

Abstract
Colorectal adenomas are common precancerous lesions with the potential for malignant transformation to colorectal adenocarcinoma. Endoscopic polypectomy provides an opportunity for cancer prevention; however, recurrence rates are high. We collected formalin-fixed paraffin-embedded tissue of fifteen primary adenomas with recurrence, fifteen adenomas without recurrence, and fourteen matched pair samples (primary adenoma and the corresponding recurrent adenoma). The samples were analysed by array-comparative genomic hybridisation (aCGH) and single-cell multiplex-interphase fluorescence in situ hybridisation (miFISH) to understand clonal evolution, to examine the dynamics of copy number alterations (CNAs) and to identify molecular markers for recurrence prediction. The miFISH probe panel consisted of fourteen colorectal carcinogenesis-relevant genes (COX2, PIK3CA, APC, CLIC1, EGFR, MYC, CCND1, CDX2, CDH1, TP53, HER2, SMAD7, SMAD4 and ZNF217), and a centromere probe (CEP10). ACGH analysis confirmed the genetic landscape typical for colorectal tumorigenesis, i.e., CNAs of chromosomes 7, 13q, 18 and 20q. Focal aberrations (≤10 Mbp) were mapped to chromosome bands 6p22.1-p21.33 (33.3%), 7q22.1 (31.4%) and 16q21 (29.4%). MiFISH detected gains of EGFR (23.6%), CDX2 (21.8%) and ZNF217 (18.2%). Most adenomas exhibited a major clone population which was accompanied by multiple smaller clone populations. Gains of CDX2 were exclusively seen in primary adenomas with recurrence (25%) compared to primary adenomas without recurrence (0%). Generation of phylogenetic trees for matched pair samples revealed four distinct patterns of clonal dynamics. In conclusion, adenoma development and recurrence are complex genetic processes driven by multiple CNAs whose evaluation by miFISH, with emphasis on CDX2, might serve as predictor of recurrence.
Keywords: clonal evolution, CDX2, colorectal adenoma, FISH, genomic instability, intra-tumour heterogeneity, recurrence
Introduction
Colorectal adenomas are common lesions which are estimated to be present in one-third to one-half of all individuals in Western countries.1,2 Although adenomas are benign they can progress to invasive colorectal adenocarcinoma (CRC). On the molecular level, this so-called adenoma-carcinoma sequence is defined by the accumulation of specific gene mutations and genomic imbalances.3 Early colorectal adenomas show gains of chromosome 7 while in more advanced adenomas genomic aberration patterns become more complex with copy number alterations (CNAs) of 13q, 18q and 20q.4–7 In CRC, additional CNAs affecting 8q, 17p and 17q emerge.7–9 Studies on the sequence of genetic changes in individual adenomas and their recurrences, however, are rare.
Up to 30% of adenomas recur after initial endoscopic polypectomy.10–12 However, underlying genetic changes and markers predicting local recurrence of colorectal adenomas remain largely elusive. By analysing colorectal adenomas, Habermann and colleagues reported the gain of 20q as an indicator of adenoma recurrence and/or synchronous carcinoma.13 To identify specific patterns of chromosomal aberrations that indicate local recurrence, we collected colorectal adenomas with and without recurrence and mapped genomic imbalances using array-comparative genomic hybridisation (aCGH). However, there is also evidence that subpopulations, or even single cells, play a role in cancer development, therapy resistance and cancer recurrence, which might be missed by aCGH.14–17 We have therefore performed single-cell genetic analyses to assess tumour phylogenies using multiplex interphase fluorescence in situ hybridisation (miFISH).18 The miFISH panel used in this study allowed to simultaneously detect copy numbers of fourteen colorectal carcinogenesis-specific and frequently altered oncogenes (COX2, PIK3CA, CLIC1, EGFR, MYC, CCND1, CDX2, HER2, ZNF217) and tumour suppressor genes (APC, CDH1, TP53, SMAD4, SMAD7).
Our study improves the understanding of adenoma recurrence, identifies a genetic marker for recurrence and provides insights into the clonal evolution of adenomatous polyps.
Material and Methods
Clinical samples
This study was approved by the local board of ethics (Medical Ethics Committee II, University of Heidelberg, ethics approval: 2012-608R-MA) and by the Office of Human Subjects Research of the National Institutes of Health (OHSR #13220). All colonoscopic samples were collected between 2002 and 2014 and stored at the tissue archive of the Institute of Pathology of the University Medical Centre Mannheim. In total, fifty-eight formalin-fixed paraffin-embedded (FFPE) specimens could be obtained after screening the electronic clinical database of the Central Interdisciplinary Endoscopy Unit of the University Medical Centre Mannheim. While pathological evaluation for in toto polypectomy was not possible due to fragmentation of the tissue, endoscopic removal was indicated as complete based on thorough clinical assessment for all lesions. Adenoma recurrence would be defined by the endoscopist (SB) if a scar was present in connection with the new adenoma formation and/or if the exact same anatomical position as described in centimetres from the anus was ensured. The median observation time after polypectomy was 19.8 months (IQR, 10.6-27.6 months). Histological classification discerned tubular, tubulo-villous or villous adenomas with low-grade dysplasia (LGD) or high-grade dysplasia (HGD), respectively (Supporting Information Table 1). Pathological classification was done in accordance with the current WHO-classification19 from 2010 by two board-certified pathologists (TG/JR) blinded to all data. Adenomas were categorised by recurrence status and two patient groups were established (Table 1). Adenoma samples for analyses comprised: (i) primary adenomas, not presenting any recurrent adenoma in the follow-up period (n=15; primary adenomas without recurrence); (ii) primary adenomas, with documented adenoma recurrence at the same location as the primary adenoma (n=29; primary adenomas with recurrence); (iii) matched pair samples (primary adenoma and the corresponding recurrent adenoma) (n=14; adenoma matched pairs).
Table 1.
Distribution of age at diagnosis, gender, adenoma location, histology, size and observation time of patients with primary adenomas without recurrence and with recurrence.
| Primary adenomas w/o recurrence | Primary adenomas w/ recurrence | ||
|---|---|---|---|
| Variable | (n=15) | (n=29) | p-value |
| Age at diagnosis (y) | |||
| Mean ± SD | 65.0 ± 6.3 | 66.6 ± 11.7 | 0.633a |
| Median (IQR) | 66.7 (63.9-68.5) | 68.2 (57.4-72.9) | |
| Gender | |||
| Female | 4 | 14 | 0.208b |
| Male | 11 | 15 | |
| Location | |||
| Right hemicolon | 9 | 14 | 0.774c |
| Left hemicolon | 3 | 6 | |
| Rectum | 3 | 9 | |
| Histology | |||
| Tubular | 5 | 3 | 0.099b |
| Tubulo-villous / Villous | 10 | 26 | |
| Size (mm) | |||
| Mean ± SD | 22.8 ± 9.7 | 32.5 ± 20.2 | 0.086a |
| Median (IQR) | 20.0 (15.0-30.0) | 27.5 (20.0-40.0) | |
| Observation time/recurrence-free time (m) | |||
| Mean ± SD | 25.4 ± 14.4 | 21.7 ± 19.5 | 0.528a |
| Median (IQR) | 21.9 (12.5-38.0) | 17.2 (8.2-25.7) |
Student t test;
Fisher exact test;
Freeman-Halton test
Array-comparative genomic hybridisation (aCGH)
Haematoxylin and eosin (H&E)-stained sections were prepared from archived FFPE tissue and the regions of interest comprising at least 70% tumour content were marked.20 Two 10 μm-thick consecutive unstained FFPE sections were used per sample, which were twice deparaffinised in xylene for 10 minutes prior to rehydration in an ethanol series for 10 minutes. Marked H&E-slides were used for guidance to macro-dissect tumour regions with a scalpel. Genomic DNA extraction was performed with Gentra Puregene Tissue Kit (Qiagen, Hilden, Germany) and isolated DNA was purified using DNA Clean & Concentrator Kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s instructions, respectively. Eluted DNA was labelled by Genomic Universal Linkage System (ULS) Labelling Kit (Agilent, Santa Clara, CA, USA) prior to hybridisation on SurePrint G3 Human CGH Microarray 8×60K (Agilent) following the manufacturer’s protocol version 3.1 (see Supporting Information Material and Methods for details). Four samples (A1, A19, P4a and P4b) were initially excluded from subsequent aCGH analysis due to not passing quality standards (insufficient DNA labelling by Cy3 resulting in poor hybridisation quality). Aberration calls were visualised by Nexus Copy Number 8.0 (BioDiscovery, El Segundo, CA, USA). Profiles were adjusted by rank segmentation algorithm and manually reviewed according to baseline noise. Aberrations <1.5 Mbp, sex-chromosomes and copy number variants (CNV)-overlapping regions were excluded from analysis. The average number of copy alterations (ANCA) was calculated by dividing the sum of observed copy number imbalances by the respective number of cases.21 Microarray data has been deposited in GEO database (data accession number: GSE110221).
Multiplex-interphase fluorescence in situ hybridisation (miFISH)
For each FISH probe, contigs consisting of three to four overlapping bacterial artificial chromosome (BAC) clones were assembled in the UCSC Genome Browser (http://genome.ucsc.edu) targeting genes frequently altered in colorectal carcinogenesis. Gene selection was based on (i) published data on chromosomal aberrations in colorectal tumorigenesis.4–9,22–24 and (ii) aCGH results of this study and included the following fourteen genes: COX2 (1q31.1), PIK3CA (3q26.32), APC (5q22.2), CLIC1 (6p21.33), EGFR (7p11.2), MYC (8q24.21), CCND1 (11q13.3), CDX2 (13q12.2), CDH1 (16q22.1), TP53 (17p13.1), HER2/ERBB2 (17q12), SMAD7 (18q21.1), SMAD4 (18q21.2), ZNF217 (20q13.2). Centromere probe CEP10 was used as ploidy reference. Clone DNA was extracted, labelled with fluorophores by nick translation and precipitated (see Supporting Information Material and Methods for details). Cytospin slides containing single-layered interphase nuclei from two 50 μm-thick unstained FFPE tissue sections per sample were prepared using a modified Hedley-method by disintegrating the tumour tissue with 0.1% proteinase (P-8038, Sigma-Aldrich, St. Louis, MO, USA) as previously published.20 Slides were pre-treated with 0.1% proteinase (Sigma-Aldrich) at 37°C for 60 min, hybridised with probe Panel 1 (CDX2, CCND1, SMAD4, PIK3CA and MYC) and detected as previously described.18,20 Slides were subsequently imaged and scanned with a custom program on a DUET automated imaging workstation (BioView, Rehovot, Israel) using a fluorescence microscope with a 40x oil immersion objective (BX63, Olympus, Tokyo, Japan) equipped with a motorized stage and custom filters (Chroma, Bellow Falls, VT, USA). After stripping with 50% formamide/2xSSC for 2 min at 80°C, slides were dehydrated by ethanol series (70%, 80% and 100% for 3 min) prior to sequential re-hybridisation and detection of Panel 2 (CLIC1, COX2, APC, SMAD7 and EGFR) and Panel 3 (HER2, CDH1, TP53, CEP10 and ZNF217), respectively. Images were automatically overlaid to collect signal counts of all fifteen probes within the same nuclei. Automated counts were manually reviewed for accuracy using the custom gallery overview of the SOLO workstation (BioView). A nucleus would be excluded from analysis if any of the following criteria applied: (i) overlapping with other nuclei, (ii) non-epithelial morphology, (iii) visible damage, (iv) indistinguishable probe signals, or (v) no centromere signal. Once 350 aberrant nuclei were reached, counting was stopped. In cases with less than 350 aberrant nuclei, counting was continued for the whole scan to sample more cells to be sure not to overlook any clonal pattern (maximum 12,000 targets per scan). On average, 181 (range, 5-350) aberrant nuclei and 341 (range, 18-867) non-aberrant nuclei were evaluated per case.
FISH data algorithm
Processing of raw data and annotation of ploidy were performed as described.20 Cellular ploidy was annotated by assessment of signal counts for CEP10 and the fourteen gene identifier probes. Gain and loss patterns were determined in relation to the estimated ploidy of the respective nucleus. Ploidy was assigned diploid (2N) for 55/58 (94.8%) adenomas, while three (5.1%) adenomas displayed tetraploid (4N) chromosome sets (cases A13, A30 and P7b) and were excluded from overall analysis due to increased genomic instability in tetraploid lesions. Copy number (CN) gains or losses were considered for statistical analyses only when the respective aberration was present in at least 10% of counted cells. For matched pairs, phylogenetic tree models were inferred by FISHtrees software (see Supporting Information Material and Methods for details).25
Statistical analysis
GraphPad Prism 6.01 (GraphPad Software, San Diego, CA, USA) was used to analyse the data by two-sided tests which were considered statistically significant if p<0.05 and not adjusted for multiple comparisons demanding cautious interpretation. Means are noted with standard deviation (± SD) while medians are annotated with interquartile range (IQR).
Tumour heterogeneity was initially analysed by computing the frequencies of signal patterns allowing to calculate and compare the samples via cellular diversity measures.18,20,26,27 To define the indices, let pi be the frequency of the ith count pattern of k loci with the total pattern count N of n nuclei. The accumulated numbers of altered and diploid probe counts were defined as XK and YK, respectively. The following four diversity measures were assessed:
(i) instability index (measure of species richness in ecology):
| (1) |
(ii) Shannon entropy (measure of richness and evenness in information theory):
| (2) |
(iii) Simpson index (measure of dominance in population genetics):
| (3) |
(iv) accumulated pairwise genetic diversity (adopted diversity metric in genetics):
| (4) |
Unsupervised clustering of adenomas specimens was performed by Gene Cluster 3.0 (Laboratory of DNA Information Analysis, University of Tokyo) using the average signal number per sample and miFISH marker.27 Genes were normalised and centred by the mean. Finally, samples were correlated (non-centred) and clustered via complete linkage. Java Tree View 1.1.6r4 was used for visualisation.
Results
Array-based CGH analysis of CNAs in colorectal adenomas without and with recurrence
CNAs were detected in 41/51 (80.4%) of colorectal adenomas. Focal CN gains of 6p22.1-p21.33 (17/51; 33.3%) and 7q22.1 (16/51; 31.4%) and focal CN loss of 16q21 (15/51; 29.4%) were the most frequent alterations (Fig. 1a). CN gains of entire chromosome arms or chromosomes were mapped to chromosomes 7 (7/51; 13.7%), 13q (7/51; 13.7%) and 20q (7/51; 13.7%). The most frequent CN loss was observed for chromosome 18 (3/51; 5.8%). Differential arm-level CNAs for primary adenomas with (n=26) and without (n=13) recurrence were observed for chr7 (11.5% versus 23.1%), chr8 (3.8% versus 15.4%), chr13 (15.4% versus 0%), 17p (7.7% versus 0%), chr18 (7.7% versus 0%), 19q (7.7% versus 15.4%), 20p (11.5% versus 7.7%) and 20q (19.2% versus 7.7%) (Supporting Information Table 2).
Figure 1.
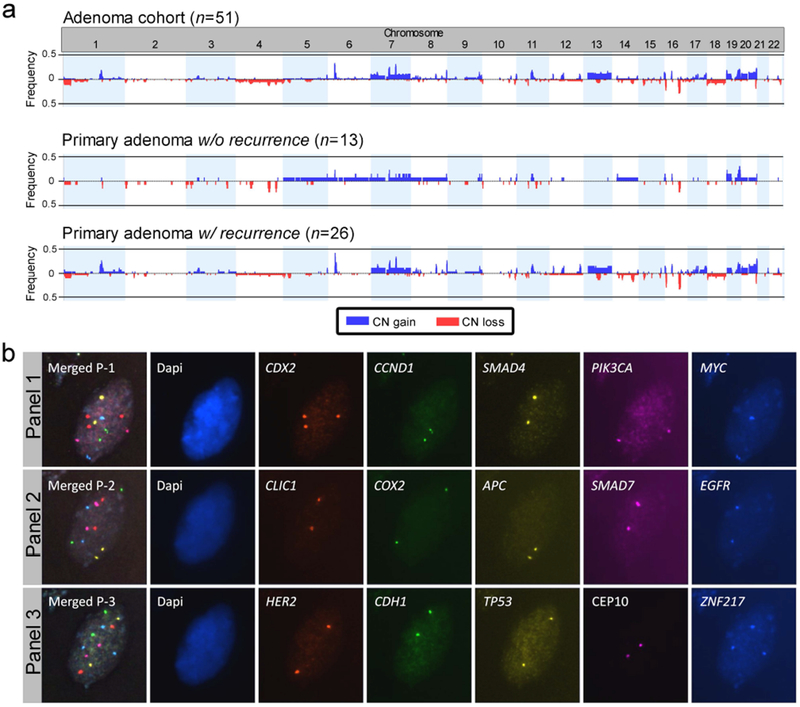
Copy number alterations in the colorectal adenoma cohort. (a) Frequency plots of copy number alterations of adenomas without and with recurrence identified by aCGH. Only adenomas with recurrence presented gains of chromosome arm 13q and losses of chromosome 18. (b) Representative image of one colorectal adenoma nucleus hybridised with miFISH probe panels 1-3. Top row shows the first hybridisation with panel 1 followed by re-hybridisation with panel 2 (middle row) and panel 3 (bottom row), respectively. The merged image in each row shows the overlay of all channels per panel. For this nucleus, only CDX2 and ZNF217 displayed a CN gain with three signals.
At least one CNA was detected in 76.9% (10/13) of primary adenomas without recurrence versus 80.7% (21/26) of primary adenomas with recurrence (p=0.955). Consequently, the average number of copy alterations (all adenomas, mean 3.5 ± 4.1) did not discriminate the groups (mean 3.3 ± 3.2 versus 3.4 ± 4.3) (Supporting Information Fig. 1).
MiFISH single-cell analysis of CNAs in colorectal adenomas without and with recurrence
Adenomas were hybridised with three miFISH probe panels allowing the enumeration of 15 gene loci per nucleus (Fig. 1b). On average, 523 nuclei (range 326–1164 nuclei) were counted per case. Gains of EGFR (13/55; 23.6%), CDX2 (12/55; 21.8%) and ZNF217 (10/55; 18.2%) were the most common alterations in the cohort while other CNAs were only rarely observed (Supporting Information Table 3). Consistent with previous results, probes targeting oncogenes on 1q, 3q, 6p, 7p, 8q, 11q, 13q, 17q, and 20q were subject to CN gains, whereas probes representing tumour suppressor genes on 5q, 16q, 17p, and 18q were subject to CN losses (Supporting Information Fig. 2a, b).5,7 The distribution of genomic imbalances was specific for CRC. CNAs detected by aCGH and miFISH were concordant expressed by κ=0.861 (CI, 0.80–0.92; Cohen’s kappa) (Supporting Information Fig. 2c).
The distribution of CNAs determined by miFISH was plotted per individual sample along with clinical data (Figure 2a). Although average signal numbers (ASN) of the miFISH markers ranged from 1.96 to 2.32 across the sample set, none of the probes was significantly predictive for adenoma recurrence (Fig. 2b). However, average signal numbers of CDX2 tended to be capable to discriminate primary adenomas with recurrence (mean 2.18 ± 0.33) from primary adenomas without recurrence (mean 2.02 ± 0.03) and recurrent adenomas (mean 2.32 ± 0.54; p=0.102; Fig. 2b). CDX2 gain indicated recurrence among primary adenomas (sensitivity 25%, specificity 100%, p=0.040, Fig. 2c). Of note, a probe set comprising CLIC1, CDX2, and ZNF217 would have detected 11 of 28 (sensitivity 39%) primary adenomas with recurrence while only one primary adenoma without recurrence (specificity 93%) would have been inadvertently identified (p=0.036; Supporting Information Fig. 3a).
Figure 2.
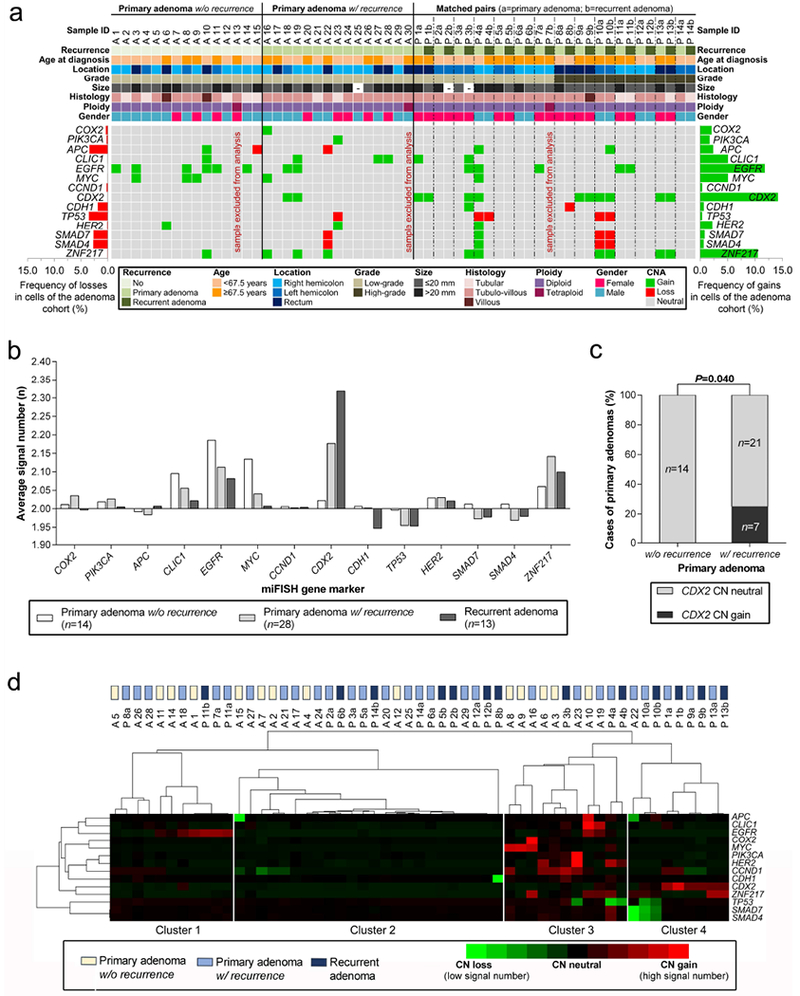
Summary of copy number alterations identified by miFISH. (a) Clinicopathological features (rows, upper panel) and miFISH gene marker status (rows, lower panel) are plotted per individual adenoma sample (columns). (b) Average signal numbers of the fourteen gene probes in primary adenomas without and with recurrence and in recurrent adenomas showed no statistically significant differences across the groups but revealed a trend for CDX2 only (p=0.102; one-way ANOVA). (c) CDX2 was exclusively gained in primary adenomas with recurrence compared to primary adenomas without recurrence (Chi square test). (d) Heat map cluster analysis of average signal numbers per marker in each patient. Genes and samples were sorted by dendrogram correlation. Adenomas were divided into four clustering groups. Cluster assignment showed a trend to separate adenomas without recurrence from primary adenomas with recurrence (p=0.193, Freeman-Halton test).
Unsupervised clustering based on average signal numbers of the fourteen FISH probes separated the adenomas (n=55) into four clusters (Fig. 2d). Cluster 1 combined adenomas with EGFR gain and cluster 2 mainly included samples without CNA. Cluster 3 comprised adenomas with multiple different CNAs and cluster 4 was linked to CDX2 gains and SMAD4/SMAD7 losses. Notably, cluster 4 did not contain any primary adenoma without recurrence. Cluster assignment revealed a trend to separate adenomas without recurrence from primary adenomas with recurrence (p=0.193).
Next, we correlated CNAs of primary adenomas with clinicopathological data. Advanced patient age correlated with increased genomic instability (p=0.026; Supporting Information Fig. 3b). Remarkably, rectal adenomas were more frequently affected by CNAs, e.g. CLIC1 gains, than colonic adenomas (p=0.036 and p=0.013; Supporting Information Fig. 3c, d). Tubulo-villous adenomas were larger compared to adenomas with tubular histology (p=0.034; Supporting Information Fig. 3e). We also noted that in paired samples, primary adenomas were on average larger than the corresponding recurrent adenomas due to tight surveillance and prompt polypectomy (p=0.003; Supporting Information Fig. 3f).
Clonal composition of primary colorectal adenomas without and with recurrence
The comparison of the clonal composition within colorectal adenomas was performed by arranging the aberrant clones per case according to their incidence and frequency (Fig. 3; Supporting Information Fig. 4-12). Major clone populations comprised on average 62.8% (IQR, 39-88%) of the aberrant cell populations (Supporting Information Table 4). No difference was found for adenomas without and with recurrence (mean 63.5% versus 60.8%; p=0.795).
Figure 3.

Colour displays of miFISH analysis of six representative primary colorectal adenomas (a) without recurrence (cases A1, A4, A15) and (b) with recurrence (cases A18, A21, A22). Gene-specific miFISH markers are plotted vertically and sorted by chromosomal location (chromosome arm) as indicated by the “Locus” column. Nuclei are arranged horizontally by the frequency of signal patterns from left to right. Vertical lines separate the clone populations and display the prevalence of these clones in the aberrant population. The pattern of the largest clone is indicated in brackets. CN gains and losses are depicted as percentages of the total cell population. Average ploidy, average signal numbers per gene marker and the number of signal patterns are shown in the respective columns. The number of signal patterns includes the amassment of clonal patterns summing up the aberrant patterns (including imbalanced patterns with the same direction but not exact signal counts) and the neutral (diploid) pattern. Green, CN gain; red, CN loss; blue, CN neutral; AP, average ploidy; ASN, average signal number; Sig. pat., number of signal patterns.
Adenomas without recurrence showed no CNA in 35.7% (5/14), a single CNA in 42.8% (6/14) and multiple CNAs in 21.4% (3/14) of cases, respectively. Only case A10 exhibited a major clone with four CNAs (i.e. APC, CLIC1, EGFR and ZNF217), all other clones had three or less CNAs (Supporting Information Fig. 5a). Primary adenomas with recurrence demonstrated no CNA in 50.0% (14/28), a single CNA in 17.9% (5/28) and two or more CNAs in 32.1% (9/28) of adenomas, respectively. However, primary adenomas with recurrence revealed, on average, a higher number of CNAs per clone than adenomas without recurrence when only cases with clonal aberrations were considered (p=0.016; Supporting Information Fig. 13a). The presence of microsatellite instability in samples without CNA (n=24) was excluded by immunohistochemistry for DNA mismatch repair proteins (Supporting Information Material and Methods and Supporting Information Fig. 13b, c).
Tumour heterogeneity was quantitatively assessed by calculating four measures of diversity (Supporting Information Table 4 and Supporting Information Fig. 14a-d). Investigated adenomas (n=55) displayed an average instability index of 4.9 (± 5.1), indicating a low-level intra-tumour heterogeneity (ITH). No detectable clone population (threshold 5%) was present in 29.1% (16/55) of adenomas, one clone population was found in 23.6% (13/55) of adenomas, while two or more clone populations were observed in 47.3% (26/55) of adenomas. However, average instability indices failed to discriminate between adenomas without and with recurrence (mean 4.8 ± 5.6 versus 4.8 ± 3.2; p=0.603; Supporting Information Fig. 14a, e). When grouped by intervals, Shannon entropy (p=0.219), Simpson index (p=0.052) and accumulated pairwise genetic diversity (p=0.156), respectively, displayed trends of heterogeneous sample distributions in primary adenomas with recurrence compared to adenomas without recurrence (Supporting Information Fig. 14f-h). Diversity indices of tetraploid adenomas differed strongly compared to diploid adenomas (p≤0.001; Supporting Information Fig. 15a-c). Fractions of tetraploid cells within the tumour amassment were also strongly correlated with increased accumulated pairwise genetic diversity (p≤0.001; Supporting Information Fig. 15d).
Clonal evolution of paired primary and recurrent adenomas
Subanalysis of miFISH ASNs did not reveal changes in paired samples (p=0.383; Supporting Information Fig. 15e), although 6/14 (43%) pairs differed in ASNs across both lesions (Supporting Information Table 5). Thus, the clonal evolution from primary adenomas towards the corresponding recurrent adenomas was visualised by constructing consensus phylogenetic trees based on the signal patterns (Fig. 4 and Supporting Information Fig. 16-18). Using the FISHtrees algorithm,25 trees were inferred by heuristically seeking to minimise the total number of CNAs across the tree which initially emanates from a diploid root cell. In-depth analyses of primary adenoma-recurrence consensus trees suggested four distinct clonal evolution patterns (Fig. 4 and Supporting Information Fig. 16-18): (1) Simplification pattern: The primary adenoma shows multiple different clones. The recurrent adenoma is dominated by a lower clone number compared to the primary adenoma, leading to decreased ITH; (2) Complexity pattern: The primary adenoma exhibits a distinct major clone which becomes a minor clone in the recurrent adenoma while multiple new clones emerge and increase clonal ITH in the recurrent adenoma; (3) Stabilisation pattern: primary and recurrent adenoma display an identical major clone population; the dominant clones from the primary adenoma remain largely unchanged and persist in the recurrent adenoma. In all three patterns described above, at least one clone fraction present in the primary adenoma is found back in the recurrent lesion; (4) “Zero” pattern: Copy number changes are non-clonal and observed in less than 10% of the population. Thus, modelling phylogenetic trees for these lesions is not possible due to the few aberrant cells present in the respective primary and recurrent adenomas. Notably, evolution patterns were tested for robustness to rare clone- and sample bias (Supporting Information Fig.19).
Figure 4.
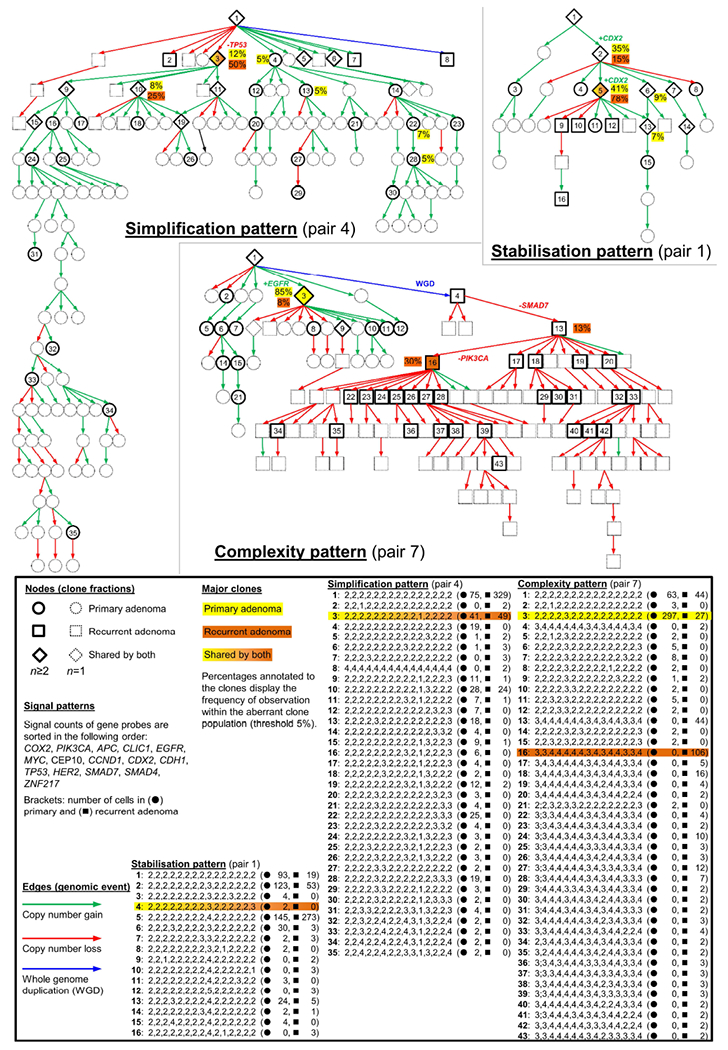
Consensus phylogenetic trees for three representative patients with adenoma recurrence. Each tree node (circle, square, rhomb) represents a distinct genetic aberration pattern based on miFISH analyses. Detailed signal patterns for each clone are displayed in the legend. Genetic events characterizing the evolutionary pathway of the major clones are annotated to the antecedent edges. Population sizes of clones exceeding 5% of the aberrant clones are shown in percentages for primary and recurrent adenoma. Pair P4 (simplification pattern) presents a shared major clone with TP53 loss (No. 3). Consistent with the term simplification pattern, the primary adenoma displayed more clones than present in the recurrent adenoma. In pair P7 (complexity pattern), the primary adenoma consisted mainly of a clone with EGFR gain. In the recurrent adenoma a whole genome duplication induced a novel branch of the tree. In pair P1 (stabilisation pattern), the tree showed a low node depth and clone populations are mainly shared by primary and recurrent adenoma, including a major clone with CDX2 gain.
Trees were rendered into graphs showing the respective relative contribution of each clone to the aberrant tumour cell population (Fig. 5a and Supporting Information Fig. 20, 21). Observed patterns were detected with different frequencies (Fig. 5b). Interestingly, CDX2 gain was only observed within the stabilisation pattern emphasizing the crucial role of this gene with respect to recurrence (p=0.005; Fig. 5c).
Figure 5.
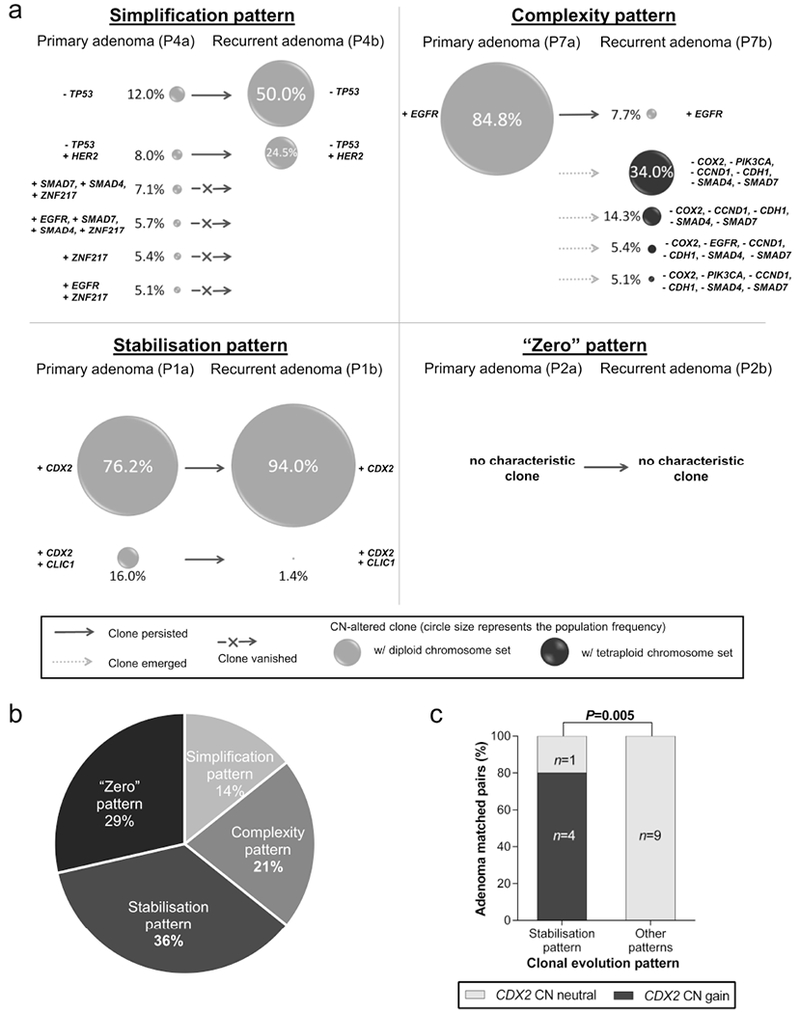
Clonal evolution patterns in colorectal adenomas. (a) Patterns display the clonal composition of the primary adenoma and the corresponding recurrent adenoma. The simplification pattern was characterised by multiple clones in the primary adenoma while the recurrent adenoma was dominated by a major clone. The complexity pattern was marked by a major clone in the primary adenoma which became a minor clone in the recurrent adenoma while multiple new clones emerged. Adenomas following the stabilisation pattern displayed similar clone populations in primary and recurrent adenoma. “Zero” pattern summarises adenoma pairs without any CNA detected. (b) Distribution of clonal patterns in matched pairs (n=14). (c) CDX2 CN gain was associated with the stabilisation pattern (Fisher exact test).
Discussion
We investigated the dynamics of chromosomal gains and losses that define adenoma recurrence. Our aim was to study clonal relationships from primary adenoma to the corresponding recurrent adenoma and to identify a predictive biomarker for recurrence. Therefore, we utilised aCGH as a global screening technique and miFISH as a single-cell cytogenetic approach on colorectal adenomas that were successfully removed by polypectomy and those that recurred.
Performing aCGH analysis of fifty-one adenomas, we observed gains of chromosomes 7, 13q and 20q and losses of chromosome 18. Together, our findings on colorectal adenomas align with published data asserting these alterations to be early genomic events in the adenoma-carcinoma sequence.22–24 Additionally, we revealed three focal CNAs being most frequently altered in colorectal adenomas: (i) gain of 6p22.1–21.33 (including CLIC1), (ii) gain of 7q22 (including a cluster of mucin-genes) and (iii) loss of 16q21 (including CDH11). CNAs in these loci were previously described in colorectal lesions28–33 and the respective genes were associated with growth advantage and tumour progression.33–35
Overall, the numbers of CNAs based on aCGH were relatively low with on average 3.5 CNAs per case, which was expected from previously published own data.6,21 Neither primary adenomas with or without recurrence nor primary adenomas and their matched recurrences differed in the average number of CNAs.
However, this did not rule out different genetic subclones since aCGH only reveals major genomic imbalances in the tumour cell population. Therefore, we determined clonal evolution by a novel single-cell genomic approach (miFISH). Genomic instability was quantitatively assessed and an instability index was calculated as published recently.18,20 In alignment with the aCGH data, instability indices in our colorectal adenomas cohort were very low (mean 4.9 patterns) compared to what was previously observed in invasive breast cancer (70.6) and its precursor lesion, ductal carcinoma in situ (62.3).20 By comparing primary adenomas with and without recurrence we did not observe a difference across genomic instability indices (4.8 versus 4.8). Apparently, the low instability index in colorectal adenomas, caused by the predominance of major clones (mean major clone size 62.8%), emphasises the stable clonal development of colorectal adenomas. This low level of clonal diversity might be the genetic correlate of the clinical observation of the long latency of adenoma progression to invasive CRC. We compared primary adenomas with and without recurrence by three additional measures of diversity among which Simpson index revealed the strongest differences. Although a wide range of sample distribution was observed across the adenomatous polyps these diversity measures seem to unveil ITH with good precision which was previously demonstrated in other cancers.18,27,28
The gain of EGFR was the most frequent CNA observed by miFISH affecting 23.6% (13/55) of colorectal adenomas consistent with the results from Habermann et al.13 The frequency of EGFR CNAs in 42.9% (6/14) of adenomas without recurrence underlines early occurrence of this CNA which is not related to recurrence. The overexpression of this transmembrane protein with intrinsic tyrosine-kinase activity promotes tumour expansion by resistance to apoptosis and increased proliferation.13,36,37 However, CNAs detected by miFISH might, in fact, resemble disruptions of chromosomal regions exceeding single genes as confirmed by aCGH.
The second most-frequent aberration occurring in colorectal adenomas was the gain of CDX2 altered in 21.8% (12/55) of the samples, which is in line with other publications listing chromosomal gain of 13q (including CDX2) as a frequent target in colorectal tumorigenesis.4–9,22–24,38 In contrast to EGFR, CDX2 CNAs occurred with significantly different percentages in primary adenomas with and without recurrence. While primary adenomas without recurrence showed no (0/14) CDX2 gain, this CNA was present in 25% (7/28) of primary adenomas with recurrence. Functionally, CDX2 regulates intestinal lineage development and differentiation. Although several publications described tumour suppressive abilities for CDX2,39–41 an amplification of CDX2 seems to confer oncogenic potential by promoting proliferation and survival of CRC cells.42,43 Tumour cells with additional copies of CDX2 might eventually gain a functional advantage being capable to proliferate independently and recur after polypectomy. In our cohort, CDX2 gain predicted recurrence with 100% specificity, however, only 25% sensitivity. In combination with CLIC1 and ZNF217 the sensitivity could be increased to 39%, although the specificity decreased to 93% making this panel clinically less valuable.
We also investigated the clonal evolution from primary adenomas to the corresponding recurrent adenomas by inferring phylogeny using the FISHtrees algorithm. We could observe four distinct clonal evolution patterns underlying adenoma recurrence: (i) simplification pattern, (ii) complexity pattern, (iii) stabilisation pattern and (iv) “zero” pattern. Mechanistically, the simplification type could be interpreted in a way that certain subclonal populations, which were able to compete in the primary adenoma, were either removed or damaged by the polypectomy or were overgrown by the dominant clonal population of the recurrent tumour. Burrell and Swanton15 defined this linear evolutionary event, i.e. one clone taking over the entire population, as ‘clonal sweep’. The complexity pattern indicates that the dominant clone population of the primary adenoma did not retain its competitive advantage in the recurrent adenoma, possibly due to a shift in the tumour environment and growth conditions, which favoured the emergence of multiple subclones resulting in an increased ITH. This phenomenon concurs with a branched evolution of the tumour44 and is suggestive to concur with the Big Bang growth model.45,46 Moreover, aneuploid clones with a near-tetraploid karyotype, indicating their emergence via whole genome duplication, may have acquired multifaceted benefits which allow them to outcompete near-diploid clone populations.47 Of note, adenoma pairs following the stabilisation pattern were exclusively clonally dominant for gains of EGFR or CDX2. These events seem to provide such a strong selection advantage that additional CNAs do not provide a further growth advantage, hence, clones with CN gains for EGFR and CDX2 persisted in the recurrent lesions.42,48 This perspective of adenomatous clonal evolution is in line with an evolutionary tempo of stasis in which the tumour reaches a fitness peak.45 Our findings of adenoma recurrence patterns arguably resemble the existence of a punctuated equilibrium in CRC attributing to variable evolutionary patterns as proposed by Cross et al.45 When we compare the patterns observed in the recurrence of adenomas with those mapped in the progression of ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) of the breast,20 we noted some overlap, i.e. both the stabilisation pattern and the complexity pattern were observed as well. However, neither the simplification nor the “zero” pattern played a role in DCIS-IDC progression. This is not surprising because, as also clearly evident from the greatly higher instability index in DCIS/IDC compared to colorectal adenomas, these lesions are far more progressed.
Arguably, shared clone populations in both the primary adenoma and the recurrent adenoma, as observed in the stabilisation pattern, are suggestive for incomplete resection. Indeed, incomplete resection cannot be entirely ruled out in our sample cohort especially since case selection was based on endoscopic complete resection, and not histopathologic complete resection, which is exceedingly difficult to assess.
In conclusion, adenoma development and recurrence are complex genetic processes driven by multiple gene copy number changes. However, adenoma recurrence followed four distinct, defined pathways: simplification-, complexity-, stabilisation- and “zero”-pattern. Importantly, the assessment of an individual risk for adenoma recurrence may possibly be improved by evaluating the CN status of CDX2 by miFISH analysis.
Supplementary Material
Novelty Statement.
We performed the very modern single-cell approach multiplex-interphase fluorescence in situ hybridisation to detect copy number changes on a rare collective of primary and recurrent colorectal adenomas. We were able to assess diverse levels of intra-tumour heterogeneity for the two groups and could identify CDX2 as a potential marker of recurrence. Clonal evolution was modelled by phylogenetic trees and revealed four distinct patterns of recurrence.
Acknowledgements
We thank Alexandra Eichhorn and Romina Laegel for excellent technical assistance, and the BioView team for the development of the custom microscopy scanning software. Jordi Camps and Isabela Quintanilla provided help to construct phylogenetic trees. This study was supported in part by the Intramural Research Program of the National Institutes of Health/NCI, USA. DF received a travel grant by the Boehringer Ingelheim Fond (BIF), Germany.
Abbreviations
- aCGH:
array-comparative genomic hybridisation
- ANCA:
average number of copy alterations
- AP:
average ploidy
- ASN:
average signal number
- BAC:
bacterial artificial chromosome
- CIN:
chromosomal instability
- CN:
copy number
- CNA:
(somatic) copy number alteration
- CNV:
copy number variant
- CRC:
colorectal cancer
- DCIS:
ductal carcinoma in situ
- FFPE:
formalin-fixed paraffin-embedded tissue
- H&E:
haematoxylin and eosin stained tissue
- HGD:
high-grade dysplasia
- IDC:
invasive ductal carcinoma
- ITH:
intra-tumour heterogeneity
- LGD:
low-grade dysplasia
- miFISH:
multiplex interphase fluorescence in situ hybridisation
- MSI:
microsatellite instable
- ULS:
universal linkage system
- WHO:
World Health Organization
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
- 1.Bond JH. Polyp guideline: diagnosis, treatment, and surveillance for patients with colorectal polyps. Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 2000;95:3053–63. [DOI] [PubMed] [Google Scholar]
- 2.Schatzkin A, Freedman LS, Dawsey SM, et al. Interpreting precursor studies: what polyp trials tell us about large-bowel cancer. J Natl Cancer Inst 1994;86:1053–7. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. [DOI] [PubMed] [Google Scholar]
- 4.Bomme L, Bardi G, Pandis N, et al. Clonal karyotypic abnormalities in colorectal adenomas: clues to the early genetic events in the adenoma-carcinoma sequence. Genes Chromosomes Cancer 1994;10:190–6. [DOI] [PubMed] [Google Scholar]
- 5.Meijer GA J A Hermsen MA, A Baak JP, et al. Progression from colorectal adenoma to carcinoma is associated with non-random chromosomal gains as detected by comparative genomic hybridisation. J Clin Pathol 1998;51:901–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirsch D, Camps J, Varma S, et al. A new whole genome amplification method for studying clonal evolution patterns in malignant colorectal polyps. Genes Chromosomes Cancer 2012;51:490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ried T, Knutzen R, Steinbeck R, et al. Comparative genomic hybridization reveals a specific pattern of chromosomal gains and losses during the genesis of colorectal tumors. Genes Chromosomes Cancer 1996;15:234–45. [DOI] [PubMed] [Google Scholar]
- 8.Bardi G, Johansson B, Pandis N, et al. Cytogenetic analysis of 52 colorectal carcinomas--non-random aberration pattern and correlation with pathologic parameters. Int J Cancer 1993;55:422–8. [DOI] [PubMed] [Google Scholar]
- 9.Hermsen M, Postma C, Baak J, et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology 2002;123:1109–19. [DOI] [PubMed] [Google Scholar]
- 10.Avidan B, Sonnenberg A, Schnell TG, et al. New occurrence and recurrence of neoplasms within 5 years of a screening colonoscopy. Am J Gastroenterol 2002;97:1524–9. [DOI] [PubMed] [Google Scholar]
- 11.Bonithon-Kopp C, Piard F, Fenger C, et al. Colorectal adenoma characteristics as predictors of recurrence. Dis Colon Rectum 2004;47:323–33. [DOI] [PubMed] [Google Scholar]
- 12.Neugut AI, Jacobson JS, Ahsan H, et al. Incidence and recurrence rates of colorectal adenomas: a prospective study. Gastroenterology 1995;108:402–8. [DOI] [PubMed] [Google Scholar]
- 13.Habermann JK, Brucker CA, Freitag-Wolf S, et al. Genomic instability and oncogene amplifications in colorectal adenomas predict recurrence and synchronous carcinoma. Mod Pathol 2011;24:542–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clevers H The cancer stem cell: premises, promises and challenges. Nat Med 2011;17:313–9. [DOI] [PubMed] [Google Scholar]
- 15.Burrell RA, Swanton C. The evolution of the unstable cancer genome. Curr Opin Genet Dev 2014;24:61–7. [DOI] [PubMed] [Google Scholar]
- 16.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008;26:2839–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Losi L, Baisse B, Bouzourene H, et al. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis 2005;26:916–22. [DOI] [PubMed] [Google Scholar]
- 18.Heselmeyer-Haddad KM, Garcia LYB, Bradley A, et al. Single-cell genetic analysis reveals insights into clonal development of prostate cancers and indicates loss of PTEN as a marker of poor prognosis. Am J Pathol 2014;184:2671–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bosman F, Carneiro F, Hruban R, et al. WHO Classification of Tumours of the Digestive System, 4th ed. IARC Press: Lyon; 2010. [Google Scholar]
- 20.Heselmeyer-Haddad K, Berroa Garcia LY, Bradley A, et al. Single-cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumor heterogeneity yet conserved genomic imbalances and gain of MYC during progression. Am J Pathol 2012;181:1807–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ried T, Heselmeyer-Haddad K, Blegen H, et al. Genomic changes defining the genesis, progression, and malignancy potential in solid human tumors: a phenotype/genotype correlation. Genes Chromosomes Cancer 1999;25:195–204. [DOI] [PubMed] [Google Scholar]
- 22.Grade M, Becker H, Liersch T, et al. Molecular cytogenetics: Genomic imbalances in colorectal cancer and their clinical impact 1. Cell Oncol 2006;28:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ried T, Hu Y, Difilippantonio MJ, et al. The consequences of chromosomal aneuploidy on the transcriptome of cancer cells. Biochim Biophys Acta 2012;1819:784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie T, D’Ario G, Lamb JR, et al. A comprehensive characterization of genome-wide copy number aberrations in colorectal cancer reveals novel oncogenes and patterns of alterations. PLoS One 2012;7:e42001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chowdhury SA, Shackney SE, Heselmeyer-Haddad K, et al. Phylogenetic analysis of multiprobe fluorescence in situ hybridization data from tumor cell populations. Bioinformatics 2013;29:189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez P, Timmer MR, Lau CT, et al. Dynamic clonal equilibrium and predetermined cancer risk in Barett’s oesophagus. Nat Commun 2016;7:12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wangsa D, Chowdhury SA, Ryott M, et al. Phylogenetic analysis of multiple FISH markers in oral tongue squamous cell carcinoma suggests that a diverse distribution of copy number changes is associated with poor prognosis. Int J Cancer 2016;138:98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brosens RPM, Haan JC, Carvalho B, et al. Candidate driver genes in focal chromosomal aberrations of stage II colon cancer. J Pathol 2010;221:411–24. [DOI] [PubMed] [Google Scholar]
- 29.Camps J, Grade M, Nguyen QT, et al. Chromosomal breakpoints in primary colon cancer cluster at sites of structural variants in the genome. Cancer Res 2008;68:1284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrova DT, Asif AR, Armstrong VW, et al. Expression of chloride intracellular channel protein 1 (CLIC1) and tumor protein D52 (TPD52) as potential biomarkers for colorectal cancer. Clin Biochem 2008;41:1224–36. [DOI] [PubMed] [Google Scholar]
- 31.Walsh MD, Young JP, Leggett BA, et al. The MUC13 cell surface mucin is highly expressed by human colorectal carcinomas. Hum Pathol 2007;38:883–92. [DOI] [PubMed] [Google Scholar]
- 32.van Roy F Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer 2014;14:121–34. [DOI] [PubMed] [Google Scholar]
- 33.Byrd JC, Bresalier RS. Mucins and mucin binding proteins in colorectal cancer. Cancer Metastasis Rev 2004;23:77–99. [DOI] [PubMed] [Google Scholar]
- 34.Santos GC, Zielenska M, Prasad M, et al. Chromosome 6p amplification and cancer progression. J Clin Pathol 2007;60:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Ying J, Li H, et al. The human cadherin 11 is a pro-apoptotic tumor suppressor modulating cell stemness through Wnt/β-catenin signaling and silenced in common carcinomas. Oncogene 2012;31:3901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts RB, Min L, Washington MK, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci USA 2002;99:1521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grünwald V, Hidalgo M. Development of the epidermal growth factor receptor inhibitor OSI-774. Semin Oncol 2003;30:23–31. [DOI] [PubMed] [Google Scholar]
- 38.Borras E, San Lucas FA, Chang K, et al. Genomic landscape of colorectal mucosa and adenomas. Cancer Prev Res 2016;9:417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bai Y-Q, Miyake S, Iwai T, et al. CDX2, a homeobox transcription factor, upregulates transcription of the p21/WAF1/CIP1 gene. Oncogene 2003;22:7942–9. [DOI] [PubMed] [Google Scholar]
- 40.Chawengsaksophak K, James R, Hammond VE, et al. Homeosis and intestinal tumours in Cdx2 mutant mice. Nature 1997;386:84–7. [DOI] [PubMed] [Google Scholar]
- 41.Choi BJ, Kim CJ, Cho YG, et al. Altered expression of CDX2 in colorectal cancers. Acta Pathol Microbiol Immunol Scand 2006;114:50–4. [DOI] [PubMed] [Google Scholar]
- 42.Salari K, Spulak ME, Cuff J, et al. CDX2 is an amplified lineage-survival oncogene in colorectal cancer. Proc Natl Acad Sci USA 2012;109:e3196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dang LH, Chen F, Ying C, et al. CDX2 has tumorigenic potential in the human colon cancer cell lines LOVO and SW48. Oncogene 2006;25:2264–72. [DOI] [PubMed] [Google Scholar]
- 44.Burrell RA, McGranahan N, Bartek J, et al. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013;501:338–45. [DOI] [PubMed] [Google Scholar]
- 45.Sottoriva A, Kang H, Ma Z, et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015;47:209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cross WC, Graham TA, Wright NA. New paradigms in clonal evolution: punctuated equilibrium in cancer. J Pathol 2016;240:126–36. [DOI] [PubMed] [Google Scholar]
- 47.Kuznetsova AY, Seget K, Moeller GK, et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015;14:2810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lockhart AC, Lockhart C, Berlin JD. The epidermal growth factor receptor as a target for colorectal cancer therapy. Semin Oncol 2005;32:52–60. [DOI] [PubMed] [Google Scholar]
- *49.Gaiser T, Meinhardt S, Hirsch D, et al. Molecular patterns in the evolution of serrated lesion of the colorectum. Int J Cancer 2013;132:1800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Cited in supporting information only.
- *50.Rüschoff J, Bocker T, Schlegel J, et al. Microsatellite instability: new aspects in the carcinogenesis of colorectal carcinoma. Virchows Arch 1995;426:215–22. [DOI] [PubMed] [Google Scholar]; * Cited in supporting information only.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.