into Th1 and Th17 subsets. Similar to Th0 cells, a greater percentage of GLSfl/flCD4-Cre+ T cells expressed IFNγ when in Th1-skewing conditions (Figures 4A and 4B). Conversely, a decreased percentage of GLS-deficient T cells expressed IL17A when in Th17-skewing conditions. Expression of effector molecules and differentiation in Th1, Th17, and Treg are regulated by Tbet, RORγt, and FoxP3, respectively, and GLS-deficient T cells showed increased Tbet under Th1 conditions and decreased RORγt under Th17 conditions (Figures 4C and 4D). FoxP3 expression was unchanged in the absence of GLS. Similar results to GLS knockout (KO) were obtained when GLS was acutely inhibited using CB839 in Th1-, Th17-, and Treg-skewing conditions (Figures S4A–S4D).
Figure 4. GLS Specifies Th1 and Th17 Differentiation and Metabolism.

(A–D) Naive CD4+ T cells from WT and GLS KO T cells differentiated in Th1-, Th17-, or Treg-skewing media over 5 days. (A) IFNγ and IL2 production in Th1-skewing conditions (top) and IL-17 production in Th17-skewing conditions (bottom). (B) Average percent change cytokine producers in Th1 and Th17 cells from WT (paired t test). (C) Transcription factor expression of Th1, Th17, and Treg cells in WT (black) and GLS KO (red). (D) Average percent change from WT of transcription factors (one-sample t test) in GLS KO T cells.
(E–K) WT CD4+ T cells differentiated in Th1 or Th17 conditions in the presence of vehicle or CB839 over 5 days. (E) Percent of Th1 cells producing IFNγ, IL2, and TNFα at day 5 (unpaired Student’s t test; NS, no stim). (F) Median fluorescence intensity of inhibitory receptors (two-way ANOVA). (G) Fold change of metabolites from T cells differentiated in Th1 and Th17 conditions in the presence of CB839 relative to vehicle by mass spectrometry over 5 days. (H) 3H-2-deoxyglucose uptake in Th1- and Th17-skewed T cells at day 3 (left) and day 5 (right) (Student’s t test). (I) Extracellular acidification rate (ECAR) of Th1- and Th17-skewed T cells at day 5 as in (H). (J) Fold change of Tbet (Th1) or RORyt (Th17) protein levels and (K) cell size in CB839-treated cells normalized to vehicle from same experiment as (G).
Also see Figure S4.
GLS deficiency promoted Th1 and suppressed Th17 differentiation and may affect plasticity and terminal fates. However, GLS-deficient T cells stimulated in Th17 conditions that failed to express RORγt and IL17 did not significantly elevate IFNγ or FoxP3 (Figures 2E, 4A, and S4A). In contrast, GLS-deficient T cells stimulated in Th1 conditions showed evidence of excessive effector function as the proportion of multifunctional Th1 cells (Figure 4E) as well as expression of KLRG1 and inhibitory receptors PD-1, Tim3, and Lag3 were elevated (Figures 4F, S4E, and S4F).
We next assessed how GLS inhibition affected Th1 and Th17 metabolism and differentiation over time. Steady-state levels of glutamine rapidly increased while glutamate and aspartate rapidly decreased in both Th1 and Th17 cells upon GLS inhibition (Figure 4G). While levels of these metabolites partially recovered in GLS inhibitor-treated Th1 cells starting on day 3, they remained low in treated Th17 cells. Likewise, oxidized glutathione (GSSG) recovered in Th1 but remained low in Th17. Similar trends of initial decrease followed by recovery in Th1 cells were observed in glycolytic and TCA cycle intermediates (Figures S4G and S4H). Consistent with impaired early metabolism, flux measurements showed that glucose uptake was reduced in both Th1 and Th17 cells on day 3 (Figure 4H). By day 5, however, Th1 cells had increased levels of glucose uptake and glycolytic flux relative to controls, while Th17 remained impaired by GLS inhibition (Figures 4H and 4I).
Changes in metabolism occurred rapidly upon GLS inhibition and preceded Th1 and Th17 differentiation. Indeed, GLS inhibition led both Th1 and Th17 to have reduced levels of subset transcription factors and prevented an increase in cell size relative to control cells on days 1 and 2 after activation (Figures 4J and 4K). By day 5, however, Th1 cells had recovered and increased both cell size and Tbet expression. These data are consistent with overall changes in biomass, as total rRNA levels per cell were similar in GLS inhibitor- or control-treated T cells on day 3, but Th1 had increased and Th17 had decreased rRNA levels by day 5 of GLS inhibition (Figure S4I).
GLS Affects Gene Expression and Chromatin Accessibility
Deficient GLS activity may alter differentiation through production of cofactors, including α-KG and 2-hydroxyglutarate (2-HG), for epigenetic marks and changes in chromatin status (Reid et al., 2017; Xu et al., 2017). Intracellular levels of α-KG were reduced in CB839-treated Th1 but not Th17 cells, while 2-HG increased in both Th1 and Th17 (Figures S5A and S5B). Reduced α-KG in CB839-treated Th1 cells suggested that α-KG may become limiting to regulate Th1 differentiation and function. A cell-permeable α-KG analog, dimethyl 2-ketogluta-rate (DMaKG), was tested to determine whether provision of α-KG could restore normal Th1 specification of CB839-treated T cells (Figures 5A–5C). DMaKG did not reduce cytokine production in Th1 cells by itself. However, DMaKG rectified IFNγ production and Tbet expression of CB839-treated Th1 cells to control levels. In contrast, Th17 cells were not rescued by DMaKG, and IL17 production and RORγt were unchanged or further decreased (Figures 5A and 5D), suggesting a distinct mechanism of regulation for Th17 cells by GLS.
Figure 5. Th17 and Th1 Cells Differentially Rely on GLS-Mediated ROS Neutralization and Production of α-Ketoglutarate to Maintain Chromatin.

(A–D) WT CD4+ T cells differentiated in Th1 or Th17 conditions in the presence of vehicle or CB839 over 5 days. (A) Cytokine production in Th1 (top)- and Th17 (bottom)-skewing conditions dosed as indicated. (B) Average IFNγ+ only producers (left) and average IFNγ+IL2+ producers (right) as in (A). (C) Average protein expression of Tbet as in (A). (D) Average IL-17A producers in Th17-skewing media (left) and average RORyt expression (right) (one-way ANOVA).
(E and F) Global H3K27 trimethylation normalized to total H3 by flow cytometry. (E) Average H3K27 trimethylation expression at day 3. (F) Same as (E), but at day 5 (Student’s t test).
(G) Average cytokine producers of skewed CD4+ T cells in the presence of CB839, JMJD3-inhibitor GSKJ4, or CB839+GSKJ4 (CB+J4) at day 5 (one-way ANOVA).
(H and I) WT CD4+ T cells differentiated in Th17 conditions as indicated. (H) Percent IL17A+ producers (left) and protein expression of RORyt (right). (I) Average expression of H3K27me3 normalized to total H3 as in (H) (one-way ANOVA).
(J) Number of loci with more (blue circles) and less accessible (orange circles) chromatin peaks with CB839 as determined by ATAC-seq.
(K) Example ATAC-seq traces of IFNγ in Th1 and IL17 gene locus in Th17-skewing conditions.
Also see Figure S5.
Changes in α-KG and 2-HG may alter histone methylation and chromatin accessibility that influence T cell differentiation (Xu et al., 2017). Histone trimethylation was globally assessed by flow cytometry. Initially, GLS inhibition led to increased H3K27 trimethylation (Figure 5E). At later time points when Th1 differentiation was enhanced, however, CB839-treated Th1 and Th17 cells were found to have decreased or increased global H3K27 trimethylation, respectively (Figure 5F). H3K4 trimethylation was similarly reduced or increased in Th1 and Th17 cells, respectively, at day 5 (Figure S5C). Consistent with altered regulation of demethylation as a cause of Th1 differentiation upon GLS inhibition, treatment of T cells with an inhibitor of the histone demethylase JMJD3 also led to increased cytokine production in Th1 but not Th17 cells at day 5 (Figure 5G).
The dependence of Th17 cells on GLS was not rescued by DMaKG, but Th17 cells can be highly sensitive to increased ROS (Gerriets et al., 2015). The glutathione mimic N-acetyl cysteine (NAC) was tested to rescue GLS-deficient Th17 cells. NAC treatment alone modestly reduced Th17 expression of IL17 and RORγt (Figure 5H) while decreasing IFNγ secretion by Th1 (Figure S5D). Th17 production of IL17 and expression of RORγt were partially restored to control levels when combined with CB839. The combination did not, however, increase Th1 production of IFNγ. Changes in Th17 inhibition by CB839 may be mediated through chromatin modifications as NAC also restored H3K27 trimethylation in GLS-deficient Th17 cells to control levels (Figure 5I) yet had no effect on H3K27 trimethylation in Th1 cells (Figure S5E).
Because multiple epigenetic marks may be altered, we performed the assay for transposase-accessible chromatin sequencing (ATAC-seq) to determine whether GLS deficiency altered chromatin accessibility after 5 days of Th1 and Th17 differentiation. CB839-treated Th1 cells had more genes with regions of increased accessibility than genes with decreased accessibility (Figure 5J). Th17 cells, however, had more genes with regions of reduced accessibility. While partially overlapping, affected genes were largely distinct for Th1 and Th17 cells (Figure S5F). Key Th1 and Th17 genes showed changes, including the Ifng and Il17a/f loci in Th1 and Th17 cells, respectively (Figure 5K). Further, Ingenuity Pathway analyses of genes with altered promoter accessibility in Th1 cells showed changes in networks of cell survival and inflammation (Figure S5G). Analysis of promoter regions with altered accessibility identified recognition motifs for canonical T cell differentiation transcription factors, including AP-1, ETS, and IRF (Figure S5H). These altered promoter regions were also enriched in CTCF recognition motifs, a DNA-binding protein recently implicated to mediate the effects of α-KG on chromatin state (Chisolm et al., 2017).
Because altered chromatin accessibility can influence gene expression and T cell differentiation, T cells were cultured in Th1 or Th17 conditions with vehicle or CB839 and examined by RNA sequencing. Of the 200 genes with the most significantly altered expression in CB839-treated Th1 cells, the majority showed increased expression (Figure 6A). Conversely, more of these genes were downregulated in Th17 cells. Functional annotation using gene set enrichment analyses showed that GLS inhibition led to upregulation of specific pathways including those related to cell cycle, mTORC1, Myc, and IL2 signaling (Table S2). Similar gene sets were downregulated in Th17 cells treated with CB839.
Figure 6. GLS Inhibition Alters Gene Expression to Sensitize Th1 Cells to IL2 Activation of mTORC1.

(A) Top 200 modified genes from RNA-seq compared to vehicle (Log2Fold > 0.5, p < 0.05) in Th1 (left) and Th17 (right).
(B and C) Phospho-S6 expression on day 5 in Th1 and Th17 conditions as indicated with or without CB839 or IL2 at concentrations shown (ng/mL) at day 3 (Student’s t test).
(D) Cytokine production in Th1-skewing conditions in the presence of vehicle (top) or CB839 (bottom) after 5 days, under no IL2 conditions or with IL2 + mTOR inhibitor rapamycin added on day 3.
(E) Phospho-S6 protein expression (left), average pS6 MFI (middle), percent IFNγ+IL2+ or IL2+ cells (right) in CD4 T cells in Th1-skewing conditions and infected with control- or PIK3IP1-expressing retrovirus with CB839-treatment. (middle, Student’s t test; right, two-way ANOVA).
(F) Protein expression of phospho-S6 (left) and IFNγ (right) in activated Cas9-transgenic CD4+ T cells transduced with retrovirus-containing control guide RNA or guide RNAs targeting PIK3IP1.
(G and H) Wild-type CD4+ T cells activated and treated with PIK3IP1 antibody or IgG control antibody over 3 days. Protein expression of phospho-S6 (left) and average MFI of pS6 (right, one-way ANOVA). (H) Protein expression of activation markers of control or PIK3IP1 antibody-treated T cells upon stimulation (no CB839). Also see Figure S6 and Table S2.
IL2 signaling activates mTORC1 to promote Myc signaling, glycolysis, and Th1 effector differentiation (Boyman and Sprent, 2012; Chisolm et al., 2017). Given enrichment in these pathways by RNA sequencing (RNA-seq), the contribution of IL2/mTORC1 signaling was tested to increase effector function of GLS-deficient Th1 cells. Levels of the mTORC1 downstream target phospho-S6 were measured in Th1 and Th17 cells differentiated in IL2 and the presence or absence of CB839. GLS inhibition led to increased phospho-S6 in Th1 and decreased phospho-S6 in Th17 cells (Figure 6B). IL2 played a key role to promote phospho-S6, as increased phospho-S6, IFNγ, and Tbet in CB839-treated Th1 were dependent on IL2 (Figures 6C and S6A). Consistent with mTOR regulation of Myc protein, GLS inhibition modestly increased Myc in Th1 but not Th17 cells (Figure S6B). Importantly, while GLS inhibition in the presence of IL2 led to enhanced differentiation and a hypomethylated state, T cells hypermethylated H3K27 upon treatment with CB839 in the absence of IL2 (Figure S6C). The role of mTORC1 signaling in GLS-mediated regulation of Th1 cells was directly tested by treatment of cells on day 3 after activation with rapamycin. While rapamycin treatment at this time had no effect on control Th1 cells, it reduced phospho-S6 and cytokine production in CB839-treated Th1 cells (Figures 6D and S6D). A similar mechanism may occur for regulation of Th0 and CTL, as GLS inhibition also led to enhanced phospho-S6 for these cells in the presence of IL2 (Figure S6E).
Several regulators of mTORC1 signaling were altered by GLS inhibition in Th1 cells by RNA-seq, including Pik3ip1, Akt, Tsc2, Sestrin2, and Castor1 (Figure S6F). Of these, Pik3ip1, which has been shown to suppress PI3K and mTORC1 in T cells (DeFrances et al., 2012; Wei et al., 2016), was most strongly downregulated in Th1 cells by GLS inhibition. Restoring PIK3IP1 in CB839-treated Th1 cells by retroviral transduction was sufficient to reduce phospho-S6 and cytokine secretion (Figures 6E and S6G). Conversely, CRISPR genetic deletion of Pik3ip1 in primary T cells led to increased phospho-S6 and IFNγ production (Figures 6F and S6H). PIK3IP1 is a transmembrane protein, and treatment of stimulated T cells with anti-PIK3IP1 antibody directed against the extracellular domain suppressed phospho-S6 (Figure 6G) and T cell activation as evidenced by down-regulation of CD25, CD44, and CD62L (Figures 6H and S6I). Together, these data suggest that PIK3IP1 levels can contribute to mTORC1 activity and effector function in Th1 cells, while Th17 cells are dependent on GLS-mediated regulation of cellular redox state.
GLS Regulates In vivo for Inflammatory Effector T Cell Responses
We next tested whether Th17 cells require GLS to elicit inflammation In vivo. Allogenic bone marrow was transplanted alone or with control and GLSfl/flCD4-Cre+ T cells to induce a model of IL17-dependent chronic graft-versus-host disease (cGvHD). Recipient mice were weighed regularly, and GLS-deficient allogenic T cells led to less weight loss than control T cells (Figure S7A). cGvHD is a multi-organ disease (Panoskaltsis-Mortari et al., 2007), and mouse models of cGvHD include lung inflammation. Histological examination showed that GLS-deficient T cells reduced lung immune infiltrate and clinical inflammation score (Figures 7A and 7B) and caused significantly less airway functional impairment than control T cells (Figure S7B). Immunologically, GLS deficiency reduced IL17-producing CD4 cells, with a trend toward reduced IFNγ (Figure 7C). GLS was also critical in an independent model of Th17-mediated lung inflammation, in which control and GLS-deficient animals were sensitized and challenged in the airway with house dust mite antigen, and lipopolysaccharide (LPS) failed to accumulate CD4 T cells and produce inflammatory cytokine in the lung (Figure S7C). Inflammatory bowel disease (IBD) also involves Th17 cells, and we found that while adoptive transfer of control T cells led to weight loss and inflammation, mice that received GLS-deficient T cells maintained weight (Figure 7D). Despite partial protection from disease, a greater percentage of GLS-deficient T cells in the mesenteric lymph nodes produced IFNγ, consistent with a preferential Th1 response (Figure S7D).
Figure 7. GLS Is Essential for T Cell-Mediated Inflammation, but Transient Inhibition Can Augment T Cell Responses.

(A–C) Airway inflammation in cGvHD following transfer of WT or GLS KO T cells. (A) Hematoxylin- and eosin-stained lung sections focusing on bronchioles.
(B) Average histopathological scores from sections from (A) (unpaired t test). (C) Percent cytokine producers from peripheral lymph node cells stimulated with PMA/ionomycin for 5 hr from GvHD mice (BM, n = 8; WT, n = 5; KO, n = 7; unpaired t test).
(D) Bodyweights from T cell adoptive transfer inflammatory bowel disease (IBD) model in which RAG1 KO mice injected with WT and GLS KO naive CD4+ T cells and induced for IBD with piroxicam (#p < 0.05, ‡p < 0.01, two-way ANOVA; WT, n = 6; GLS KO, n = 8; data presented as SEM).
(E and F) T cells from WT and GLS KO infected to express CAR T cell constructs and injected into recipient mice. CAR- no infection; Δ, m19-delta-ζ; 28-ζ, m19–28-ζ. (E) CD19+ B cells per μL of blood at day 14 (left) and day 28 (right). (F) Same as in (E), but at day 42 (one-way ANOVA).
(G) CAR T cell numbers on day 28 following transfer of CAR T cells treated with vehicle or CB839 prior to transfer to recipient mice (WT no CAR, n = 2 animals; all others, n = 5–6 animals; one-way ANOVA).
(H) Number of CD19+ Eμ-Myc lymphoma cells after 48 hr culture with indicated ratios of CAR T cells (Student’s t test).
(I) Counts of total CD8+ cells in response to hgp10025–33-expressing vaccinia virus collected from tail vein after indicated time (two-way ANOVA).
(J) Counts of total CD8+ T cells in spleen (left) and lymph node (right) after 38 days and re-challenge with hgp10025–33-expressing vaccinia virus (Vehicle, n = 5 animals; GLS inhibitor, n = 4 animals; two-way ANOVA).
Also see Figure S7.
The role of GLS deficiency to enhance Th1 and CTL function was next tested In vivo. Control and GLSfl/flCD4-Cre+ T cells were evaluated in a murine chimeric antigen receptor (CAR) model for the ability to eliminate endogenous target B cells and persist In vivo. T cells were in vitro transduced with CAR T expression vectors either lacking a cytoplasmic tail (Δ) or with a CD3ζ-CD28 (28-ζ) intracellular tail and adoptively transferred into animals conditioned with cyclophosphamide. 14 days after T cell transfer, endogenous CD19-expressing B cells were significantly reduced by both control and GLSfl/flCD4-Cre+ CAR T cells (Figure 7E). After 28 days, however, B cells had accumulated in recipients of GLSfl/flCD4-Cre+ CAR T cells and were fully recovered in lymph nodes by day 42 (Figures 7E and 7F). Consistent with upregulation of inhibitory receptors upon activation, GLS-deficient T cells appeared unable to sustain an effector response In vivo.
Because GLS inhibition altered chromatin accessibility in Th1 cells in vitro, it was possible that transient treatment with CB839 could induce long-lasting effect. T cells were treated with vehicle or CB839 during in vitro transduction to express CARs and tested for subsequent In vivo function. Vehicle-and CB839-treated CAR T cells were equally capable of eliminating CD19+ targets In vivo (Figure S7E). Importantly, in vitro CB839-treated 28-ζ CAR T cells accumulated In vivo to a greater extent than untreated CAR T cells (Figure 7G) and showed greater ability to eliminate B cell leukemia cells in vitro (Figure 7H). This increased ability of Th1 and CD8 effector T cells to proliferate or persist following transient GLS inhibition was not specific to CAR T cells. CD8 T cells bearing a Pmel-specific T cell receptor (TCR) transgene treated with CB839 in vitro prior to adoptive transfer also accumulated to greater numbers In vivo by day 7 when challenged with an antigen-expressing vaccinia virus (Figure 7I), and increased cell numbers persisted for greater than 5 weeks (Figure 7J). Thus, chronic or complete GLS deficiency impairs T cell responses In vivo, while transient in vitro inhibition may enhance subsequent Th1 and CD8 CTL effector function and long-lasting cell numbers In vivo.
DISCUSSION
T cell activation and specification into functional subsets requires increased biosynthesis and establishment of the appropriate gene expression program. Importantly, the metabolic requirements of each subset are distinct, and Th1, Th17, and Treg have critical metabolic differences that influence their differentiation and fate. While glucose metabolism has been extensively studied, here we show that glutamine metabolism and GLS activity regulate T cell activation to promote Th17 while impairing Th1 differentiation and effector function.
Glutamine and generation of α-KG through GLS and glutaminolysis can play a key role to maintain levels of TCA intermediates. Indeed, acute GLS inhibition lowered T cell respiration and abundance of TCA intermediates while increasing glucose contribution to these pathways. In effector T cell populations, however, this association becomes context dependent as IL2 can promote a GLS-independent Th1-like phenotype in CD4 T cells and enhance CD8 CTL effector function. Th17 cells, however, remain GLS dependent. For Th1 and cytotoxic CD8 T cells to adapt to GLS deficiency, an alternate anaplerotic source is essential and pyruvate carboxylase may generate oxaloacetate from glycolysis-derived pyruvate, as can occur in glutamine-independent cancers (Cheng et al., 2011).
While GLS acts on glutamine to facilitate the first event of glutaminolysis, modulation of glutamine metabolism at different steps can lead to strikingly different phenotypes. The absence of glutamine prevented cytokine production and proliferation of both Th1 and Th17 cells and instead promoted Treg generation. Deficiency of the glutamine transporter ASCT2 suppressed proliferation, impaired both Th1 and Th17 T cell specification, and enhanced Treg generation (Nakaya et al., 2014). Suppression of GOT1, which converts aspartate and α-KG to glutamate and oxaloacetate, impaired Th17 with increased Treg differentiation (Xu et al., 2017). GLS deficiency differs to selectively promote Th1 and impair Th17 differentiation while not affecting Treg. Subset-specific protein expression and levels of different metabolites in glutamine-related pathways may mediate these distinct responses. ASCT2 is one of multiple glutamine transporters and may interact with the CARMA1 complex upon T cell activation to regulate NF-κB signaling (Nakaya et al., 2014). GOT1 deficiency may restrict glutamate production from aspartate while leaving glutamine conversion to glutamate and α-KG intact. GLS deficiency, in contrast, allows glutamine uptake but leads to both glutamate and aspartate deficiencies and increased ROS. Glutamate and glutamine can be generated from other sources, such as aspartate, which can be p53 dependent and induce cell survival in glutamine-limiting conditions (Tajan et al., 2018). Given the interconnections of each metabolite in these pathways, it is likely that inhibition of this metabolic network leads to context-specific outcomes.
Glutamine-dependent metabolites may influence T cells through alteration of epigenetic marks and chromatin accessibility. Increased cytosolic acetyl-CoA levels can modulate his-tone acetylation and can regulate IFNγ expression (Peng et al., 2016), and α-KG can play an important role in histone and DNA methylation. Individual histone marks have distinct regulatory roles, and while we found consistent changes in global H3K4 and H3K27 trimethylation that differentially influenced gene expression, other marks likely also affect chromatin status as a whole to determine gene expression. Changes in the abundance of α-KG, TCA cycle intermediates, and 2-HG likely contribute to altered chromatin state and gene expression. The initial hyper-methylation phenotype continued in Th17 but reversed Th1 cells by IL2, suggesting that IL2-induced signals and Th1 differentiation ultimately dominate the overall chromatin accessibility and gene expression patterns. While inhibition of JMJD3 demethylase by GSKJ4 phenocopies CB839 treatment in Th1 cells, other epigenetic modifiers also likely contribute to this differential response. ROS also promoted increased histone methylation in GLS-inhibited Th17 cells and influenced cytokine production by Th1 cells, although the mechanisms are unclear. One protein that may play a central role in GLS regulation of chromatin is CTCF, which was shown to regulate IL-2-dependent gene programming in T cells in response to α-KG (Chisolm et al., 2017). While Ctcf mRNA levels were not strongly affected in this study by either Th1 or Th17 GLS-deficient T cells, CTCF consensus-binding sequences were significantly enriched in chromatin sites identified as altered in ATAC-seq studies. The shared occur-rence of CTCF-binding sites may represent an initial α-KG-dependent response and lead to more open or closed chromatin based on other DNA-binding factors and transcription factor expression differences in Th1 and Th17 cells.
Opposing effects of GLS inhibition on mTORC1 signaling in Th1 and Th17 and the ability of mTORC1 to promote a glycolytic and effector differentiation program suggest that this pathway plays a key role to mediate the effects of GLS on T cell fate. IL2 was essential to stimulate mTORC1 activity that enhanced differentiation of GLS-deficient Th1 cells. GLS inhibition induced several changes in IL2 and mTORC1 signaling that may contribute to the differential response of Th1 and Th17. In addition to increased Sestrin2 and Castor1 that are involved in amino acid sensing (Chantranupong et al., 2014; Wolfson and Sabatini, 2017) and Akt1, PIK3IP1 was sharply downregulated in GLS-deficient Th1 cells. This transmembrane protein was expressed at a low level in Th17 cells and is inhibitory to the mTOR signaling pathway by binding PI3K p110 and inhibiting PI3K activity. It has been shown to suppress liver carcinomas (He et al., 2008) and inhibit T cell activation (DeFrances et al., 2012; Wei et al., 2016). Manipulation of PIK3IP1 expression and deletion here demonstrates that this protein may also contribute to GLS inhibition to modulate mTORC1 signaling in Th1 cells. Conversely, Th17 cells can respond poorly to IL2 (Quintana et al., 2012), and reduced IL2 response or altered PI3K/Akt/mTORC1 activating signal may also impair differentiation in GLS-deficient Th17 cells.
Targeting GLS may selectively impair Th17 cells while having the potential to enhance Th1 responses In vivo. Th17 cells were GLS dependent and failed to induce lung inflammation in either cGvHD or acute airway inflammation models, and GLS deficiency protected against inflammatory bowel disease. In addition, inhibition of GLS was recently shown to suppress a model of rheumatoid arthritis (Takahashi et al., 2017), although mechanisms were uncertain. Th1 cells were also impaired in these models, potentially due to the lower levels of inflammation or the eventual impairment of Th1 cells due to induction of inhibitory receptors. Similarly, GLS-deficient CAR T cells were initially competent to eliminate B cell targets In vivo but failed over time. Transient in vitro GLS inhibition, however, enhanced effector activity and allowed CAR T cells and antiviral T cells to accumulate to greater numbers In vivo. This effect is likely due to GLS-inhibition-induced changes to chromatin accessibility and sensitization to cytokine signals that promote sustained function in a setting where T cells are capable of performing glutaminolysis.
Subset-specific integration of glucose and glutamine metabolism may now offer new opportunities to modulate immunity. We propose that reduced αKG upon GLS inhibition leads to an initial increase in histone methylation that contributes to decreased PIK3IP1 expression and sensitization to IL2 signals in Th0, Th1, and CTL. Increased IL2 activation of mTORC1 signaling then promotes metabolic adaptation and results in a cumulative shift in epigenetic marks and chromatin status consistent with effector differentiation. In contrast, increased ROS caused by GLS inhibition promotes closed chromatin to prevent Th17 cell differentiation, which cannot be rescued by IL2, although molecular mechanisms remain uncertain. Glutaminolysis thus both promotes Th17 and suppresses Th1 responses, and GLS may provide a target to treat inflammation In vivo with continuous inhibition. Conversely, transient inhibition may program T cells for enhanced IFNγ-specific effector responses. GLS is a promising candidate to inhibit cancer cell metabolism, and it may be important to consider transient or episodic GLS inhibition to epigenetically reprogram T cells for enhanced effector function and immunotherapy.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Jeffrey Rathmell (Jeff.rathmell@vumc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
With exception of GLSfl/fl mice, which were generated as described below, all mice were obtained from the Jackson laboratory or described previously. All mouse procedures were performed under Institutional Animal Care and Utilization Committee (IACUC)-approved protocols from Duke University, Vanderbilt University (2W tetramer, IBD, homeostatic proliferation, and asthma models), the National Cancer Institute (Vaccinia model), the Moffitt Cancer Center and Research Institute (CAR T cell In vivo model), and the University of Minnesota (Graft versus Host Disease model) and conformed to all relevant regulatory standards. Mice were housed in specific pathogen-free facilities in ventilated cages with at most 5 animals per cage and provided ad libitum food and water. With the exception of homeostatic proliferation studies, which used RAG1−/− immunodeficient C57BL/6J mice, all studies used immunocompetent C57BL/6J mice that were treatment-naive until the start of study.
GLS1fl/fl animals were generated from Glstm1a(KOMP)Mbp embryonic stem cells (Project ID: CSD29307) from the KOMP that were blastocyst microinjected to generate mice (Duke University Transgenic and Knockout Shared Resource) and crossed to FLP transgenic animals. Progeny were then crossed with CD4-CRE transgenic mice to develop the GLS1fl/fl CD4-CRE (GLS KO). In all cases comparing wild-type to GLS KO, healthy, sex-matched and age-matched littermates were used (male and female, 8 to 14 weeks of age unless otherwise stated). No sex differences in phenotype were noted in WT and GLS KO CD4 T cells. Animals were genotyped for floxed alleles and CRE allele.
Male C57BL/6J mice aged 8–16 weeks were used for in vitro CB839 experiments (RRID: IMSR_JAX:000664).
Cell Lines and In Vitro cultures
T cell in vitro experiments were carried out using primary mouse T cell cultures from male and female mice at 37°C with 5% CO2 in RPMI media (CAT#10–40-CV) supplemented with HEPES, β-mercaptoethanol, Pen/Strep, and glutamine, unless otherwise stated. Generation of viral particles was performed in PLAT-E retroviral packing cell lines (Cell Biolabs). T cell killing assays were performed on CD19 expressing Emu cell lines (Generous gift from Dr. Davila Lab).
METHOD DETAILS
T cell in vitro activation and skew experiments
CB839 was dosed at 1 μM (activation) or 500 nM (differentiation), GSKJ4 (Cat#:S7070) at 1 μM, dimethyl-2-oxoglutarate (DMaKG) (Cat#: 349631) at 1.5 mM. and/or rapamycin (Cat#: 553210) at 5 nM. Briefly, CD4+ T cells were isolated (Cat#130-104-454) from wild-type animals (WT) and GLS1fl/fl CD4-CRE+ mice (GLS KO) and activated over various time points via 5 ug/mL anti-CD3/anti-CD28 antibodies plate bound (CD3: Cat # 16-0031-85, CD28: Cat # 16-0281-85). Non-stimulated CD4 samples were maintained using 10 ng/mL IL-7 (Cat#: 217–17). For skewing experiments, naive CD4 T cells from WT or KO animals were plated with αCD3 (as above) and subset-specific cytokines and antibodies (Th1: IL-12p70, αIFNy, αIL4; Th17: IL6, αIFNy, TGFβ, Treg: TGFβ and stimulated with feeder layer of irradiated splenocytes. Th0 experiments were run in skewing condition (+αCD3 antibody) without additional cytokines. After 3 days, cells were split with fresh media and stimulated with or without 10 ng/mL IL-2 (Cat#:14-8021-64) for a further 2 days. For intracellular cytokine stains, cells were re-stimulated using PMA/ionomycin in the presence of GolgiPlug (Cat#: 555029) for 4 hours, then fixed and stained for intracellular subset-specific cytokines using fix/perm kit (Cat#: 554714). For all other intracellular or intranuclear stains such as transcription factor, pS6, C-MYC, H3K4me3, H3K27me3, and total H3 protein, cells were removed from media, stained for surface markers (See Key Resources antibodies table), fixed, then stained for intracellular proteins using fix/perm kit (Cat# 00-5223-56, 00-5123-43). Cell proliferation was assessed by staining naive CD4+ cells with Cell Trace Violet proliferative dye at 5 μM (Cat#: c34557).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-IFNy functional | ThermoFisher | Cat#116-7311-38; RRID:AB_2637490 |
| Anti-IL4 functional | ThermoFisher | Cat#16-7041-95; RRID:AB_2573101 |
| Anti-CD3 functional | ThermoFisher | Cat#16-0038-85; RRID:AB_468857 |
| Anti-CD28 functional | ThermoFisher | Cat#16-0289-85; RRID:AB_468927 |
| Anti-PIK3IP1 | ProteinTech | Cat#16826-1-AP; RRID:AB_2163333 |
| Anti-GLS1 antibody | GeneTex | Cat#GTX81012; RRID:AB_11162809 |
| Anti-IFNy APC | BD | Cat#554413; RRID:AB_398551 |
| Anti-IL17APE | ThermoFisher | Cat#12-7177-81; RRID:AB_763582 |
| Anti-IL2 PE | BD | Cat#554428; RRID:AB_395386 |
| Anti-CD4 e450 | ThermoFisher | Cat#48-0041-82; RRID:AB_10718983 |
| Anti-CD8BV510 | BD | Cat#563068; RRID:AB_2687548 |
| Anti-Thy1.1 e450 | ThermoFisher | Cat#48-0900-82; RRID:AB_1272254 |
| Anti-CD25 e450 | ThermoFisher | Cat#48-0251-82; RRID:AB_10671550 |
| Anti-CD44 PECy5 | ThermoFisher | Cat#15-0441 −82; RRID:AB_468749 |
| Anti-CD62L APC | ThermoFisher | Cat#17-0621-82; RRID:AB_469410 |
| Anti-FOXP3 PE | ThermoFisher | Cat#12-5773-82; RRID:AB_465936 |
| Anti-Tbet PE | ThermoFisher | Cat#12-5825-82; RRID:AB_925761 |
| Anti-Tbet PECy7 | ThermoFisher | Cat#25-5825-82; RRID:AB_11042699 |
| Anti-RORyt APC | ThermoFisher | Cat#12-6988-82; RRID:AB_1834470 |
| Anti-CD19APC | Biolegend | Cat#115512; RRID:AB_313647 |
| Anti-CD45RB | Biolegend | Cat#103320; RRID:AB_2565229 |
| Anti-pS6 PE | Cell Signaling Technology | Cat#5316S; RRID:AB_10694989 |
| Anti-c-Myc PE | Cell Signaling Technology | Cat#14819S |
| Anti-H3K27me3 af647 | Cell Signaling Technology | Cat#12158S |
| Anti-H3K4me3 af647 | Cell Signaling Technology | Cat#12064S |
| Anti-H3 total protein af647 | Cell Signaling Technology | Cat#12230S |
| Isotype PE control | ThermoFisher | Cat#12-4714-42; RRID:AB_1944423 |
| APC-conjugated Tetramers | (Moon et al., 2007) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse IL2 | ThermoFisher | Cat#14-8021-64 |
| Recombinant Mouse IL-7 | Peprotech | Cat#217-17 |
| Recombinant IL12p70 | ThermoFisher | Cat#14-8121-62 |
| Recombinant TGFβ | R&D systems | Cat#100-B-001 |
| Recombinant IL6 | ThermoFisher | Cat#14-8061-80 |
| 2-deoxyglucose | Sigma-Aldrich | Cat#D6134; CAS 154-17-6 |
| Dimethyl 2-oxoglutarate | Sigma-Aldrich | Cat#349631; CAS 13192-04-6 |
| D-Glucose(U-13C6) | Cambridge Isotope Labs | Cat#CLM-1396-1 |
| Deoxy-D-glucose 2-[1,2,3H(N)] | Perkin Elmer | Cat#NET549001MC |
| phase Dinonyl phthalate | Sigma-Aldrich | Cat#80151 |
| Dow Corning Silicon 550 oil | Motion Industries, INC | Cat#784198 |
| Phloretin | Millipore | Cat#524488; CAS 60-82-2 |
| GSKJ4 | Selleckchem | Cat#S7070 |
| UK5099 | Sigma-Aldrich | Cat#PZ0160; CAS 56396-35-1 |
| CB839 | Calithera Biosciences | N/A |
| Gls1 inihibitor III | Millipore | Cat# 533717; CAS 1439399-58-2 |
| Hgp100\/25-33 KVPRNQDWL | (Gattinoni et al., 2009) | N/A |
| Gp100-W vaccinia virus vector | (Overwijk et al., 1998) | N/A |
| 2W peptide EAWGALANWAVDSA | GenScript | N/A |
| Rapamycin | Sigma Aldrich | Cat#553210 |
| GolgiPlug | BD | Cat#555029 |
| PMA | Sigma Aldrich | Cat#P8139 |
| lonomycin | Sigma Aldrich | Cat#IO634 |
| Cell Trace Violet | Invitrogen | Cat#c34557 |
| ACK Lysing Buffer | VWR | Cat#10–548E |
| Cyclophosphamide | Sigma Aldrich | Cat#C0768 |
| Complete Freund’s Adjuvant | Sigma Aldrich | Cat#F5881 |
| Critical Commercial Assays | ||
| Genomic DNA clean & concentrator | Zymo Research | Cat#D4011 |
| Nextera DNA prep kit | Illumina | Cat#FC-121-1030 |
| Nextera index kit | Illumina | Cat#FC-121-1011 |
| CD4+ isolation kit | Miltenyi | Cat#130-104-454 |
| CD8+ isolation kit | Miltenyi | Cat#130-095-236 |
| Pan T cell isolation kit | Miltenyi | Cat#130-095-130 |
| Fix/perm kit | ThermoFisher | Cat#88-8824-00 |
| Fixation/permeabilization kit | BD | Cat#554715 |
| NEBNext High-fidelity 2x PCR mix | New England Biolabs | Cat#M0541 |
| RNeasy Mini Kit | QIAGEN | Cat#:74104 |
| NEBNext Ultra RNA Library Kit | New England Biolabs | Cat#E7530 |
| XF Mito Stress test kit | Agilent | Cat#103015-100 |
| KAPA express Extraction Kit | KAPA | Cat#KK5121 |
| Deposited Data | ||
| RNA Seq data | This paper | GEO: GSE112244; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112244 |
| ATAC Seq data | This paper | ArrayExpress: E-MTAB-6648 https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6648/ |
| Experimental Models: Cell Lines | ||
| CD19+ Eμ-ALL01 cells (Emu) | (Davila et al., 2013) | N/A |
| Plat-E retroviral packaging cell line | Cell Biolabs | Cat#RV-101 |
| E. Coli LPS | Sigma | Cat#L4391 |
| Dust Mite Antigen | Greer | Cat#XPB70D3A2.5 |
| Experimental Models: Organisms/Strains | ||
| Mouse: GLS KO: GLSfl/fl CD4-CRE mice | This paper | N/A |
| Mouse: pmel Tg mouse: B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J | (Sukumar et al., 2013) | RRID: IMSR_JAX:005023 |
| Mouse: C57BL6 | Jackson Labs | RRID: IMSR_JAX:000664 |
| Mouse: RAG1 KO | Jackson Labs | RRID: IMSR_JAX:002216 |
| Mouse: CAS9 | Jackson Labs | RRID: IMSR_JAX:024858 |
| Oligonucleotides | ||
| GLS KO Forward ACGAGAAAGTGGAGATCG | This paper | N/A |
| GLS KO reverse GCCTTCTGGAAAACA | This paper | N/A |
| PIK3IP1 guide RNA 1 AGACCGGCGTCCCTGAAAAG | This paper | N/A |
| PIK3IP1 guide RNA 2 TCGTGGGCTACACTTACAAG | This paper | N/A |
| Recombinant DNA | ||
| Plasmid MSCV-IRES-Thy1.1 | (Wu et al., 2006) | Addgene Cat#17442 |
| Plasmid MSCV-Pik3ip1-IRES-Thy1.1 | (Wei etal., 2016) | N/A |
| Plasmid pMx-U6-empty-GFP | (Toffalini et al., 2009) | N/A |
| Plasmid pMx-U6-PIK3IP1gRNA1-GFP | This paper | N/A |
| Plasmid pMx-U6-PIK3IP1gRNA2-GFP | This paper | N/A |
| Software and Algorithms | ||
| Flowjo version V10 | Flowjo | https://www.flowjo.com; RRID: SCRJD08520 |
| GraphPad Prism v6.0 | GraphPad Software | https://www.graphpad.com; RRID: SCR_002798 |
| R statistical software | R Foundation | https://www.r-project.org/ |
| STAR software | (Mudge and Harrow, 2015) | https://github.com/alexdobin/STAR |
| SAMtools | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| featureCounts | (Liao etal., 2014) | http://subread.sourceforge.net/ |
| DESeq2 Sequencing software | (Love etal., 2014) | N/A |
Homeostatic Proliferation
Homeostatic proliferation was measured as previously described (Jacobs et al., 2010). Briefly, naive CD4+ and CD8+ T cells were isolated from male GLSfl/flCD4-Cre and wild-type Thy1.1+ mice. Cells were mixed in a 1:1 ratio and stained with proliferative dye Cell-Trace Violet (Cat#: c34557). Cells were transplanted by i.v. injection into recipient RAG knockout mice (RRID:IMSR_JAX:002216) 8 weeks of age. Five days after injection, spleen and mesenteric lymph node were collected, homogenized, and stained with antibodies against CD4, CD8, and Thy1.1 for flow cytometry analysis.
ATAC-Sequencing Experiments
Crude nuclei pellets for ATAC-seq were isolated according to Buenrostro et. al (Buenrostro et al., 2013) with modifications. Briefly, naive CD4 T cells from C57BL/6J mice were skewed to Th1 and Th17 subsets in vitro with vehicle or in the presence of 0.5 μM CB839. At Day 5, T cells were re-isolated for CD4+ cells using CD4+ negative selection kit (Cat#: 130-104-454). 1×105 cells were removed for nuclei extraction in ATAC-Seq lysing buffer. Cells were exposed to Tn5+adaptor proteins from Nextera prep kit (Cat#: FC-121–1030) for 30 min at 37°C and immediately placed on ice. Transposed eluate was amplified via PCR using NEBNext High-fidelity 2x PCR mix (Cat#:M0541) and multiplexed (Cat#: FC-121–1011). Samples were purified using Zymo DNA cleanup kit (Cat#: D4011). QC of samples was run on bioanalyzer before being sent for sequencing.
RNA Sequencing Experiments
Naive CD4+ T cells from C57BL/6J mice were skewed to Th1 and Th17 subsets with or without CB839 over 5 days and total RNA extracted for RNaseq (Cat#: 74104). RNA was sent to VANderbilt Technologies for Advanced GEnomics (VANTAGE) core at Vanderbilt University. Libraries were prepared using 50ng of total RNA using the NEBNext Ultra RNA Library Kit for Illumina (Cat# E7530) and sequenced on HiSeq3000 at 75bp paired-end. Each sample was analyzed in triplicate. Sequencing reads were aligned against the Mouse GENCODE genome, Version M14 (January 2017 freeze, GRCm38, Ensembl 89) using the Spliced Transcripts Alignment to a Reference (STAR) software (Mudge and Harrow, 2015). Reads were preprocessed and index using SAMtools (Li et al., 2009). Mapped reads were assigned to gene features and quantified using featureCounts (Liao et al., 2014). Normalization and differential expression was performed using DESeq2 (Love et al., 2014). Skewed lymphocytes with and without CB839 were compared in both Th1 and Th17 groups. The top most significantly differentially expressed genes (FDR < 0.01 and Log2 difference greater than 0.5 in magnitude) were considered for subsequent functional enrichment using Geneset Enrichment Analysis. The top 200 most differentially expressed genes were used for unsupervised hierarchical cluster analysis and visualized using heatmap representations.
PCR
Pan T cells from WT or GLS KO mice were isolated and purified using Miltenyi isolation kit (Cat#: 130-095-130). Genomic DNA was generated using Kapa express Extract kit (Cat#: KR0370). Primers targeted over exon 10 and exon 11 were generated for wild-type band with a melting temperature of 54°C:
Forward: ACGAGAAAGTGGAGATCG
Reverse: GCCTTCTGGAAAACA
PCR product was then run on a 1% agarose gel with ethidium bromide and visualized by GelDoc XR (Cat#: 1708195).
Glucose Uptake
Glucose uptake assays were performed as previously described (Macintyre et al., 2014). Naive CD4+ T cells from C57BL/6J mice were differentiated into Th1 and Th17 cells, in triplicate, in the presence or absence of CB839 over five days and spun down after reisolation using CD4 kit as previously described. At day 3 and 5, cells were removed, washed twice in PBS, counted, then rested in 1 mL Kreb’s Ringers HEPES (KRH) for at least 10 minutes. Cells were spun and resuspended to 5×105 cells/50 μL KRH for glucose uptake assay. Briefly, 3H-2-deoxyglucose (Cat#: NET549001MC) was suspended in KRH bubble layered in oil (50:50 Dinonyl phthalate:Silicon 550, Cat#: 80151, Cat#: 784198), and cells were added to this bubble. Cells were incubated for 10 minutes at 37°C. Immediately after incubation, reaction was quenched with 200 μM phloretin (Cat#: 524488). Cells were spun, washed, and then resuspended in scintillation fluid for counting on Beckman-Coulter scintillation counter (3H, 1 min/sample read).
Extracellular Flux Analyses (Seahorse)
Experiments were carried out on Agilent Seahorse XF96 bioanalyzer (Agilent). Briefly, wild-type CD4+ cells from C57BL/6J mice were isolated as previous and activated for 3 days on αCD3/CD28 coated plates as previously described, or skewed to Th1 and Th17 subsets as described above. T cells were isolated and spun onto XF96 Cell-Tak (BD Bioscience, Cat#: 354240) coated plates and rested in Seahorse XF RPMI 1640 media supplemented with glutamine, sodium pyruvate, and glucose. For immediate metabolic response, 1 μM CB839 and 5 μM UK5099 (Cat#: PZ0160–5MG) were injected separately or in combination, and OCR and ECAR measured. For activation response, 1 μM CB839 was injected into IL-7 maintained naive CD4+ T cells in seahorse medium and allowed to incubate for 20 minutes, followed by soluble αCD3/CD28 injection. Mito Stress assay was performed using kit (Cat#103015–100).
Mass Spectrometry
13C Tracing
To measure 13C-Glucose tracing in T cell activation, CD4+ cells from C57BL/6J mice were stimulated on 5 μg/mL anti-CD3/CD28 for 3 days. At day 3, cells were pooled, washed 3x in PBS, and re-stimulated in presence of 1 μM CB839 or Vehicle (DMSO) and 11 mM 13C glucose (Cambridge Isotope Labs, Cat#CLM-1396–1). Cells were incubated for 24 hours at 37°C, then scraped and combined in triplicate. Cells were rinsed with 0.9% saline and metabolites were extracted in methanol. Metabolites measured by LC-High-Resolution Mass Spectrometer (LC-HRMS) using a Q-Exactive machine as previously described(Liberti et al., 2017). The time-dependent glucose labeling pattern was modeled as with the following equation:
In which [X*] is the concentration of labeled glucose, XT is the total concentration (both labeled and unlabeled) of glucose, fX is the glucose production flux. This model was fit to glucose MIDs using the fit() function in MATLAB to determine relative glucose production fluxes. Relative glucose pool sizes were estimated from MS signal intensities.
Differentiation
CD4+ cells were isolated as previously described and differentiated in subset-specific medium in the presence of vehicle or CB839 (in triplicate) for 3 days, split at day 3 with new media and IL-2, then allowed to incubate a further 2 days. At day 5, wells were combined, cells washed 1x in MACS buffer and re-isolated for CD4 via AutoMACS Pro automated magnetic separator (Miltenyi, Cat#: 130-092-545). Metabolites from Th1 and Th17 cells were extracted and analyzed by LC-High-Resolution Mass Spectrometer (LC-HRMS) using a Q-Exactive as described previously (Gerriets et al., 2015). Data were range scaled and analyzed using Metaboanalyst 3.5(Xia and Wishart, 2002) (http://www.metaboanalyst.ca/faces/home.xhtml) to generate heatmaps and for principle component analyses.
Immunoblotting
Immunoblots were performed as previously described (Jacobs et al., 2008) with the following modifications. Cells lysed with RIPA buffer and Halt protease/phosphatase cocktail inhibitors (Life Tech, Cat#: 78443). Protein was quantified by Pierce BCA kit II (Cat#: 23227). Actin blots were visualized by near infrared fluorescence via Li-Cor Odyssey imager. GLS blots were visualized by chemiluminescence using anti-rabbit conjugated horseradish peroxidase. The antibodies used for westerns were: GLS (Cat#: GTX81012, 1:1000), β-Actin (Cat#: 8226, 1:10,000).
Viral Infection with PIK3IP1
Naive CD4+ T cells were isolated from wild-type C57BL/6J mice. T cells were stimulated in Th1 and Th17 skewing conditions plus vehicle of CB839 as previously described. These were incubated for 16 hours with a feeder layer of irradiated splenocytes. Plasmid constructs MSCV-PIK3IP-IRES-Thy1.1 (“PIK3IP1”) and control vector MSCV-IRES-Thy1.1 (“Control”) were used to transfect Plat-E cells (Cat#RV-101). T cells were then infected with cell supernatant containing retrovirus and polybrene and rested for 48 hours. Cells were split at Day 3 in new media containing 10 ng/mL IL-2 (Cat#14-8021-64) and then incubated for 48 hours before removing for intracellular cytokine and transcription factor staining by flow cytometry as described above.
CRISPR/CAS9 PIK3IP1
Naive CD4+ T cells were isolated from Cas9 transgenic mice (RRID:IMSR_JAX:024858)) aged 10–12 weeks old. T cells were plated on an αCD3/CD28 coated 24-well plate and one day after activation, cells were transduced with viral supernatant prepared from PLAT-E cells (Cat#RV-101) transfected with a solution of 2000μg DNA (empty vector pMx-U6-empty-GFP or two different PIK3IP1 targeting guide RNA containing vectors pMx-U6-PIK3IP1-GFP). T cells with the viral particles were centrifuged at 2000rpm for 2 hours at 37°C, followed by incubation for 2 hours at 37°C and 5% CO2. The media was then replaced with 1mL fresh Th1 skewing media and incubated overnight. This was repeated a second time on day 2 of T cell activation. Cells were collected ten days post activation for pS6, intracellular cytokine production, and transcription factor staining by flow cytometry as described.
PIK3IP1 Antibody in vitro
Naive CD4+ T cells were isolated from C57BL6 mice and activated on aCD3/CD28-coated 24 well plates at 1×106 cells/well with either control antibody (Cat#bs-0295P) or PIK3IP1 antibody (Cat#16826–1-AP) at 0.5 μg/mL. Cells were incubated at 37°C for 72 hours and cells removed at 24, 48, and 72 hours for flow cytometry analysis of activation.
In vivo Graft Versus Host Disease
Induction of Graft versus Host Disease (cGVHD) was performed as previously described (Panoskaltsis-Mortari et al., 2007). Briefly, female mice aged 13–14 weeks were lethally irradiated the day before bone marrow transplant. Mice were dosed with cyclophosphamide (Cytoxan, Bristol Myers Squibb) at 120 mg/kg/day on days −3 and −2. Recipient irradiated mice were transplanted via caudal vein with 15 × 106 T cell depleted allogeneic marrow with 1 × 106 cells splenic CD4+ cells from WT or GLS KO mice, or control (no CD4+ T cells). Mice were assessed for lung elasticity, resistance, and compliance at Day 28 by whole body plethysmography using the Flexivent system (Scireq, Montreal, PQ, Canada). Histological assessment of GVHD was assessed as previously described (Blazar et al., 1998).
Asthma Model
Female WT or GLS KO mice at 24 weeks of age were administered intranasal sensitization of either PBS alone or a combination of 100 μg house dust mite extract (Cat#XPB70D3A2.5) and 0.1 ug LPS from Escherichia coli 0111:B4 (Cat#L4391) in 50 μL of PBS. Sensitizations were performed on Day 0, 7, and 14. Mice were harvested 24 hours post-challenge, and lung homogenates were digested to single cells and analyzed for cytokine production and transcription factors by flow cytometry.
In vivo Vaccinia Viral Response
Spleens from pmel-1 Ly5.1 (B6.Cg-Thy-1a/Cy Tg [TcraTcrb] 8Rest/J) mice were used to generate a single cell suspension and treated with ACK buffer to lyse red blood cells. Splenocytes were stimulated in vitro with 1 μM human glycoprotein 100 nine-mer peptide (hgp10025–33) and expanded in culture medium containing IL-2 for 7 days along with 1 μM CB839 or DMSO vehicle. Subsequently, one million CD8+ cells from each condition were transferred by IV injection into recipient 6 week old female Ly5.2 C57BL/6 mice. Immediately following transfer, mice were infected with rhgp100 vaccinina virus (1 × 107 plaque-forming units (PFU)). At the indicated time points following transfer, recipient mouse blood or tissues were collected for analysis.
Immunization with 2W peptide
10–14 week old male GLS WT and KO animals were injected with 10 μg 2W peptide (Genscript) emulsified with Complete Freunds Adjuvant (Cat#F5881) or PBS control and injected subcutaneously in the rear flank as previously described (Moon et al., 2007) and rested for 8 days. At day 8, inguinal lymph nodes and spleens were removed and isolated. MHCII-specific CD4 cells were isolated and purified with APC-conjugated tetramers (generously provided by Dr. Marc Jenkins laboratory, Minneapolis, MN) using Miltenyi LS magnetic columns (Cat#130-042-401) and stained for extracellular and intracellular targets. Intracellular IFNγ was measured in a separate experiment on day 15 after immunization.
In vitro CAR T cell co-culture with target Eμ B ALL cells
T cells were isolated from wild-type C57BL6 spleens using the Pan T Cell isolation kit (Cat#: 130-095-130) and were activated on anti-CD3 anti-CD28 coated plates with IL2 for four days with or without CB839. On days 1 and 2, T cells were transduced with retrovirus produced by Plat-E cells carrying the CAR 28-ζ construct targeting CD19 with GFP reporter. On day 4, CAR T cells were washed three times to remove any drug remnants and plated to equal concentrations on a 96 well plate at 5×105 cells per well and serial dilutions thereof. 5×105 Emu cells, a CD19+ B cell acute lymphoblastic leukemia cell line (Generously provided by Dr. Davila Lab) were then added to every well to assay cell numbers. CD19+ and GFP+ events were stained and counted by flow for each well after 72 hours.
In vivo CAR T cells
CAR T cells were produced as previously described (Li et al., 2017). Briefly, spleen T cells were isolated from wild-type B6, Thy1.1, or GLS KO mice at day 0. Cells were then activated with mouse CD3/CD28 Dynabeads and 30 IU/mL recombinant human IL2. At day 1 and 2, cells were spin transduced twice with retrovirus carrying CARs. At day 3, cells were fed with fresh medium. At day 4, transduced T cells were harvested, beads removed, evaluated for viability, transduction efficiency, immune phenotype and ready for use. For CB839 treated CAR T cells, compound was added to the culture at day 1, 2 and 3. For In vivo study, C57B6 mice (n = 25) were i.p. injected with cyclophosphamide (Cat#C0768) at 300 mg/kg. Mice were i.v. injected with 3×105 CAR T cells one day after CTX injection. Peripheral blood (PB) samples were collected after CAR T injection, stained with B cell and T cell antibodies and subjected to flow cytometry. CountBright beads were added to measure B and T cell numbers.
Colitis/IBD Induction
Colitis was induced by adoptive transfer of 0.4×106 purified (> 99% purity) CD4+CD25−CD45RBhi cells i.p. into recipient male RAG KO mice (RRID: IMSR_JAX:002216). Spleen and lymph node suspensions were used first to purify CD4+ cells using magnetic bead cell separation with a StemCell Kit and these cells were stained with anti-CD4, anti-CD25 and anti-CD45RB for further flow sorting using a FACS Diva flow cytometer (Becton Dickinson) with purities over 95% of the indicated populations. Mice that received adoptive transfers of different cell genotypes were always cohoused in the same cages to avoid differences due to microbiota composition divergence during colitis development. Mice were treated with the NSAID Piroxicam to induce gut damage and initiate disease and animals were weighed over time. Mice that reached humane endpoints and were euthanized were maintained in the analysis at the final weight. At the end of the experiment, mesenteric lymph nodes were isolated and single cell suspensions were analyzed for cytokine production.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed with Prism software version 7.01 (GraphPad Software, La Jolla California, USA, https://www.graphpad.com) using the Student’s t test, one-way ANOVA, or one-sample t test. Longitudinal data was analyzed by two-way ANOVA followed by Tukey’s test and followed up with one-way ANOVA or t test at one specific time point as specified. Statistically significant results are indicated (* p < 0.05, ** p < 0.01, *** p < 0.001) and ns indicates select non-significant data. Error bars show mean ± Standard Deviation unless otherwise indicated. All experiments were carried out in triplicate technical and biological replicates unless otherwise stated. FACs plots shown are representative of n = 3 replicates. RNA-Seq data were analyzed by DESeq2 (Love et al., 2014) in R (Team, 2017).
DATA AND SOFTWARE AVAILABILITY
RNASeq data have been deposited in the GEO database under accession number GEO: GSE112244. ATACSeq data have been deposited in the ArrayExpress database under accession number ArrayExpress: E-MTAB-6648.
Supplementary Material
Highlights.
T cells utilize GLS to support glutaminolysis that integrates with glycolysis
GLS promotes differentiation and function of Th17 cells yet restrains Th1 cells
GLS alters chromatin and gene expression to enhance IL2 and mTORC1 signaling
Targeting GLS protects from Th17 and enhances Th1 cells but can lead to exhaustion
ACKNOWLEDGMENTS
We thank members of the Rathmell lab and H. Hu (University of Alabama, Birmingham) for providing MSCV-PIK3IP-IRES-Thy1.1 and control retroviral constructs, T. Dileepan and M.K. Jenkins (University of Minnesota) for providing reagents for 2W immunization and tetramer staining, and J. Cools (VIB) for providing pMx-U6 plasmids. Some CB839 for in vitro studies was generously provided by Calithera Biosciences. This research was funded by the National Institutes of Health R01 HL136664 (J.C.R.), R01 DK105550 (J.C.R.), R01 CA217987 (J.C.R., W.K.R.), R01CA193256 (J.W.L.), and P30CA01423 (J.W.L.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.10.001.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Blazar BR, Taylor PA, McElmurry R, Tian L, Panoskaltsis-Mortari A, Lam S, Lees C, Waldschmidt T, and Vallera DA (1998). Engraftment of severe combined immune deficient mice receiving allogeneic bone marrow via In utero or postnatal transfer. Blood 92, 3949–3959. [PubMed] [Google Scholar]
- Boyman O, and Sprent J (2012). The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol 12, 180–190. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, and Frauwirth KA (2010). Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol 185, 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerione JWE, and Richard A (2010). Glutaminase: A Hot Spot For Regulation Of Cancer Cell Metabolism? Oncotarget 1, 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Post-transcriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, and Sabatini DM (2014). The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Cheng T (2010). Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Matés JM, and DeBerardinis RJ (2011). Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 108, 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, Leon B, Cross-man DK, Murre C, Myers RM, and Weinmann AS (2017). CCCTC-Binding Factor Translates Interleukin 2- and alpha-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity 47, 251–267.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curthoys NP, and Watford M (1995). Regulation of glutaminase activity and glutamine metabolism. Annu. Rev. Nutr 15, 133–159. [DOI] [PubMed] [Google Scholar]
- Davila ML, Kloss CC, Gunset G, and Sadelain M (2013). CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immuno-competent mouse model of B cell acute lymphoblastic leukemia. PLoS ONE 8, e61338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, and Thompson CB (2008). The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20. [DOI] [PubMed] [Google Scholar]
- DeFrances MC, Debelius DR, Cheng J, and Kane LP (2012). Inhibition of T-cell activation by PIK3IP1. Eur. J. Immunol 42, 2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Zhong X-S, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et al. (2009). Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med 15, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. (2015). Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest 125, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zhu Z, Johnson C, Stoops J, Eaker AE, Bowen W, and DeFrances MC (2008). PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res. 68, 5591–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ, and Rathmell JC (2008). Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J Immunol 180, 4476–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SR, Michalek RD, and Rathmell JC (2010). IL-7 Is Essential for Homeostatic Control of T Cell Metabolism In vivo. J Immunol 184, 3461–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, et al. (2015). Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal 8, ra97. [DOI] [PubMed] [Google Scholar]
- Knox JJ, Cosma GL, Betts MR, and McLane LM (2014). Characterization of T-bet and eomes in peripheral human immune cells. Front. Immunol 5, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Park K, and Davila ML (2017). Gammaretroviral Production and T Cell Transduction to Genetically Retarget Primary T Cells Against Cancer In T-Cell Differentiation: Methods and Protocols, Lugli E, ed. (New York, NY: Springer New York; ), pp. 111–118. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, et al. (2017). A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 26, 648–659.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, and Rathmell JC (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol 186, 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, and Jenkins MK (2007). Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudge JM, and Harrow J (2015). Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm. Genome 26, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, and Kunimoto H (2014). TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci. 105, 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X, and Sun SC (2014). Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, and Restifo NP (1998). gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med 188, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panoskaltsis-Mortari A, Tram KV, Price AP, Wendt CH, and Blazar BR (2007). A new murine model for bronchiolitis obliterans post-bone marrow transplant. Am. J. Respir. Crit. Care Med 176, 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, et al. (2012). Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat. Immunol 13, 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MA, Dai Z, and Locasale JW (2017). The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol 19, 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, and Cantrell DA (2013). Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol 14, 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska PJ, Kim B, Ji X, Hoeksema MD, Massion PP, Beckermann KE, Wu J, Chi J-T, Hong J, and Rathmell JC (2016). Fluorescence-based measurement of cystine uptake through xCT shows requirement for ROS detoxification in activated lymphocytes. J. Immunol. Methods 438, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. (2013). Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest 123, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajan M, Hock AK, Blagih J, Robertson NA, Labuschagne CF, Kruiswijk F, Humpton TJ, Adams PD, and Vousden KH (2018). A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab 10.1016/j.cmet.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Saegusa J, Sendo S, Okano T, Akashi K, Irino Y, and Morinobu A (2017). Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther 19, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC (2017). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Toffalini F, Kallin A, Vandenberghe P, Pierre P, Michaux L, Cools J, and Demoulin J-B (2009). The fusion proteins TEL-PDGFRbeta and FIP1L1-PDGFRalpha escape ubiquitination and degradation. Haematologica 94, 1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, and Cerione RA (2010). Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, and Green DR (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Geng J, Shi B, Liu Z, Wang YH, Stevens AC, Sprout SL, Yao M, Wang H, and Hu H (2016). Cutting Edge: Foxp1 Controls Naive CD8+ T Cell Quiescence by Simultaneously Repressing Key Pathways in Cellular Metabolism and Cell Cycle Progression. J. Immunol 196, 3537–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, and Thompson CB (2010). Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci 35, 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, and Sabatini DM (2017). The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 26, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. (2006). FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126, 375–387. [DOI] [PubMed] [Google Scholar]
- Xia J, and Wishart DS (2002). Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr Protoc Bioinformatics. 10.1002/cpbi.11. [DOI] [PubMed] [Google Scholar]
- Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, Li K, Ma T, Wang H, Ni L, et al. (2017). Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 548, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Choi S-C, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, and Morel L (2015). Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med 7, 274ra218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Song Y, Yang CH, Jan LY, and Jan YN (2014). Female contact modulates male aggression via a sexually dimorphic GABAergic circuit in Drosophila. Nat. Neurosci 17, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNASeq data have been deposited in the GEO database under accession number GEO: GSE112244. ATACSeq data have been deposited in the ArrayExpress database under accession number ArrayExpress: E-MTAB-6648.