

Abstract
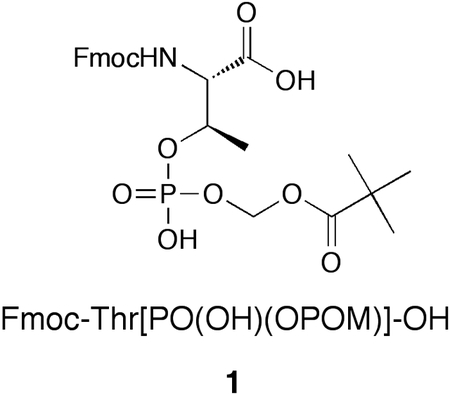
The design and efficient synthesis of N-Fmoc phosphothreonine bearing mono-pivaloyloxymethyl (POM) protection of its phosphoryl group [Fmoc-Thr[PO(OH)(OPOM)]-OH, (1)] is reported. This reagent is suitable for use in solid-phase synthesis employing acid-labile resin and Fmoc-based protocols. It allows the preparation of phosphothreonine (pThr)-containing peptides bearing bis-POM phosphoryl protection. The methodology allows the first reported synthesis of pThr-containing polypeptides having bio-reversible prodrug protection and as such it should be useful in a variety of biological applications.
Keywords: phosphothreonine, prodrug, solid-phase, peptide synthesis, signal transduction
Graphical Abstract
Introduction. –
Post-translational modification by phosphorylation plays central roles in cellular signaling. Phosphoryl esterification of proteins on the side chain hydroxyls of threonine, serine and tyrosine residues to yield the corresponding phosphothreonine (pThr), phosphoserine (pSer) and phosphotyrosine (pTyr) species can introduce unique molecular features that facilitate signaling cascades, often by promoting specific protein-protein interactions (PPIs) [1–3]. The contributions that aberrations in protein phosphorylation can make to the etiology of a number of diseases, including cancers [4] [5], have rendered synthetic phosphopeptides as valuable pharmacological tools for studying both normal and pathological processes [6] [7]. However, in cellular systems the bioavailabilty of such phosphopeptides may be limited by poor membrane transport due to the di-anionic character of the phosphoryl species [8–10]. Many approaches have been investigated to enhance cellular bioavailability of organic phosphates. The most common methodologies involve masking the phosphoryl moiety with “prodrug” protecting groups that can be removed biologically within the cell [11]. The pivaloyloxymethyl (POM) moiety is an esterase-cleavable group that has found wide utility in phosphoryl prodrug protection, particularly in nucleotides [12] and in phosphate and phosphonic acid functionality in peptide mimetics [13–16]. In spite of its usefulness, there have been no reports of pThr or pSer reagents that utilize phosphoryl-POM protection in forms suitable for direct solid-phase synthesis. This may be one reason why there have been no reports of pThr or pSer-containing polypeptides bearing POM phosphoryl protection. Furthermore, to our knowledge, there are no reports of polypeptides containing pThr bearing bio-reversible protection of any kind. Given the physiological importance of pThr-containing sequences [3], there is a considerable need for a reagent that would permit the solid-phase synthesis of peptides containing POM-protected pThr residues. Accordingly, we report herein the preparation of Fmoc-Thr[PO(OH)(OPOM)]-OH (1) as a new reagent for the synthesis of peptides containing pThr with its phosphoryl group in fully protected bis-POM prodrug form, i.e. Thr[PO3(POM)2].
Results and Discussion. –
Reagent Design.
When considering protocols for the incorporation of Thr[PO3(POM)2] into peptides, we noted that the solid-phase synthesis of pTyr-containing peptides can be achieved in the absence of phosphoryl protection using Fmoc-Tyr(PO3H2)-OH [17]. Accordingly, we envisioned that one potential route to the synthesis of Thr[PO3(POM)2]-containing peptides could involve using unprotected Fmoc-Thr(PO3H2)-OH, which would result in resin-bound peptides bearing full side chain protection except for a free pThr phosphoryl group. In this approach, in situ introduction of phosphoryl POM-protection (‘POMylation’) would be carried out immediately prior to cleavage of the peptide from the resin. However, when we investigated this approach, we found that although initial coupling of unprotected Fmoc-Thr(PO3H2)-OH occurred cleanly, further peptide synthesis through the introduction of additional amino acid residues followed by resin cleavage, led to very low yields of isolated pThr-containing peptides. This indicated that peptide chain elongation in the presence of unprotected Thr(PO3H2) was problematic.
In further studies we observed that treatment of resin-bound Fmoc-Thr(PO3H2) with excess pivaloyloxymethyl iodide (POMI) [18] and diisopropyl(ethyl)amine (DIPEA) followed by resin cleavage (1% TFA in CH2Cl2) cleanly provided Fmoc-Thr[PO3(POM)2]-amide. This demonstrated both the ability to perform on-resin POM-protection of the phosphoryl group and the stability of the resulting Thr[PO3(POM)2] residue to the conditions of resin cleavage. However, we found that piperidine-mediated Fmoc-deprotection of resin-bound Fmoc-Thr[PO3(POM)2] followed by 1% TFA resin cleavage gave the Thr[PO(OH)(OPOM)]-containing peptide, indicating that mono-POM deprotection had occurred in the presence of piperidine. This is consistent with the reported ability of piperidine to selectively cleave a single POM group from the triester PO(OPOM)3 to yield PO(OH)(POM)2 [19]. The instability of pThr phosphoryl bis-esters to piperidine treatment is one reason why incorporation of pThr and pSer residues on acid-labile resins is normally done using commercially-available Fmoc derivatives bearing mono-benzyl-protection of the phosphoryl group [Fmoc-Thr[PO(OH)(OBn)]-OH and Fmoc-Ser[PO(OH)(OBn)]-OH, respectively (reviewed in [20–22]). Based on this information, we concluded that synthesis of Thr[PO3(POM)2]-containing peptides could best be achieved using Fmoc-Thr[PO(OH)(OPOM)]-OH (1) as a reagent, with conversion of the Thr[PO(OH)(OPOM)] residue to the desired fully-protected Thr[PO3(POM)2] form by POMI/DIPEA treatment after completion of peptide synthesis and Fmoc deprotection but immediately prior to final resin cleavage. It should be noted that the current protocol provides N-terminally acetylated peptides. To obtain peptides having a free amine at the N-terminus, the procedure can be altered through the use of terminal N-Boc-protection, in which ‘POMylation’ is undertaken with maintenance of Boc protection. Desired peptides bearing a free terminal amine can then be obtained by cleavage under conditions that are compatible with Boc removal and retention of phosphoryl POM protection (95% TFA, unreported results).
Synthesis of Fmoc-Thr[PO(OH)(OPOM)]-OH (1) from Commercially-Available Fmoc-Thr[PO(OH)(OBn)]-OH.
Benzyl esterification of the carboxylic acid of commercially available Fmoc-Thr[PO(OH)(OBn)]-OH was achieved by treatment with benzyl alcohol and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) in the presence of 4-dimethylaminopyridine (DMAP) (Scheme 1). Reaction of the resulting Fmoc-Thr[PO(OH)(OBn)]-OBn (2) with excess POMI (formed from the corresponding commercially-available chloride by halide exchange using NaI in MeCN [18]) and DIPEA resulted in esterification of the free phosphoryl OH-group to yield Fmoc-Thr[PO(OBn)(OPOM)]-OBn (3) as a 1:1 mixture of phosphoryl diastereomers. Subsequent hydrogenolytic cleavage of all benzyl groups cleanly yielded the desired reagent 1 (Scheme 1).
Scheme 1.

Reagents and conditions: (i) BnOH (1.5 equiv), DMAP (1.0 equiv), EDCI (2.5 equiv), CH2Cl2, 0° to r.t., 2 h, 70% yield; (ii) iodomethylpivalate (POMI) (2.0 equiv), DIPEA (2.0 equiv), DMF, r.t., 12 h, 60% yield; (iii) 10% Pd•C, MeOH, r.t., 1 h, 68% yield.
Utilization of Fmoc-Thr[PO(OH)(OPOM)]-OH (1) for the Synthesis of Ac-Pro-Leu-His-Ser-Thr[PO3(POM)2]-amide (4).
In order to demonstrate the applicability of 1 for the synthesis of Thr[PO3(POM)2]-containing peptides, we chose as a target the pentapeptide “Pro-Leu-His-Ser-pThr”, which has been reported as a high affinity ligand of the polo-like kinase 1 (Plk) polo box domain (PBD) [23–25]. PBD-binding ligands may potentially serve as anti-cancer agents by blocking the spatial organization required for Plk1 to function in oncogenic processes [26]. The synthesis of Ac-Pro-Leu-His-Ser-Thr[PO3(POM)2]-amide (4) was accomplished on NovaSyn TG Sieber resin using reagent 1 and standard Fmoc protocols. Histidine and serine were employed in their methyltrityl (Mtt) and trityl side chain-protected forms, respectively, to allow selective on-resin cleavage using 1% TFA (Scheme 2). Following peptide formation and amino-terminal acetylation of the Pro residue, the resin 5 was treated with POMI (10 equiv.) and DIPEA (10 equiv.) in DMF (4 h). Reaction products were then cleaved from the resin (1% TFA) and purified by reversed phase HPLC to provide two peptides with retention times of t = 18.8 min (peptide 6) and t = 20.9 min (peptide 4), respectively. Both peptides exhibited molecular weights and elemental compositions (HRMS) corresponding to the desired product, Ac-Pro-Leu-His-Ser-Thr(PO3POM2)-amide (4).
Scheme 2.

Reagents and conditions: NovaSyn®TG Sieber resin; (i) POMI, DIPEA, DMF, 4 h; (ii) 1% TFA, CH2Cl2.
MS/MS Analysis of Peptides 4 and 6.
High resolution MS/MS analysis of the MH+ (m/z 903.4) ions of peptides 4 and 6 indicated that, while both peptides contained two POM groups, the locations of one of the POM moieties differed for each. The MS/MS fragmentation pattern of the later eluting peptide (Fig. 1a) was consistent with product 4, in which both POM groups are on the phosphoryl moiety. With this peptide, the primary MS/MS product ion at m/z 559.2997 resulted from concerted loss of the elements of (HO)PO3(POM)2 and H2O from MH+, which can only occur if the phosphate is di-POMylated. In addition, b3 and b4 product ions were also observed that provided strong evidence for an unaltered Ac-Pro-Leu-His-Ser-sequence (Fig. 1). The MS/MS spectrum of the faster eluting peptide (Fig. 1b) strongly suggested that both the phosphoryl group and histidine residue each contained a single POM adduct (6, Scheme 2). In this spectrum, the most abundant MS/MS product ion at m/z 771.3430 resulted from loss of t-C4H9CO2H and CH2O from MH+, which can only occur for a ‘POMylated’ phosphate and not for a ‘POMylated’ histidine. The subsequent loss of HPO3 (m/z 691.3774) and the elements of HPO3 and H2O (m/z 673.3670) further corroborated the presence of only one POM moiety on the phosphate (Fig. 1). The absence of b3 and b4 product ions expected for an unaltered Ac-Pro-Leu-His-Ser-sequence was additional evidence that the histidine residue had been modified.
Fig. 1.

MS/MS spectra of peptides 4 [spectrum (a)] and 6 [spectrum (b)] obtained by collision-induced dissociation (CID) of the MH+ (m/z 903.4, 3.0 Da selection window) of each peptide.
The ‘POMylation’ of histidine to yield peptide 6 was not unexpected, since we had previously observed alkylation of the imidazole N(π) atom in histidine residues bearing standard trityl protection of the N(τ) atom [24] [27]. While alkylation of histidine does represent an unwanted source of side product formation, a search of the literature reveals that in general, alkyl halide-mediated modification of side chain-protected peptides on solid support is not a significant concern. Of greater consideration regarding the chemistry reported herein is that it presents the first and so far the only method of preparing polypeptides having pThr residues in fully-protected bio-cleavable form. This uniqueness overcomes limitation associated with the methodology.
Conclusions. –
Our current work reports the design, synthesis and application of Fmoc-Thr[PO(OH)(OPOM)]-OH (1) as a reagent for the facile solid-phase preparation of peptides bearing pThr residues in the fully-protected Thr[PO3(POM)2] form. To our knowledge, this represents the first methodology for preparing biolabile-protected pThr residues within a polypeptide platform. Our approach should have significant utility in a variety of biological applications.
Experimental Part
General Methods.
All experiments involving moisture-sensitive compounds were conducted under anhydrous conditions (positive Ar pressure) using standard syringe, cannula, and septa apparatus. Fmoc-Ser(Trt)-OH, Fmoc-His(Mtt)-OH, and Fmoc-Thr[PO(OH)((OBn)]-OH were purchased from Novabiochem. All solvents were purchased in anhydrous form (Aldrich) and used directly. HPLC-grade hexanes, AcOEt, CH2Cl2, MeCN and MeOH were used for both column chromatography (CC) and preparative high performance chromatograhy (HPLC). LC/MS-grade solvents (Burdick and Jackson Brand, Honeywell), ultra-high purity acids (HCO2H, AcOH) and buffers (NH4HCO2, NH4OAc) were employed for all LC/MS analyses. Analytical TLCs were performed using Analtech precoated plates (Uniplate, silica gel GHLF, 250 nm) containing a fluorescence indicator. NMR spectra were recorded using a Varian Inova 400 MHz spectrometer. Coupling constants are reported in Hz, and chemical shifts are ppm relative to TMS. Low resolution, electrospray ionization (ESI) mass spectra were obtained on an Agilent 1200/1100 LC/MSD single quadrupole system, equipped with an in-line UV diode-array detector (DAD), to assess compound identity and homogeneity. HR-MS analyses were conducted on a Thermo-Fisher LTQ-Orbitrap-XL hybrid mass spectrometer system operated at a resolution of 30,0000 (FWHM) in either positive or negative ion mode, depending on which mode was the most suitable based on previous low resolution analyses. For LC/MS analyses on the Orbitrap, a narrow-bore (50 × 2.1 mm), Zorbax Rapid-Resolution reversed-phase C18 column coupled with a C18 guard column (12.5 × 2.1 mm) was eluted at 250 μl/min. with a 5–90% gradient of MeCN/H2O containing either 0.1% HCOOH or 1 mM NH4HCO2. The resulting accurate mass measurement of a molecular species ([M+H]+ or [M-H]¯) was then used to determine a unique elemental composition for each particular compound. Where appropriate, 1H and 13C NMR data were used to set elemental constraints for this calculation. For peptides 4 and 6, MS/MS spectra of MH+ ions were obtained at a resolution of 15,000 (FWHM) by both CID and HCD fragmentation. The resulting high resolution product ion spectra were used to confirm peptide sequences and determine structural assignments.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-L-threonine [Phenylmethyl Hydrogen Phosphate (Ester)] Phenylmethyl Ester (2).
Commercially available Fmoc-Thr[PO(OH)((OBn)]-OH (1.0 g, 1.96 mmol), BnOH (0.30 ml, 2.93 mmol), and DMAP (0.24 g, 1.96 mmol) were dissolved in CH2Cl2 (20 ml) and the mixture was cooled to 0°. To the cold sol. was added EDCI (0.94 g, 4.89 mmol) and the mixture was warmed to r.t. and stirred (2 h). The reaction was quenched by the addition of aqueous 1N HCl (10 ml) and extracted with CH2Cl2. The combined org. layer was washed with 1 N HCl and brine, dried (MgSO4), concentrated and the residue was purified by CC, SiO2 (CH2Cl2/MeOH from 20/1 to 4/1) to afford 2 as a white semisolid (0.82 g, 70% yield). [α]D 24.8 (c 1.65, CHCl3). UV (DAD, MeCN/H2O) λmax = 264 nm, 289 nm, 299 nm (Fmoc moiety). 1H – NMR (400 MHz, CDCl3): 8.81 (brs, 1H), 7.78 (d, J = 8.0, 2H), 7.62 (dd, J = 12.0, 8.0, 2H); 7.41 (t, J = 8.0, 2H); 7.38 – 7.27 (m, 12H); 5.91 (d, J = 8.0, 1H); 5.26 (d, J = 12.0, 1H); 5.11 – 4.94 (m, 4H); 4.54 (d, J = 12.0, 1H); 4.44 – 4.33 (m, 2H); 4.22 (t, J = 8.0, 1H); 1.38 (d, J = 8.0, 3H). 13C – NMR (100 MHz, CDCl3): 169.7, 156.9, 144.1, 143.7, 141.4, 135.74, 135.68, 135.1, 128.8, 128.7, 128.0, 127.9, 127.3, 125.3, 120.1, 75.6, 69.5, 68.1, 67.6, 58.7, 47.2, 18.7. ESI-HRMS: calcd for C33H31NO8P [M-H]−: 600.1787, found: 600.1782.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-L-threonine [(2,2-Dimethyl-1-oxopropoxy)methyl Phenylmethyl Phosphate (Ester)] Phenylmethyl Ester (3).
Compound 2 (0.3 g, 0. 5 mmol) in DMF (5.0 ml) under Ar was treated with DIPEA (0.174 ml, 1.00 mmol) followed by iodomethylpivalate (POMI) [18] (0.241 g, 1.00 mmol) and the mixture was stirred at r.t. (overnight). The mixture was partitioned (H2O/AcOEt) and the organic layer was washed with H2O and brine, dried (MgSO4), concentrated and the residue was purified by CC, SiO2 (AcOEt: hexanes from 1/2 to 2/1) to afford 3 as a white semi-solid (0.21 g, 60% yield) as a mixture of two diastereoisomers (ratio 1:1 as determined by 1H – NMR).
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-L-threonine [(2,2-Dimethyl-1-oxopropoxy)methyl Hydrogen Phosphate (Ester)] [Fmoc-Thr[PO(OH)(OPOM)]-OH] (1).
A sol. of 3 (45 mg, 0.063 mmol) in CH3OH (1.25 ml) was hydrogenated over 10% Pd•C (5 mg, 1 h), then filtered and concentrated and the residue was purified by CC, SiO2 (CH2Cl2/MeOH = 10/1 to 4/1) to afford 1 as a colorless oil (23 mg, 68% yield). [α]D 4.98 (c 0.34, MeOH). UV (DAD, MeCN/H2O) λmax = 264 nm, 289 nm, 299 nm (Fmoc moiety). 1H – NMR (400 MHz, CD3OD): 7.80 (d, J = 8.0, 2H); 7.70 (t, J = 8.0, 2H); 7.39 (t, J = 8.0, 2H); 7.31 (tt, J = 8.0 Hz, 4.0, 2H); 5.58 (d, J = 16.0, 2H); 5.04 – 4.96 (m, 1H); 4.43 – 4.35 (m, 3H); 4.26 (t, J = 4.0, 1H); 1.39 (d, J = 8.0, 3H); 1.23 (s, 9H). 13C – NMR (100 MHz, CD3OD): 178.4, 172.7, 159.1, 145.4, 142.7, 128.9, 128.3, 126.4, 121.1, 84.0, 76.4, 68.4, 60.1, 60.0, 39.9, 27.4, 19.0. ESI-HRMS: calcd for C25H29NO10P [M-H]−: 534.1529, found: 534.1524.
Synthesis of Ac-Pro-Leu-His-Ser-Thr(PO2POM2)-amide (4).
Peptide 4 was synthesized on NovaSyn®TG Sieber resin (Novabiochem, cat. no. 01-64-0092) using reagent 1 under standard Fmoc-based protocols employing N-methyl-2-pyrrolidone (NMP) as solvent and 1-O-benzotriazole-N,N,N’,N’-tetramethyluronium hexafluoro-phosphate (HBTU) (5.0 equiv), hydroxybenzotriazole (HOBT) (5.0 equiv) and N,N-diisopropylethylamine (DIPEA) (10.0 equiv) as coupling reagents. Amino terminal acetylation was achieved using 1-acetylimidazole. Following acetylation the resin (0.1 mmol) was treated with POMI (242 mg, 1.0 mmol) and DIPEA (0.174 ml, 1.0 ml) in DMF (3 ml) (4 h). The resulting resin (0.1 mmol) was washed with DMF, MeOH, CH2Cl2 and Et2O and then dried under vacuum (overnight). The peptide was cleaved from the resin using 1% TFA in CH2Cl2 (10 min; repeated five times). Resin was removed by filtration, the filtrate was concentrated under vacuum, precipitated with Et2O and the precipitate was washed with cold Et2O. The resulting solid was dissolved in 50% aq. MeCN (5 ml) and purified by reverse phase preparative HPLC using a Phenomenex C18 column (250 × 21 mm) with a linear gradient from 20% aq. MeCN (0.1% TFA) to 90% CH3CN (0.1% trifluoroacetic acid) over 30 min at a flow rate of 10.0 ml/min. Lyophilization provided 4 as a white powder (4.8 mg) (>99% pure by analytical HPLC). ESI-HRMS: calcd for C38H64N8O15P (M+H)+: 903.4229, found: 903.4227. Byproduct 6 was also isolated as a white powder (7.0 mg) (>99% purity by analytical). ESI-HRMS m/z calcd for C38H64N8O15P [M+H]+: 903.4229, found: 903.4227.
MS/MS Analysis of Peptides 4 and 6.
Collision-induced dissociation (CID) product ion mass spectra for the MH+ (m/z 903.4, 3.0 Da selection window) of peptide isomers 4 and 6 were generated by fragmentation in the LTQ at an energy of 30 and analysis in the Orbitrap at a resolution of 15,000 (FWHM) over the maximum allowable mass range. Each spectrum shown in Fig. 1 was the average of 10 or more scans and resulted from an independent LC/MS/MS analysis of individual, HPLC-purified pentapeptide. A shallow linear MeCN/H2O gradient (2 – 47% MeCN in 20 min) followed by a steeper linear gradient (47 – 90% in 10 min) was employed on a 3-μm, 150 × 2.0 mm Cadenza CD-C18 HPLC column (Imtakt USA) at a flow rate of 250 μl/min for the separation and elution of these POM-peptide derivatives.
Acknowledgements
This work was supported by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health.
REFERENCES
- [1].Yaffe MB, Nat. Rev. Mol. Cell Biol 2002, 3, 177. [DOI] [PubMed] [Google Scholar]
- [2].Ladbury JE, Protein Rev 2005, 3, 165. [Google Scholar]
- [3].Elia AEH, Yaffe MB. In Modular Protein Domains; Cesare G, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; p. 163. [Google Scholar]
- [4].Chong P-K, Lee H, Kong JW-F, Loh MC-S, Wong C-H, Lim Y-P, Proteomics 2008, 8, 4370. [DOI] [PubMed] [Google Scholar]
- [5].Pawson T, Kofler M, Curr. Opin. Cell Biol 2009, 21, 147. [DOI] [PubMed] [Google Scholar]
- [6].Eisele F, Owen DJ, Waldmann H, Bioorg. Med. Chem 1999, 7, 193. [DOI] [PubMed] [Google Scholar]
- [7].Lu CHS, Liu K, Tan LP, Yao SQ, Chem. Eur. J 2012, 18, 28. [DOI] [PubMed] [Google Scholar]
- [8].Allentoff A, Mandiyan S, Liang H, Yuryev A, Vlattas I, Duelfer T, Sytwu I-I, Wennogle L, Cell Biochem. Biophys 1999, 31, 129. [DOI] [PubMed] [Google Scholar]
- [9].Dunican DJ, Doherty P, Biopolymers 2001, 60, 45. [DOI] [PubMed] [Google Scholar]
- [10].Richter S, Bergmann R, Pietzsch J, Ramenda T, Steinbach J, Wuest F, Biopolymers 2009, 92, 479. [DOI] [PubMed] [Google Scholar]
- [11].Schultz C, Bioorg. Med. Chem 2003, 11, 885. [DOI] [PubMed] [Google Scholar]
- [12].Hecker SJ, Erion MD, J. Med. Chem 2008, 51, 2328. [DOI] [PubMed] [Google Scholar]
- [13].Stankovic CJ, Surendran N, Lunney EA, Plummer MS, Para KS, Shahripour A, Fergus JH, Marks JS, Herrera R, Hubbell SE, Humblet C, Saltiel AR, Stewart BH, Sawyer TK, Bioorg. Med. Chem. Lett 1997, 7, 1909. [DOI] [PubMed] [Google Scholar]
- [14].Mandal PK, Liao WSL, McMurray JS, Org. Lett 2009, 11, 3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC, Liao WS, McMurray JS, J. Med. Chem 2011, 54, 3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhao S, Etzkorn FA, Bioorg. Med. Chem. Lett 2007, 17, 6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ottinger EA, Shekels LL, Bernlohr DA, Barany G, Biochemistry 1993, 32, 4354. [DOI] [PubMed] [Google Scholar]
- [18].Bandgar BP, Sarangdhar RJ, Viswakarma S, Ahamed FA, J. Med. Chem 2011, 54, 1191. [DOI] [PubMed] [Google Scholar]
- [19].Hwang Y, Cole PA, Org. Lett 2004, 6, 1555. [DOI] [PubMed] [Google Scholar]
- [20].McMurray JS, Coleman DRIV, Wang W, Campbell ML, Biopolymers 2001, 60, 3. [DOI] [PubMed] [Google Scholar]
- [21].Attard TJ, O’Brien-Simpson N, Reynolds EC, Int. J. Pept. Res. Ther 2007, 13, 447. [Google Scholar]
- [22].Toth GK, Kele Z, Varadi G, Curr. Org. Chem 2007, 11, 409. [Google Scholar]
- [23].Yun S-M, Moulaei T, Lim D, Bang JK, Park J-E, Shenoy SR, Liu F, Kang YH, Liao C, Soung N-K, Lee S, Yoon D-Y, Lim Y, Lee D-H, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR Jr., Yaffe MB, Wlodawer A, Lee KS, Nat. Struct. Mol. Biol 2009, 16, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu F, Park J-E, Qian W-J, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR Jr., Nat. Chem. Biol 2011, 7, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu F, Park J-E, Qian W-J, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR Jr., ChemBioChem 2012, 13, 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].van de Weerdt BCM, Littler DR, Klompmaker R, Huseinovic A, Fish A, Perrakis A, Medema RH, Biochim. Biophys. Acta, Mol. Cell Res 2008, 1783, 1015. [DOI] [PubMed] [Google Scholar]
- [27].Qian W, Liu F, Burke TR Jr., J. Org. Chem 2011, 76, 8885. [DOI] [PMC free article] [PubMed] [Google Scholar]