

Abstract
Lysosomal phospholipase A2 (PLA2G15) is a ubiquitous enzyme uniquely characterized by a subcellular localization to the lysosome and late endosome. PLA2G15 has an acidic pH optimum, is calcium independent, and acts as a transacylase in the presence of N-acetyl-sphingosine as an acceptor. Recent studies aided by the delineation of the crystal structure of PLA2G15 have clarified further the catalytic mechanism, sn-1 versus sn-2 specificity, and the basis whereby cationic amphiphilic drugs inhibit its activity. PLA2G15 has recently been shown to hydrolyze short chain oxidized phospholipids which access the catalytic site directly based on their aqueous solubility. Studies on the PLA2G15 null mouse suggest a role for the enzyme in the catabolism of pulmonary surfactant. PLA2G15 may also have a role in host defense and in the processing of lipid antigens for presentation by CD1 proteins.
Keywords: antigen presentation, ceramide, lysosome, phospholipase A2, phospholipidosis, tuberculosis
1.1. Introduction
Phospholipases are hydrolases, enzymes that use a molecule of water to degrade phospholipid substrates (1). Phospholipases are named based on their ability to hydrolyze either phosphate esters or acyl esters. Thus PLA1 and PLA2 are acyl esterases and hydrolyze the sn-1 and sn-2 positions of glycerophospholipids respectively; phospholipases C and D are phosphate esterases and are defined based on hydrolysis on the glycerol or distal side of the phosphate group (figure 1).
Figure 1.

Sites of action of phospholipases on glycerophospholipids.
Among phospholipases, the family of phospholipase A2s are the most intensively studied reflecting their biological relevance. There are at least sixteen groups of phospholipase A2s. Dennis and coworkers have categorized these into six groups based on their properties: secreted phospholipase A2 (sPLA2 Groups I, II, III, V, IX, X, XI, XII, XIII, and XIV); cytosolic phospholipase A2 (Group IV cPLA2); calciumindependent phospholipase A2 (Group VI iPLA2); PAF acetylhydrolases (GVII and GVIII PAF-AH PLA2s); lysosomal phospholipase A2 (Group XV LPLA2); and adipose-specific phospholipase A2 (GXVI AdPLA) (2).
PLA2 group XV, the subject of this review, is one of the most recently identified PLA2s (3). It is unique member of the PLA2 family based on its cellular localization to the lysosome and acidic pH optimum. This enzyme like many other enzymes named based on the initial assay used in its discovery has additional activities that may be different than the name implies. This enzyme was co-discovered as a lysophospholipase (4) and 1-O-acyl-ceramide synthase (5) reflecting the initial assays used to discover their activities. In the former case, the enzyme was identified from a screen of foam cell formation in a macrophage cell line and initially assayed at neutral pH. In the latter case, the enzyme was discovered as an off target site of action for glucosylceramide synthase inhibitors and is a transacylase that uses ceramide as an acceptor. Subsequent work revealed that the enzyme, localized within cells to lysosomes and late endosomes, has an acid pH optimum and acts as a PLA2 (6).
In this regard, features considered pathognomonic of phospholipases may not strictly apply to PLA2G15. These features include the use of water as an acceptor for the scissile fatty acyl group and the strict need for the enzyme to locate its substrate within a lipid bilayer or micelle. With respect to the former feature, PLA2G15 can act as a transacylase, recognizing short chain lipophilic alcohols such as N-acetyl-sphingosine as acceptors (7). The PLA2GVI group (iPLA2ϵ, iPLA2ζ, and iPLA2η) of phospholipase A2s are also transacylases (8,9). With respect to the absolute requirement that a phospholipase act on a substrate within a lipid bilayer, short chain oxidized phospholipids have been recently reported to be substrates for the enzyme (10). These phospholipids access the catalytic site in the absence of liposomes. In this regard, the PLA2G15 is similar to platelet activating factor acetylhydrolase (PAF-AH) (11). These and several other features including structure, function, cellular regulation, and role of PLA2G15 in normal biology and disease are discussed in detail below.
2.1. Discovery and identification
An effort to target the inhibition of glucosylceramide synthase was first proposed by Normal Radin as a strategy for the treatment of Gaucher disease as well as other glycosphingolipidoses (12). This enzyme catalyzes the synthesis of glucosylceramide using UDP-glucose and ceramide as substrates. D-threo-1-phenyl-2-decanoylamino-3morpholino-propanol (D-threo-PDMP) was identified as a reversible inhibitor of this cerebroside synthase (13). This lead compound demonstrated an IC50 of 20 μM. Initial concerns about the viability of this strategy were focused on the possibility that the inhibition of this pathway would result in the accumulation of ceramide, a potential mediator of cell death (14).
Early work with D-threo-PDMP appeared to validate this concern (15). The depletion of glucosylceramide in cultured MDCK cells was associated with a parallel increase in ceramide levels. At inhibitor concentrations that did not result in overt cell death, cell cycle arrest at G2/M was observed (16). Thus while secondary increases in ceramide by D-threo-PDMP treatment might have utility as a means for blocking cell growth, the prospect of chronic treatment of Gaucher patients appeared to be poorly justified. However, the interpretation that glucosylceramide synthase inhibition resulted in ceramide accumulation secondary to substrate accumulation was not supported by further study of the structure activity relationships of analogues of D-threo-PDMP (17,18). Specifically, three observations were not consistent with the interpretation that increased ceramide would follow from specific inhibition of glucosylceramide synthase. First, PDMP the pharmacophore contained two chiral carbons. Thus four possible enantiomers of PDMP could exist including the D-threo-, D-erythro-, L-threo- and Lerythro- forms. When these were studied in cultured MDCK cells, only the D-threo enantiomer inhibited glucosylceramide synthase and lowered glucosylceramide. However, all four enantiomers were equally potent in elevating cell ceramide levels (19). Second, an alternative glucosylceramide synthase was identified containing a scaffold that differed from PDMP, 1-pyrrolidino-1-deoxyceramide. This compound inhibited glucosylceramide synthase activity without any change in cell ceramide levels even at micromolar levels (20). Finally, analogues of PDMP were identified that were greater than one thousand times more potent in inhibiting glucosylceramide synthase, including those with ethylenedioxyphenyl-substitutions. These compounds lowered cell glucosylceramide at low nanomolar concentrations but raised ceramide levels in the mid micromolar range (18).
These findings were more consistent with the existence of a second intracellular site of action for PDMP resulting in increased ceramide. A search was initiated to determine whether PDMP or its more active analogue D-threo-1-phenyl-2palmitoylamino-3-pyrrolidino-propanol (P4) activated or inhibited known enzymes associated with ceramide metabolism including ceramide synthase, acid and neutral ceramidase, sphingomyelin synthase, and sphingomyelinase. No changes in the activities of these enzymes in the presence of D-threo-PDMP was observed (17). Employing a different strategy, MDCK cells were incubated with a C2-ceramide radiolabeled in the long chain base. This membrane permeant short chain ceramide was avidly metabolized to free sphingosine, short chain and long chain glucosylceramide and sphingomyelin. Additionally, a highly lipophilic radiolabeled product was formed. This metabolite was sensitive to alkaline methanolysis yielding the C2-ceramide precursor consistent with the product representing an acylated form of ceramide (5).
To differentiate between the 3-hydroxyl and 1-hydroxyl groups on ceramide as the site of acylation, C2-ceramide was pretreated with 2,3-dichloro-5,6dicyanobenzoquinone which oxidizes hydroxyls that are vicinal to a double bond (5). Acylation of this product occurred consistent with acylation occurring at the C1 hydroxyl. This product was thus identified as 1-O-acylceramide. We next determined the source of the fatty acid of the 1-O-acylceramide. When enzyme activity was assayed in cell homogenates and free fatty acids or fatty acyl-CoAs were used as substrate, the enzyme was either inactive or inhibited. However, enhanced activity was observed in the presence of liposomes containing phosphatidylcholine or phosphatidylethanolamine consistent with the enzyme acting as a transacylase (figure 2) (21).
Figure 2.

1-O-Acylceramide synthase reaction scheme. Phosphatidylcholine (PC) is shown as the acyl donor and N-acetylsphingosine (NAS) as the acceptor molecule.
The identification of what appeared to be novel pathway for ceramide metabolism provided the incentive to pursue the purification of the 1-O-acylceramide synthase (6). The specificity of the reaction product provided an unequivocal readout of activity during the enrichment steps employed. These steps included ammonium sulfate fractionation, DEAE-Sephacel, phenyl-Sepharose, Sephadex G-75, concanavalin A-agarose, and heparin Sepharose chromatography. Starting with bovine brain a greater than 190,000fold increase in specific activity was achieved. Importantly, the most significant enrichment of the enzyme occurred by use of a concanavalin A column in which the enzyme was eluted with α-methyl mannoside, suggesting that the enzyme was mannose rich. This was the earliest clue that the 1-O-acylceramide synthase might have a subcellular lysosomal site of action, an observation later confirmed in a proteomics analysis of the lysosome (22).
SDS-polyacrylamide gel electrophoresis yielded a single band which retained 1O-acylceramide synthase activity. However, when the assay reaction was performed using L-α−1-acyl-2-[1-14C]arachidonyl-phoshatidylethanolamine as substrate in the absence of N-acetylsphingosine as the acceptor, the enzyme acted as a phospholipase A2 releasing the free radiolabeled arachidonic acid. This observation raised the possibility that the enzyme was in fact a bone fide phospholipase A2 (6).
Early characterization of the purified enzyme was performed to ascertain whether the enzyme was a novel member of the phospholipase A2 family. Gel filtration revealed that the purified enzyme was a glycoprotein consisting of a single polypeptide chain of approximately 40 kDa. The phospholipase A2 and 1-O-acylceramide synthase activities assayed with liposomes had pH optima of 4.5. These properties distinguished the enzyme from the ten small molecular weight secreted PLA2s (molecular mass 12–19 kDa) and from group PLA2GIV (molecular mass 60–114 kDa) and group PLA2GVI (molecular mass 84–90). Although the enzyme was modestly activated by calcium and magnesium, neither divalent cation was required for activity. In addition, AACOF3 and bromoenollactone, inhibitors of PLA2GIVA and PLA2VI, respectively, were inactive against 1-O-acylceramide synthase (6).
The evaluation of the effect of inhibitors and activators on the 1-O-acylceramide synthase activity identified additional features that distinguished the enzyme from known phospholipase A2s. For example, exposure of the purified enzyme to dithiothreitol was not associated with significant loss of activity in contrast to groups I, II, and III sPLA2s in which disulfide bonds are essential for activity. Neither calcium or magnesium ions were required for activity, although a modest enhancement in activity was observed in the presence of both ions (6).
2.2. Enzymology and sequence identification
The protein was subjected to tryptic digestion and the partial amino acid sequences were analyzed by mass spectrometry. A BLAST search suggested that the protein was homologous to lecithin-cholesterol acyltransferase like lysophospholipase (LLPL) (23). Taniyama and colleagues had originally identified LLPL in a screen of the THP-1 macrophage cell line following exposure to phorbol ester and β-VLDL (4). This group identified the presence of lipase activity in the gene product. When the enzyme was assayed at pH 7.5 against dioleoyl-phosphatidylcholine and lysophosphatidylcholine, they observed modest (12 nmol/min/mg protein) but higher activity against the lyso-phosphatidylcholine substrate with hydrolysis of the sn-1 fatty acyl group of the lyso-phosphatidylcholine.
A TBLAST N data base search of GenBank was used to find expressed sequence tags that aligned with the LLPL sequence. Subsequently mouse, bovine and human EST clones were used to design primers and obtain the entire coding sequence from each species. The PCR products of mouse and bovine kidney cDNAs gave rise to single bands of 1.3 kb in length. The PCR products were subcloned into pCR4-TOPO and sequenced in both directions. This sequence was identical to that of the LLPL gene previously reported. To confirm that this gene retained the observed acyltransferase and acidic phospholipase activities, the open reading frame was cloned into pcDNA3 to generate proteins that were carboxyl-terminally tagged with FLAG, HA, or c-myc proteins and transfected into COS-7 cells. Using antibodies to the carboxyl-tagged sequences, the expressed protein from the soluble fractions from each of these cells were recovered and assayed for 1-O-acylceramide synthase activity and phospholipase A2 activity. These experiments confirmed that the purified synthase and LLPL proteins were in fact identical and retained their respective activities (23).
The primary structure of the PLA2G15 was highly conserved between the three species. Six exons are present in the gene. The primary structure of the human and mouse enzyme consists of 412 amino acids (407 for the bovine enzyme). The enzymes contain consensus sequences that include a signal peptide cleavage site and a lipase motif, AXSXG that is characteristic of serine hydrolases. The serine is part of a catalytic triad that also includes aspartic acid and histidine. An amino terminal 33 amino acid signal peptide is present with a cleavage site between proline 33 and alanine 34 on the mouse and human peptide. In addition, four N-linked glycosylation sites are present in the mouse and human protein (three in the bovine protein) (24).
Remarkably, PLA2G15 shares 49 percent sequence identity with lecithincholesterol acyl transferase (LCAT). LCAT, a secreted serum protein, cleaves the sn-2 acyl ester of phosphatidylcholine and acts as an acyltransferase with cholesterol as its acceptor (25). LCAT has a similar catalytic motif (GCSXG) with a catalytic serine. Both enzymes are members of the αβ-hydrolase superfamily of proteins. The human genes for PLA2G15 and LCAT map to 16q22.1. Phylogenetic analysis demonstrates significant relatedness between PLA2G15, LCAT, and phospholipid:diacylglycerol acyltransferases (PDATs) (3). Members of this enzyme family are characterized by phospholipase A and transacylase activities, the latter in the presence of specific acceptor molecules.
Following the identification of the primary sequence and predicted structure of PLA2G15, a series of studies were pursued to validate the importance of these structural elements. Site directed substitutions of alanine for the catalytic amino acids serine 198, aspartate 351 and histidine 392 were made. In each case there was loss of 1-O-acylceramide synthase activity (24). Four cysteine residues are conserved between LCAT and PLA2G15. In LCAT these cysteines form disulfide bonds that are required for activity. Single or double substitutions at C65 and C89 resulted in complete loss of enzyme activity. However, similar substitutions at C330 and C371 resulted in only partial loss of activity. These findings were consistent with the presence of one disulfide bond between C65 and C89 in PLA2G15. This finding was confirmed when the crystal structure of PLA2G15 was solved (26). Given that dithiothreitol did not significantly inhibit PLA2G15 purified from bovine brain, it is reasonable to presume that the disulfide bond at C65 and C89 is required for proper folding of the enzyme.
The binding of crude bovine PLA2G15 to concanavalin A-agarose beads and elution with methyl-α-D-mannopyranoside was consistent with high mannose containing glycans. Treatment of the expressed mouse and human PLA2G15 from COS-7 cells with endoglycosidase F1, a glycosidase that is active against asparagine-linked oligomannose groups, resulted in a shift in molecular weight consistent with hydrolysis of the glycan groups. The deglycosylated enzyme retained activity. However, in a subsequent study the N-glycosylation sites were modified by replacement of the asparagine with alanine. A single substitution at the first asparagine (N99 with the leader sequence present) resulted in complete loss of activity as did quadruple mutations that included all four glycosylation sites. The N99 mutation resulted in the retention of the enzyme in the membrane fraction of COS-7 cells. Substitutions at N273, N289, and N398 resulted in only partial loss of activity (27). These findings are consistent with a role for glycosylation in the proper synthesis and folding of a catalytically active enzyme. Mannose 6-phosphate groups also function as sorting ligands for lysosomal proteins and can mediate the cellular uptake of lysosomal enzymes that have been secreted. Mannose receptors mediate the uptake of PLA2G15 by macrophages (28). The phospholipid accumulation in macrophages from PLA2G15 knockout mice was lowered following treatment with recombinant PLA2G15. These observations suggest that mannose terminated PLA2G15 might be an effective form of enzyme replacement therapy in the setting of PLA2G15 deficiency, a strategy that has been employed for other lysosomal hydrolases (29).
2.3. Substrate and acceptor specificity
Determining the phospholipid substrate specificity of PLA2G15 has been an ongoing effort since it was first identified. The uniqueness of the enzyme was established in large part due to its ability to transacylate N-acetyl-sphingosine in an assay in which phospholipid substrate was presented in liposomes. Liposomes typically are comprised of phospholipid/sulfatide/N-acetyl-sphingosine in a molar ratio of 10:1:3. The presence of sulfatide, a non-hydrolyzed lipid, to enhance electrostatic charge interactions between the liposome and enzyme enhances activity. Substitution of sulfatide with galactosylceramide is associated with markedly lower specific activity of PLA2G15 (30). This assay has the advantage of conferring specificity to the result since many enzymes display lipase activity but no other enzyme has been reported to transacylate ceramide at the C-1 hydroxyl position. Potential disadvantages include the ability to incorporate the substrate into the liposome in a manner that is accessible to the enzyme, physical changes in the liposomes that occur secondary to the phospholipid present but which may not be applicable to endogenously presented substrate, and the possibility that the enzyme might act on the substrate as a phospholipase but not as a transacylase. For example, cardiolipin is not easily incorporated into these liposomes and has no detectable activity as a substrate. Phosphatidylglycerol containing liposomes have markedly higher specific activities probably due to their anionic properties. Many truncated oxidized phospholipids are not retained in liposomes but are readily hydrolyzed as water soluble substrates (discussed in greater detail below).
Based on the use of liposomes as a measure of substrate specificity, phosphatidylcholine, phosphatidylethanolamine, phosphatidylglycerol, and phosphatidylserine are recognized substrates. Phosphatidylinositol and sphingomyelin are not recognized as substrates. The absence of activity toward sphingomyelin suggests that the enzyme does not recognize fatty acids in amide linkage. Neither ether linked nor vinyl ether linked fatty acids are recognized as scissile groups as well.
These observations led to a more comprehensive study of the positional specificity of the enzyme. Using the novel property of PLA2G15 to form 1-Oacylceramides, alveolar macrophages from wild type and Pla2g15 knockout mice were used as a source of enzyme. Using 1-palmitoyl-2-[14C]oleoyl-sn-glycero-3phosphocholine as substrate, both 2-[14C]oleoyl-lyso-phosphatidylcholine and [14C]oleic acid were formed. This suggested that the enzyme could deacylate groups at both the sn-1 and sn-2 positions (31). Additional experiments in PLA2G15 transfected MDCK cells comparing phosphatidylcholines and phosphatidylethanolamines with saturated and unsaturated acyl groups at either the sn-1 or sn-2 positions demonstrated that unsaturated fatty acyl groups were preferred as acyl donors regardless of their position. A notable exception was for arachidonoyl substitutions in which the sn-2 position was highly preferred to the sn-1 positon. These studies indicated that the enzyme had much broader positional specificity for sn-1 and sn-2 acyl groups than initially believed (32).
Although PLA2G15 is unique in transacylating ceramide at the C-1 hydroxyl group, PNPLA1, a member of the iPLA2 family, has recently been reported to catalyze the transacylation of linoleate to omega-hydroxy ceramide giving rise to omega-Oacylceramide (33,34).
The unique and specific formation of 1-O-acyl-ceramides has also made possible a fuller exploration of the acceptor specificity of the phospholipase reaction. A series of lipophilic alcohols were evaluated as acyl acceptors (7). These included 1-O-hexadecyl-1-acetyl-sn-glycerol, 1-O-hexadecyl-glycerol, 1-O-palmityl-2-O-methyl-sn-glycerol, various monoacylglycerols, and natural lipophilic alcohols including anandamide and oleoylethanolamide. We generally observed that PLA2G15 preferred acceptor molecules that were primary alcohols with one long carbon chain and one small nonpolar residue linked to the C2 position of ethanol. These findings raise the possibility that the enzyme could function to remodel acyl groups and also regulate the biological activities of biologically active lipophilic alcohols.
An alternative approach for determining the substrate specificity was the measurement of changes in phospholipids in tissues in which the enzyme has been deleted. PLA2G15 deficient mice were created using Cre/loxP and Flp/FRT recombination systems (35). Exon 5 of the PLA2G15 gene, which encodes the lipase motif essential for lysosomal phospholipase activity, was systemically deleted. The resultant PLA2G15−/− mice showed no phospholipase A2 or 1-O-acylceramide synthase activity, developed normally, and were characterized by a marked accumulation of phospholipid in their alveolar macrophages, peritoneal macrophages, and spleens at an early age. Increased lung surfactant phospholipid and splenomegaly were observed in mice by 1 year of age. The pulmonary phenotype, specifically the transformation of alveolar macrophages into foam cells, was predicted based on the observation that the LPLA2 activity was greater than 50 fold higher in these cells than in any other tissue or cell type, including monocytes. Bronchoalveolar lavage of one year-old mice demonstrated increased surfactant phospholipid levels, consistent with a specific role for PLA2G15 in alveolar macrophages (36).
However, the phospholipid changes were specific and reflected known substrate specificities of the enzyme. Thus phosphatidylcholine, phosphatidylethanolamine, and phosphatidylglycerol were significantly increased in the knockout macrophages. Sphingomyelin, ceramide, and phosphatidylinositol levels were not, largely consistent with in vitro measurements of enzyme activity. Lyso-phosphatidylcholine, measured at less than one percent the levels of phosphatidylcholine, was also increased consistent with a potential role for PLA2G15 as a lyso-phospholipase (table 1). Specific species of individual phospholipids containing fatty acyl groups of varying chain length and saturation accumulated to different degrees, suggesting that substrate selectivity was not only based on the phospholipid head group but also on the fatty acyl groups present. The observed conversion of alveolar macrophages to foam cells coupled with observed lysosomal phospholipid accumulation led us to consider whether inherited or acquired forms of lysosomal phospholipidosis might track to deficiencies or inhibition of PLA2G15. The knockout mouse lungs demonstrated histological changes that were reminiscent of amiodarone induced drug toxicity. Amiodarone is a potent anti-arrhythmic the use of which is often limited by its long term toxicity (37,38). Structurally, amiodarone is a cationic amphiphile, akin to other small molecules that cause phospholipidosis. Long term exposure of MDCK cells was associated with lysosomal phospholipid accumulation and inhibition of PLA2G15 activity, raising the possibility that the lysosomal phospholipase was the target of amiodarone induced phospholipidosis (39).
Table 1.
Phospholipid content in alveolar lavage fluid from wild type and PLA2G15 knockout mice.
| Lipid | Lipid level (nmol/mg protein) by phenotype |
|
|---|---|---|
| PLA2G15+/+ | PLA2G15−/− | |
| Phosphatidylcholine | 283 | 538 |
| 16:0 Phosphatidylcholine | 184 | 374 |
| Lysophosphatidylcholine | 1.2 | 2.3 |
| Phosphatidylethanolamine | 250 | 524 |
| Plasmalogen phosphatidylethanolamines | 173 | 414 |
| Sphingomyelin | 87.4 | 76.0 |
| Phosphatidic acid | 0.74 | 0.26 |
| Phosphatidylglycerol | 79.8 | 129.0 |
| Ceramide | 1.87 | 2.24 |
| Free fatty acid | 124 | 155 |
Lipid levels are derived from pooled samples of six 12 month-old mice (three male and three female) from each group. 16:0 designates the presence of palmitoyl containing phosphatidylcholine (35).
This hypothesis was explored in greater depth by first probing the role of zwitterionic phospholipid liposomes on enzyme activity measured by the transacylation of C2-ceramide (30). As mentioned previously, incorporation of sulfatide but not galactosylceramide into substrate containing liposomes significantly increased the enzyme activity. Anionic phospholipids including cardiolipin, phosphatidylglycerol, phosphatidic acid, phosphatidylinositol, phosphatidylserine, and phosphatidylethanol all increased activity and in the case of phosphatidylglycerol, phosphatidic acid, and phosphatidylethanol were themselves substrates for the transacylation reaction. We next determined that in addition to pH, increasing the ionic strength of the reaction buffer with sodium chloride significantly inhibited the enzyme activity. However, when pnitrophenylbutyrate, a water soluble substrate was used, transacylation of C2-ceramide occurred, but there was no longer an effect of pH. In addition, the inhibitory effect of amiodarone was lost. These findings suggested that the electrostatic charge interaction between the liposomes and PLA2G15 was an important determinant of enzyme activity when the substrate was a phospholipid but not when the substrate could readily access the catalytic site (39).
This model was further supported by studies in which the co-sedimentation of enzyme with liposomes was measured. Inclusion of sulfatide but not galactosylceramide in the liposomes increased the co-sedimentation and recovery of enzyme/liposome complexes. Co-sedimentation was impaired by raising the pH from 4.5 to 7.4, increasing the sodium chloride content of the incubation buffer, or incorporating amiodarone into the liposomes.
Two classes of PLA2G15 inhibitors have been identified. These include cationic amphiphilic drugs exemplified by amiodarone and fluorophosphonates that bind irreversibly to the catalytic serine of the enzyme (Table 2).
2.4. Oxidized phospholipids as substrates for PLA2G15
Up to this point, studies on the substrate specificity of PLA2G15 were focused on long-chain phospholipids. In this model, a critical first step is the binding of the enzyme to the phospholipid containing membrane based on the electrostatic interactions. PLA2G15 has an estimated isoelectric point of 5.9, sufficient for binding to membranes containing anionic phospholipids. We considered whether there were endogenous substrates that could access the catalytic domain of the enzyme but did not require presentation within a membrane, specifically whether oxidized phospholipids might be such substrates. Such lipids are often truncated and are characterized by the presence of exposed carboxyl or formyl groups at the sn-2 position. Furthermore, they are water soluble and can spontaneously distribute from a lipid bilayer into the aqueous phase (40,41).
A series of oxidized phosphatidylcholines were assayed in liposomes containing the non-hydrolyzable 1,2-O-octadecenyl-sn-glycero-3-phosphocholine, including 1palmitoyl-2-(5′-oxo-valeroyl)-sn-glycero-3-phosphocholine (POVPC), 1-palmitoyl-2glutaryl-sn-glycero-3-phosphocholine (PGPC), 1-palmitoyl-2-azelaoyl-sn-glycero-3phosphocholine (PAzPC), and 1-palmitoyl-2-(9′-oxo-nonanoyl)-sn-glycero-3phosphocholine (PONPC). Remarkably, the preferential deacylation of the oxidized substrate at the sn-1 position was observed (10). The hydrolysis of the sn-1 fatty acyl group was preferred to the formation of the corresponding 1-O-acylceramide. This reaction was observed at neutral pH, albeit at a lower activity than at pH 4.5. Finally, when the enzyme activity was measured in the presence of oxidized and non-oxidized substrates together, the catabolism of the oxidized phospholipid substrate was greatly preferred to that of the non-oxidized phosphatidylcholine. Exposure to PAzPC resulted in growth inhibition of CHO cells. When transfected with PLA2G15, the CHO cells were protected from growth inhibition.
These findings raise the possibility that PLA2G15 may catabolize oxidized phospholipids that are formed intracellularly but also traffic to the lysosome. In addition, secreted PLA2G15 that circulates in the plasma may potentially degrade such lipids as well (42). The selective hydrolysis of the sn-1 fatty acyl group on the oxidized phospholipids also raises the possibility that PLA2G15 is complementary to PAF-AH in the catabolism of oxidized phospholipids since the latter enzyme acts specifically at the sn-2 fatty acyl group. The basis on which PLA2G15 is able to recognize the sn-1 fatty acyl group of both oxidized and non-oxidized phospholipids was clarified when the structure of the enzyme was determined.
2.5. Structural characterization of PLA2G15
In an effort to confirm the prior studies on the structure and function of PLA2G15 as well as to better understand the mechanism of catalysis, the crystal structure of the human enzyme was determined. The human protein was expressed in HEK293S GnTI- cells in which N-acetyl-glucosaminyltransferase I was absent and therefore lack the ability to make complex glycans. The secreted protein was subsequently treated with endoglycosidase F1 yielding crystals with single wavelength diffraction of 1.83 Å. Selenomethionine incorporation into the recombinant protein was used for structure determination.
The protein structure was more similar to Family I bacterial triacylglycerol lipases than to other phospholipase A2 family members. The presence of a six-stranded α/β hydrolase fold domain and the catalytic triad were confirmed. More specifically, Ser165 was located in a nucleophile elbow between β5 and αC; Asp327 in a loop before αE; and His359 in a loop following β8. Also confirmed was the presence of a single disulfide bond found in the loop between β3 and αA’ and asparagine linked sugars at residues 66, 240, 256, and 365. By analogy to the bacterial lipases, three distinct domains were identified, a cap domain, membrane binding domain, and α/β hydrolase domain (figure 3).
Figure 3.

Left panel. The structure of human PLA2G15 resolved to 1.83 Å resolution using single wavelength anomalous diffraction and seleno-methionine labeling. PLA2G15 has a core α/β hydrolase domain featuring a catalytic triad consisting of Ser165, His359 and Asp327 (inset). Right lower panel. Inserted into loops of the α/β domain are membrane binding and cap domains of novel structure responsible for membrane targeting and framing the active site. (Reprinted by permission from Nature communications 6: 6250 (26)).
Attempts to co-crystallize PLA2G15 and the catalytically inactive S165A form with phospholipids were unsuccessful. Fluorophosphonate inhibitors that bind covalently to the catalytic serine were used instead. The structures of apo-PLA2G15 were compared to those of PLA2G15 bound to methyl arachidonyl fluorophosphonate (MAFP) and isopropyldodec-11-enyl fluorophosphonate (IDFP). Omit map density demonstrated high density for the head groups bound to Ser165 and occupying the oxyanion hole formed by the backbone amides of Asp13 and Met166. The hydrophobic tails of the inhibitors displayed heterogeneous densities after the C3 consistent with two hydrophobic tracks, termed tracks A and B. These tracks were proposed to be binding sites for the sn-1 and sn-2 fatty acyl groups of the phospholipid substrates (figure 4).
Figure 4.

Left upper panel. Surface hydrophobic residues Y30, L31, L50 and V52 constitute part of the PLA2G15 membrane binding surface. Left lower panel. Surface electrostatic potential of PLA2G15, calculated at pH 5. Mutation of hydrophobic residues to hydrophilic serines reduces PLA2G15 activity on liposomes (right upper panel) and liposome binding (right lower panel). PLA2G15 ability to form an acyl-intermediate is crucial for stable membrane binding. Liposome co-sedimentation is completely abolished in S165A LPLA2 mutant, while co-sedimentation of PLA2G15 coupled to IDFP or MAFP correlates with the length of their aliphatic chains. The error bars represent the standard deviation of three independent experiments. (** 0.001<p<0.01, ***p<0.001. na, not assayed due to poor protein expression; Student’s t-test)
The role of membrane binding of PLA2G15 to membranes containing substrate was studied by the mutation of residues present on the hydrophobic patch of the membrane binding domain. Specifically, Tyr30, Leu31, Leu50, and Val52 were R260/263A, L336A, and K383A were used as controls. Hydrolysis of the water soluble substrate p-nitro-phenylbutyrate was unaffected by any of the mutations consistent with a properly folded enzyme and intact catalytic site. However, the Y30S, L31S, L50S, and V52S substitutions all led to a loss of transacylase activity in concert with impaired cosedimentation of enzyme and liposomes.
An unexpected finding from these studies was the loss of co-sedimentation between liposomes and enzyme following a S165A substitution (figure 4). While no hydrolytic activity to p-nitro-phenylbutyrate and transacylation activity were expected, the dependence of the catalytic serine for liposome binding was surprising. Subsequently, co-sedimentation was measured in wild type enzyme exposed to IDFP and MAFP. IDFP partially and MAFP fully restored the binding interaction. The restoration of binding was correlated with the length of the alkyl arm of the respective phosphonate inhibitors. These experiments led to a revised model of the association between PLA2G15 and membrane (figure 5). In this model transient membrane binding is driven by complementary electrostatic charges and the hydrophobic patch on the membrane binding domain. This is followed by the formation of a covalent acyl intermediate with the enzyme that tethers the enzyme at the membrane surface (26).
Figure 5.

Proposed two-step mechanism for PLA2G15 membrane binding: 1. transient interaction via surface hydrophobic residues. 2. tight tethering via acyl-intermediate for more efficient turnover. (Reprinted by permission from Nature communications 6: 6250 (26)).
More recently, a comprehensive study was conducted to ascertain the role of the atypical acidic residue, Asp13, that is present in the hydrophobic active site cleft of tract A (43). The potential role of Asp13 in determining the pH profile and substrate preference for unsaturated fatty acyl groups was studied. Substitutions at Asp13 led to a significant increase in enzyme activity at pH 7.4 and perturbed the selectivity of the enzyme for unsaturated acyl chains. No changes in selectivity for the phospholipid substrate head group or acceptor was seen as a result of these substitutions. Modeling from these studies was consistent with the scissile fatty acyl group always residing in track A. these findings are thus consistent with PLA2G15 having both PLA1 and PLA2 activities.
2.6. Ascertaining a biological role for PLA2G15
While much as been learned about PLA2G15 following its discovery and early characterization, there is to date no fully defined role for PLA2G15 in any human disease. Hopefully establishing such a role will only be a matter of time. PLA2G15 is first and foremost a lysosomal enzyme. Greater than 80 genes encoding lysosomal proteins have been identified and variants in more than two thirds of these genes result in monogenetic disorders of lysosomal dysfunction (44). Approximately 0.5 percent of the general population carries mutations in the PLA2G15 gene that truncate encoded protein prior to the translation of exon 5 which encodes the catalytic domain. Thus if loss of PLA2G15 function is compatible with survival, then there ought to be some albeit rare individuals who are homozygous null for the phospholipase. Alternatively, haploinsufficiency could be associated with increased disease risk.
Based on the high expression of PLA2G15 in terminally differentiated macrophages and the early formation of a foam cell phenotype in the Pla2g15 null mice, disorders of pulmonary surfactant metabolism may be one expression of deficiency. A loss of PLA2G15 function might result in the upregulation of alternative pathways for surfactant clearance. Whether such a PLA2G15 deficiency would be clinically manifest as neonatal lung disease or a later onset phenotype akin to pulmonary alveolar proteinosis would only be speculative at this point (28).
An active focus of work on the role of PLA2G15 in disease has been in the areas of inflammation and host defense. Phospholipase A2s in general play multiple roles in host response to pulmonary infection and inflammation. They have been implicated in the production of eicosanoids, cytokines, and in direct bacteriocidal activities. Mycobacterium tuberculosis enters the lung as an aerosol where the bacteria is taken up by resident macrophages. The infected macrophages in concert with dendritic cells initiate a local inflammatory response. These dendritic cells subsequently transport the Mycobacterium tuberculosis to draining lymph nodes leading to the priming of CD4+ and CD8+ T cells. Schaible and colleagues studied this response in wild type and Pla2g15 knockout mice (45). The infected knockout mice had lower survival compared to wild type mice in association with higher numbers of colony forming units in the lung but less T-cell recruitment and activation. T-cell priming was abolished in the mediastinal lymph nodes of the knockout mice. The Pla2g15 deficient mice also failed to secrete interferon-gamma. They concluded that PLA2G15 is required for the induction of adaptive T-cell immunity to Mycobacterium tuberculosis.
The specific mechanism by which for PLA2G15 mediates the adaptive T-cell response is not known. One possibility is that apoptotic bodies from alveolar macrophages containing tuberculosis antigens are endocytosed by the dendritic cells and trafficked to the lysosomes where PLA2G15 is required for the degradation of the apoptotic body membrane (46).
An alternative possibility is that the phospholipase A2 actually degrades the bacterial membrane. The cell wall of Mycobacterium tuberculosis contain phosphatidylmyo-inositol mannosides (PIMs) which are important in homeostasis and pathogenesis. These structures are composed of a phosphatidylinositol unit, one to six a-Dmannopyranosyl residues and up to four acyl chains. An additional mannose-phosphate residue glycosylates either the O-2 or O-6 position on the inositol. These lipids are presented as lipid antigens to T-cells on CD1 molecules after partial deglycosylation by α-mannosidase. The human genome has five distinct CD1 genes (CD1a through CD1e) with CD1b having the most complex lipid binding domain. CD1b is thought to present PIM6 to T cells. Gilleron and colleagues studied the role of PLA2G15 and a second phospholipase, pancreatic lipase related protein 2 (PLRP2) in Mycobacterium tuberculosis PIM antigen processing. They observed that PLRP2 and PLA2G15, which deacylate at the sn-1 and sn-2 postions, respectively are required for PIM presentation to T cells (47).
The potential role for PLA2G15 in the presentation of self-lipid antigens by CD1d to invariant natural killer T (iNKT) cells has also been studied. Lysophospholipids are thought to be a class of such self-lipid antigens. PLA2G15 null mice displayed decreased numbers of iNKT cells, but this was neither the result of decreased CD1d expression nor a defect in lymphocyte development. Rather endogenous lipid antigen presentation by CD1d was reduced in the absence of the phospholipase. Thus PLA2G15 may play a role in the generation of CD1d/lipid complexes required for either thymic selection or maturation of iNKT cells.
Autoimmune uveitis in the mouse has been used to probe the role of PLA2G15 during an acute inflammatory response. Hiraoka, Abe, and co-workers induced autoimmune uveitis in the Lewis rat by injection of lipopolysaccharide. They noted that the PLA2G15 activity was significantly elevated in the aqueous humor and confirmed by western blotting. They subsequently measured enzyme activity in aqueous humor samples from patients with active uveitis and noted that it was higher compared to samples from patients with other ocular diseases. This finding was subsequently confirmed in a mouse model in which intraocular pressures were measured as well. PLA2G15 knockout mice displayed higher intraocular pressures following inflammation suggesting that the lipase might function to prevent a glaucoma like phenotype in the presence of ocular inflammation (48).
Finally, because the PLA2G15 gene was originally identified through a foam cell model of macrophages, Taniyama and colleagues probed a possible relationship between atherosclerosis and PLA2G15 function (49). They employed the apoE null mouse as a model of atherosclerosis, first demonstrating that PLA2G15 protein was present in atherosclerotic lesions of the apoE null mice. Next they crossed the apoE −/− mice with Pla2g15 −/− mice and measured atherosclerotic lesion areas over the aortic tree. Although lesion area was no greater in Pla2g15 null mice compared to controls, apoE null mice bred on the Pla2g15 null background had significantly greater lesion areas than did those bred on a wild type background. Finally, peritoneal macrophages from the Pla2g15 null mice were highly susceptible to apoptosis following exposure to oxidized LDL as measured by phosphatidylserine externalization compared to wild type macrophages exposed to oxidized LDL. They concluded that PLA2G15 has a protective effect in preventing atherosclerosis in this mouse model.
3.1. Summary and future directions
PLA2G15 is to date the first and only identified lysosomal phospholipase A. Although originally characterized as a member of the PLA2 family, recent work supported by its unique property as a transacylase has clearly demonstrated PLA1 as well as PLA2 activity. This is supported by the recent delineation of its structure and modeling that identifies track A of the catalytic domain as the site in which either an sn-1 or sn-2 fatty acyl group can be hydrolyzed. Membrane associated phospholipid substrates are hydrolyzed at acidic pH consistent with its function as a lysosomal enzyme. However, other natural substrates, notably oxidized phospholipids can access the catalytic site in an aqueous phase and are readily metabolized at neutral pH. This latter property raises the possibility that secreted enzyme circulating in the plasma or alveolar space may have an independent role in phospholipid metabolism.
Establishing a clear functional role for PLA2G15 in normal biology and disease remains a work in progress. A particular role for PLA2G15 in the pathobiology of a disease may result from studies that logically extend observations on its catalytic mechanisms, substrate specificity, subcellular, or organ specific distribution. Such work may logically extend from initial studies on immunity, surfactant metabolism, or the clearance of oxidized phospholipids. Alternatively, facile approaches to gene sequencing and RNAseq may result in the identification of individual patients with clinically important phenotypes that are associated with functioning significant variants in the PLA2G15 gene. At this point in time PLA2G15 may cease to be a protein in search of a disease.
Table 2.
Inhibitors of PLA2G15.
| Class | Examples | Structure | Mechanism |
|---|---|---|---|
| Suicide inhibitors | methoxy arachidonyl fluorophosphonate (MAFP) | 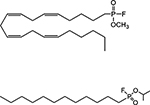 |
covalent binding to catalytic serine |
| isopropyl dodecylfluorophosphonate (IDFP) | |||
| cationic amphiphiles | amiodarone | 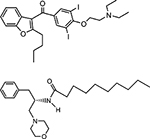 |
inhibition of electrostatic charge interaction between cationic residues and anionic phospholipids |
| D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) | |||
Highlights.
Lysosomal phospholipase A2 (PLA2G15) is a transacylase recognizing short chain ceramides as fatty acyl acceptors.
PLA2G15 is 50 percent identical to lecithin cholesterol acyltransferase; these enzymes bear close structural homology.
PLA2G15 demonstrates broad substrate recognition toward multiple glycerophospholipids including oxidized phospholipids.
PLA2G15 catabolizes pulmonary surfactant and processes lipid antigens for presentation by CD1 proteins.
Acknowledgements
This work was supported by grants from the National Institutes of Health (1RO1HL22416) and Department of Veterans Affairs (1I01BX002021).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burke JE, and Dennis EA (2009) Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res 50 Suppl, S237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dennis EA, Cao J, Hsu YH, Magrioti V, and Kokotos G (2011) Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev 111, 6130–6185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shayman JA, Kelly R, Kollmeyer J, He Y, and Abe A (2011) Group XV phospholipase A(2), a lysosomal phospholipase A(2). Prog Lipid Res 50, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taniyama Y, Shibata S, Kita S, Horikoshi K, Fuse H, Shirafuji H, Sumino Y, and Fujino M (1999) Cloning and expression of a novel lysophospholipase which structurally resembles lecithin cholesterol acyltransferase. Biochem Biophys Res Commun 257, 50–56 [DOI] [PubMed] [Google Scholar]
- 5.Abe A, Shayman JA, and Radin NS (1996) A novel enzyme that catalyzes the esterification of N-acetylsphingosine. Metabolism of C2-ceramides. J Biol Chem 271, 14383–14389 [DOI] [PubMed] [Google Scholar]
- 6.Abe A, and Shayman JA (1998) Purification and characterization of 1-Oacylceramide synthase, a novel phospholipase A2 with transacylase activity. J Biol Chem 273, 8467–8474 [DOI] [PubMed] [Google Scholar]
- 7.Abe A, Hiraoka M, and Shayman JA (2007) The acylation of lipophilic alcohols by lysosomal phospholipase A2. J Lipid Res 48, 2255–2263 [DOI] [PubMed] [Google Scholar]
- 8.Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, and Gross RW (2007) Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J Biol Chem 282, 34611–34622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramanadham S, Ali T, Ashley JW, Bone RN, Hancock WD, and Lei X (2015) Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J Lipid Res 56, 1643–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abe A, Hiraoka M, Ohguro H, Tesmer JJ, and Shayman JA (2017) Preferential hydrolysis of truncated oxidized glycerophospholipids by lysosomal phospholipase A2. J Lipid Res 58, 339–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenson RS, and Stafforini DM (2012) Modulation of oxidative stress, inflammation, and atherosclerosis by lipoprotein-associated phospholipase A2. J Lipid Res 53, 1767–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radin NS (1996) Treatment of Gaucher disease with an enzyme inhibitor. Glycoconj J 13, 153–157 [DOI] [PubMed] [Google Scholar]
- 13.Inokuchi J, and Radin NS (1987) Preparation of the active isomer of 1phenyl-2-decanoylamino-3-morpholino-1-propanol, inhibitor of murine glucocerebroside synthetase. J Lipid Res 28, 565–571 [PubMed] [Google Scholar]
- 14.Young MM, Kester M, and Wang HG (2013) Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res 54, 5–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shayman JA, Deshmukh GD, Mahdiyoun S, Thomas TP, Wu D, Barcelon FS, and Radin NS (1991) Modulation of renal epithelial cell growth by glucosylceramide. Association with protein kinase C, sphingosine, and diacylglycerol. J Biol Chem 266, 22968–22974 [PubMed] [Google Scholar]
- 16.Rani CS, Abe A, Chang Y, Rosenzweig N, Saltiel AR, Radin NS, and Shayman JA (1995) Cell cycle arrest induced by an inhibitor of glucosylceramide synthase. Correlation with cyclin-dependent kinases. J Biol Chem 270, 2859–2867 [DOI] [PubMed] [Google Scholar]
- 17.Shayman JA, Lee L, Abe A, and Shu L (2000) Inhibitors of glucosylceramide synthase. Methods Enzymol 311, 373–387 [DOI] [PubMed] [Google Scholar]
- 18.Lee L, Abe A, and Shayman JA (1999) Improved inhibitors of glucosylceramide synthase. J Biol Chem 274, 14662–14669 [DOI] [PubMed] [Google Scholar]
- 19.Abe A, Wild SR, Lee WL, and Shayman JA (2001) Agents for the treatment of glycosphingolipid storage disorders. Curr Drug Metab 2, 331–338 [DOI] [PubMed] [Google Scholar]
- 20.Carson KG, Ganem B, Radin NS, Abe A, and Shayman JA (1994) Studies on Morpholinosphingolipids - Potent Inhibitors of Glucosylceramide Synthase. Tetrahedron Lett 35, 2659–2662 [Google Scholar]
- 21.Abe A, Shayman JA, and Radin NS (1996) A novel enzyme that catalyzes the esterification of N-acetylsphingosine - Metabolism of C-2-ceramides. Journal of Biological Chemistry 271, 14383–14389 [DOI] [PubMed] [Google Scholar]
- 22.Lubke T, Lobel P, and Sleat DE (2009) Proteomics of the lysosome. Biochim Biophys Acta 1793, 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiraoka M, Abe A, and Shayman JA (2002) Cloning and characterization of a lysosomal phospholipase A2, 1-O-acylceramide synthase. J Biol Chem 277, 10090–10099 [DOI] [PubMed] [Google Scholar]
- 24.Hiraoka M, Abe A, and Shayman JA (2005) Structure and function of lysosomal phospholipase A2: identification of the catalytic triad and the role of cysteine residues. J Lipid Res 46, 2441–2447 [DOI] [PubMed] [Google Scholar]
- 25.Piper DE, Romanow WG, Gunawardane RN, Fordstrom P, Masterman S, Pan O, Thibault ST, Zhang R, Meininger D, Schwarz M, Wang Z, King C, Zhou M, and Walker NP (2015) The high-resolution crystal structure of human LCAT. J Lipid Res 56, 1711–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glukhova A, Hinkovska-Galcheva V, Kelly R, Abe A, Shayman JA, and Tesmer JJ (2015) Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase. Nature communications 6, 6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiraoka M, Okamoto K, Ohguro H, and Abe A (2013) Role of Nglycosylation of human lysosomal phospholipase A2 for the formation of catalytically active enzyme. J Lipid Res 54, 3098–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abe A, Hiraoka M, Wild S, Wilcoxen SE, Paine R 3rd, and Shayman JA (2004) Lysosomal phospholipase A2 is selectively expressed in alveolar macrophages. J Biol Chem 279, 42605–42611 [DOI] [PubMed] [Google Scholar]
- 29.Ferreira CR, and Gahl WA (2017) Lysosomal storage diseases. Transl Sci Rare Dis 2, 1–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abe A, and Shayman JA (2009) The role of negatively charged lipids in lysosomal phospholipase A2 function. J Lipid Res 50, 2027–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abe A, Hiraoka M, and Shayman JA (2006) Positional specificity of lysosomal phospholipase A2. J Lipid Res 47, 2268–2279 [DOI] [PubMed] [Google Scholar]
- 32.Abe A, Hiraoka M, and Shayman JA (2006) Positional specificity of lysosomal phospholipase A(2). Journal of Lipid Research 47, 2268–2279 [DOI] [PubMed] [Google Scholar]
- 33.Ohno Y, Kamiyama N, Nakamichi S, and Kihara A (2017) PNPLA1 is a transacylase essential for the generation of the skin barrier lipid omega-Oacylceramide. Nature communications 8, 14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirabayashi T, Anjo T, Kaneko A, Senoo Y, Shibata A, Takama H, Yokoyama K, Nishito Y, Ono T, Taya C, Muramatsu K, Fukami K, Munoz-Garcia A, Brash AR, Ikeda K, Arita M, Akiyama M, and Murakami M (2017) PNPLA1 has a crucial role in skin barrier function by directing acylceramide biosynthesis. Nature communications 8, 14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiraoka M, Abe A, Lu Y, Yang K, Han X, Gross RW, and Shayman JA (2006) Lysosomal phospholipase A2 and phospholipidosis. Mol Cell Biol 26, 6139–6148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abe A, Kelly R, Kollmeyer J, Hiraoka M, Lu Y, and Shayman JA (2008) The secretion and uptake of lysosomal phospholipase A2 by alveolar macrophages. J Immunol 181, 7873–7881 [DOI] [PubMed] [Google Scholar]
- 37.Reasor MJ, Ogle CL, Walker ER, and Kacew S (1988) Amiodaroneinduced phospholipidosis in rat alveolar macrophages. Am Rev Respir Dis 137, 510–518 [DOI] [PubMed] [Google Scholar]
- 38.Martin WJ 2nd, and Standing JE (1988) Amiodarone pulmonary toxicity: biochemical evidence for a cellular phospholipidosis in the bronchoalveolar lavage of human subjects. The Journal of pharmacology and experimental therapeutics 244, 774–779 [PubMed] [Google Scholar]
- 39.Abe A, Hiraoka M, and Shayman JA (2007) A role for lysosomal phospholipase A2 in drug induced phospholipidosis. Drug Metab Lett 1, 49–53 [DOI] [PubMed] [Google Scholar]
- 40.Catala A (2012) Lipid peroxidation modifies the picture of membranes from the “Fluid Mosaic Model” to the “Lipid Whisker Model”. Biochimie 94, 101–109 [DOI] [PubMed] [Google Scholar]
- 41.Greenberg ME, Li XM, Gugiu BG, Gu X, Qin J, Salomon RG, and Hazen SL (2008) The lipid whisker model of the structure of oxidized cell membranes. J Biol Chem 283, 2385–2396 [DOI] [PubMed] [Google Scholar]
- 42.Abe A, Kelly R, and Shayman JA (2010) The measurement of lysosomal phospholipase A2 activity in plasma. J Lipid Res 51, 2464–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinkovska-Galcheva V, Kelly RJ, Manthei KA, Bouley R, Yuan W, Schwendeman A, Tesmer JJG, and Shayman JA (2018) Determinants of pH profile and acyl chain selectivity in lysosomal phospholipase A2. J Lipid Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Futerman AH, and van Meer G (2004) The cell biology of lysosomal storage disorders. Nature reviews. Molecular cell biology 5, 554–565 [DOI] [PubMed] [Google Scholar]
- 45.Schneider BE, Behrends J, Hagens K, Harmel N, Shayman JA, and Schaible UE (2014) Lysosomal phospholipase A : A novel player in host immunity to Mycobacterium tuberculosis. European journal of immunology [DOI] [PubMed] [Google Scholar]
- 46.Kolter T, Winau F, Schaible UE, Leippe M, and Sandhoff K (2005) Lipidbinding proteins in membrane digestion, antigen presentation, and antimicrobial defense. J Biol Chem 280, 41125–41128 [DOI] [PubMed] [Google Scholar]
- 47.Gilleron M, Lepore M, Layre E, Cala-De Paepe D, Mebarek N, Shayman JA, Canaan S, Mori L, Carriere F, Puzo G, and De Libero G (2016) Lysosomal Lipases PLRP2 and LPLA2 Process Mycobacterial Multi-acylated Lipids and Generate T Cell Stimulatory Antigens. Cell Chem Biol 23, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 48.Sawada K, Hiraoka M, Abe A, Kelly R, Shayman JA, and Ohguro H (2017) Prolonged Ocular Inflammation in Endotoxin-Induced Uveitis in Lysosomal Phospholipase A2-Deficient Mice. Current eye research 42, 611–616 [DOI] [PubMed] [Google Scholar]
- 49.Taniyama Y, Fuse H, Satomi T, Tozawa R, Yasuhara Y, Shimakawa K, Shibata S, Hattori M, Nakata M, and Taketomi S (2005) Loss of lysophospholipase 3 increases atherosclerosis in apolipoprotein E-deficient mice. Biochem Biophys Res Commun 330, 104–110 [DOI] [PubMed] [Google Scholar]