

Abstract
To analyze the possible clonal origin of a part of Synchronous colorectal cancer (SCRC), we studied 104 paired-SCRCs from 52 consecutive patients without hereditary forms of CRC. We used a Single-Nucleotide Polymorphism array to characterize the genomic profiles, and subsequently used a statistical application to define them according to clonality within the same individual. We categorized the ensuing groups according to colonic location to identify differential phenotypes. The SCRC Monoclonal group (M) (19 cases) was divided into Monosegmental (MM) and Pancolonic (MP) groups. The SCRC Polyclonal group (P) (33 cases) was also divided into Monosegmental (PM) and Pancolonic (PP), the first exhibiting preference for left colon. The MM group showed a high rate of mucinous tumors, the lowest mean-number of tumors and associated-polyps, and the worst prognosis. The MP group included the largest mean-number of associated-polyps, best prognosis and familial cancer component. The PM group seemed to be a “frontier” group. Finally, the PP group also exhibited a mucin component, the highest mean-number of tumors (4.6) compared with the mean-number of polyps (7.7), poor prognosis and sporadic cases. Most relevant differential genomic regions within M groups were gains on 1q24 and 8q24, and deletions on 1p21 and 1p23 for MM, while within P were the gains on 7q36 and deletions on 1p36 for PM. The statistical application employed seems to define clonality more accurately in SCRC -more likely to be polyclonal in origin-, and together with the tumor locations, helped us to configure a classification with prognostic and clinical value.
Keywords: Synchronous colorectal cancer, clonality, Single-Nucleotide Polymorphism array (SNP array), colon location, monoclonal, polyclonal
INTRODUCTION
Synchronous colorectal cancer (SCRC) refers to more than one primary colorectal carcinoma (CRC) detected in a single patient at the time of diagnosis (1). These cancers are different from metachronous CRCs (MCRCs), defined as CRCs with a second tumor diagnosed at least 6 months after the initial cancer diagnosis (2). SCRC accounts for about 1.1–8.1% of all newly diagnosed CRCs (1). There are some well-known cancer syndromes and hereditary forms of CRC such as Lynch syndrome (LS) or familial adenomatous polyposis (FAP), in which tumor co-occurrence happens frequently observed; patients with inflammatory bowel disease and serrated polyps/hyperplastic polyposis are also known to have a higher risk of developing SCRC (1). Nevertheless, these situations only account for around ~10% of SCRC patients (3).
To date, specific approaches to understanding the molecular basis of SCRC have primarily focused on the key colorectal cancer pathways. SCRC patients seem to have a higher proportion of microsatellite instability (MSI) as well as CpG island methylator phenotype (CIMP) tumors, via-a-vis patients with singular tumors (4–6). It has been hypothesized that there may be a correlation between the molecular basis of SCRC and the tumor location in the colon. The right-sided SCRC may be related to a CIMP-High genotypes and LS, whereas left-sided SCRC seems to associate with the chromosomal instability (CIN) pathway and low-penetrance genes; SCRCs throughout the entire colon seems to relate to the CIN pathway and germline mutations in APC or MUTYH (7). Our group has recently defined 4 molecular groups according to the MSI/CIMP statuses of SCRC that show certain parallels with the molecular classification of CRC described by Ogino and Goel (8), displaying also a relationship with the location of the tumors in the same individual (paired tumors) (9).
We speculate that a subset of paired SCRC tumors could share the same somatic mutations and abnormal genetic pathways, thus indicating a likely clonal origin. Comparative analyses of SCRCs have been carried out frequently, especially focused on MSI or CIMP, or somatic mutations, with conflicting results (10–11). Some recent studies addressed the idea of a possible clonal origin of some subsets of tumors such as bilateral breast cancer or SCRC using mutational concordance or clonality analysis by means of copy number profiles and concluded that paired tumors were different in origin (13–15). However, this conclusion should be considered cautiously given the limited sample size of these studies. Nevertheless, if a subset of SCRC fulfil clonality features, this should have not only therapeutic implications (treating different or similar paired tumors in the same patient), but it could also imply changes in the intrinsic definition of SCRC.
In the present study, we tried to find a clonal origin in cases of paired SCRCs without a hereditary molecular basis (sporadic or familial aggregation forms). We used a Single-Nucleotide Polymorphism array (SNP array) to characterize the genomic profiles and confirmed the results by using next generation sequencing (NGS) of the key genes related to CRC. Moreover, we categorized the ensuing groups according to colonic location, in an effort to identify differential phenotypes with clinical implications.
MATERIAL AND METHODS
Patients, samples and data collection
A total of 53 individuals diagnosed with SCRC were consecutively collected from January 2006 at the 12 de Octubre University Hospital (Madrid). We defined a CRC as SCRC when 2 or more histologically distinct colorectal tumors were identified in the same patient at the same time or in a period less than six month after the first diagnosis. Tumor relapse was defined either as regrowth at the anastomosis site (± 5cm) or as the detection of new metastatic disease. MCRC was defined as a secondary tumor occurring outside the anastomosis area more than 6 months after surgery. For each patient, we performed molecular analysis of the two tumors with the highest percentage of neoplastic material and used the most advanced neoplasia for staging. Tumor location was defined as previously published (9), and thus patients were initially classified into 3 categories according to the anatomical location of the tumors. Thus, “right colon” was defined as the colocation of the synchronous tumors at the right side of the large intestine (cecum, ascending colon, hepatic flexure, and first portion of the transverse colon); “left colon” was defined as the colocation at the left side of the large intestine (second portion of the transverse colon, splenic flexure, descending colon, sigmoid colon, and rectum); and “entire colon” was defined as the location of the synchronous tumors at different sides of the colon. Finally, “right colon” and “left colon” cases were defined as monosegmental, while the other cases were defined as pancolonic.
Family history of cancer and clinicopathological information was obtained for each patient with a mean follow-up of 74 months after surgery. We considered local recurrence, severely dysplastic tumors and a diagnosis of inflammatory bowel disease, LS, or FAP as exclusion criteria. All patients (or a first-degree relative in case of death of the index case) provided written consent, and the study was approved by the Ethics Committee of our Institution.
DNA isolation and MSI and CIMP characterization of the tumors
Two tissue specimens were obtained from each index case (the two highest stages at diagnosis). Microscopic inspection of paraffin-embedded samples was performed by a pathologist, and samples with more than 70% of tumor cells in the neoplastic material were considered adequate for further analysis. The protocol for DNA isolation was as previously reported (16).
We used the Bethesda panel to assess the MSI status and considered two or more altered markers as a positive result (17). MSI tumors were first analyzed for the BRAF V600E mutation and hypermethylation of the MLH1 gene promoter to confirm their sporadic nature and, when negative, they were subsequently screened for germline mutations in the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6 and PMS2 (18). For the assessment of CIMP, we investigated the methylation status of the promoter regions of CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1. Each patient was categorized as CIMP-High, CIMP-Low or CIMP-0 depending on whether their simultaneous tumors showed ≥5/8, 2/8 to 4/8, or 0/8 to 1/8 methylated promoters, respectively. Patients with different CIMP status in their paired tumors were categorized as CIMP-MM (mismatching). The procedures for the evaluation of MSI and CIMP have been previously described (18).
MUTYH analysis
MUTYH hotspots analysis included the three most common mutations in our population: c.536A>G, p.(Tyr179Cys), rs34612342; c.1187G>A, p.(Gly396Asp), rs36053993; and c.1227_1228dupGG, p.(Glu410Glyfs*43), rs587780078 (RefSeq NM_001128425.1, NP_001121897.1, dbSNP). This screening was carried out using high resolution melting (HRM) analysis on a LightCycler 96 System (Roche). Positive profiles were sequenced using the Sanger dideoxy method to identify the variant.
Analysis of copy number alterations by SNP array and analysis of clonality
We performed the OncoScan Formalin-Fixed, Paraffin-Embedded (FFPE) Assay to asses copy number and loss of heterozygosity (LOH) on the paired samples obtained from patients with SCRC. The OncoScan FFPE Assay (Affymetrix Inc) is a platform based on Molecular Inversion Probe technology which uses small amounts of DNA from FFPE samples (19, 20). Genomic DNA was quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, P-7589). The OncoScan FFPE Assay Kit was used according to the manufacturer’s instructions. A GeneAmp PCR system 9700 Thermal Cycler (Life Technologies) was used from the annealing stage to the denaturation stage. The digested DNA target was hybridized onto the OncoScan array and incubated at 49ºC in a Genechip Hybridization oven 640 (Affymetrix) for 17h at 60rpm. OncoScan arrays were then washed and stained in a GeneChips Fluidics Station 450 (Affymetrix) using OncoScan staining and wash reagents according to the manufacturer’s instructions. The microarrays were finally scanned with a GeneChip scanner 3,000 (Affymetrix). Data QC analysis was performed with the OncoScan Console software (Affymetrix). Copy Number Alterations (CAN) events were called using the normalized data using Nexus Express for OncoScan 3.1 (Affymetrix). Applying the Affymetrix OSCHP-TruScan- allowed to identify the percentage of aberrant cells and overall ploidy, as well as copy number events and the percentage of LOH for each sample. Data were included in Gene Expression Omnibus (GEO) database (GSE110026).
Weighted log2 ratios from the ChAS console (Affymetrix) were also processed using the copy number R package (21). This package performs a pre-processing step of detection and modification of extreme values through a method called Winsorization (22), and a single sample segmentation step using Piecewise Constant Fitting (PCF) algorithms. The Gamma value, which is the penalty for each discontinuity in the curve, was set to 40, and the minimum number of probes allowed in each segment was set to 5. Copy Number frequency plots were constructed using this package, setting the log2 ratio threshold for gains and losses to 0.1 and −0.1, respectively. The grade of Genome Instability was assessed as previously reported (16).
With respect to CNA, we used the R software package Clonality (23), which uses tumor copy number profiles at the probe level, to determine whether two tumors from the same patient were clonal or origin-independent using a likelihood ratio 2 (LR2) statistic (quantifying the odds that the two tumors are clonal). To run Clonality, we used the DNA-copy package to create a copy number array object. The copy number array object was used as input for Clonality. What we observe during our clonality tests based on CNAs is that independent additional changes would take place, as paired tumors develop thereafter separately. Moreover, two monoclonal paired tumors could involve a combination of alterations, some of which will be identical (clonal), some independent, arising from the beginning (non-clonal). Observed genomic changes and matching with higher frequency have more probability to be clonal. The concordance of the genomic alterations between two paired tumors is compared using a LR, which allows us to quantify the probability that both tumors would be clonal. For every partial gain or loss, we also calculate the probability rate that both paired tumors have the same change. These two LR are multiplied by LR1 to get our statistic, LR2. If this is much higher than 1, it indicates clonality; if it is slighter small than 1, it shows independence.
We further enhanced the analysis by using a second algorithm termed GISTIC (Genomic Identification of Significant Targets in Cancer), which identifies functionally significant CNAs by giving more weight to high copy gains and homozygous losses that may be functionally relevant (24, 25). Parameters used to run the analysis are described in Supplementary Material and Methods.
Next Generation Sequencing
Ion torrent PGM library preparation.
An Ion Torrent adapter-ligated library was generated using the Ion AmpliSeq Library Kit 2.0 and the Ion AmpliSeq Cancer Hotspot Panel v2 (Thermo Fisher Scientific, Rev. B.0; MAN0006735). Briefly, 2μL of 5X Ion AmpliSeq™ HiFi mix, 2μL of 5X Ion AmpliSeq™ Primer Pool and 5ng of gDNA per 10μL reaction were mixed together and amplified following the temperature conditions provided by the manufacturer. Then, primer sequences were partially digested by adding 1μL of FuPa Reagent and loaded in a thermal cycler under the conditions detailed in the user guide. Finally, each library was labeled with a unique adapter provided in the Ion Xpress™ barcode adapters 1–96 Kit (Life Technologies) by adding 2μL of Switch Solution, 1μL of diluted barcode and 1μL of DNA Ligase to the reaction mixture, also under the temperature conditions recommended by the manufacturer.
After AMPure bead (Beckman Coulter, Brea, CA, USA) purification, the concentration of the library (in a 100-fold dilution) was determined using the Ion Library TaqMan quantitation kit (Thermo Fisher Scientific) in a 7500 Real-Time PCR System (Thermo Fisher Scientific, Foster City, CA). A minimum of two replicates were run of each sample.
Emulsion PCR
Emulsion PCR and enrichment were performed using the Ion PGM™ Template OT2 200 Kit and the Ion One TouchTM 2 System (Life Technologies). We followed the manufacturer’s instructions except for the concentration of the pooled libraries, which was set at 9 pM.
Sequencing on the Ion torrent PGM platform
All barcoded samples were sequenced using the Ion PGM™ Hi-Q™ Sequencing Kit (Life Technologies) in an Ion Torrent PGM instrument (Life Technologies) with Ion 318™ v2 chips (Life Technologies).
Chip loading was performed according to the user guide for the PGM™ Hi-Q™ Sequencing Kit (Life Technologies). A maximum of 16 samples were loaded on a single chip per sequencing run.
Bioinformatics processing and data analysis
Base calling and alignment to the human genome (hg19) were executed with Torrent Suite Software v.4.0 using the variant caller plugin. Variants were annotated using Ion Reporter and each mutation was verified in the Integrative genome viewer (IGV) from the Broad Institute (http://www.broadinstitute.org/igv/) (26). Only pathogenic variants were considered, being thus excluded any polymorphism or benign variant for the study.
Statistical Analysis
Continuous variables were expressed as mean values plus/minus standard deviation (SD), and categorical variables were expressed as number of cases and their percentage. Comparison of categorical variables was done using Pearson’s Chi Square (χ2) test, and Student’s t-test for independent samples and Mann-Whitney U tests were used for continuous variables, as appropriate. For comparisons between more than two groups, analysis of variance (ANOVA) (for normal distributions) or the Kruskal-Wallis test (for nonparametric distributions) were used. The Kaplan-Meier method (log-rank test) was used to assess the relationship between CNAs and overall survival (OS) and disease-free survival (DFS). Statistical analysis was performed using SPSS version 23.0 (IBM), and differences were considered statistically significant when the p-value was <0.05.
RESULTS
Clonality analysis.
Two tumors clones that evolved from the same cancer cell will show a number of somatic changes that are identical. Consequently, comparison of the DNA profiles for the extent of similarities in the patterns of somatic changes between two synchronous tumors is a powerful strategy for determining if they are independent or derive from the same clone (27). We carried out the analysis by using an SNP array to study the CNA landscape of paired SCRCs. We analyzed the clonal relatedness of 53 paired tumors (106 pairs) and found a significantly higher likelihood ratio 2 (LR2) value in 20 SCRC pairs (40 paired tumors). No relationships were observed for the other 33 SCRCs (66 paired tumors) (P value <0.05) (Supplementary Table 1). Thus, we defined the first group as Monoclonal (M), and the second as Polyclonal (P) (an example of each is depicted in Figures 1a and 1b). One case, classified as monoclonal, had to be removed because it was a newly-diagnosed LS case. Clinico-pathological and familial features of the 52 cases are shown in Table 1.
Figure 1a.
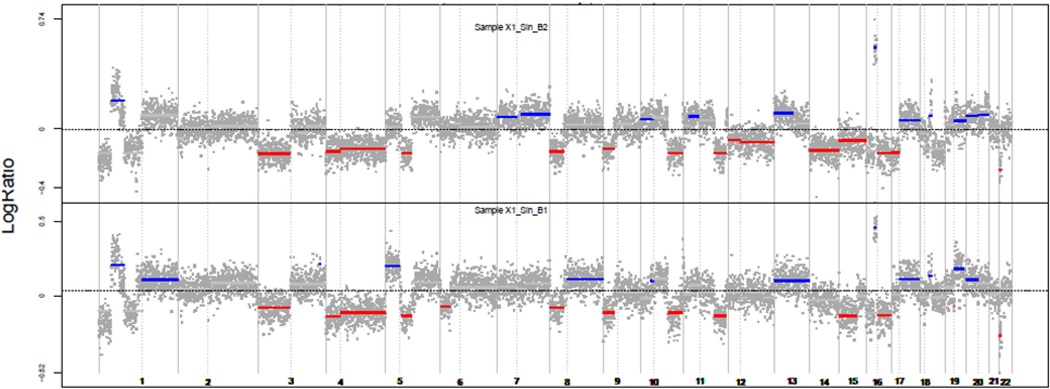
Two paired tumors of a case defined as monoclonal. Red: losses; Blue: gains
Figure 1b.
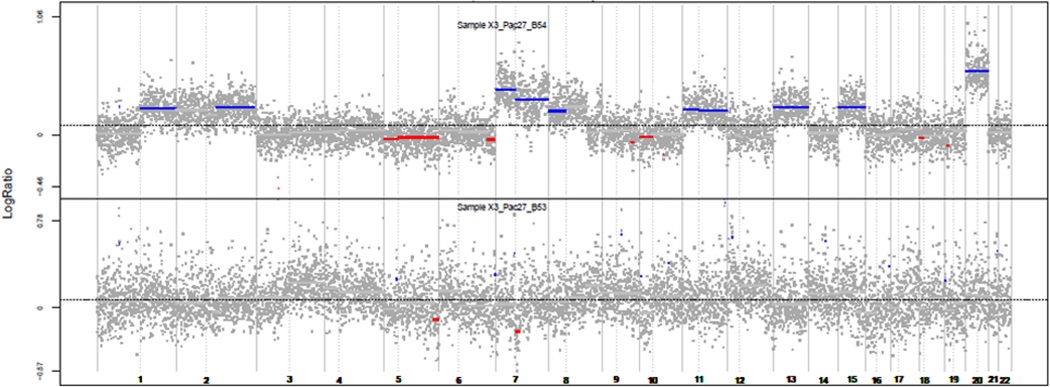
Two paired tumors of a case defined as polyclonal. Red: losses; Blue: gains.
Table 1.
Comparison and description of the clinicopathological and molecular features of the subgroups categorized according to the clonality and the anatomic location of the Synchronous Colorectal Cancers in the colon.
| Total SCRC | MM | MP | PM | PP | p- value1 |
|
|---|---|---|---|---|---|---|
| Number of patients | 52 (100) | 10 (19.2) | 9 (17.3) | 19 (36.5) | 14 (27) | - |
| Average age of onset | 71.1 [9.9] | 73.5 [8.5] | 65 [12.6] | 69.6 [9.6] | 72.4 [9.8] | 0.22 |
|
Sex: Male Female |
35 (67.2) 17 (32.8) |
8 (80) 2 (20) |
4 (44.4) 5 (55.6) |
13 (68.4) 6 (31.6) |
10 (71.4) 4 (28.6) |
0.4 |
|
Colon location: Right colon left colon Entire colon |
6 (11.5) 23 (44.2) 23 (44.2) |
4 (40) 6 (60) |
2 (10.5) 17 (89.5) |
0.08* |
||
|
Tumor differentiation3 High Moderate Low |
22 (52.4) 18 (42.8) 2 (4.8) |
3 (37.5) 5 (62.5) 0 |
3 (75) 1 (25) 0 |
9 (55.6) 8 (44.4) 0 |
7 (53.8) 4 (30.8) 2 (15.4) |
0.3 |
|
Mucin production3 “Signet ring” cells3 |
9/42 (21.4) 2/42 (4.8) |
3/8 (37.5) 0 (0) |
0/4 (0) 0 (0) |
1/17 (6) 1/1 (6) |
5/13 (38.5) 1/13 (8) |
0.07 0.8 |
|
Astler-Coller stage A B C D |
16 (30.8) 23 (44.2) 10 (19.2) 3 (5.8) |
2 (20) 6 (60) 1 (10) 1 (10) |
6 (66.6) 2 (22.2) 1 (11.) 0 (0) |
5 (26.3) 9 (47.4) 4 (21.1) 1 (5.3) |
3 (21.4)) 6 (42.9) 4 (28.6) 1 (7.1) |
0.5 |
|
Number of SCRCs Associated polyps Yes Mean. Type: Adenomatous. Hyperplastic/mixed |
3.2 [2.7] 49 (94.2) 10.1 [12.4] 27 (65.1) 22 (44.9) |
2.4 [0.5] 8 (80) 5.3 [5.3] 5 (62.5) 3 (37.5) |
3.2 [1.5] 9 (100) 15.6 [16.8] 5 (55.5) 4 (44.4) |
2.5 [0.7] 18 (95) 11.7 [15.2] 8 (44.4) 10 (55.5) |
4.6 [4.7] 14 (100) 7.7 [6.3] 9 (64.3) 5 (35.7) |
0.092 0.2 0.22 0.7 |
|
Metachronous CRC. Recurrence Global Mortality/CRM |
8 (15.7) 8 (15.7) 24 (46)/7 (13) |
3 (30) 3 (30) 8 (80)/ 3 (30) |
0 (0) 1 (9) 1 (9)/ 0 |
4 (22) 4 (28.6) 6 (43)/ 2(14) |
1 (7) 0 (0) 9 (63)/ 2 (14) |
0.2 0.2 0.006 |
|
DFS (months) OS (months) |
68.1 [41.1] 75.3 [37.2] |
51.4 [39.1] 61.1 [38.5] |
99.2 [18.7] 100.2 [19.2] |
68.7 [39] 78.5 [33.1] |
59.4 [47.6] 65.1 [44.1] |
0.052 0.092 |
|
Familial cancer history Sporadic Familial aggregation Amsterdam II positive |
43 (82.7) 8 (15.4) 1 (1.9) |
7 (70) 3 (30) 0 (0) |
4 (44.4) 4 (44.4) 1 (11.2) |
18 (94.7) 1 (5.3) 0 (0) |
14 (100) 0 (0) 0 (0) |
0.009 |
Data shown in parenthesis represent percentages. Data shown in brackets represent standard deviation
Statistical comparison was performed using Pearson’s Chi Square test (χ2)
Statistical comparison was performed using analysis of variance (ANOVA)
Percentages shown are based on varying total numbers as some cases were excluded because only one biopsy was taken (stage D), or because tumors were severely dysplastic with “in situ” carcinoma and it was not possible to study any other characteristic. DFS: Disease-free survival. MM: Monoclonal Monosegmental. MP: Monoclonal Pancolonic. PM: Polyclonal Monosegmental. PP: Polyclonal Pancolonic. OS: Overall survival. SCRC: Synchronous colorectal cancer. CRM: Cancer-related Mortality.
The most frequent changes detected in the 19 monoclonal cases (38 samples) that were present in more than 50% of the cases were: gains on chromosomes 20q13.2 (71%), 12p13.1 (66%), 8q24.3, 12p13.2, 13q22.1, 13q34 and 20p11.2 (all 61%), and losses on 1p36.2, 1p35.3, 18p11.32 (all 61%), 1p21.3 (58%), 1p13.2, 9q34, and 16p13.3 (all 53%) (Supplementary Table 2). In the 33 polyclonal SCRC cases (66 samples) the most frequent gains were on chromosomes 8q24, 20q13 (all 67%), 7q36, 20q12 (both 64%), 7p11.2, 8q23 (both 62%), 8q12, 20p11 (61%), 12p13 (all 59%), 7q31.2, 13q34 (both 56%) 13q22 (55%), 3q36.31, 7p21.1 and 12q14.1 (all 50%), and losses on 18p11.32 (62%), 1p35.3 (58%), 1p36.21 (53%) 1p35.1 and 15q25 (both 50%) (Supplementary Table 3). Monoclonal and polyclonal SCRC showed differential somatic copy number alteration (SCNA) profiles: gains on 3p13 (55%), 5p15, 6p25, 18p11.31 (all 53%), and losses on 9q34 (53%) were more frequent in monoclonal SCRC, while gains on 8q22.2 (50%), 8q13.3 and 13q32.2 (47%) were most frequent in polyclonal SCRC (p<0.05 Fischer test) (Supplementary Table 4). Corresponding putative genes and micro-RNAs mapped to the identified altered chromosomal regions defined by GISTIC are shown in Supplementary Tables 2 to 4.
We used the OncoScan® assay to simultaneously analyze CNA and Loss of Heterozygosity (LOH) regions. Comparing both SCRC groups, no significant differences were found between the ratio of LOH, nor did we find differences regarding the degree of genomic instability (using the median value of all the changes) were found comparing the four groups with each other (data not shown).
Confirmation of clonality using NGS and other indirect features.
We used mutational concordance to confirm our clonality results, as other publications used such an approach (14). In Table 2A the mutational status of all cases is shown, with paired cases possessing the same pathogenic mutations shown in “blue”. In the monoclonal group, 9 cases (47%) showed at least one set of paired tumors with the same pathogenic mutation: 4 cases with three different genes having the same-mutations in the paired tumors; 1 case with two genes having identical mutations; and 4 cases with one gene with the same mutations. Apart from KRAS, present with the same mutation in 4 cases, the most frequently mutated genes were APC and SMAD4, which presented the same mutation in 4 and 3 cases, respectively. Only 4 polyclonal cases showed the same mutation in paired tumors (12%), three of them in KRAS and one in BRAF.
Table 2A and 2B. Clonality and colon location categories within SCRC (both concordant in order).
2A. Next Generation Sequencing of the selected gene panel. 2B: CIMP and stages of the four different groups.
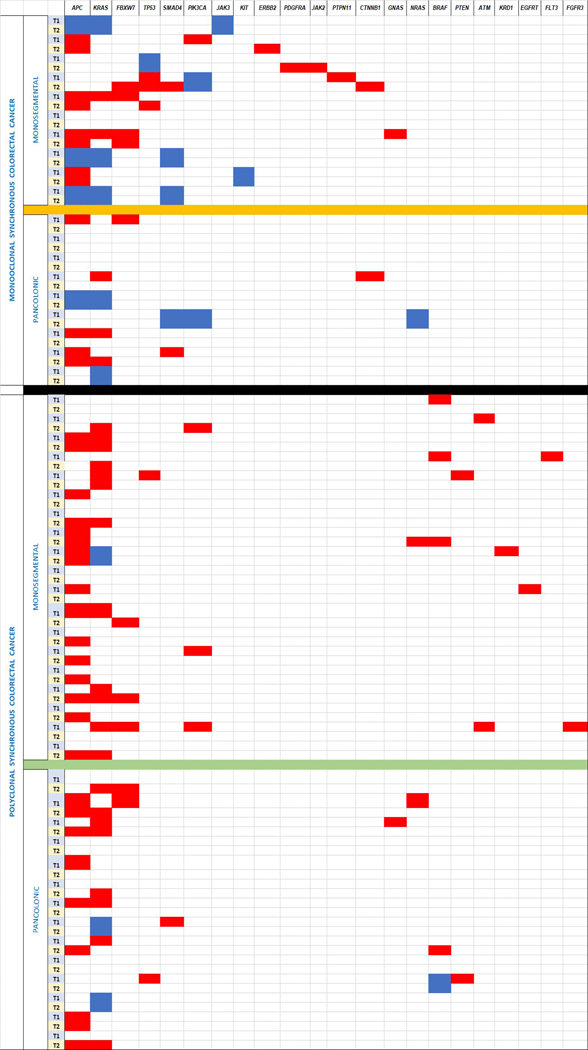
|
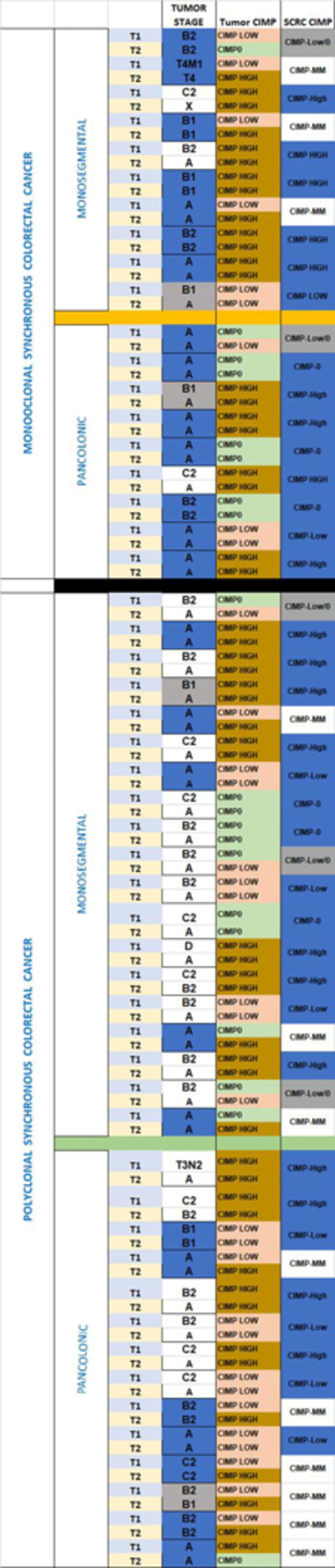
|
Table foot: Blue boxes: Same mutation in the same gene. Red boxes: Different mutations in the same gene.
Table foot: CIMP: CpG island methylator phenotype. SCRC: Synchronous colorectal cancer. Blue boxes: Coincident values. Grey boxes: Proximal values.
Other possible indirect features that can be used as a marker of clonality are CIMP and stage-at-diagnosis (Table 2B). Fourteen out of 19 monoclonal cases exhibited CIMP in the paired tumors (74%), with the other five showing adjacent CIMP values (one tumor CIMP-0 and the paired one CIMP-low, or one tumor CIMP-low and the paired one CIMP-high). Polyclonal cases showed a different distribution: 21 out of 33 showed CIMP concordance between paired tumors (64%), nine had adjacent CIMP values, and 3 cases showed a high CIMP-mismatch.
Analysis of tumors staging revealed that in the monoclonal group, 14 of the 19 cases (74%) presented the same stage at diagnosis (blue boxes) and another 2 showed adjacent stages (grey boxes). However, in the polyclonal group only 12 out 33 cases (36%) had the same stage.
Relationship between SCRC clonality and location.
Main groups and correlative regions.
The SCRC Monoclonal group (M) was divided in Monosegmental (MM) and Pancolonic (MP) subgroups. The MM subgroup was composed of 10 cases (19% of the total), while the MP subgroup contained 9 cases (17%). The colon distribution in MM was: 4 cases in the right colon and 6 cases in the left colon. The SCRC Polyclonal group (P) was also divided into Monosegmental (PM) and Pancolonic (PP) subgroups: 19 cases were PM (37%), showing preference for left-colon location (17 cases, 89%), whereas 14 cases were PP (27%).
Within the MM group the most frequent changes were gains on 20q13 (85%) and 12p13 (75%), and deletions on 1p21 (85%), 1p36 and 1p13 (both 75%) (Supplementary Table 5). The MP group was characterized by gains on 13q22 (67%) 4p16 and 8q24.3 (both 61%). This latter group presented the lowest number of genomic alterations within four groups (Supplementary Table 6). Relevant differential genomic regions in both monoclonal categories were gains on 1q24 and 8q24, and deletions on 1p21 and 1p13 in the MM group (Supplementary Table 7).
The PM group was characterized by gains on 7q36 and 20q13, and deletion on 18p11 (all 68%) (Supplementary Table 8), while in the PP group among the most frequent changes were gains on 8q24 (71–75%) and 7p11 (68%)(Supplementary Table 9). Most frequent differential regions in the polyclonal cases were gain on 7q36 and deletion on 1p36, for PM group. (Supplementary Table 10). Corresponding putative genes and micro-RNAs codified in the identified altered chromosomal regions defined by GISTIC are shown in Supplementary Tables 5 to 10.
The genomic profiles of the four different categories based on clonality and colon location of the tumors are shown in Figure 2.
Figure 2.
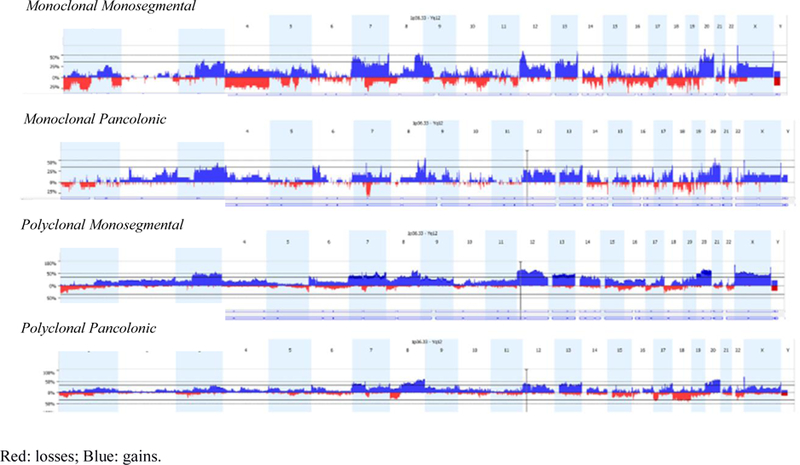
Genomic profiles of the four different categories according to clonality and location of the tumors in the colon.
Mutational status.
As mentioned above, monoclonal cases showed most paired tumors with genes carrying the same mutation (Table 2A). Six cases of the MM subgroup (60% of the group) exhibited same-mutation genes in paired tumors, three of them in three different genes. In the MP group there were 3 such cases (33%). After KRAS gene, APC was the gene most frequently observed to have the same mutation in paired tumors (4 cases).
It is noteworthy that there was a substantianl proportion of APC mutations in the MM and PM groups (70% and 42% of all tumors, respectively), when compared with the pancolonic categories (33% in MP and 32% in PP). Remarkably, TP53 mutations were not as frequent and were mostly observed in MM cases (25%).
Distribution of the main carcinogenetic pathways and analysis of hereditary CRC forms.
Only two monosegmental cases (one monoclonal and one polyclonal) showed MSI based on MLH1 promoter hypermethylation and/or BRAF mutations. CIMP-High status showed a high prevalence among our SCRC cases (Table 2B). The proportion of CIMP-High tumors was 65% in MM, 44% in MP, 45% in PM, and 50% in PP groups, but focusing on paired CIMP-High tumors, a decreasing order was observed: 50% in MM, 44% in MP, 37% in PM and 29% in PP.
None of the four cases with the same APC mutation in paired tumors showed pathogenic germ-line mutations. MUTYH germ-line mutations analysis was carried out in 40 out of the 52 cases (77%) (due to the death of some patients, it was not possible to extract blood samples, while in others this was already done because of previous hereditary CRC screening strategies). Only two cases showed the c.118 7G>A (p. Gly396Asp) mutation. One of them presented the mutation in a homozygous manner; the SCRC was diagnosed at an age of 71 and was defined as PM (left colon), with 41 mixed-type associated polyps during follow-up, and no familial cancer history. The patient also developed an MCRC one year later. The second case showed the mutation as heterozygotic; the SCRC was diagnosed at an age of 77, classified as MM (left colon), with 17 also mixed-type associated polyps. Although no familial cancer history was apparent, the patient developed multiple primary neoplasms: a urothelial tumor (diagnosed at 71 y/o) and a MCRC (diagnosed at 80 y/o), in addition to the SCRC.
Phenotypes and clinical implications.
Comparative results regarding clinico-pathological and familial features of the four defined categories are shown in Table 1. Specific differences were observed for each category. Firstly, the MM group showed a high proportion of mucinous tumors (37.5%), the lowest mean number of SCRCs as well as associated polyps, and displayed the worst prognosis related to recurrence and mortality (Figures 3A and 3B). The MP group included the youngest age at diagnosis and the largest mean-number of associated polyps, with a comparatively intermediate mean number of SCRCs, and showed the best prognosis and an important familial cancer component. The PM group seemed to be a “frontier” group, with features similar to MP except for a high sporadic component. Lastly, the PP group also exhibited a mucin component (almost 40%), had the highest mean-number of SCRCs (4.6) compared with the mean-number of polyps (7.7), had a poor prognosis and consisted entirely of sporadic cases.
Figure 3A and 3B.
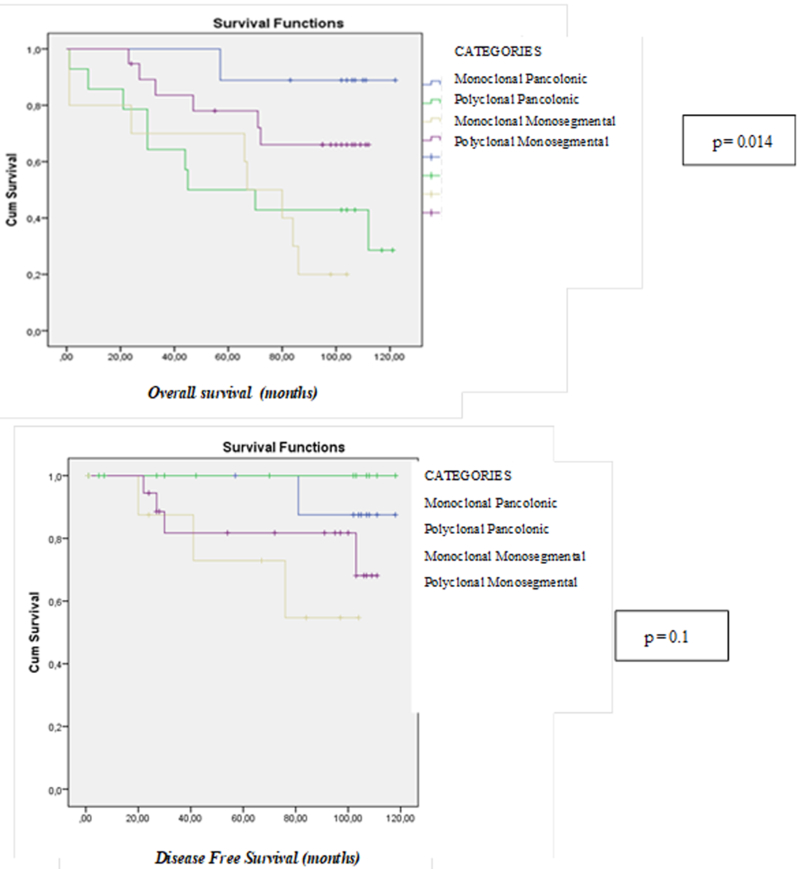
Overal survival and Disease-Free Survival curves of the four different categories according to clonality and colon location of the tumors.
DISCUSSION
Tumor multiplicity is widely recognized as a feature of genetic predisposition for the development of neoplasms (28). Although the heterogeneous phenotype of SCRC has been extensively discussed, the disease remains poorly understood. Comparing the genetic patterns of synchronous lesions may provide important knowledge about the biology of these tumors. Moreover, it addresses the additional possibility that the paired tumors share genetic features and the same genetic origin, acquiring at the beginning similar driver somatic mutations, and therefore having a likely clonal origin.
Thirty-six per cent of our SCRCs fulfilled clonality features. There are only two recent studies analyzing clonality in SCRC, and both showed an apparent intertumor heterogeneity within the same patient (14, 15). However, these studies analyzed a rather smaller patient cohort (10 and 15 cases, respectively, vs our 52) from a genetic point of view (concordance of paired-mutated genes or whole exome sequencing), whereas we developed a different strategy by comparing the genomic profiles for similarities in the patterns of somatic changes. Moreover, both previous studies tried to define clonality as concordance in high proportions of gene mutations - a condition that we believe is problematic due to the high degree of heterogeneity that CRCs acquire during development. Copy number variation data can pinpoint the loci in the genome where allelic gains and losses begin and end and allow for a comprehensive comparison of different mutational profiles, thus offering the potential for identifying exact matches that are the hallmark of clonal allelic changes (29). The optimal situation would be that two monoclonal SCRCs are derived from a single “clonal” cell that experienced the pivotal mutations that led to tumor development. In practice however, since additional independent mutations are likely to occur subsequently in the two tumors as they develop separately, two clonal tumors will consist of a mixture of identical (clonal) mutations and independent mutations, while tumors that arise independently will consist solely of independently occurring mutations (29). Our strategy involves comparing the mutational patterns in two tumors to determine whether they are sufficiently alike to conclude that they are indeed clonal. The concordance of gene mutations in paired tumors, together with some other indirect parameters, such as CIMP concordance or an identical or similar stage of the paired tumors, has helped us to outline the clonality between paired tumors.
The relationship between colonic location and differential clinical and molecular features of CRC has been widely established (30–32), and the same seems to apply to SCRC (2, 7, 9). Our division of SCRC into categories according to clonality and colon location of the tumors within the same patient showed a smaller proportion of SCRC cases in the right colon that were mainly monoclonal, as opposed to the higher rate of SCRCs in the left colon, which were mainly polyclonal. This seems to confirm the predisposition to establish a certain type of colon cancer in the right colon different from the usual sporadic-isolated CRC, characteristic of left colon cancers. In addition, we were able to define prognostic subclasses, highlighting MM and PP, with an important mucinous component, and also to observe a possible differential influence of the field effect within SCRC (33, 34). When we highlight the concept of such clonality, current techniques do not allow to distinguish between multicentric occurrence and local metastasis, and it is possible that the MM SCRC group, which has the worst prognostic features, will arise as an aggressive category of CRC, with early local metastasis in the colon. In this direction, some of the relevant differential altered genomic regions in this group, gain on 1q24 and deletion on 1p13, have been also associated with worse prognosis within CRC (35, 36). Moreover, the majority of the specific altered chromosomal regions related with the four defined categories have been related with CRC and/or a higher risk of its development as well (e.g. gain on 8q24 or deletions on 1p21, 1p13 or 1p36) (37–39). But more remarkable are gains on 20q13 and deletions on 18p11, two of the most frequently altered regions in PM group, that have been already described by Di et al. (15), presenting similarly within their sample of 15 SCRC patients, in which they defined that synchronous tumors were of different genetic origins, therefore polyclonal.
Many studies point out the importance of an environmental field effect that promotes multiple colorectal tumors, with likely causes as an important exposure to a carcinogen or genetic predisposition for cancer development (4, 14, 15, 33). Cereda et al. showed a possible combination of both exposure and genetic predisposition underlying SCRC, joining inherited damaging alterations of immune-related genes with the consequent inflammatory conditions, all of which stimulate tumorigenesis (14). One interesting aspect of our results, in agreement with the field effect, is the different frequency of malignancy associated with polyps in each category. While the MM and PP groups showed a proportion of around one of SCRC for each of the two polyps, in the MP and PM groups there was around one SCRC for each 5 polyps. This, together with the fact that the MP group presented an important familial cancer component, may indicate that the MP cases are the ones with the lowest influence of a field effect, and an unidentified genetic predisposition may lead to them as occur also within a background of a high number of polyps and a younger age at diagnosis. Although the existence of clonality throughout the entire colon together with the fact that most of this group were stages A for both paired-tumors could be a contradiction, not only the likelihood of a genetic predisposition in these cases suggests the possibility of clonality, but also the fact that almost half of the cases within MP group had already developed polyps before the SCRC (data not shown). On the other hand, the PM category, with an important predisposition for the left-sided colon, should be due to a possible environmental field effect. In the PP cases, the field effect also appears to have a high impact throughout the entire colon developing heterogeneous tumors, while in MM cases a mixture of all the factors, appears to play a role. Some of the mechanisms that have been related with field effect are increased genomic instability or via cytokine and growth factor secretion, epigenetic alterations or increased mutation rate (14, 40–42).
The existence of clonality within SCRC has important consequences at the time of diagnosis and throughout therapeutic management of the patient, and calls for an analysis of both tumors, especially in cases of polyclonal SCRC. The statistical package we used seems to asses more accurately the probability of clonality in SCRC, and this, together with the tumor locations, helped us to configure a novel classification approach which also has prognostic and clinical consequences. Our series is the largest until now about SCRC and clonality analysis by genomic profiles approach, and our results should be validated by other groups to confirm our findings. Assessing the microbiome and molecular alterations in normal mucosa in SCRC patients should be next steps to clarify the exact roles of environment and individual predisposition in SCRC. Moreover, the growing importance that clonality analysis is acquiring within SCRC calls for more extensive studies, and if the existence of clonal cases is confirmed, SCRC may need to be redefined not only from a clinical or chronological point of view.
Supplementary Material
What´s new:
To our knowledge, this is the largest series of Synchronous Colorectal Cancer (SCRC) in which clonality is analyzed, with a Single-Nucleotide Polymorphism array and the subsequent statistical application, and the first to correlate it with clinical phenotypes. The existence of clonality within SCRC has important consequences throughout therapeutic management. These categories may also serve as a starting point to analyze more selectively the molecular basis of SCRC and its relationship with environmental factors.
Acknowledgement and GRANT SUPPORT.
This work was funded by Projects PI10/00683 and PI16/01650 to J.P, and PI16/01920 to R.G.S, from the Spanish Ministry of Health and Consumer Affairs and FEDER, by Project 2012–0036 from the Mutua Madrileña Foundation, and supported by the CA72851, CA181572, CA184792, CA187956 and CA202797 grants from the National Cancer Institute, National Institute of Health; RP140784 from the Cancer Prevention Research Institute of Texas; grants from the Sammons Cancer Center and Baylor Foundation, as well as funds from the Baylor Scott & White Research Institute, Dallas, TX, USA. We thank the Tumor Registry of the Pathology Department of the 12 de Octubre University Hospital for providing the paraffin-embedded tissues, and Ron Hartong for his help with the English revision of this manuscript.
Footnotes
Disclosures:
The authors declare that they have no competing interests.
REFERENCES
- 1.Lam AK- Y, Chan SS- Y, Leung M. Synchronous colorectal cancer: Clinical, pathological and molecular implications. World J Gastroenterol 2014; 20(22):6815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moertel CG, Bargen JA, Dockerty MB. Multiple carcinomas of the large intestine: a review of the literature and a study of 261 cases. Gastroenterology 1958; 34: 85–98. [PubMed] [Google Scholar]
- 3.Lam AK, Carmichael R, Gertraud Buettner P, Gopalan V, Ho YH, Siu S. Clinicopathological significance of synchronous carcinoma in colorectal cancer. Am. J. Surg 2011; 202, 39–44. [DOI] [PubMed] [Google Scholar]
- 4.Nosho K, Kure S, Irahara N, Shima K, Baba Y, Spiegelman D, et al. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology 2009;137(5):1609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pajares JA, Perea J. Multiple primary colorectal cancer: Individual or familial predisposition? World J Gastrointest Oncol 2015; 7(12):434–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Macedo MP, de Melo FM, Ribeiro Jda S, de Mello CA, de Souza Begnami MD, Soares FA, et al. RAS mutations vary between lesions in synchronous primary colorectal cancer: testing only one lesion is not sufficient to guide anti-EGFR treatment decisions. Oncoscience 2015;2(2):125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leggett BA, Worthley DL. Synchronous colorectal cancer: not just bad luck? Gastroenterology 2009; 137: 1559–1562. doi: 10.1053/j.gastro.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 8.Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn 2008;10:13–27. doi: 10.2353/jmoldx.2008.070082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arriba M, Sánchez R, Rueda D, Gómez L, García JL, Rodríguez Y, et al. Toward a Molecular Classification of Synchronous Colorectal Cancer: Clinical and Molecular Characterization. Clin Colorectal Cancer 2017;16(1):31–37. [DOI] [PubMed] [Google Scholar]
- 10.Eguchi K, Yao T, Konomoto T, Hayashi K, Fujishima M, Tsuneyoshi M. Discordance of p53 mutations of synchronous colorectal carcinomas. Mod. Pathol 13, 131–139 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Dykes SL, Qui H, Rothenberger DA, Garcia-Aguilar J. Evidence of a preferred molecular pathway in patients with synchronous colorectal cancer. Cancer 2003; 98, 48–54. [DOI] [PubMed] [Google Scholar]
- 12.Song F, Li X, Song F, Zhao Y, Li H, Zheng H, et al. Comparative genomic analysis reveals bilateral breast cancers are genetically independent. Oncotarget 2015; 6 (31): 31820–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cereda M, Gambardella G, Benedetti L, Iannelli F, Patel D, Basso G, et al. Patients with genetically heterogeneous synchronous colorectal cancer carry rare damaging germline mutations in immune-related genes. Nat Commun 2016; July 5;7:12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di J, Yang H, Jiang B, Wang Z, Ji J, Su X. Whole exome sequencing reveals intertumor heterogeneity and distinct genetic origins of sporadic synchronous colorectal cancer. Int J Cancer 2017. November 6. doi: 10.1002/ijc.31140. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15.Arriba M, García JL, Inglada-Pérez L, Rueda D, Osorio I, Rodríguez Y, et al. DNA copy number profiling reveals different patterns of chromosomal instability within colorectal cancer according to the age of onset. Mol Carcinog 2016; 55:705–16. [DOI] [PubMed] [Google Scholar]
- 16.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96(4):261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perea J, Rueda D, Canal A, Rodríguez Y, Álvaro E, Osorio I, et al. Age at onset should be a major criterion for subclassification of colorectal cancer. J Mol Diagn 2014; 16:116–26. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Cottman M, Schiffman JD. Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer Genet 2012; 205: 341–355. [DOI] [PubMed] [Google Scholar]
- 19.Singh RR, Mehrotra M, Chen H, Almohammedsalim AA, Sahin A, Bosamra A, et al. Comprehensive Screening of Gene Copy Number Aberrations in Formalin-Fixed, Paraffin-Embedded Solid Tumors Using Molecular Inversion Probe-Based Single-Nucleotide Polymorphism Array. J Mol Diagn 2016. September;18(5):676–687. [DOI] [PubMed] [Google Scholar]
- 20.Nilsen G, Liestøl K, Van Loo P, Moen Vollan HK, Eide MB, Rueda OM, et al. Copynumber: Efficient algorithms for single- and multi-track copy number segmentation. BMC Genomics 2012;13: 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon WJ. Simplified estimation from censored normal samples. Ann Math Statist 1960;31:385–91. [Google Scholar]
- 22.Ostrovnaya I, Seshan VE, Olshen AB, Begg CB. Clonality: An R package for testing clonal relatedness of two tumors from the same patient based on their genomic profiles. Bioinformatics 2011; 27: 1698–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers Genome Biol 2011;12(4):R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics 2012;14(2):178–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ostrovnaya I, Seshan VE and Begg CB. Using somatic mutation data to test tumors for clonal relatedness. Ann Appl Stat 2015; 9(3): 1533–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedroni M, Pedroni M, , Tamassia MG, Percesepe A, Roncucci L, Benatti P et al. Microsatellite instability in multiple colorectal tumors. Int J Cancer 1999;81(1):1–5. [DOI] [PubMed] [Google Scholar]
- 28.Ostrovnaya I Olshen AB, Seshan VE, Orlow I, Albertson DG, Begg CB. A metastasis or a second independent cancer? Evaluating the clonal origin of tumors using array copy number data. Stat Med 2010; 29(15):1608–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamauchi M, Morikawa T, Kuchiba A, Imamura Y, Qian ZR, Nishihara R, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012;61(6):847–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perea J, Cano JM, Rueda D, García JL, Inglada L, Osorio I, et al. Classifying early-onset colorectal cancer according to tumor location: new potential subcategories to explore. Am J Cancer Res 2015; 5(7): 2308–2313. [PMC free article] [PubMed] [Google Scholar]
- 31.Shimada Y, Kameyama H, Nagahashi M, Ichikawa H, Muneoka Y, Yagi R, et al. Comprehensive genomic sequencing detects important genetic differences between right-sided and left-sided colorectal cancer. Oncotarget 2017; 8:93567–93579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giovannucci E, Ogino S. DNA methylation, field effects, and colorectal cancer. J Natl Cancer Inst 2005;97(18):1317–9. [DOI] [PubMed] [Google Scholar]
- 33.Lochhead P, Chan AT, Nishihara R, Fuchs CS, Beck AH, Giovannucci E, Ogino S. Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression. Mod Pathol 2015;28(1):14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugai T, Takahashi Y, Eizuka M, Sugimoto R, Fujita Y, Habano W, et al. Molecular profiling and genome‐wide analysis based on somatic copy number alterations in advanced colorectal cancers. Mol Carcinog 2018; 57(3): 451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parada LA, Marañon A, Hallén M, Tranberg KG, Stenram U, Bardi G, et al. Cytogenetic analyses of secondary liver tumors reveal significant differences in genomic imbalances between primary and metastatic colon carcinomas. Clin Exp Metastasis 1999;17(6):471–9. [DOI] [PubMed] [Google Scholar]
- 36.Loo LW, Tiirikainen M, Cheng I, Lum-Jones A, Seifried A, Church JM, et al. Integrated analysis of genome-wide copy number alterations and gene expression in microsatellite stable, CpG island methylator phenotype-negative colon cancer. Genes Chromosomes Cancer 2013; 52(5): 450–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alonso MH, Aussó S, Lopez-Doriga A, Cordero D, Guinó E, Solé X, et al. Comprehensive analysis of copy number aberrations in microsatellite stable colon cancer in view of stromal component. Br J Cancer 2017. July 25;117(3):421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanskanen T, van den Berg L, Välimäki N, Aavikko M, Ness-Jensen E, Hveem K, et al. , Genome-wide association study and meta-analysis in Northern European populations replicate multiple colorectal cancer risk loci. Int J Cancer 2018; 142(3): 540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suter CM, Martin DI, Ward RL. Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int J Colorectal Dis 2004;19:95–101. [DOI] [PubMed] [Google Scholar]
- 41.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 2005;97: 1330–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.