

Abstract
The molecular mechanisms underlying various types of synaptic plasticity are historically regarded as separate processes involved in independent cellular events. However, recent progress in our molecular understanding of Hebbian and homeostatic synaptic plasticity supports the observation that these two types of plasticity share common cellular events, and are often altered together in neurological diseases. Here, we discuss the emerging concept of homeostatic synaptic plasticity as a metaplasticity mechanism with a focus on cellular signaling processes that enables a direct interaction between Hebbian and homeostatic plasticity. We also identify distinct and shared molecular players involved in these cellular processes that may be explored experimentally in future studies to test the hypothesis that homeostatic synaptic plasticity serves as a metaplasticity mechanism to integrate changes in neuronal activity and support optimal Hebbian learning.
Introduction
One of the defining features of the nervous systems in both invertebrate and vertebrate animals is that they are plastic – changes in the activity and connectivity of the various circuits within the nervous system enable learning, encode memory, and drive behavior. Hebbian and non-Hebbian types of synaptic plasticity have been described as two major mechanisms driving synaptic connectivity changes as a result of synaptic activity experience. Hebbian plasticity, referred to here as input-specific synaptic modifications in the forms of long-term potentiation (LTP) and long-term depression (LTD), is thought to underlie associative learning through bidirectional modification of synaptic strength [1]. The direction of modification is determined by the levels of the postsynaptic responses relative to a “modification threshold” (θm), which itself is subject to modification (the sliding threshold model or BCM model) [2,3]. Hebbian plasticity is thought to be self-reinforcing with a tendency to run away if left unchecked (e.g. LTP leads to synaptic strengthening and more correlated pre- and post-synaptic activity, which facilitates additional LTP). The non-Hebbian (sometimes also called anti-Hebbian) plasticity was brought into the picture as a “corrective” mechanism to prevent runaway Hebbian plasticity.
Significant progress has been made in the past two decades toward a molecular understanding of the mechanisms underlying Hebbian plasticity. It becomes clear that although synaptic strengthening (LTP) and weakening (LTD) push synaptic weight changes toward opposite directions by primarily up- or down-regulating presynaptic release probability and moving postsynaptic neurotransmitter receptors in or out of the synaptic membranes, the molecular signaling pathways leading to these changes are quite distinct – instead of sliding on the scale of activation of the same signaling pathway (one end of the scale being LTP and the other LTD), different molecular players are recruited to support opposite changes at synapses. What are the possible mechanisms that mediate the crosstalk between these distinct LTP- and LTD-driving signaling mechanisms in such a manner that they can be engaged in a coordinated fashion to achieve the “sliding” of the θm (i.e. metaplasticity)?
Homeostatic synaptic plasticity, as a major form of non-Hebbian plasticity, has been studied historically in a slightly different context. It is thought to stabilize neural networks through negative feedback-based modifications, thus countering the self-reinforcing nature of Hebbian plasticity [4]. Although Hebbian and homeostatic plasticity are believed to achieve distinct purposes (associative learning versus network stability), the biological parameters they modify are often the same. These parameters include, but are not limited to, neuronal excitability [5,6], synaptic strength [7–9], and – on a longer time scale – changes in the number of synaptic contacts [10,11]. A recent review on a similar topic did an excellent job covering in vivo evidence supporting the interactions between synaptic homeostatic and Hebbian mechanisms [12]. We have previously proposed the idea that local homeostatic synaptic plasticity, which potentially maintains activity stability in a neuronal subcompartment (e.g. a segment of dendrite or even single synapses) instead of the entire neuron [13–17], may function as a type of metaplasticity to modulate Hebbian plasticity [18]. In this article, we will first focus on the regulation of postsynaptic AMPA receptor (AMPAR) abundance as an example of a major converging point between Hebbian and homeostatic plasticity. We will discuss the cellular processes and the molecular players involved in each process to identify potential cellular ‘nodes’ through which homeostatic synaptic plasticity may act to impact Hebbian plasticity. In addition, we discuss possibilities that homeostatic plasticity deficit may contribute to impaired cognitive functions through changes that drive synaptic excitation/inhibition (E/I) imbalance in animal models of neuropsychiatric disorders
Synaptic retinoic acid (RA) signaling, homeostatic synaptic plasticity and their interaction with Hebbian plasticity
The discovery of RA’s involvement in homeostatic synaptic plasticity was a bit serendipitous. Acute RA treatment in cultured hippocampal neurons increases excitatory synaptic transmission with a mechanism that does not require its genomic action [19]. The search for RA synthesis mechanism revealed that it is suppressed by intracellular Ca2+ and thus de-repression occurs during prolonged blockade of synaptic activity [19,20]. Putting the two findings together allowed us to uncover the central role of RA and its action on the RA receptor RARα [21] in synaptic silencing-induced homeostatic upregulation of excitatory synaptic strength (Figure 1). RA signaling also modulates inhibitory synapses in the opposite direction [22], thus acting as a master organizer to coordinate synaptic E/I balance in the context of homeostatic synaptic plasticity.
Figure 1. Molecular pathways involved in RA-dependent homeostatic synaptic plasticity and its interaction with Hebbian plasticity.
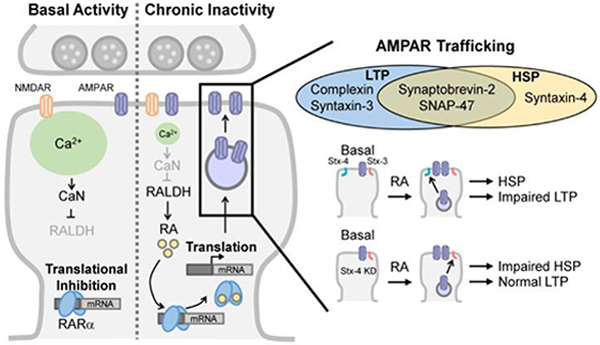
Left: Molecular pathways involved in RA-dependent homeostatic synaptic plasticity (HSP). A reduction in postsynaptic Ca2+ levels resulted from synaptic inactivity triggers RA synthesis, which disinhibits local protein synthesis and promotes synaptic insertion of AMPARs. CaN: calcineurin; RALDH: retinal dehydrogenase. Right: Exocytosis of AMPAR-containing vesicles into synaptic membranes during LTP and HSP is mediated by partially overlapping SNARE components. Postsynaptic deletion of Q-SNARE syntaxin-4 (Stx-4), which is uniquely required for HSP, prevents the impairment of LTP following RA treatment by blocking RA-induced HSP.
Aside from RA-dependent homeostatic synaptic plasticity, synaptic strength can also be modified homeostatically by RA-independent mechanisms. For example, Homeostatic synaptic plasticity induced by prolonged blockade of neuronal firing alone (e.g. tetrodotoxin (TTX) treatment to block voltage-gated sodium channels) does not require RA or local protein synthesis and therefore is not affected by RA synthesis blockers [20,23]. Synaptic activity blockade with co-treatment of glutamate receptor antagonists and TTX reduces dendritic Ca2+ concentration further below a critical level, triggers RA synthesis, and induces rapid compensation of synaptic strength by engaging local protein synthesis [13,19,20,24,25] (Figure 1). Thus, an important distinction between RA-dependent and RA-independent homeostatic plasticity is that local protein synthesis activated by RA in neuronal dendrites allows homeostatic modulation of synaptic strength in potentially discrete subcellular compartments. As we discuss below, these different forms of homeostatic plasticity have differential impacts on subsequent Hebbian plasticity.
In the case of RA-dependent homeostatic plasticity, although synaptic RA signaling is not directly involved in Hebbian plasticity, altering synaptic strength with RA suppresses subsequent LTP expression [26] (Figure 1). RA-induced impairment of LTP can be rescued by protein synthesis inhibitors or RARα deletion, indicating RARα signaling and local protein synthesis play significant roles in the interaction between LTP and RA-dependent homeostatic plasticity [26]. Interestingly, chronic TTX treatment, which induces homeostatic upregulation of mEPSC amplitude in an RA-independent manner, has the opposite effect on LTP – it promotes greater LTP [27]. This is because in addition to upregulating strength of existing synapses, TTX treatment also increases the relative proportion of silent synapses by significantly increasing the formation of a number of new synapses that are functionally silent (e.g. AMPAR-lacking). LTP induction in TTX-treated slices leads to AMPAR insertion into and functional activation of these silent synapses, thus generating greater LTP than control slices [27]. A similar study using a longer (3–4 days) TTX treatment conducted in slightly older rat organotypic hippocampal slices concluded that the effect of circuit inactivity on silent synapse formation and subsequent LTP induction is age-dependent, and that TTX treatment reduces fidelity of presynaptic release and thus compromises LTP [28]. Further study is needed to resolve the exact mechanisms underlying the opposite outcomes from these seemingly similar treatments. However, it is worth noting that although RA-treatment and TTX-treatment both induce homeostatic upregulation of the strength of existing excitatory synapses, their impact on subsequent LTP induction is diverse due to their differential effect on silent synapse formation and/or presynaptic release, revealing the complex nature of the interaction between Hebbian and homeostatic plasticity.
In some of the studies discussed below probing molecular mechanisms of homeostatic synaptic plasticity downstream of RA synthesis, RA treatment was used as a proxy for homeostatic synaptic plasticity induction, much similar to the use of DHPG to induce LTD in studies probing mechanisms of mGluR-dependent LTD. In this review, we will cover cellular events and molecular mechanisms involved in regulating excitatory synaptic strength in, but not limited to, RA-dependent homeostatic synaptic plasticity.
AMPA receptor trafficking – the final common pathway for excitatory postsynaptic modification
Similar to Hebbian plasticity, homeostatic synaptic plasticity is expressed post-synaptically as modulation of synaptic strength by up- or down-regulation of AMPA receptor (AMPAR) abundance in the postsynaptic density (PSD). Regulation of AMPAR-containing trafficking vesicles can occur at multiple stages of exocytosis and endocytosis.
Within the exocytosis process, the SNARE complex molecules for AMPAR-vesicle fusion has been studied in both LTP and RA-dependent homeostatic synaptic plasticity, and was shown that the SNARE components required for both types of plasticity are partially overlapping. Both processes require the R-SNARE synaptobrevin-2 (Syb-2) and the Q-SNARE SNAP-47 [26,29]. However, the dependence on complexin and the other Q-SNARE syntaxins are different: LTP requires complexin and syntaxin-3 [29,30], while RA-induced AMPA receptor exocytosis requires syntaxin-4 and does not involve complexin [26]. Importantly, direct acute treatment with RA increases excitatory synaptic strength and blocks subsequent induction of LTP. The RA blockade of LTP can be reversed by syntaxin-4 knockdown and prevention of RA-induced AMPA receptor insertion [26], indicating that the step of AMPA receptor exocytosis may act as a critical interaction point between some forms of Hebbian and homeostatic synaptic plasticity (Figure 1).
Another key step in regulated exocytosis is Ca2+-trigger vesicle fusion (e.g. Ca2+-dependent neurotransmitter release). Both RA- and LTP-mediated AMPAR exocytosis requires NMDA receptor (NMDAR) activation (we will come back to the specific point of NMDAR-dependence later), suggesting that a Ca2+-triggered fusion process may be involved. Indeed, postsynaptic synaptotagmin-1 and synaptotagmin-7 act as redundant Ca2+-sensors for activity-regulated AMPAR exocytosis during LTP [31]. Curiously, RA-dependent AMPAR insertion is intact in synaptotagmin-1/7 double knockout neurons, indicating a yet-to-be-identified Ca2+-sensor is involved for AMPAR exocytosis during homeostatic plasticity [31].
Down-regulation of excitatory synaptic strength in Hebbian (i.e. LTD) and homeostatic plasticity both involve endocytosis of AMPARs. The immediate early gene Arc/Arg3.1, upregulated by elevated synaptic activity [32], seems to be a central node in AMPAR endocytosis in both processes by recruiting clathrin-dependent endocytosis machinery to AMPARs and mediating their removal from the synapse [33–35]. Interestingly, aside from modulating AMPAR endocytosis, Arc also localizes to nucleus and suppresses GluA1 transcription through expression of promyelocytic leukemia nuclear bodies [36], thus reinforcing the changes at synapse to weaken synaptic strength during homeostatic down scaling. Additionally, it was proposed that clathrin-independent constitutive endocytosis of AMPARs may be involved in homeostatic downscaling, which requires small GTPase Rac1 and F-actin [37].
In addition to components directly related to vesicular trafficking, posttranslational modification of AMPA receptors, in particular phosphorylation of AMPARs, also influence the trafficking pathways involved in synaptic plasticity. AMPAR synaptic targeting and channel properties are largely affected by phosphorylation of its C-terminal sequences [38]. Among the most studied, phosphorylation of the two serine residues in the C-terminal sequence of GluA1 (S831 and S845) appear to govern the conductance and trafficking of AMPARs in and out of synaptic membranes during LTP and LTD [39]. PKA-mediated phosphorylation of GluA1 S845 has been shown to promote plasma membrane insertion of GluA1 and synaptic retention, thereby facilitating LTP [40–43], whereas dephosphorylation of S845 by calcineurin (CaN) and other phosphatases has been correlated with AMPAR endocytosis and LTD [39,42,44]. Additionally, it has been suggested that regulation of GluA1 S845 phosphorylation by PKA and CaN is involved in AMPAR trafficking during bidirectional homeostatic synaptic plasticity in cortical neurons [45,46], but not in hippocampal neurons [47]. More recent evidence using GluA1 knockin mice lacking the two phosphorylation sites [48] further support the notion that the involvement of phosphorylation-dependent AMPAR trafficking in homeostatic synaptic plasticity may not be as universal but is brain region/neuronal type-specific [24].
Synaptic scaffold proteins – modulators of AMPAR trafficking
It is probably not surprising that many of the synaptic scaffold proteins are involved in synaptic plasticity as their roles in trafficking and synaptic stabilization of AMPARs are well established. PSD-95 and PSD-93 are membrane‐associated guanylate kinase (MAGUK) family proteins that have been shown to be involved in Hebbian and homeostatic synaptic plasticity [49–52]. MAGUK family proteins interact with many transmembrane proteins, including the transmembrane AMPAR regulatory proteins (TARPs) that are considered AMPAR auxiliary subunit instrumental for AMPAR surface and synaptic targeting [53,54]. TARPs are also known to participate in both Hebbian and homeostatic synaptic plasticity [55–58].
Given the significant roles of AMPAR C-terminal phosphorylation in its surface trafficking, scaffold proteins that anchor relevant kinases (e.g. PKA) and phosphatases (e.g. calcineurin) were also studied for their roles in synaptic plasticity. Indeed, non-MAGUK scaffold proteins such as PICK1, GRIP and AKAP150 contribute to Hebbian plasticity regulation [59–61] and homeostatic synaptic plasticity [62–65]. It is worth noting that in addition to posttranslational modification of AMPARs, kinases and phosphatases may also be recruited to dendrites and postsynaptic density to regulate other critical steps of homeostatic synaptic plasticity [24,66].
The Homer protein family is another class of postsynaptic density scaffold protein that supports postsynaptic structure and mediates postsynaptic signaling. Homer-1a, a short variant of homer-1 encoded by an immediate-early gene, was first identified as a mGluR-binding protein [67]. Regulation of mGluR1/5 trafficking by Homer1a not only affects mGluR activation, but also has been shown to be involved in Hebbian plasticity [68,69] and homeostatic plasticity [70] in in vitro studies. A recent study further explored the Homer-1a-mGluR1/5 interaction in an in vivo model of homeostatic synaptic plasticity, namely excitatory synapse weakening during sleep. In this context, synaptic targeting of Homer-1a, which removes AMPARs from postsynaptic density by activating mGluR5 and downstream signaling pathways, is coupled to the state of arousal via wake- and sleep-promoting neuromodulators [71]. Thus, the same Homer1a/mGluR-mediated signaling cascade may be used in both Hebbian and homeostatic synaptic plasticity to regulate synaptic AMPAR removal.
Metaplasticity – a mechanism that integrates homeostatic and Hebbian plasticity in health and disease?
As summarized above, various cellular processes activated during homeostatic synaptic plasticity adjust the state of the synapses (e.g. modification of synaptic AMPAR abundance) in response to activity experience. Multiple signaling pathways are altered in the process of achieving this new status quo, including the activation states of various kinases and phosphatases, the phosphorylation states of AMPARs, and the availability of synaptic slots through addition or removal of synaptic scaffolds, etc. Changes in one or a combination of these signaling pathways may impose a limitation/new constrains onto subsequent Hebbian plasticity (Figure 2). For example, it is conceivable that homeostatic upregulation of the strength of existing synapses through AMPAR insertion will constrain the ability of the affected synapses to undergo LTP but facilitate their ability for LTD within a certain time window – a process that may contribute mechanistically to the sliding threshold model. Meanwhile, the phosphorylation states of AMPARs, the availability of synaptic slots for AMPAR anchoring, and the abundance of silent synapses could also change as a result of homeostatic signaling pathway activation, adding additional complexity to the sliding threshold mechanisms. More importantly, numerous in vitro and in vivo studies show that homeostatic modifications occur at both excitatory and inhibitory synapses (reviewed in [72,73]). Synaptic inhibition is known to modulate Hebbian plasticity through regulating integration of dendritic excitatory inputs both temporally and spatially [74–79]. Homeostatic modulation of synaptic E/I balance in local dendrites may push the θm of a particular excitatory synapse in either directions depending on the state the synapse. In the case of increased E/I after homeostatic plasticity, for example, LTP may be facilitated at unsaturated synapses due to reduced local inhibition but may be constrained if synaptic excitation is already saturated. Thus, homeostatic modification of synaptic E/I ratio may be another metaplasticity mechanism through which sliding threshold for Hebbian plasticity may be achieved at individual synapses.
Figure 2. A schematic diagram depicting the relationship between Hebbian and homeostatic plasticity.
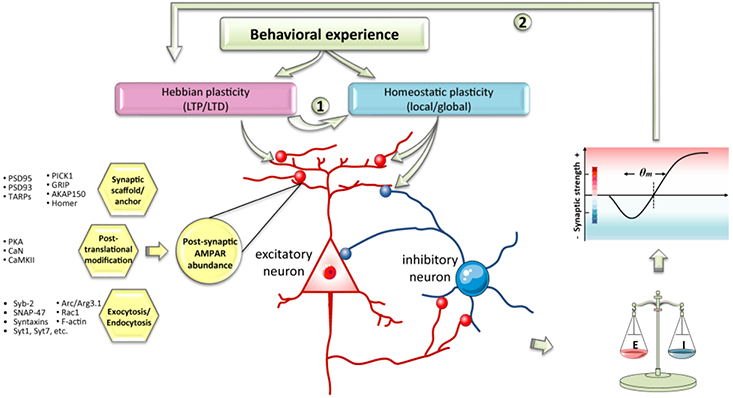
It is traditionally believed that network activity drifts as a result of Hebbian plasticity, which drives homeostatic synaptic plasticity (①). Here we propose the possibility that an animal’s behavioral experience may directly lead to homeostatic plasticity at both excitatory and inhibitory synapses that results in a shift in synaptic E/I balance and modification of the BCM curve. In this context, homeostatic plasticity plays the role of a metaplasticity mechanism, which in turn affects Hebbian plasticity (②).
Given the accumulating knowledge on molecular players specifically involved in homeostatic plasticity, direct testing of the hypothesis that homeostatic plasticity acts as a metaplasticity mechanism for the sliding threshold model is becoming possible and will likely be a major future direction for the synaptic plasticity field. Currently, observations from studies investigating the pathophysiology of neurological diseases provided indirect support for this hypothesis. Among these observations, a disrupted synaptic E/I balance appears to be a common theme in many neurological disorders, including but not limited to autism spectrum disorders (ASDs), schizophrenia, epilepsy and neurodegenerative disorders such as Alzheimer’s disease and Huntington’s disease [80–84]. In extreme cases, E/I imbalance leads to severe network instability, which is consistent with the fact that many neurological disorders have a comorbidity of epileptic activity in subsets of patients [85]. By contrast, intellectual disability is a much more common symptom affecting most patients with ASDs, Schizophrenia and degenerative disorders. How may impaired homeostatic plasticity and defective E/I balance contribute to cognitive dysfunctions?
Homeostatic synaptic plasticity has been relatively more extensively studied in ASDs probably because as a form of neurodevelopmental disorders, ASDs provide an attractive model system for studying long-term functional consequences of defective synaptic plasticity and circuit remodeling without apparent synapse loss or neurodegeneration. For example, mutations in Mecp2 gene, which encodes the transcriptional regulator methyl-CpG-binding protein 2 (MeCP2), leads to Rett syndrome and shows high co-morbidity with ASDs [86]. Mecp2 deletion not only leads to defective Hebbian plasticity and learning [87], but also causes impaired excitatory synaptic up-scaling in visual cortex [88] and down-scaling in hippocampus [89]. Fragile X syndrome (FXS) is another neuropsychiatric disorder characterized by developmental problems including intellectual disability, deficits in communication and social interaction, and in some cases seizures. FXS, in most cases, is caused by silencing of the Fmr1 gene and a complete loss of expression of its protein product FMRP, an RNA-binding protein known to regulate protein synthesis of a subset of neuronal transcripts [90–92]. Hyperactivity of neural network [93] and altered Hebbian plasticity [94] have been described in FXS model mice. Moreover, a complete absence of RA-dependent homeostatic synaptic plasticity at both excitatory and inhibitory synapses have been reported in both FXS mouse [22,23] and human neurons differentiated from FXS patients [95].
Defective learning and memory in various disease models are often attributed to impaired Hebbian plasticity. Altered homeostatic synaptic plasticity found in these disease models is usually considered synaptic phenotypes independent of impaired Hebbian plasticity due to the multifaceted functions of the mutated genes. However, we would like to posit here that these disease phenotypes may be more connected than expected. Most in vivo homeostatic synaptic plasticity studies have been carried out in sensory cortices, which have the advantage of being easily accessible for activity perturbation via sensory modality-specific input manipulations [96–98]. The demonstration of homeostatic plasticity in various in vivo systems not only allows validation of molecular contributors to homeostatic mechanisms in intact circuits, but more importantly, permits further exploration of functional significance of homeostatic plasticity in intact circuits in vivo. In other words, if the signaling pathway known to be specific for homeostatic synaptic plasticity is disrupted, what would be the impact on subsequent Hebbian plasticity and behavior?
A recent study using conditional RARα knockout mice investigated the role of RA-dependent synaptic signaling in whisker-based sensory processing in the barrel cortex [99]. Expression of RARα in layer 5 (L5) pyramidal neurons in the somatosensory cortex was found necessary for normal tactile sensory processing. Transcranial two-photon imaging revealed a significant increase in elimination of more mature-looking dendritic spines on apical dendrites of L5 pyramidal neurons in the absence of RARα. Consistent with RARα’s role in homeostatic plasticity, the enhancement of spine elimination was whisker experience-dependent as whisker trimming rescued the spine elimination phenotype [99]. Although mechanisms underlying whisker-dependent texture encoding remain largely unexplored, it is conceivable that RARα deletion in L5 pyramidal neurons impairs experience-dependent homeostatic synaptic plasticity that fine-tunes the strength of active synapses (i.e. a balance maintained through elimination of immature thin spines and maintenance of mature mushroom-type spines), and negatively impacts sensory information integration from L2/3 neurons to L5 neurons. Future studies are required to investigate how impaired RA signaling and homeostatic plasticity affects whisker experience-dependent Hebbian plasticity, and more broadly speaking, any experience-dependent plasticity, in the context of local circuit wiring and behavioral output.
Although often studied separately, homeostatic synaptic plasticity-inducing sensory manipulations have been shown to affect Hebbian plasticity within the same circuit. Arguably the best examples come from studies in the visual system where prolonged visual deprivation, which is known to induce homeostatic plasticity in the V1 cortical synapses, shifts the modification threshold θm of the Hebbian plasticity BCM curve (reviewed in [100]). Most strikingly, in adult animals, prolonged visual deprivation through dark rearing reopens visual cortical critical period and restores ocular dominance plasticity [101]. These modifications of Hebbian plasticity is thought to be achieved through experience-dependent GluN2A/GluN2B ratio shift [102–104]. Dark rearing, a widely used visual deprivation approach to study visual cortical plasticity, is known to upregulate GluN2B expression and shifts GluN2A/GluN2B ratio [105] toward favoring LTP by lowering LTP threshold [103,106]. Increases in miniature excitatory postsynaptic current (mEPSC) amplitude after dark rearing has been characterized as a typical homeostatic upregulation of excitatory synapses [107]. Intriguingly, a recent study demonstrated that mEPSC amplitude is actually potentiated through a Hebbian mechanism mediated by increased GluN2B-containing NMDAR at synapses, not by a synaptic scaling mechanism (i.e. lowering spontaneous firing) although NMDAR-independent synaptic scaling may still be induced by extreme reduction of activity [108]. This seemingly blurry distinction between ‘true’ homeostatic mechanisms and Hebbian mechanisms may reflect our incomplete understanding of biological processes involved in these different forms of plasticity, it also serves as a reminder that Hebbian and homeostatic plasticity are not two separate entities co-exist within the same system, but are intertwined and even coupled to adjust synaptic and network activity for optimal function.
Conclusions
In this short article, we summarized cellular processes and signaling molecules involved in these processes that are shared between Hebbian and homeostatic synaptic plasticity, and outlined synaptic modifications and behavioral outcomes that are likely the consequence of the interaction between these two forms of synaptic plasticity. Behavioral experiences in an animal drive network activity that may lead to Hebbian or homeostatic plasticity, or both. The latter, through its impact on synaptic excitation and inhibition, alters the modification threshold of Hebbian plasticity and its subsequent involvement in future behavior (Figure 2). The synaptic plasticity field is at an exciting time when we are equipped with rapidly growing insight on the molecular basis of plasticity processes, in conjunction with cutting-edge technologies allowing sophisticated genetic and circuit manipulations. Future experiments will be possible to systematically explore how homeostatic synaptic plasticity, as a mechanism of metaplasticity, impacts Hebbian plasticity and cognitive function.
Highlights.
Hebbian and homeostatic synaptic plasticity converge on shared cellular processes
Homeostatic plasticity adjusts the state of synapses to impact Hebbian plasticity
Homeostatic plasticity alters synaptic E/I and drives the Hebbian sliding threshold
Impaired homeostatic plasticity may be linked to cognitive deficits in ASDs
Acknowledgment
We thank Omid Miry for comments on the manuscript. This work was supported by NIH grants MH086403 (L.C.), MH091193 (L.C.), and HD084215 (L.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Hebb DO: The organization of behavior : a neuropsychological theory. New York: Wiley; 1949. [Google Scholar]
- 2.Bienenstock EL, Cooper LN, Munro PW: Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 1982, 2:32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper LN, Liberman F, Oja E: A theory for the acquisition and loss of neuron specificity in visual cortex. Biological cybernetics 1979, 33:9–28. [DOI] [PubMed] [Google Scholar]
- 4.Watt AJ, Desai NS: Homeostatic Plasticity and STDP: Keeping a Neuron’s Cool in a Fluctuating World. Frontiers in synaptic neuroscience 2010, 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campanac E, Debanne D: Plasticity of neuronal excitability: Hebbian rules beyond the synapse. Archives italiennes de biologie 2007, 145:277–287. [PubMed] [Google Scholar]
- 6.Magee JC, Johnston D: Plasticity of dendritic function. Current opinion in neurobiology 2005, 15:334–342. [DOI] [PubMed] [Google Scholar]
- 7.Huganir RL, Nicoll RA: AMPARs and synaptic plasticity: the last 25 years. Neuron 2013, 80:704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grasselli G, Hansel C: Cerebellar long-term potentiation: cellular mechanisms and role in learning. International review of neurobiology 2014, 117:39–51. [DOI] [PubMed] [Google Scholar]
- 9.Davis GW, Muller M: Homeostatic Control of Presynaptic Neurotransmitter Release. Annual review of physiology 2014. [DOI] [PubMed] [Google Scholar]
- 10.Bourne JN, Harris KM: Balancing structure and function at hippocampal dendritic spines. Annual review of neuroscience 2008, 31:47–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu X, Zuo Y: Spine plasticity in the motor cortex. Current opinion in neurobiology 2011, 21:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keck T, Hubener M, Bonhoeffer T: Interactions between synaptic homeostatic mechanisms: an attempt to reconcile BCM theory, synaptic scaling, and changing excitation/inhibition balance. Current opinion in neurobiology 2017, 43:87–93. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Lau AG, Sarti F: Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology 2014, 78:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Branco T, Staras K, Darcy KJ, Goda Y: Local dendritic activity sets release probability at hippocampal synapses. Neuron 2008, 59:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY: Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc Natl Acad Sci U S A 2008, 105:775–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC: Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci 2004, 7:244–253. [DOI] [PubMed] [Google Scholar]
- 17.Sutton MA, Schuman EM: Dendritic protein synthesis, synaptic plasticity, and memory. Cell 2006, 127:49–58. [DOI] [PubMed] [Google Scholar]
- 18.Yee AX, Hsu YT, Chen L: A metaplasticity view of the interaction between homeostatic and Hebbian plasticity. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 2017, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoto J, Nam CI, Poon MM, Ting P, Chen L: Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 2008, 60:308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang HL, Zhang Z, Hintze M, Chen L: Decrease in calcium concentration triggers neuronal retinoic acid synthesis during homeostatic synaptic plasticity. The Journal of neuroscience : the official journal of the Society for Neuroscience 2011, 31:17764–17771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarti F, Schroeder J, Aoto J, Chen L: Conditional RARalpha knockout mice reveal acute requirement for retinoic acid and RARalpha in homeostatic plasticity. Front Mol Neurosci 2012, 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarti F, Zhang Z, Schroeder J, Chen L: Rapid suppression of inhibitory synaptic transmission by retinoic acid. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013, 33:11440–11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soden ME, Chen L: Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010, 30:16910–16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arendt KL, Zhang Z, Ganesan S, Hintze M, Shin MM, Tang Y, Cho A, Graef IA, Chen L: Calcineurin mediates homeostatic synaptic plasticity by regulating retinoic acid synthesis. Proceedings of the National Academy of Sciences of the United States of America 2015, 112:E5744–5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poon MM, Chen L: Retinoic acid-gated sequence-specific translational control by RARalpha. Proc Natl Acad Sci U S A 2008, 105:20303–20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••26.Arendt KL, Zhang Y, Jurado S, Malenka RC, Sudhof TC, Chen L: Retinoic Acid and LTP Recruit Postsynaptic AMPA Receptors Using Distinct SNARE-Dependent Mechanisms. Neuron 2015, 86:442–456.This study demonstrates that synaptic insertion of AMPAR-containing vesicles during RA-dependent homeostatic plasticity requires postsynaptic SNARE fusion machinery, some of which are distinct from those required for AMPAR insertion during LTP.
- ••27.Arendt KL, Sarti F, Chen L: Chronic inactivation of a neural circuit enhances LTP by inducing silent synapse formation. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013, 33:2087–2096.In a mouse organotypic hippocampal slice culture system, the authors explored the impact of chronic TTX treatment on LTP, and found that counterintuitively, LTP was enhanced despite the already elevated basal synaptic transmission as a result of TTX-induced synaptic scaling. Further experiments revealed increased AMPAR-lacking silent synapse formation following TTX treatment, and activation of these silent synapses during LTP induction contributed to greater LTP.
- •28.Soares C, Lee KFH, Beique JC: Metaplasticity at CA1 Synapses by Homeostatic Control of Presynaptic Release Dynamics. Cell reports 2017, 21:1293–1303.This is a study conducted in rat organotypic hippocampal slices, where prolonged TTX treatment was found to decrease the reliability of presynaptic release, thus contributing to impaired LTP after TTX treatment. This study, togetehr with the one described in reference #27, reveal the complex nature of the interactions between Hebbian and homeostatic plasticity.
- •29.Jurado S, Goswami D, Zhang Y, Molina AJ, Sudhof TC, Malenka RC: LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 2013, 77:542–558.Using shRNA and genetic manipulations, this study explored the requirement of SNARE fusion machninery in postsynaptic vesicle fusion events. In particular, postsynapitc exocytosis of AMPAR-containing vesicles during LTP was investigated. The results from this study show that SNARE-mediated vesicule fusion is essential to LTP expression, and that the fusion machninery is distinct from that used during presynaptic neurotransmitter release.
- 30.Ahmad M, Polepalli JS, Goswami D, Yang X, Kaeser-Woo YJ, Sudhof TC, Malenka RC: Postsynaptic complexin controls AMPA receptor exocytosis during LTP. Neuron 2012, 73:260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •31.Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, Xu W, Chen L, Malenka RC, Sudhof TC: Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 2017, 544:316–321.This study explores the function of postsynaptic calcium during LTP beyond its well-known action on protein kinase and/or phophatase activation. The results suggest that postsynaptic synaptotagmin-1 and synaptotagmin-7 act as redundant calcium-sensors for AMPA receptor exocytosis during LTP, thus formally establishing calcium-triggered AMPA receptor-containing vesicle fusion as a mechanism for AMPA receptor recruitment during LTP.
- 32.Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, Worley PF: Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 1995, 14:433–445. [DOI] [PubMed] [Google Scholar]
- 33.Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM: Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 2008, 59:84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF: Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 2006, 52:475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao M, Sossa K, Song L, Errington L, Cummings L, Hwang H, Kuhl D, Worley P, Lee HK: A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010, 30:7168–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korb E, Wilkinson CL, Delgado RN, Lovero KL, Finkbeiner S: Arc in the nucleus regulates PML-dependent GluA1 transcription and homeostatic plasticity. Nature neuroscience 2013, 16:874–U266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glebov OO, Tigaret CM, Mellor JR, Henley JM: Clathrin-independent trafficking of AMPA receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience 2015, 35:4830–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shepherd JD, Huganir RL: The cell biology of synaptic plasticity: AMPA receptor trafficking. Annual review of cell and developmental biology 2007, 23:613–643. [DOI] [PubMed] [Google Scholar]
- •39.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL: Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 2000, 405:955–959.This is a seminal study establishing the biochemical link between synaptic activity history and AMPA receptor trafficking during Hebbian plasticity. The authors showed that different from common wisdom that LTP and LTD are simply the inverse of each other, they are associated with phosphorylation and dephosphorylation, respectively, of distinct GluA1 phosphorylation sites. Thus, with identical stimulation conditions, the history of synapses, refelcted in the AMPA receptor phosphorylation states, would dictate the recruitment of different signal-transduction pathways during synaptic plasticity.
- 40.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF: Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. The Journal of neuroscience : the official journal of the Society for Neuroscience 2000, 20:89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R: PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nature neuroscience 2003, 6:136–143. [DOI] [PubMed] [Google Scholar]
- 42.Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, et al. : Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 2003, 112:631–643. [DOI] [PubMed] [Google Scholar]
- 43.Oh MC, Derkach VA, Guire ES, Soderling TR: Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. The Journal of biological chemistry 2006, 281:752–758. [DOI] [PubMed] [Google Scholar]
- 44.Man HY, Sekine-Aizawa Y, Huganir RL: Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proceedings of the National Academy of Sciences of the United States of America 2007, 104:3579–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diering GH, Gustina AS, Huganir RL: PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 2014, 84:790–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, Ziff EB: Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS biology 2014, 12:e1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He K, Goel A, Ciarkowski CE, Song L, Lee HK: Brain area specific regulation of synaptic AMPA receptors by phosphorylation. Communicative & integrative biology 2011, 4:569–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HK, Takamiya K, He K, Song L, Huganir RL: Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. Journal of neurophysiology 2010, 103:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehrlich I, Malinow R: Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. The Journal of neuroscience : the official journal of the Society for Neuroscience 2004, 24:916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stein V, House DR, Bredt DS, Nicoll RA: Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience 2003, 23:5503–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun Q, Turrigiano GG: PSD-95 and PSD-93 play critical but distinct roles in synaptic scaling up and down. The Journal of neuroscience : the official journal of the Society for Neuroscience 2011, 31:6800–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlisle HJ, Fink AE, Grant SG, O’Dell TJ: Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike timing-dependent plasticity. J Physiol 2008, 586:5885–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA: Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000, 408:936–943. [DOI] [PubMed] [Google Scholar]
- 54.Bats C, Groc L, Choquet D: The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron 2007, 53:719–734. [DOI] [PubMed] [Google Scholar]
- 55.Louros SR, Hooks BM, Litvina L, Carvalho AL, Chen C: A role for stargazin in experience-dependent plasticity. Cell reports 2014, 7:1614–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsuda S, Kakegawa W, Budisantoso T, Nomura T, Kohda K, Yuzaki M: Stargazin regulates AMPA receptor trafficking through adaptor protein complexes during long-term depression. Nat Commun 2013, 4:2759. [DOI] [PubMed] [Google Scholar]
- 57.Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D: CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 2010, 67:239–252. [DOI] [PubMed] [Google Scholar]
- 58.Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS: Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron 2005, 45:269–277. [DOI] [PubMed] [Google Scholar]
- 59.Sanderson JL, Gorski JA, Dell’Acqua ML: NMDA Receptor-Dependent LTD Requires Transient Synaptic Incorporation of Ca(2)(+)-Permeable AMPARs Mediated by AKAP150-Anchored PKA and Calcineurin. Neuron 2016, 89:1000–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL: Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron 2006, 49:845–860. [DOI] [PubMed] [Google Scholar]
- 61.Pfennig S, Foss F, Bissen D, Harde E, Treeck JC, Segarra M, Acker-Palmer A: GRIP1 Binds to ApoER2 and EphrinB2 to Induce Activity-Dependent AMPA Receptor Insertion at the Synapse. Cell reports 2017, 21:84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gainey MA, Tatavarty V, Nahmani M, Lin H, Turrigiano GG: Activity-dependent synaptic GRIP1 accumulation drives synaptic scaling up in response to action potential blockade. Proceedings of the National Academy of Sciences of the United States of America 2015, 112:E3590–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tan HL, Queenan BN, Huganir RL: GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proceedings of the National Academy of Sciences of the United States of America 2015, 112:10026–10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anggono V, Clem RL, Huganir RL: PICK1 loss of function occludes homeostatic synaptic scaling. The Journal of neuroscience : the official journal of the Society for Neuroscience 2011, 31:2188–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanderson JL, Scott JD, Dell’Acqua ML: Control of Homeostatic Synaptic Plasticity by AKAP-Anchored Kinase and Phosphatase Regulation of Ca(2+)-Permeable AMPA Receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience 2018, 38:2863–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thiagarajan TC, Piedras-Renteria ES, Tsien RW: alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 2002, 36:1103–1114. [DOI] [PubMed] [Google Scholar]
- 67.Brakeman PR, Lanahan AA, O’Brien R, Roche K, Barnes CA, Huganir RL, Worley PF: Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 1997, 386:284–288. [DOI] [PubMed] [Google Scholar]
- 68.Ueta Y, Yamamoto R, Sugiura S, Inokuchi K, Kato N: Homer 1a suppresses neocortex long-term depression in a cortical layer-specific manner. Journal of neurophysiology 2008, 99:950–957. [DOI] [PubMed] [Google Scholar]
- 69.Ronesi JA, Huber KM: Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. The Journal of neuroscience : the official journal of the Society for Neuroscience 2008, 28:543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu JH, Park JM, Park S, Xiao B, Dehoff MH, Kim S, Hayashi T, Schwarz MK, Huganir RL, Seeburg PH, et al. : Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron 2010, 68:1128–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••71.Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A, Huganir RL: Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 2017, 355:511–515.Different from most homeostatic plasticity studies conducted in vivo, which involves sensory deprivation, this study examines homeostatic plasticity during a natural state of the organism – sleep. The results show that Homer 1a, as a molecular integrator of arousal and sleep, mediates synaptic weakening during sleep.
- 72.Fox K, Stryker M: Integrating Hebbian and homeostatic plasticity: introduction. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 2017, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keck T, Toyoizumi T, Chen L, Doiron B, Feldman DE, Fox K, Gerstner W, Haydon PG, Hubener M, Lee HK, et al. : Integrating Hebbian and homeostatic plasticity: the current state of the field and future research directions. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 2017, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bar-Ilan L, Gidon A, Segev I: The role of dendritic inhibition in shaping the plasticity of excitatory synapses. Frontiers in neural circuits 2012, 6:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.D’Amour JA, Froemke RC: Inhibitory and excitatory spike-timing-dependent plasticity in the auditory cortex. Neuron 2015, 86:514–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu Y, Kaneko M, Tang Y, Alvarez-Buylla A, Stryker MP: A cortical disinhibitory circuit for enhancing adult plasticity. eLife 2015, 4:e05558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF: Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 1998, 282:1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Letzkus JJ, Wolff SB, Meyer EM, Tovote P, Courtin J, Herry C, Luthi A: A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 2011, 480:331–335. [DOI] [PubMed] [Google Scholar]
- 79.Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GC, Matsuzaki M, Kasai H: GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nature neuroscience 2013, 16:1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nelson SB, Valakh V: Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87:684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao R, Penzes P: Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Current molecular medicine 2015, 15:146–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bonansco C, Fuenzalida M: Plasticity of Hippocampal Excitatory-Inhibitory Balance: Missing the Synaptic Control in the Epileptic Brain. Neural plasticity 2016, 2016:8607038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Styr B, Slutsky I: Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nature neuroscience 2018, 21:463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cao Y, Bartolome-Martin D, Rotem N, Rozas C, Dellal SS, Chacon MA, Kadriu B, Gulinello M, Khodakhah K, Faber DS: Rescue of homeostatic regulation of striatal excitability and locomotor activity in a mouse model of Huntington’s disease. Proceedings of the National Academy of Sciences of the United States of America 2015, 112:2239–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bozzi Y, Provenzano G, Casarosa S: Neurobiological bases of autism-epilepsy comorbidity: a focus on excitation/inhibition imbalance. The European journal of neuroscience 2018, 47:534–548. [DOI] [PubMed] [Google Scholar]
- 86.Moretti P, Zoghbi HY: MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev 2006, 16:276–281. [DOI] [PubMed] [Google Scholar]
- 87.Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY: Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. The Journal of neuroscience : the official journal of the Society for Neuroscience 2006, 26:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blackman MP, Djukic B, Nelson SB, Turrigiano GG: A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J Neurosci 2012, 32:13529–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qiu Z, Sylwestrak EL, Lieberman DN, Zhang Y, Liu XY, Ghosh A: The Rett syndrome protein MeCP2 regulates synaptic scaling. J Neurosci 2012, 32:989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. : FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146:247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Santoro MR, Bray SM, Warren ST: Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annual review of pathology 2012, 7:219–245. [DOI] [PubMed] [Google Scholar]
- 92.Ascano M Jr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. : FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Contractor A, Klyachko VA, Portera-Cailliau C: Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 2015, 87:699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pfeiffer BE, Huber KM: The state of synapses in fragile X syndrome. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 2009, 15:549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, Fair T, Yang N, Sudhof TC, Wernig M, et al. : Fragile-X Mutation Impairs Homeostatic Plasticity in Human Neurons by Blocking Synaptic Retinoic-Acid Signaling. Science Translational Medicine (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gainey MA, Feldman DE: Multiple shared mechanisms for homeostatic plasticity in rodent somatosensory and visual cortex. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 2017, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaneko M, Stryker MP: Homeostatic plasticity mechanisms in mouse V1. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 2017, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Whitt JL, Petrus E, Lee HK: Experience-dependent homeostatic synaptic plasticity in neocortex. Neuropharmacology 2014, 78:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••99.Park E, Tjia M, Zuo Y, Chen L: Postnatal ablation of synaptic retinoic acid signaling impairs cortical information processing and sensory discrimination in mice. The Journal of Neuroscience 2018.This is the first study establishing the role of retinoic acid receptor RARα in mature circuits in vivo. Using different genetic manipulations, the authors show that RARα expression in layer 5 pyramidal neurons of the barrel cortex is essential for normal spine dynamics and whisker-dependent texture discrimination.
- 100.Cooper LN, Bear MF: The BCM theory of synapse modification at 30: interaction of theory with experiment. Nature reviews. Neuroscience 2012, 13:798–810. [DOI] [PubMed] [Google Scholar]
- •101.He HY, Hodos W, Quinlan EM: Visual deprivation reactivates rapid ocular dominance plasticity in adult visual cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 2006, 26:2951–2955.Using visual deprivation as a means to manipulate sensory experience, the authors show that critical period plasticity in visual cortex can be reactivated in adulthood by prolonged changes in the neuronal activity. This study is highly significant because it demonstrates, in an in vivo setting, a clear interaction between Hebbian plasticity rules and homeostatic plasticity-inducing activity manipulations.
- 102.Cho KKA, Khibnik L, Philpot BD, Bear MF: The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proceedings of the National Academy of Sciences 2009, 106:5377–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Philpot BD, Espinosa JS, Bear MF: Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 2003, 23:5583–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sawtell NB, Frenkel MY, Philpot BD, Nakazawa K, Tonegawa S, Bear MF: NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron 2003, 38:977–985. [DOI] [PubMed] [Google Scholar]
- 105.Philpot BD, Sekhar AK, Shouval HZ, Bear MF: Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 2001, 29:157–169. [DOI] [PubMed] [Google Scholar]
- 106.Kirkwood A, Rioult MC, Bear MF: Experience-dependent modification of synaptic plasticity in visual cortex. Nature 1996, 381:526–528. [DOI] [PubMed] [Google Scholar]
- 107.Goel A, Lee HK: Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J Neurosci 2007, 27:6692–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••108.Bridi MCD, de Pasquale R, Lantz CL, Gu Y, Borrell A, Choi S-Y, He K, Tran T, Hong SZ, Dykman A, et al. : Two distinct mechanisms for experience-dependent homeostasis. Nature neuroscience 2018, 21:843–850.Homeostatic synaptic plasticity in the visual cortex induced by dark rearing or binocular lid suture, signified as an increase in mEPSC amplitude, is thought to be triggered by the reduction in spontaneous firing as a result of visual deprivation. This study show that contrary to this well-accepted notion, spontaneous firing together with loss of visual input causes a shift in GluN2A/2B ratio, which lowers the threshold for LTP, and potentiates mEPSC amplitude through a Hebbian mechanism. Only during extreme reductions of acivity that a NMDA receptor-independent synaptic scalig mechanism is engaged to increase mEPSC amplitude homeostatically.