

Abstract
Acyclic nucleoside phosphonates represent a well-defined class of clinically used nucleoside analogs. All acyclic nucleoside phosphonates need intracellular phosphorylation before they can bind viral DNA polymerases. Recently, a novel class of alpha-carboxynucleoside phosphonates have been designed to mimic the natural 2′-deoxynucleotide 5′-triphosphate substrates of DNA polymerases. They contain a carboxyl group in the phosphonate moiety linked to the nucleobase through a cyclic or acyclic bridge. Alpha-carboxynucleoside phosphonates act as viral DNA polymerase inhibitors without any prior requirement of metabolic conversion. Selective inhibitory activity against retroviral reverse transcriptase and herpesvirus DNA polymerases have been demonstrated. These compounds have a unique mechanism of inhibition of viral DNA polymerases, and provide possibilities for further modifications to optimize and fine tune their antiviral DNA polymerase spectrum.
Keywords: : α-carboxynucleoside phosphonate (α-CNP), acyclic nucleoside phosphonate (ANP), chemotherapeutics, herpesviruses, HIV, nucleoside-analog inhibitor, reverse transcriptase (RT), viral DNA polymerase
Graphical abstract
Acyclic nucleoside phosphonates (ANPs) represent a well-defined structural class of compounds that were originally reported in the 1980s [1]. Several members of this class of compounds were shown to have antiviral or anticancer activity. Distinct subclasses of (acyclic) nucleoside analogs have been identified depending on the molecular nature of the acyclic entity linked to the nucleobase. Each of them possesses well-defined antiviral efficacies. The first compound reported to be inhibitory to a broad range of DNA viruses, predominantly including herpesviruses, poxviruses and adenoviruses, was (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine (designated as (S)-HPMPA and belonging to the subclass of 3-hydroxy-2-phosphonylmethoxypropyl [HPMP] derivatives) (1) (Figure 1). Its cytosine counterpart (S)-HPMPC [2] was the first ANP licensed for clinical use (designated as cidofovir or Vistide®). It has a similar broad anti-DNA virus spectrum as (S)-HPMPA but it was particularly active against human cytomegalovirus (HCMV) [2,3]. It is, therefore, clinically used for the treatment of HCMV retinitis in AIDS patients, although this drug is currently also used off-label for the treatment of human papilloma virus, poxvirus and adenovirus infections [4].
Figure 1. . Structural formulae of the prototypes of subclasses of acyclic nucleoside phosphonates and their corresponding clinically used lipophilic prodrugs.

HDP: Histamine biphosphate; HPMPC: Cidofovir; PMEA: Adefovir; PMPA: Tenofovir; TAF: Tenofovir aladenamide; TDF: Tenogovir disoproxil fumarate.
A second subclass of ANPs has a 2-phosphonylmethoxyethyl (PME) moiety linked to the nucleobase (Figure 1) [5]. The prototype compound, designated PMEA [9-(2-phosphonylmethoxyethyl)adenine] proved particularly active against herpesviruses but also retroviruses (i.e., HIV) and hepadnaviruses, represented by hepatitis B virus (HBV) [5–7]. Although active in HIV-infected individuals, the compound proved toxic at the indicated efficacy dose. However, it was also found active against HBV-infected individuals at markedly lower doses, and therefore, the compound has been approved for HBV treatment (designated as adefovir, Hepsera®) [8]. Whereas within the HPMP subclass of ANPs both purine (i.e., adenine, 2,6-diaminopurine, guanine) and pyrimidine (i.e., cytosine) derivatives showed antiviral activity, only purine nucleobase derivatives, such as adenine, 2,6-diaminopurine, guanine and 6-cyclopropyladenine, but not pyrimidine nucleobase derivatives, showed antiviral activity within the subclass of PME derivatives.
A third subclass of ANPs consists of a 2-phosphonylmethoxypropyl (PMP) entity linked to the nucleobase [9] (Figure 1). As observed for the PME derivatives, only PMP derivatives containing a purine nucleobase are antivirally active. The adenine derivative (R)-PMPA (tenofovir, Viread®) was selected within the PMP subclass of ANPs as a clinical candidate for the treatment of retrovirus (i.e., HIV) and HBV infections, and currently acts as a cornerstone for HIV (and HBV) treatment.
Several other distinct groups of ANPs have been reported to be endowed with antiviral activity in cell culture and in animal models but have not yet been advanced for clinical use. They contain either a (S)-HPMP, a PME or a (R)-PMP structural moiety linked to a modified (non-natural) nucleobase. One of such well-defined group of compounds contain a 2,4-diaminopyrimidine linked through an oxygen atom to the HPMP, PME or PMP part of the molecule (designated HPMP-DAPym, PME-DAPym or PMP-DAPym; Figure 2) [10,11]. The DAPym derivatives display an antiviral activity spectrum predominantly similar to their corresponding adenine derivatives, probably because the DAPym configuration molecularly mimics an (incomplete) adenine nucleobase [12] allowing hydrogen pairing to a pyrimidine nucleobase in the viral nucleic acid chain.
Figure 2. . Structural formulae of acyclic nucleoside phosphonates with modified nucleobase.

Another well-defined group of compounds contain a triazine (i.e., 5-azacytosine as the nucleobase) linked to the HPMP moiety [(S)-HPMP-5-azaC] [13,14]. The antiviral activity spectrum of the HPMP-5-azaC derivative parallels that of (S)-HPMPC [15].
Only a few cyclic nucleoside phosphonates have been reported to be endowed with pronounced antiviral potential. The phosphonylmethoxy-2′-fluoro-2′,3′-didehydro-2′,3′-dideoxyadenosine (Fd4A-phosphonate; GS-9148; Figure 3) has shown to possess interesting anti-HIV activity in cell culture [16]. An investigational new drug (IND) was filed in December 2016 for treatment of patients with nucleoside reverse transcriptase inhibitor (NRTI) mutations in combination with other antiretroviral drugs [17,18].
Figure 3. . Structural formulae of a cyclic nucleoside phosphonate and the corresponding lipophilic phosphonoamidate prodrug.

In conclusion, a variety of ANPs have been demonstrated to be particularly successful and/or promising to be developed for clinical use. The market for the currently available clinically used ANPs against HIV and HBV is tremendous. According to the WHO, it is estimated that around 37 million individuals are infected with HIV, and at least 2 billion individuals are infected with HBV worldwide of which 240 million people are chronically infected. Product gross sales for the treatment of HIV and HBV infections by the ANP drug tenofovir, its prodrugs tenofovir disoproxil fumarate (TDF) and tenofovir alafenamide (TAF), and its combinatory formulations amounted up to 18 billion US dollars in 2016.
The mechanism of antiviral action of ANPs
The most important feature of the ANPs is the presence of a phosphonate entity that is isosteric with the phosphate group of the natural nucleoside 5′-monophosphates but consists of a P-C-O bond instead of a P-O-C ester bond. In this respect, the phosphonate entity in the ANPs is highly stable and does not easily hydrolyze. Therefore, the ANPs can be considered as highly stable dNMP derivatives.
All ANPs require cellular uptake, and obligatory metabolic conversion to their corresponding diphosphate derivatives (the equivalent of the natural 2′-deoxynucleoside 5′-triphosphates) to be antivirally active [19,20]. The metabolic activation is catalyzed by cellular enzymes and the conversion of the ANPs to their active diphosphates occurs in two separate steps. Only under this metabolically activated form, they compete with the natural 2′-deoxyucleotide-5′-triphosphates (dNTPs) for interaction with the viral (and cellular) DNA polymerases. In this respect, they not only inhibit the normal catalytic function of these DNA polymerases, but additionally, the activated ANPs are also incorporated into the viral DNA chain. Such incorporation eventually results in obligatory viral DNA chain termination and thus in premature abortion of the viral DNA chain elongation [21,22].
Due to their anionic nature, ANPs are not very efficiently taken up by the (virus-infected) cells and display rather poor oral bioavailability. Therefore, a number of prodrugs have been designed to increase the clinical efficacy of these molecules (Figures 1–3). In this respect, alkyloxyalkyl derivatives of (S)-HPMPC (i.e., HDP-(S)-HPMPC) [23], the bis(pivaloyloxymethyl) ester of PMEA (i.e., adefovir dipivoxil) [24], the bis(isopropyloxycarbonyloxymethyl) ester of (R)-PMPA formulated as its fumarate salt (TDF) [25], the alaninyl phenylphosphonoamidate isopropyl ester of tenofovir (TAF) [26] and the hexadecyloxypropyl derivative of tenofovir (CMX-157) [27] have been successfully designed to afford a more efficient cellular uptake, increased oral bioavailability and, in case of TAF, also prolonged retention of the drug in peripheral lymphocytes [28].
Design of the alpha-carboxynucleoside phosphonates
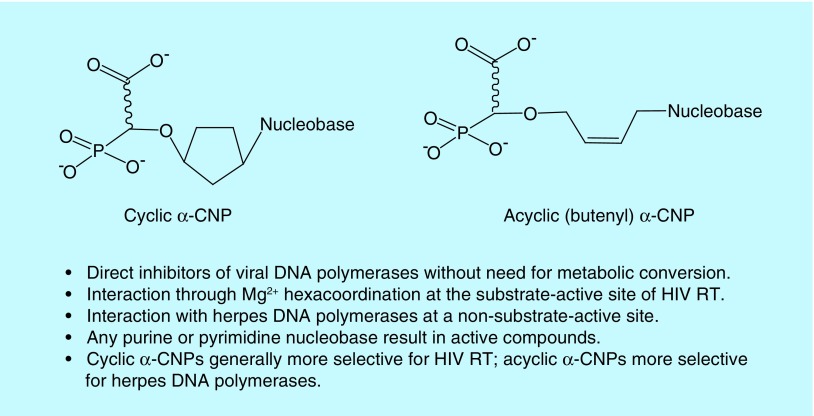
Most recently, an entirely different structural class of (cyclic and acyclic) nucleoside phosphonates has been designed, synthesized and thoroughly investigated [29–32]. It consists of a nucleobase part to allow Watson–Crick base pairing with the template strand in the viral DNA polymerase, a phosphonate moiety containing a carboxylate function in addition to the phosphonate group and a variable linker entity between the nucleobase and the carboxy phosphonate part (Figure 4). This linker originally consisted of a cyclopentyl entity (resulting in the prototype of the subclass of the cyclic alpha-carboxynucleoside phosphonates [α-CNPs]) [30], but later on, it was replaced by a more flexible acyclic aliphatic chain (i.e., butenyl; resulting in the prototype of the subclass of the acyclic α-CNPs; Figure 4) [32].
Figure 4. . Structural formulae of cyclic and acyclic alpha-carboxynucleoside phosphonates.

To avoid the requirement for metabolic activation, the carboxy phosphonate moiety was designed to potentially act as a mimic for the triphosphate present in the natural dNTP substrates of DNA polymerases. The requirements for such a triphosphate mimic were multiple: efficient Mg2+ hexacoordination between the triphosphate mimic and active-site amino acid residues of the DNA polymerase must be feasible as also required for efficient interaction between the dNTPs and the DNA polymerase enzyme active site. It was assumed that the carboxylate in the phosphonate moiety might contribute to coordinate Mg++; the α-carboxy phosphonate entity must be chemically stable under physiological conditions and should be tightly linked to the other part of the nucleoside molecule. The molecule must enable efficient canonical Watson–Crick base pairing with the template overhang, the distance between, and the positions of, the nucleobase and the triphosphate mimic. This should resemble as closely as possible the natural situation where a 2-deoxyribose is bridged between both parts of the molecule. However, the 2-deoxyribose could not be incorporated in the inhibitor structures because of the intrinsic lability of the ether oxygen that links the pentose to the phosphonate. Therefore, a cyclopentyl ring was used in the first syntheses to represent the linker moiety. This cyclopentyl entity was earlier proven to fulfill these criteria in the NRTI carbovir and abacavir [33,34]. Later on, compounds were also designed to replace the cyclopentyl by an acyclic aliphatic entity, preferably a butenyl group since this moiety, which replaces the 2-deoxyribose in nucleotide analogs, had previously been shown to generate compounds with antiherpetic activity (Figure 4) [35]. All natural nucleobases, in addition to several unnatural nucleobases and modifications in the carboxy phosphonate part, have been designed and synthesized using the α-CNP concept [30,32,36–38].
Synthesis of carbocyclic α-CNPs
The synthesis of the first-generation series of carbocyclic α-CNPs [30] involved two key transition-metal-catalyzed steps: first, rhodium-catalyzed carbenoid O–H insertion to install the carboxyphosphonate (building on our earlier work [29] attaching this moiety to protected nucleoside derivatives), and then palladium-catalyzed Tsuji–Trost allylation to install the nucleobase. The remaining manipulations required to afford the desired products were catalytic hydrogenation and final deprotection using TMSBr to cleave the methyl phosphonate and then NaOH to cleave the carboxylate ester (Figure 5). The highly polar α-CNPs were purified using charcoal chromatography. This sequence is advantageous since the common building block 2 can be converted into several α-CNP derivatives by varying the nucleobase used in the allylation step. By this method, derivatives of nucleobases U, T, C, A and 5-FU were synthesized.
Figure 5. . First-generation synthesis of cyclic alpha-carboxynucleoside phosphonate compounds.

As the first-generation route was not applicable to the synthesis of the guanine α-CNP derivative, other routes were developed to allow the preparation of this compound [38], thereby increasing the scope and applicability of the synthetic methodology.
Racemic G-α-CNP was obtained from a cyclopentylamine via initial base construction followed by O–H insertion (Figure 6). In the last step, TMSBr conveniently removed Boc protection as well as cleaving the methyl phosphonate and aqueous NaOH cleaved the carboxylate ester and converted the chloropurine moiety to guanine. The racemic G-α-CNP was also prepared from allylic acetate 1 by introducing the base first and then installing the phosphonate (Figure 7), effectively resulting in a reversal of the key steps in the first-generation synthesis. D and L forms of G-α-CNP were prepared using this route by employing the appropriate enantiomer of 1.
Figure 6. . Synthesis of G-alpha-carboxynucleoside phosphonate by base construction.

Figure 7. . Synthesis of G-alpha-carboxynucleoside phosphonate by Tsuji–Trost allylation.

All of the cyclic α-CNP compounds were prepared in racemic DL form, and separately as D and L forms by using the appropriate enantiopure precursor 1 in each instance. In all cases, the α-CNPs were formed as mixtures of diastereomers at the carboxyphosphonate group.
By omitting the NaOH hydrolysis step during the deprotection sequence, it was possible to prepare the partially deprotected esters 4, and also the unsaturated carbocyclic α-CNP 5 was prepared from the intermediate 3 with a slight modification of the deprotection reaction conditions.
Other variations on the CNP motif that were prepared (Figure 8) include the bisphosphonate 6, malonate 7 and the carboxylic acid compound without the phosphonate function 8; these derivatives were prepared using the same synthetic approach, by substituting the appropriate diazo precursor into the rhodium-catalyzed O–H insertion reaction [36].
Figure 8. . Variations on the alpha-carboxynucleoside phosphonate motif.

Synthesis of acyclic (butenyl) α-CNPs
Based on the synthetic approach described above for the cyclic α-CNPs, the synthesis of acyclic (butenyl) α-CNPs involved three steps starting with the O-H insertion of the rhodium carbenoid generated from trimethyl phosphonodiazoacetate. The rhodium-catalyzed (1 mol% of Rh2(OAc)4) reaction of but-2-ene-1,4-diol 9 with trimethyl phosphonodiazoacetate 10 was carried out in refluxing benzene (Figure 9). Upon completion of the reaction, mono- (11) and di- (12) O–H inserted compounds were isolated in 58 and 19% yield, respectively.
Figure 9. . O-H insertion of rhodium carbenoid.

The second step of the synthetic route involved the Mitsunobu reaction for the attachment of different nucleobases to the butenyl carboxyphosphonate moiety 11. The Mitsunobu reaction was initiated by generating the betaine (PPh3:DIAD). This could be achieved by treating DIAD and PPh3 at 0°C in dry THF. Betaine was added to another reaction vessel at -40°C. This vessel contained the O–H inserted compound 3 and the corresponding protected nucleobases 12a–12d in THF. The reaction mixture was warmed up to room temperature while stirring was continued for 24 h. The expected nucleobase-substituted carboxyphosphonates 13a–13d were isolated in moderate to good yields (Figure 10).
Figure 10. . Mitsunobu reaction for the attachment of the nucleobase.

Finally, a two step-deprotection of compounds 13a–13d was carried out in one pot. An excess of TMSBr followed by water was used to deprotect the dimethoxyphosphonate entity. An excess of aqueous 1 M NaOH was used to saponificate the carboxyester and to debenzoylate the N-3-benzoylthymine. A charcoal column was used for purification of the phosphononucleosides 14a–14d with a 1:1 mixture of EtOH and 20% aqueous ammonia (Figure 11).
Figure 11. . Deprotection of α-carboxy nucleoside phosphonates 13a–13d.

Reaction conditions: (i) TMSBr (7.0 equiv) CH3CN, 0°C-rt, 16 h. (ii) H2O, 30 min, rotary evaporation (iii) aq. NaOH, 50°C, 12 h and then aq. NH3, charcoal column.
Anti-viral DNA polymerase activity of α-CNPs
The cyclic α-CNPs containing a natural pyrimidine or purine nucleobase were invariably inhibitory against HIV-1 reverse transcriptase (RT) in the presence of their corresponding homopolymeric template/primer (Table 1). Thus, the thymine-containing T-α-CNP inhibited HIV-1 RT in the nanomolar range in the presence of poly rA.dT, but not in the presence of other homopolymeric template/primers, such as poly rI.dC, poly rU.dA or poly rC.dG. These findings point to selective recognition and canonical base-pairing of the T-α-CNP analog, with an adenine base present in the template overhang. In this respect, the α-CNPs behave similarly as the NRTIs and N(t)RTIs with respect of HIV RT recognition and inhibition [31,37]. The α-CNPs generally proved equally potent inhibitors of HIV-1 RT as their corresponding 2′,3′-dideoxynucleotide analogs, such as AZT-TP, ddCTP, ddATP and ddGTP. It was also shown that the α-CNPs inhibited other retrovirus-encoded RTs, such as those derived from HIV-2, SIV, FIV and Visna virus [31].
Table 1. . Inhibitory activity of the L-enantiomeric alpha-carboxynucleoside phosphonates against HIV-1 reverse transcriptase in the presence of different homopolymeric template/primers.
| Compound† | IC50‡,§ HIV-1 RT (μM) | |||
|---|---|---|---|---|
| Poly rA.dT ([3H]dTTP) | Poly rI.dC ([3H]dCTP) | Poly rU.dA ([3H]dATP) | Poly rC.dG ([3H]dGTP) | |
| T-α-CNP | 0.41# | >500 | 155 | >100 |
| U-α-CNP | 3.0# | >500 | 208 | – |
| C-α-CNP | 164 | 4.3# | 45 | – |
| A-α-CNP | >500 | >500 | 0.19# | – |
| G-α-CNP | 396 | – | – | 0.43# |
| AZT-TP | 0.11 | – | – | – |
| ddCTP | – | 14 | – | – |
| ddATP | – | – | 1.5 | – |
| ddGTP | – | – | – | 0.41 |
Inhibition of RTs by T-, U-, C-, A- and G-α-CNP analogs was evaluated using the respective homopolymeric template/primers poly rA.dT (for T- and U-α-CNP), poly rI.dC (for C-α-CNP), poly rU.dA (for A-α-CNP) and poly rC.dG (for G-α-CNP) and ∼1.25–2.5 μM of their appropriate corresponding dNTP substrates [CH3-3H]dTTP (for T- and U-α-CNP), [5-3H]dCTP (for C-α-CNP), [2,8-3H]dATP (for A-α-CNP) and [2,8-3H]dGTP (for G-α-CNP).
†Each α-CNP represents a pair of diastereomers at the alpha-carboxy stereocenter.
‡50% inhibitory concentration or compound concentration required to inhibit DNA polymerase activity by 50%.
#Pronounced activities of the alpha-CNPs.
α-CNP: Alpha-carboxynucleoside phosphonate; ATP: Adenine triphosphate; AZT-TP: Zidovudine triphosphate; CTP: Cytosine triphosphate; dNTP: 2’-Deoxyucleotide-5’-triphosphate; GTP: Guanine triphosphate; RT: Reverse transcriptase.
In contrast to HIV RT inhibition, the cyclic α-CNPs were much less potent inhibitors of herpetic DNA polymerases derived from herpes simplex virus type 1 (HSV-1), HCMV or varicella-zoster virus (VZV; Table 2) [31]. Their IC50 values were in the middle micromolar range. Only the G-α-CNP analog had an IC50 in the low micromolar range [37]. Strikingly, both the cellular DNA polymerases α and β were poorly inhibited by the cyclic α-CNPs (IC50 >100 μM). So far, the potential inhibitory activity of the α-CNPs against other cellular DNA polymerases, including the mitochondrial DNA polymerase γ and the DNA polymerases δ, ε and ζ have not been investigated. It cannot be excluded that the α-CNPs are recognized as inhibitors of one or several of these enzymes as well, which may result in potential side effects. Therefore, it would be warranted to investigate the potential interaction of α-CNPs with these enzymes and defining the structure–activity relationships of the inhibitors for these enzymes in order to identify and select the most specific inhibitors of the viral DNA polymerases for further clinical development.
Table 2. . Inhibitory activity of the L-enantiomeric alpha-carboxynucleoside phosphonates against viral and cellular DNA polymerases.
| Enzyme source | IC50† DNA polymerase (μM) | |||
|---|---|---|---|---|
| T-α-CNP | C-α-CNP | A-α-CNP | G-α-CNP | |
| HIV-1 RT | 0.41 | 4.3 | 0.19 | 0.43 |
| HSV-1 DNA pol | 26 | 19 | 3.5 | 4.2 |
| HCMV DNA pol | 38 | 17 | 23 | 20 |
| VZV DNA pol | 38 | – | – | – |
| DNA pol α | 229 | 269 | 171 | 121 |
| DNA pol β | >200 | ≥200 | >200 | 522 |
The acyclic (butenyl) α-CNPs showed an opposite inhibitory selectivity [32]; in general, they poorly inhibited HIV-1 RT (IC50 >100 μM in virtually all cases; Table 3), but they markedly gained inhibitory activity against the herpetic DNA polymerases (Table 4). All acyclic (butenyl) α-CNPs inhibited the herpetic DNA polymerases in the lower micromolar range, irrespective of the nature of the nucleobase (only the C-α-CNP analog was slightly less effective against HSV-1 and HCMV DNA polymerases, but kept pronounced inhibitory activity against VZV DNA polymerase). As also observed for all cyclic α-CNPs, the acyclic α-CNPs poorly inhibited the cellular DNA polymerases α and β (IC50 ≥ 500 μM) with the exception of moderate inhibition of DNA polymerase-α by the butenyl T-α-CNP (Table 4).
Table 3. . Inhibitory activity of acyclic (butenyl) alpha-carboxynucleoside phosphonates against HIV-1 reverse transcriptase in the presence of different homopolymeric template/primers.
| Compound | IC50† HIV-1 RT (μM) | |||
|---|---|---|---|---|
| Poly rA.dT ([3H]dTTP) | Poly rI.dC ([3H]dCTP) | Poly rU.dA ([3H]dATP) | Poly rC.dG ([3H]dGTP) | |
| Butenyl T-α-CNP | 235 | >500 | ≥500 | >500 |
| Butenyl C-α-CNP | 270 | >500 | 273 | >500 |
| Butenyl A-α-CNP | 404 | >500 | 40 | >500 |
| Butenyl G-α-CNP | ≥500 | >500 | 369 | >500 |
†Data taken from [32].
α-CNP: Alpha-carboxynucleoside phosphonate; IC50: 50% inhibitory concentration; RT: Reverse transcriptase.
Table 4. . Inhibitory activity of acyclic (butenyl) alpha-carboxynucleoside phosphonates against viral and cellular DNA polymerases.
| Enzyme source | IC50† DNA polymerase (μM) | |||
|---|---|---|---|---|
| Butenyl T-α-CNP | Butenyl C-α-CNP | Butenyl A-α-CNP | Butenyl G-α-CNP | |
| HSV-1 DNA pol | 3.7 | 12 | 2.7 | 2.1 |
| HCMV DNA pol | 2.1 | 16 | 5.5 | 3.6 |
| VZV DNA pol | 2.2 | 2.9 | 3.3 | 2.9 |
| DNA pol α | 14 | 414 | ≥500 | 365 |
| DNA pol β | >500 | >500 | 454 | >500 |
†Data taken from [32].
α-CNP: Alpha-carboxynucleoside phosphonate; HCMV: Human cytomegalovirus; HSV-1: Herpes simplex virus type 1; IC50: 50% inhibitory concentration; RT: Reverse transcriptase; VZV: Varicella-zoster virus.
An interesting feature was observed for the acyclic (butenyl) α-CNPs. Whereas the cyclic α-CNPs inhibited HIV-1 RT in a highly selective manner (specific competition in the presence of a natural dNTP containing a similar nucleobase as the α-CNP), the acyclic (butenyl) α-CNPs behaved clearly differently [37]. They efficiently inhibited the herpetic DNA polymerases (i.e., HCMV and VZV DNA polymerases) irrespective of the nature of the nucleobase in the competing dNTP (Table 5). Therefore, it could be concluded that the acyclic (butenyl) α-CNPs must interact with the herpetic DNA polymerases in a kinetic manner that is clearly different than observed for the cyclic α-CNPs against HIV-1 RT. An overview of the most important kinetic properties of the α-CNPs against HIV RT is provided in Table 6.
Table 5. . Inhibitory activity of acyclic (butenyl) alpha-carboxynucleoside phosphonates in herpetic DNA polymerase assays using different 2’-deoxynucleotide-5’-triphosphate substrates.
| Nucleobase | IC50 (μM)† | ||||
|---|---|---|---|---|---|
| HCMV DNA polymerase‡ | VZV DNA polymerase§ | ||||
| [3H]dTTP | [3H]dCTP | [3H]dATP | [3H]dGTP | [3H]dTTP | |
| Butenyl T-α-CNP | 2.1 | 3.0 | 3.1 | 2.5 | 2.2 |
| Butenyl A-α-CNP | 5.5 | 2.9 | 3.0 | 3.2 | 3.3 |
| Butenyl G-α-CNP | 3.6 | 11 | 10 | 14 | 2.9 |
| Butenyl C-α-CNP | 16 | 20 | 33 | 30 | 2.9 |
†Data taken from [32].
‡Template/primer: activated calf thymus DNA, [3H]dNTP substrate and three nonradiolabeled dNTPs
§Template/primer: poly dA.oligo dT, [3H]dTTP substrate.
α-CNP: Alpha-carboxynucleoside phosphonate; dNTP: 2’-Deoxyucleotide-5’-triphosphate; HCMV: Human cytomegalovirus; IC50: 50% inhibitory concentration; VZV: Varicella-zoster virus.
Table 6. . Comparative overview of the properties of alpha-carboxynucleoside phosphonates and other HIV reverse transcriptase inhibitor classes.
| Properties | α-CNP† | NRTI‡ | NtRTI§ | NNRTI¶ |
|---|---|---|---|---|
| Nucleoside structure? | No | Yes | No | No |
| Nucleotide structure? | Yes | No | Yes | No |
| Metabolic activation required? | No | Yes (three steps) | Yes (two steps) | No |
| Enzyme inhibition versus dNTP substrate | Competitive | Competitive | Competitive | Noncompetitive |
| Binding to RT substrate-binding site? | Yes | Yes | Yes | No |
| Nature of the RT binding | Mutually exclusive with dNTP, but coordinating with Mg2+ | Mutually exclusive with dNTP, but coordinating with Mg2+ | Mutually exclusive with dNTP, but coordinating with Mg2+ | Mutually nonexclusive with dNTP and Mg2+ |
| Nucleobase-specific binding to template? | Yes | Yes | Yes | No |
| DNA chain termination? | No | Yes | Yes | No |
| Selective for HIV-1 RT? | No | No | No | Yes |
†α-Carboxynucleoside phosphonate.
‡NRTI: Nucleoside RT inhibitor (e.g., zidovudine triphosphate).
§NtRTI: Nucleotide RT inhibitor (e.g., tenofovir diphosphate).
¶NNRTI: Non-nucleoside RT inhibitor (e.g., nevirapine or rilpivirine).
dNTP: 2’-Deoxyucleotide-5’-triphosphate; RT: Reverse transcriptase.
Molecular interactions of α-CNP in the substrate-binding site of HIV-1 RT
Structural studies were performed to ascertain the molecular interactions of T-α-CNP with HIV-1 RT. Crystal structures of the ternary RT–dsDNA–T-α-CNP and RT–DNA-aptamer–T-α-CNP complexes were determined [31,39]. These investigations clearly showed that the thymine nucleobase of T-α-CNP base-paired with the adenine overhang of the template, the cyclopentyl ring interacted with active site amino acids in a similar manner as the 2-deoxyribose does in the dNTP/enzyme interactions, only the L form of the synthesized T-α-CNP racemic mixture (L/D) is the active constituent, and the α-carboxymethylphosphonate part of the molecule actively participated in the hexacoordination of both catalytic Mg2+ ions at the polymerase active site, mimicking the coordination of a dNTP substrate. One carboxy oxygen and two phosphonate oxygens of T-α-CNP chelate the Mg2+ ions in a manner analogous to the chelation of an oxygen atom from the alpha, beta and γ phosphates of dNTP by the Mg2+ ions (Figure 12), however, spatial positions of the three chelating atoms of T-α-CNP are different from those of a dNTP. The chelated carboxyphosphonate entity is not susceptible to hydrolysis by the RT enzyme and thus, forms a nonproductive ternary complex with RT, leading to inhibition of the catalytic function of the enzyme as a competitive inhibitor. These structural findings explain the direct-acting inhibitory activity of the α-CNPs against HIV RT and defined the unique characteristics of this class of molecules. The concomitant presence and availability of three negatively charged oxygens in the carboxyphosphonate part of the α-CNP molecule effectively compensate for the chelation of the phosphates of a dNTP. In agreement with the crystal structures, substitutions of the negative charge of the carboxy group or the two negative charges of the phosphonate group by methyl ester groups [31], caused a complete loss of their inhibitory potential against HIV-1 RT. Also, the bisphosphonate 6, the malonate 7 and the carboxylic acid compound without the phosphonate function 8 showed poor, if any inhibitory activity against HIV RT. These findings also demonstrate that any synthesis of novel α-CNP derivatives should take these requirements into consideration while designing and generating active molecules.
Figure 12. . Structural basis for T-alpha-carboxynucleoside phosphonate inhibition of HIV reverse transcriptase.

Mode of binding of T-α-CNP to RT–dsDNA and RT–DNA-aptamer complexes shown in yellow and cyan, respectively (left); two catalytic ions (A & B) are presented in the RT–DNA-aptamer–T-α-CNP complex. A schematic comparison of the structural interactions of T-α-CNP (center) and the natural dTTP (right) with HIV-1 RT and the template–primer. The hexacoordination of metal ions A and B (Mg2+) is shown. The contribution of the oxygens of the carboxylic acid and the phosphonate groups in α-CNP are shown (center), to be compared with the contribution of the oxygens of the triphosphate group in the natural dNTP substrate (right).
α-CNP: Alpha-carboxynucleoside phosphonate; dNTP: 2’-Deoxyucleotide-5’-triphosphate; RT: Reverse transcriptase.
Similarities between α-CNPs and NcRTIs for interaction with HIV RT
Beside the nucleoside RT inhibitors (NRTIs), non-nucleoside RT inhibitors (NNRTIs), pyrophosphate inhibitors and RNAseH inhibitors of HIV RT, nucleotide-competing RT inhibitors (NcRTIs) have been reported. INDOPY (indolopyridinone) is the prototype compound of this peculiar class of RT inhibitors (Figure 13) [40] and several other structures have since then been reported as NcRTIs [41,42]. Although the NcRTIs have a structure that is entirely different from the classical nucleotide structure, they interact, alike the NRTIs, with the substrate-binding site of HIV RT in a competitive manner with the natural dNTP substrates. However, unlike the NRTIs, the NcRTIs do not incorporate into the viral growing DNA chain. Thus, in this respect, α-CNPs are reminiscent of the mechanism of action reported for the NcRTIs in that they reversibly interact with the substrate-binding site of RT in a competitive manner with the incoming dNTPs and bind to the post-translocated RT–nucleic acid complex conformation. However, whereas the α-CNPs can only form stable ternary complexes with HIV RT when the nucleobase in the α-CNP is complementary to the templating nucleobase (indicative for Watson–Crick base pairing), the NcRTIs form a stable ternary complex irrespective of the nature of the templating nucleobase. The α-CNPs also differ from the NcRTIs in that they obligatorily require hexacoordination with Mg2+ (like NRTIs and the natural dNTPs), whereas Mg2+ is not required for the binding of the NcRTIs with HIV RT. A last striking difference between α-CNPs and the NcRTIs is the fact that α-CNPs can also interact with other viral DNA polymerases (i.e., herpetic DNA polymerases) and even with dNTP-interacting enzymes different from DNA polymerases (see Inhibitory activity of α-CNPs against enzymes other than viral DBA polymerases), whereas NcRTIs are solely and exclusively specific for reverse transcriptases.
Figure 13. . Structural formula of the nucleotide-competing reverse transcriptase inhibitor indolopyridinone-1 (5-methyl-1-(4-nitrophenyl)-2-oxopyrido[3,2-b]indole-3-carbonitrile).

Different kinetic interactions of α-CNPs with viral DNA polymerases depending the nature of the enzyme
When cyclic α-CNPs were investigated for their interaction with HIV-1 RT in kinetic and biochemical assays, it was demonstrated that they showed competitive inhibition with respect of the natural dNTP substrate, and obligatory binding of the template–primer to the enzyme prior to efficient binding of the α-CNP to the enzyme [31,37]. The α-CNPs reversibly inhibited the RT enzyme. Specific Watson–Crick base pairing was required for inhibition, but there was no indication of incorporation of the α-CNP into the growing DNA chain (in contrast with NRTIs and N(t)RTIs). It should also be noticed that solely the L-enantiomer but not the D-enantiomer of α-CNPs showed significant activity against HIV-1 RT [31,37].
The kinetic picture seems entirely different for the interaction of the cyclic α-CNPs and the acyclic (butenyl) α-CNPs with the herpetic DNA polymerases. First of all, both the L- and D-enantiomers of the cyclic G-α-CNP equally inhibited herpetic (i.e., HSV-1, HCMV) DNA polymerases [37]. They did so in a non- or uncompetitive manner. These findings indicate that the L- and D-α-CNPs must bind to the herpetic DNA polymerase at a nonsubstrate active site. Also, the acyclic (butenyl) α-CNPs (as demonstrated for the T analog) noncompetitively inhibited the VZV DNA polymerase, showing identical (noncompetitive) kinetics as the cyclic T-α-CNP. The inhibition proved independent of the nature of the nucleobase (Table 5). Moreover, evidence was provided that inhibition of HCMV DNA polymerase occurred independent of Watson–Crick base pairing. Taken all available data together, α-CNPs seem to bind to an enzyme site that differs from the active substrate-binding site of the DNA polymerase. It is possible that these compounds selectively bind to the 3′,5′-exonuclease active center of the herpetic DNA polymerases or to a yet unknown binding site. For inhibition of the herpetic DNA polymerases Watson–Crick base pairing was not strictly required. Therefore, the aromatic ring system might be further modified and optimized in the cyclic and acyclic α-CNPs to allow inhibitor structures that are markedly different from the natural nucleobase entity and nucleotide structure.
Drug resistance characteristics of α-CNPs
The cyclic α-CNPs containing the four natural nucleobases have been explored for their inhibitory potential against HIV RT that contain amino acid mutations that are characteristically selected by established clinically used drugs [37] (Table 7). The highest resistance profile for the G-α-CNP derivative was observed for the F61A (79-fold), V75I (8.7-fold), M184I (19-fold) and M184V (8.7-fold) mutant enzymes. However, in some cases, sensitization was observed (i.e., K70E [0.7-fold] and K70Q [0.7-fold], but not K70R [2.6-fold]). The structural and biochemical data show that α-CNPs bind HIV-1 RT like a nucleoside triphosphate mimic. As expected, the dNTP-binding pocket mutations have varying degrees of effect on the binding, and consequently on the inhibitory potency of α-CNPs (Table 7). The mutations that have the highest effect on the efficacy of α-CNPs are M184V/I and F61A. The M184V/I mutation appears to discriminate the L-nucleoside inhibitors lamivudine (3TC) and emtricitabine (FTC) by a steric hindrance mechanism [43,44]. The analogous mutation in HBV DNA polymerase also develops resistance to 3TC and FTC by a similar mechanism [45]. The crystal structure of the RT–DNA–T-α-CNP complex [22] suggests a similar mechanism of resistance to α-CNPs that contain a cyclopentyl ring extending toward the branched side chain of the M184V/I mutation in HIV-1 RT. Instead, the F61A mutation in the fingers subdomain of HIV-1 RT may affect the positioning of the nucleobase of α-CNPs due to loss of interaction between F61 and V74.
Table 7. . Fold-resistance of the alpha-carboxynucleoside phosphonates against mutant HIV-1 reverse transcriptases compared with wild-type (control).
| Mutation in HIV-1 RT | G-α-CNP Poly rC.dG [3H]dGTP | A-α-CNP Poly rU.dA [3H]dATP | C-α-CNP Poly rI.dC [3H]dCTP | T-α-CNP Poly rA.dT [3H]dTTP |
|---|---|---|---|---|
| F61A | 79 | 3.9 | 5.2 | 31 |
| K65R | 2.1 | 0.8 | 1.6 | 1.5 |
| K70E | 0.7 | 3.0 | 0.2 | 0.8 |
| K70Q | 0.7 | 3.7 | 0.3 | 1.1 |
| K70R | 2.6 | 3.3 | 2.3 | 0.9 |
| V75I | 8.7 | 7.0 | 6.0 | 2.6 |
| L100I | 5.2 | 3.0 | 0.5 | 1.4 |
| K103N | 1.1 | 0.6 | 1.5 | 0.6 |
| V106A | 1.6 | 0.8 | 0.2 | 1.1 |
| Y115F | 2.3 | 0.8 | 1.1 | 2.4 |
| E138K | 0.9 | 0.7 | 1.5 | 1.0 |
| Q151M | 3.1 | 0.8 | 0.4 | 3.1 |
| Y181C | 5.2 | 1.8 | 1.5 | 0.9 |
| M184I | 19 | 2.8 | 12 | 36 |
| M184V | 8.7 | 1.7 | 11 | 16 |
Data taken from [37].
α-CNP: Alpha-carboxynucleoside phosphonate; RT: Reverse transcriptase.
Interestingly, the fold-resistance/sensitization against a well-defined amino acid mutation was shown to differ depending on the nature of the nucleobase in the α-CNP molecule. For example, whereas G-α-CNP showed 79-fold resistance to F61A, the A-α-CNP derivative was only 3.9-fold resistant. Also, the G-α-CNP proved 19- and 8.7-fold less sensitive to M184I and M184V RT, respectively, whereas the A-α-CNP was only 2.8- and 1.7-fold less sensitive to these RT mutations. Similar observations could be made for the degree of sensitization for K65E and K65Q RT, which showed three- to 3.7-fold resistance for A-α-CNP but three- to fivefold sensitization for C-α-CNP. The observed differences in resistance/sensitivity profile of α-CNPs depending on the nature of both the mutations in the enzyme and the nature of the nucleobase in the α-CNP molecule opens interesting perspectives for the potential of combination therapy within the α-CNP class of compounds.
Lipophilic prodrugs of α-CNPs
So far, no antiviral activity (i.e., HIV, herpesviruses) has been observed for the α-CNPs in cell culture. This may most likely be due to the highly anionic nature of the compounds. In comparison to the classical ANPs who have two negative charges exposed in the molecule at the phosphonate site, the α-CNPs contain even an additional negative charge, derived from the carboxy group in the phosphonate part. This makes these molecules rather unlikely candidates for efficiently penetrating membranes of their target cells. To increase cellular uptake and oral bioavailability in vivo, the clinical candidates of the acyclic nucleotide phosphonates have been converted to lipophilic prodrugs before administration. Their structures are shown in Figures 1 & 3.
Synthesis and investigation of the antiviral activity of prodrugs of the α-CNPs, using known and novel prodrug approaches, is underway in an attempt to deliver the free α-CNPs into the intact cells. In fact, the most obvious prodrug derivatives to be made for the α-CNPs to increase lipophilicity and cellular uptake are those that have been currently used to increase cellular uptake and oral bioavailability of the clinically approved ANPs. They include a hexadecyloxypropyl (HDP) group as in HDP-(S)-HPMPC, a BisPOM (bis((pivaloyloxy)methoxy)) entity as in Adefovir dipivoxil, a BisPOC (bis(isopropoxycarbonyloxymethyl)) moiety as in TDF or an arylisopropylalaninylphosphoramidate configuration as in Tenofovir alafenamide (Figure 1). Covering the anionic charge of the alpha-carboxyl in α-CNPs by a methyl group has been tried but proven unsuccessful to generate antiviral activity, most likely due to the lack of efficient release of the methyl group after exposure of the prodrug to the cellular environment. Generation of an α-CNP with a free negatively charged carboxyl group has indeed earlier been shown to be crucial for efficient interaction with the DNA polymerases. Alternatively, other types of drug formulations, including liposome preparations containing the α-CNPs should be explored, both in vitro and in vivo.
Inhibitory activity of α-CNPs against enzymes other than viral DNA polymerases
DNA polymerases are enzymes for which the dNTPs act as substrates for incorporation into the growing DNA chain. However, many enzymes are also feedback-regulated by dNTPs under the conditions in which dNTPs are not to be hydrolyzed but act as such. In nucleotide metabolism, there are numerous examples of such feedback mechanisms.
Two of such well-known interactions are dTTP feedback inhibition of thymidine kinase activity, and dCTP-triggered stimulation of dCMP deaminase. Interestingly, it could be demonstrated that T-α-CNP-containing thymine as nucleobase (not α-CNPs with other nucleobases) was able to inhibit the activity of mitochondrial TK-2 and to a somewhat lesser extent the herpesvirus-encoded TKs [31]. The cytosolic TK-1 seemed not to be affected by T-α-CNP. These data demonstrate again that subtle differences between enzymes eventually determine the potential of interaction with α-CNPs. Also for dCMP deaminase, it was found that C-α-CNP, which contains cytosine as the nucleobase, but not the other α-CNPs, was able to inhibit this enzyme in the presence of the natural dCTP stimulator [31]. The C-α-CNP derivative as such had no stimulatory activity. The findings that enzymes other than DNA polymerases interact with dNTPs for regulatory purposes, may be subject of inhibition by α-CNPs. It is of interest to notice that dCMP deaminase plays a role in the conversion of anticancer drugs, such as araC and gemcitabine to their (deaminated) inactive form, and thus, it may be speculated that in such case C-α-CNPs may have a potentiating effect on these anticancer compounds if combined. Thus, these findings may open new perspectives for interfering with cellular functions by α-CNPs, and such possibilities are worth to be further explored. It may broaden the potential application of the α-CNPs to other fields in addition to the antiviral domain.
Conclusion
The α-carboxynucleoside phosphonates mimic the natural 2′-deoxynucleotide 5′-triphosphate substrates of viral DNA polymerases. They were shown to inhibit retroviral reverse transcriptase and herpetic DNA polymerases without prior requirement of metabolic conversion. The α-CNPs have a unique kinetic mechanism of inhibition of these viral DNA polymerases. Efficient uptake by virus-infected cells likely seems a major bottleneck for pronounced biological activity.
Future perspective
The α-CNPs represent a novel class of stable CNPs that directly interact with their target (viral DNA polymerases) without the requirement of prior metabolic activation (phosphorylation). The lack of obligatory metabolic conversion prior to become active against its target avoid variability in activity due to different metabolic properties inherent to the phase of the cell cycle and different cell types to which the α-CNPs are exposed. They also distinguish themselves among the nucleoside phosphonates in that any nucleobase shows inhibitory activity, whereas it is usually only the adenine derivative within the PME and PMPA, and adenine and cytosine derivatives within the HPMP subclass of ANPs that show pronounced antiviral activity. Therefore, the structure–activity relationship is considerably broader than the current ANPs, which allow a better fine-tuning of the optimal structure of the α-CNPs and thus a higher selectivity for each of its potential targets.
The α-CNPs have so far only been explored for their inhibitory activity against retroviral and herpetic DNA polymerases. It may be assumed that they should also be active against HBV DNA polymerase because this enzyme has also an RT function. However, beside HBV DNA polymerase, other viral DNA polymerases, including poxvirus and adenovirus DNA polymerases, should be preferentially investigated for their sensitivity to the inhibitory activity of α-CNPs.
Given the importance of the current configuration of the α-carboxy phosphonate part of the molecule to act as dNTP mimics and direct inhibitors of DNA polymerases, pronounced modifications at this part of the α-CNP molecule might compromise their inhibitory potential. The bridge between the α-carboxy phosphonate and the nucleobase should also be the subject of further modifications. Given the fact that replacement of the cyclopentyl by the aliphatic butenyl entity shifts the selectivity spectrum of the α-CNPs from HIV- to herpesvirus-encoded DNA polymerases, a broad variety of novel cyclic and acyclic bridges should be explored to further fine-tune their observed inhibitory activities and also to define the structure–activity relationship of the α-CNPs against other DNA polymerases. In addition, the search for other DNA polymerases should be further extended to viral RNA polymerases as well, since the principle of required Mg2+-hexacoordination for efficient dNTP (α-CNP) binding to the substrate-active polymerase site also accounts for NTP binding to RNA polymerases.
Our findings that the nature of the nucleobase in the α-CNPs is not crucial for recognition by herpetic DNA polymerases (in contrast with HIV RTs) open interesting perspectives for replacing the natural nucleobases, such as thymine, cytosine, adenine and guanine by other heterocycles (i.e., substituted purines and pyrimidines, pyridines, pyrroles, thiophenes, etc.) or even nonheterocycles with or without the presence of functional groups. Such novel α-CNP derivatives may make the molecules more simple and straightforward in terms of synthesis and possibly more inhibitory.
The major drawback so far seems to be efficient delivery of the α-CNPs into their target (virus-infected) cells to enable intracellular inhibition of the viral DNA polymerases. Therefore, synthetic efforts are currently devoted to the synthesis of prodrugs that may shield part or all of the negative charges of these molecules to allow efficient cellular uptake and intracellular delivery of the free α-CNPs. Alternatively, physical technologies to deliver anionic molecules into cells (i.e., liposome formulations) should be explored.
Executive summary.
Design of the alpha-carboxynucleoside phosphonates
Alpha-carboxynucleoside phosphonates (α-CNPs) represent a novel synthetic scaffold of viral DNA polymerase (i.e., HIV reverse transcriptase [RT] and herpetic DNA polymerase) inhibitors. They are structurally simple nucleoside monophosphonate analogs that contain an additional carboxyl group in the phosphonate moiety and a cyclic or acyclic entity that bridges the carboxy phosphonate with the nucleobase.
Antiviral DNA polymerase activity of α-CNPs
In contrast with the classical acyclic nucleoside phosphonates, the α-CNPs do not require any prior metabolic conversion (phosphorylation) to exert their inhibitory activity against viral DNA polymerases. As a consequence, the α-CNPs function as direct-acting inhibitors of viral DNA polymerases.
Binding of the α-CNPs in the HIV RT substrate (2’-deoxyucleotide-5’-triphosphate [dNTP]) binding site is nucleobase-specific and selectively competes with an incoming dNTP containing a similar nucleobase as the α-CNP.
Binding of the α-CNPs to the herpesvirus DNA polymerases is not nucleobase-specific and does not compete efficiently with an incoming dNTP.
In contrast with nucleoside RT inhibitors and nucleotide RT inhibitors, α-CNPs are not incorporated into the growing viral DNA chain.
Molecular interaction of α-CNP in the substrate-binding site of HIV-1 RT
The α-carboxy group on the phosphonate provides an oxygen that is equivalent to an oxygen of the α-phosphate moiety in natural dNTPs for the obligatory Mg2+-hexacoordination in the substrate-binding site of HIV RT. Two oxygens of the phosphonate group of the α-CNPs have a similar function as the oxygens of the β- and γ-phosphates in dNTPs for efficient octahedral coordination of both Mg2+ ions.
Different kinetic interactions of α-CNP with viral DNA polymerases depending on the nature of the enzyme
Whereas cyclic α-CNPs preferentially inhibit retroviral (i.e., HIV) RTs, the acyclic (butenyl) α-CNPs shift their inhibitory selectivity spectrum to herpetic DNA polymerases. Both cyclic and acyclic α-CNPs show poor, if any, inhibitory action against cellular DNA polymerases α and β.
Inhibitory activity of α-CNPs against enzymes other than viral DNA polymerases
Specific interactions of α-CNPs with enzymes that do not use dNTPs as a substrate but rather as a regulatory (feedback) mechanism have been shown and open new perspectives to broaden the biological fields of application of the α-CNPs.
Lipophilic prodrugs of α-CNPs
None of the α-CNPs show antiviral activity in cell culture, presumably due to the overall strong negative charge of the α-CNP molecules and consequently lack of efficient uptake by virus-infected cells. Initial attempts of the synthesis of α-CNP prodrugs that have been designed to cover (neutralize) the negative charges of the carboxy-phosphonate moiety has not yet resulted in active antiviral compounds in cell culture; further prodrug approaches are under investigation.
Adapted prodrug moieties or other technologies may be required to efficiently internalize the α-CNPs into their target cells.
Acknowledgements
The authors thank Lizette van Berckelaer, Ria Van Berwaer and Kristien Minner for excellent technical assistance, Matthias Gotte for providing biochemical data and Christiane Callebaut for dedicated editorial help.
Footnotes
Financial & competing interests disclosure
The research that led to the data shown in the manuscript was supported by Grants PF 10/018 and GOA 10/14 from the KU Leuven (to J Balzarini), Grant P50 GM103368 and Grant R37 AI027690 MERIT Award (to E Arnold) from The NIH, and The Science Foundation Ireland Grant 05/PICA/B802 and Grant 14/TIDA/2402 (to A Maguire). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.De Clercq E, Holý A, Rosenberg T, Sakuma T, Balzarini J, Maudgal PC. A novel selective broad-spectrum anti-DNA virus agent. Nature. 1986;323:464–467. doi: 10.1038/323464a0. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E, Sakuma T, Baba M, et al. Antiviral activity of phosphonylmethoxyalkyl derivatives of purine and pyrimidines. Antiviral Res. 1987;8:261–272. doi: 10.1016/s0166-3542(87)80004-9. [DOI] [PubMed] [Google Scholar]
- 3.Snoeck R, Sakuma T, De Clercq E, Rosenberg I, Holý A. (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, a potent and selective inhibitor of human cytomegalovirus replication. Antimicrob. Agents Chemother. 1988;32:1839–1844. doi: 10.1128/aac.32.12.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Clercq E. The next ten stories on antiviral drug discovery (part E): advents, advances, and adventures. Med. Res. Rev. 2011;31:118–160. doi: 10.1002/med.20179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pauwels R, Balzarini J, Schols D, et al. Phosphonylmethoxyethyl purine derivatives, a new class of anti-human immunodeficiency virus agents. Antimicrob. Agents Chemother. 1988;32:1025–1030. doi: 10.1128/aac.32.7.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Clercq E, Holý A, Rosenberg I. Efficacy of phosphonylmethoxyalkyl derivatives of adenine in experimental herpes simplex virus and vaccinia virus infections in vivo . Antimicrob. Agents Chemother. 1989;33:185–191. doi: 10.1128/aac.33.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heijtink RA, Kruining J, de Wilde GA, Balzarini J, De Clercq E, Schalm SW. Inhibitory effects of acyclic nucleoside phosphonates on human hepatitis B virus and duck hepatitis B virus infections in tissue culture. Antimicrob. Agents Chemother. 1994;38:2180–2182. doi: 10.1128/aac.38.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N. Engl. J. Med. 2003;348:800–807. doi: 10.1056/NEJMoa021812. [DOI] [PubMed] [Google Scholar]
- 9.Balzarini J, Holý A, Jindrich J, et al. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob. Agents Chemother. 1993;37:332–338. doi: 10.1128/aac.37.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holý A, Votruba I, Masojídková M, et al. 6-[2-(Phosphonomethoxy)alkoxy]pyrimidines with antiviral activity. J. Med. Chem. 2002;45:1918–1929. doi: 10.1021/jm011095y. [DOI] [PubMed] [Google Scholar]
- 11.Balzarini J, Pannecouque C, De Clercq E, et al. Antiretrovirus activity of a novel class of acyclic pyrimidine nucleoside phosphonates. Antimicrob. Agents Chemother. 2002;46:2185–2193. doi: 10.1128/AAC.46.7.2185-2193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman BD, Votruba I, Holý A, Sluis-Cremer N, Balzarini J. The acyclic 2,4-diaminopyrimidine nucleoside phosphonate acts as a purine mimetic in HIV-1 reverse transcriptase DNA polymerization. J. Biol. Chem. 2010;285:12101–12108. doi: 10.1074/jbc.M109.096529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dracínský M, Krecmerová M, Holý A. Study of chemical stability of antivirally active 5-azacytosine acyclic nucleoside phosphonates using NMR spectroscopy. Bioorg. Med. Chem. 2008;16:6778–6782. doi: 10.1016/j.bmc.2008.05.058. [DOI] [PubMed] [Google Scholar]
- 14.Naesens L, Andrei G, Votruba I, et al. Intracellular metabolism of the new antiviral compound 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine. Biochem. Pharmacol. 2008;76:997–1005. doi: 10.1016/j.bcp.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Krecmerová M, Holý A, Pískala A, et al. Antiviral activity of triazine analogues of 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine (cidofovir) and related compounds. J. Med. Chem. 2007;50:1069–1077. doi: 10.1021/jm061281+. [DOI] [PubMed] [Google Scholar]
- 16.Cihlar T, Ray AS, Boojamra CG, et al. Design and profiling of GS-9148, a novel nucleotide analog active against nucleoside-resistant variants of human immunodeficiency virus type 1, and its orally bioavailable phosphonoamidate prodrug, GS-9131. Antimicrob. Agents Chemother. 2008;52:655–665. doi: 10.1128/AAC.01215-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Song Z, Yu H, Liu K, Ma X. Adenine: an important drug scaffold for the design of antiviral agents. Acta Pharm. Sin. B. 2015;5:431–441. doi: 10.1016/j.apsb.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White K, Margot N, Stray K, et al. Presented at: The Conference on Retroviruses and Opportunistic Infections. WA, USA: 13–16 February 2017. GS-9131 is a novel NRTI with activity against NRTI-resistant HIV-1. [Google Scholar]
- 19.Merta A, Votruba I, Jindrich J, et al. Phosphorylation of 9-(2-phosphonomethoxyethyl)adenine and 9-(S)-(3-hydroxy-2-phosphonomethoxypropyl)adenine by AMP(dAMP) kinase from L1210 cells. Biochem. Pharmacol. 1992;44:2067–2077. doi: 10.1016/0006-2952(92)90110-5. [DOI] [PubMed] [Google Scholar]
- 20.Cihlar T, Chen MS. Identification of enzymes catalyzing two-step phosphorylation of cidofovir and the effect of cytomegalovirus infection on their activities in host cells. Mol. Pharmacol. 1996;50:1502–1510. [PubMed] [Google Scholar]
- 21.Xiong X, Smith JL, Chen MS. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation. Antimicrob. Agents Chemother. 1997;41:594–599. doi: 10.1128/aac.41.3.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balzarini J, Hao Z, Herdewijn P, Johns DG, De Clercq E. Intracellular metabolism and mechanism of anti-retrovirus action of 9-(2-phosphonylmethoxyethyl)adenine, a potent anti-human immunodeficiency virus compound. Proc. Natl Acad. Sci. USA. 1991;88:1499–1503. doi: 10.1073/pnas.88.4.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hostetler KY. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: current state of the art. Antiviral Res. 2009;82:A84–A98. doi: 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starrett JE, Jr, Tortolani DR, Hitchcock MJ, Martin JC, Mansuri MM. Synthesis and in vitro evaluation of a phosphonate prodrug: bis(pivaloyloxymethyl) 9-(2-phosphonylmethoxyethyl)adenine. Antiviral Res. 1992;19:267–273. doi: 10.1016/0166-3542(92)90084-i. [DOI] [PubMed] [Google Scholar]
- 25.Robbins BL, Srinivas RV, Kim C, Bischofberger N, Fridland A. Anti-human immunodeficiency virus activity and cellular metabolism of a potential prodrug of the acyclic nucleoside phosphonate 9-R-(2-phosphonomethoxypropyl)adenine (PMPA), bis(isopropyloxymethylcarbonyl)-PMPA. Antimicrob. Agents Chemother. 1998;42:612–617. doi: 10.1128/aac.42.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee WA, He GX, Eisenberg E, et al. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob. Agents Chemother. 2005;49:1898–1906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Painter GR, Almond MR, Trost LC, et al. Evaluation of hexadecyloxypropyl-9-R-[2-(Phosphonomethoxy)propyl]- adenine, CMX157, as a potential treatment for human immunodeficiency virus type 1 and hepatitis B virus infections. Antimicrob. Agents Chemother. 2007;51:3505–3509. doi: 10.1128/AAC.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Callebaut C, Stepan G, Tian Y, Miller MD. In vitro virology profile of tenofovir alafenamide, a novel oral prodrug of tenofovir with improved antiviral activity compared to that of tenofovir disoproxil fumarate. Antimicrob. Agents Chemother. 2015;59:5909–5916. doi: 10.1128/AAC.01152-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Debarge S, Balzarini J, Maguire AR. Design and synthesis of α-carboxy phosphononucleosides. J. Org. Chem. 2011;76:105–126. doi: 10.1021/jo101738e. [DOI] [PubMed] [Google Scholar]
- 30.Keane SJ, Ford A, Mullins ND, et al. Design and synthesis of α-carboxy nucleoside phosphonate analogues and evaluation as HIV-1 reverse transcriptase-targeting agents. J. Org. Chem. 2015;80:2479–2493. doi: 10.1021/jo502549y. [DOI] [PubMed] [Google Scholar]; • First report on the chemical synthesis of the cyclic α-carboxynucleoside phosphonates (α-CNP).
- 31.Balzarini J, Das K, Bernatchez JA, et al. Alpha-carboxy nucleoside phosphonates as universal nucleoside triphosphate mimics. Proc. Natl Acad. Sci. USA. 2015;112:3475–3480. doi: 10.1073/pnas.1420233112. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First report on the kinetics and detailed structural mechanisms of the direct-acting CNPs.
- 32.John J, Kim Y, Bennett N, et al. Pronounced inhibition shift from HIV reverse transcriptase to herpetic DNA polymerases by increasing the flexibility of α-carboxy nucleoside phosphonates. J. Med. Chem. 2015;58:8110–8127. doi: 10.1021/acs.jmedchem.5b01180. [DOI] [PMC free article] [PubMed] [Google Scholar]; • First report on the chemical synthesis of the acyclic α-CNPs.
- 33.Vince R, Hua M, Brownell J, et al. Potent and selective activity of a new carbocyclic nucleoside analog (carbovir: NSC 614846) against human immunodeficiency virus in vitro . Biochem. Biophys. Res. Commun. 1988;156:1046–1053. doi: 10.1016/s0006-291x(88)80950-1. [DOI] [PubMed] [Google Scholar]
- 34.Daluge SM, Good SS, Faletto MB, et al. 1592U89, a novel carbocyclic nucleoside analog with potent, selective anti-human immunodeficiency virus activity. Antimicrob. Agents Chemother. 1997;41:1082–1093. doi: 10.1128/aac.41.5.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pradère U, Roy V, Montagu A, et al. Synthesis and antiviral evaluation of bis(POM) prodrugs of (E)-[4’-phosphono-but-2’-en-1’-yl]purine nucleosides. Eur. J. Med. Chem. 2012;57:126–133. doi: 10.1016/j.ejmech.2012.08.042. [DOI] [PubMed] [Google Scholar]
- 36.Mullins ND, Maguire NM, Ford A, et al. Exploring the role of the α-carboxyphosphonate moiety in the HIV-RT activity of α-carboxy nucleoside phosphonates. Org. Biomol. Chem. 2016;14:2454–2465. doi: 10.1039/c5ob02507a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balzarini J, Menni M, Das K, et al. Guanine α-carboxy nucleoside phosphonate (G-α-CNP) shows a different inhibitory kinetic profile against the DNA polymerases of human immunodeficiency virus (HIV) and herpes viruses. Biochem. Pharmacol. 2017;136:51–61. doi: 10.1016/j.bcp.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First demonstration of the different nature of kinetics of α-CNPs against retroviral versus herpetic DNA polymerases.
- 38.Maguire NM, Ford A, Balzarini J, Maguire AR. Synthesis of Guanine α-carboxy nucleoside phosphonate (G-α-CNP), a direct inhibitor of multiple viral DNA polymerases. J. Org. Chem. 2018;83:10510–10517. doi: 10.1021/acs.joc.8b01124. [DOI] [PubMed] [Google Scholar]; • First report on an alternative synthetic pathway to synthesize the guanine α-CNP analog.
- 39.Das K, Balzarini J, Miller MT, Maguire AR, DeStefano JJ, Arnold E. Conformational states of HIV-1 reverse transcriptase for nucleotide incorporation vs pyrophosphorolysis-binding of foscarnet. ACS Chem. Biol. 2016;11:2158–2164. doi: 10.1021/acschembio.6b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides the structural information on how a pyrophosphate analog would bind relative to α-CNP.
- 40.Jochmans D, Deval J, Kesteleyn B, et al. Indolopyridones inhibit human immunodeficiency virus reverse transcriptase with a novel mechanism of action. J. Virol. 2006;80:12283–12292. doi: 10.1128/JVI.00889-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajotte D, Tremblay S, Pelletier A, et al. Identification and characterization of a novel HIV-1 nucleotide-competing reverse transcriptase inhibitor series. Antimicrob. Agents Chemother. 2013;57:2712–2718. doi: 10.1128/AAC.00113-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tremblay M, Bethell RC, Cordingley MG, et al. Identification of benzofurano[3,2-d]pyrimidin-2-ones, a new series of HIV-1 nucleotide-competing reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2013;23:2775–2780. doi: 10.1016/j.bmcl.2013.02.042. [DOI] [PubMed] [Google Scholar]
- 43.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 44.Sarafianos SG, Das K, Clark AD, Jr, et al. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc. Natl Acad. Sci. USA. 1999;96:10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das K, Xiong X, Yang H, et al. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC) J. Virol. 2001;75:4771–4779. doi: 10.1128/JVI.75.10.4771-4779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]