

Abstract
Ischemic stroke destroys blood–brain barrier (BBB) integrity. There are currently no effective treatments available in the clinical setting. Post-ischemia treatment with phenothiazine drugs [combined chlorpromazine and promethazine (C+P)] has been shown to be neuroprotective in stroke. The present study determined the effect of C+P in BBB integrity. Sprague-Dawley rats were divided into the following groups (n=8 each): (1) stroke, (2) stroke treated by C+P with temperature control, and (3) stroke treated by C+P without temperature control. Infarct volume and neurological deficits were measured to assess the neuroprotective effect of C+P. BBB permeability was determined by brain edema and Evans blue leakage. Expression of BBB integral molecules, including proteins of aquaporin-4 and -9 (AQP-4, AQP-9), matrix metalloproteinase-2 and -9 (MMP-2, MMP-9), zonula occludens-1 (ZO-1), claudin-1/5, occludin, and laminin were determined by Western blot. Stroke caused brain infarction and neurological deficits, as well as BBB damage, which were all attenuated by C+P through drug-induced hypothermia. When the reduced temperature was controlled to physiological levels, C+P still conferred neuroprotection, suggesting a therapeutic effect independent of hypothermia. Furthermore, C+P significantly attenuated the increase in AQP-4, AQP-9, MMP-2, and MMP-9 levels after stroke, and reversed the decrease in tight junction protein (ZO-1, claudin-1/5, occludin) and basal laminar protein (laminin) levels. This study clearly indicates a beneficial effect of C+P on BBB integrity after stroke, which may be independent of drug-induced hypothermia. These findings further prove the clinical target and cell-signal communication of C+P treatment, which may direct us closer toward the development of an efficacious neuroprotective therapy.
Keywords: hibernation-like therapeutic effect, ischemia/reperfusion, AQP-4, AQP-9, MMP-2, MMP-9, ZO-1, claudin-1, claudin-5, occludin and laminin
Introduction
The blood–brain barrier (BBB) is composed of endothelial cells interconnected by tight junctions (TJs) and intervening basal lamina (BL), segregating cerebral blood flow from the peripheral circulation to regulate the brain microenvironment1. It is well known that ischemic stroke causes brain damage by destroying BBB integrity2,3. As a consequence, this altered BBB allows for a stepwise sequence of different edema mechanisms that ultimately leads to BBB leakage4. As the BBB plays such a key role in providing selectivity toward the material transferred between the brain parenchyma and bloodstream, its disruption directly relates to negative neurological outcomes. However, no treatment has been proven effective in maintaining the BBB intact after stroke. Our group previously showed that following 2 h ischemia, phenothiazine drugs (chlorpromazine and promethazine (C+P)) reduced brain injury in ischemic rats5. However, whether C+P reduces BBB damage remains unknown.
Matrix metalloproteinases (MMPs) and aquaporins (AQPs) are known as important components in the pathophysiology of BBB integrity and have become viable candidates for potential pharmacological targets6,7. Members of the MMP family, especially MMP-2 and MMP-9, play critical roles in BBB disruption during the acute phase of ischemic stroke8. Studies have shown that brain ischemia cleaves the extracellular matrix, including major components of the BL and TJs (claudin-1/5, ZO-1, and occludin) between endothelial cells, by increasing the expression of MMPs that lead to a loss of junctional integrity and BBB disruption9. These changes together cause brain vasogenic edema. In addition, the AQP family (known as water channels) has also been proven to play a key role in brain edema progression after stroke by acting as a direct passageway for fluid movement between the brain and blood vessels10. Studies have shown that brain ischemia enhanced the expression of AQP-4 and AQP-9 in the edematous rodent brain11. Furthermore, our previous studies found an association between infarct volume and AQP-4/AQP-9 expression12.
Many studies have shown neuroprotective effects when animals with ischemia/reperfusion injury were maintained in a state of hibernation13. The “hibernation-like” state with decreased energy utilization by using anesthetics has drawn much interest as a potential neuroprotective strategy13,14. In our previous studies, hibernation-like therapy with the FDA-approved C+P was proven to be neuroprotective. In the present study, we further determined whether the effects of C+P on infarct volume reduction and neurological deficits were due to reduction in BBB damage and changes in associated molecules, including AQPs, MMPs, and TJs in ischemia/reperfusion injury.
Materials and Methods
Subjects
A total of 120 adult male Sprague-Dawley rats (Vital River Laboratory Animal Technology Co. Ltd., Beijing, China) were used in this study. An ischemic stroke model was generated by occluding the right middle cerebral artery (MCA) for 2 h. Rats were sacrificed 24 h after reperfusion for edema measurement and 48 h for evaluation of BBB integrity. An additional 24 rats were used for protein expression studies. The animal care and the experiments described here were carried out in agreement with the guidelines set by the Institutional Animal Investigation Committee of the Capital Medical University. Animals were housed under a 12-hour light/dark cycle, and were kept in the same animal care facility for the duration of the study. All efforts were made to minimize any suffering and to reduce the total number of animals used.
Focal Cerebral Ischemia
This model has been described previously by us. Briefly, male Sprague-Dawley rats were initially anesthetized in a chamber with 1–3% isoflurane along with a mixture of 70% nitrous oxide and 30% oxygen. The rats were transferred to a surgical table and anesthesia was maintained with a facemask using 1% isoflurane delivered from a calibrated precision vaporizer. Poly-L-lysine-coated intraluminal nylon (4.0) sutures were used to occlude the MCA and yield consistent infarcts and greatly reduce inter-animal variability.
Chlorpromazine and Promethazine Administration
In 2 h MCA occlusion with reperfusion, the combination of chlorpromazine and promethazine (1:1) at doses of 8 mg/kg in 3 ml saline (as determined by a preliminary study to induce significant neuroprotection) was injected IP at 2 h after the onset of ischemia. A second injection with one-third of the original dose was added 1–2 h later to enhance the drugs’ effects. Two sets of animals were used: one with body temperature maintained at physiological levels (rectal temperature at 36.5–37.5°C) and one without body temperature control. Because of the close correlation between body and brain temperatures15 and increased risk of intracranial hemorrhage, rectal temperature was monitored instead of brain temperature. During the recovery period (24–28 h), in all temperature-controlled groups, rats were placed on a 37°C insulation blanket as well as under a warm light to maintain their temperature. In groups without temperature control, rats were placed in a 25°C environment.
Neurological Deficit
The modified scoring systems (5 and 12 scores) proposed by Longa et al.16 and Belayev et al.17 were used to examine the severity of neurological deficits in rats before surgery, during MCA occlusion, and after 24 h reperfusion. The severity of brain damage and consistency in each group are highly important in this study. After MCA occlusion, the modified scoring systems (five scores) for neurological deficits were used to confirm brain injury. If the scores were 1 or below, the MCA occlusions were considered unsuccessful and the rats were excluded from further studies. In our study, about 10% of animals with MCA occlusion were discarded for this reason.
Cerebral Infarct Volume
After 24 h of reperfusion in rats with 2 h MCA occlusion, the brains were resected from ischemic rats and cut into 2 mm-thick slices (brain matrix) and treated with 2, 3, 5- triphenyltetrazolium chloride (TTC, Sigma, St. Louis, MO, USA) for staining. An indirect method for calculating infarct volume was used to minimize error caused by edema. We also measured and compared infarct size of the cortex and striatum at three different levels from anterior +1.00 mm to posterior –4.8 mm to the bregma of the brain18.
Brain Edema and Brain Barrier Integrity Measurements
Brain edema was evaluated through measurements of tissue water content. Following animal sacrifice, separated right and left hemispheres were immediately weighed to obtain wet weight (WW). The tissue was then dried in an oven at 70°C for 72 h and weighed again to obtain the dry weight (DW). The formula (WW–DW)/WW×100% was used to calculate the water content and is expressed here as a percentage of WW. BBB permeability was determined by measuring the amount of Evans blue extravasation from the ipsilateral and contralateral hemispheres of the brain. Evans blue dye (2%, 4 ml/kg body weight) was slowly administered intravenously 46 h after reperfusion and allowed to circulate for 2 h. The rats were then perfused with normal saline to wash away any remaining dye in the blood vessels, and the two hemispheres of the brain were dissected and frozen at –80°C for later analysis. The Evans blue dye was extracted from tissues by homogenizing and incubating the samples in 2 ml formamide at 54°C for 2 h. The solutions were then centrifuged for 15 min at 12,000 g. The absorption of each supernatant was measured at 620 nm with a spectrophotometer (Agilent/HP 8453, Agilent/HP, San Diego, CA, USA). The content of Evans blue in each sample was expressed as μg/g of brain tissue using a standardized curve19.
Protein Expression
Western blot analysis was used to detect protein expression in the ischemic tissue, as described previously18. Protein extracted from rat brain isolates in the control group and C+P treatment groups with or without temperature control was loaded onto gel for electrophoresis. Upon conclusion of electrophoresis, proteins were transferred to a polyvinylidene fluoride membrane. Membranes were incubated with a primary antibody (rabbit anti-rat MMP-2 antibody, 87809, CST; rabbit anti-rat MMP-9 antibody, AV33090, (Sigma); rabbit anti-rat AQP-4, HPA014784, (Sigma); rabbit anti-rat AQP-9, sc-28623, (Santa Cruz, Dallas, TX, USA); rabbit anti-rat claudin-1, SAB4503545, (Sigma); rabbit anti-rat claudin-5, SAB4502981, (Sigma); rabbit anti-rat occludin, sc-5562, (Santa Cruz); rabbit anti-rat ZO-1, sc-10804, (Santa Cruz)) for 24 h at 4°C. Next, membranes were washed three times with phosphate-buffered saline for 6 minutes each, and re-incubated with a secondary antibody (goat anti-rabbit IgG, (Santa Cruz)) for 1 h at room temperature. An enhanced chemiluminescence system was used to detect immune-reactive bands by luminescence. Western blot images for each antibody, including β-actin, were analyzed using an image analysis program (Image J 1.42, National Institutes of Health, USA) to quantify protein expression in terms of relative image density. The mean amount of protein expression from the control group after stroke was assigned a value of 1 to serve as reference.
Statistical Analysis
Statistical analysis was performed with SPSS for Windows, version 17.0 (SPSS, Inc., Chicago, IL, USA). The differences among groups were assessed using one-way ANOVA with a significance level at P<0.05. Post hoc comparison among groups was further performed using the least significant difference.
Results
Physiological Parameters
There were no significant differences in blood pH, pO2, pCO2, or blood glucose between the groups (data not shown).
Body Temperature
In wild-type (data not shown) or 2 h MCA occlusion rats, body temperatures were significantly reduced by 1–2°C after C+P administration, with 8 mg/kg doses achieving this value within as early as 5 min (Fig. 1A). At about 2 h after administration, body temperatures reached 33–34°C. These temperatures remained significantly low for up to 6 h and subsequently returned to normal levels.
Fig. 1.

(A) In 2 h MCA occlusion groups, body temperatures (n=8 per group) were measured in a time-dependent manner. ANOVA analyses indicated that C+P significantly (##P<0.01) reduced body temperature as early as within 5 min and lasting up to 12 h after stroke onset. (B) Infarct volume reduction by drug-induced hypothermia from C+P. TTC histology demonstrating infarct volume reduction in the penumbra region of ischemic territory supplied by MCA with C+P± temperature control after 2 h MCA occlusion. (C) Quantification of infarct volume reduction by C+P. Treatments without temperature control were allowed to reach drug-induced hypothermia as indicated in Fig. 1A and demonstrated slightly greater but not significant infarct volume reduction than subjects maintained at 37°C.
Infarct Volume
A large infarct volume (50.3%) was seen after 2 h MCA occlusion and 48 h reperfusion. The 8 mg/kg doses of C + P significantly (#p<0.05) decreased infarct volumes (35.7%) when body temperatures were maintained at 37°C (Fig. 1B, C). If body temperature was not controlled and allowed to reach a hypothermic state after C+P administration, brain infarct volumes were slightly (#p<0.05) further reduced further (29.8 vs. 35.7%).
Neurological Deficits
Neurological deficits were detected 24 h after reperfusion, determined by the 5 (Fig. 2A) or 12 (Fig. 2B) score systems. Compared with no treatment stroke groups, neurological deficits were decreased significantly with or without temperature control.
Fig. 2.

Neurological deficits after C+P therapy with or without body temperature control in 2 h MCA occlusion, using the 5 score system (A) and 12 score system (B). ANOVA analyses indicated that both C+P therapy with no temp control (##p<0.01) or temp control (#p<0.05) reduced neurological deficits.
Brain Edema and BBB Integrity
Brain edema was quantified 24 h after reperfusion was established (Fig. 3A). On the ipsilateral side, a significant (**p<0.01) increase in water content was observed in ischemic rats (84.9%), as compared with control rats (74.8%). C+P treatment significantly (##p<0.01) reduced tissue water content with no temperature control (79.5%) or with temperature control (79.9%). C+P treatment made no significant difference to the water levels when compared with the control group in the contralateral hemisphere of ischemic rats. BBB permeability was quantified by Evans blue dye extravasation at 48 h after reperfusion (Fig. 3B). On the ipsilateral side, a significant (**p<0.01) increase in BBB permeability was observed in ischemic rats (9.3 μg/g), as compared with control rats (1.8 μg/g). C+P treatment reduced (##p<0.01) tissue water content under either hypothermia (2.2 μg/g) or normothermia (4.5 μg/g). C+P treatment made no significant difference to the BBB permeability when compared with the control group in the contralateral hemisphere of ischemic rats.
Fig. 3.

C+P reduced brain edema and BBB leakage. (A) C+P treatment attenuated ipsilateral stroke-induced brain edema significantly versus ipsilateral stroke group without treatment (##P<0.01). (B) C+P reduced BBB leakage significantly on the ipsilateral side in comparison to the ipsilateral stroke group without treatment (##p<0.01), but unrelated to temperature.
Expression of AQP-4/9 and MMP-2/9
Ischemic rats with 2 h MCA occlusion showed a significant (*p<0.05) increase in MMP-2 and MMP-9 levels at 6 and 24 h of reperfusion. Compared with stroke groups without treatment, levels of MMP-2 (1.2 vs. 1.8 at 6 h, #p<0.05; 1.4 vs. 2.0 at 24 h, #p<0.05, Fig. 4A) and MMP-9 (1.1 vs. 1.5 at 6 h, #p<0.05; 1.1 vs. 2.1 at 24 h, #p<0.05, Fig. 4B) in C+P treatment without temperature control groups were significantly decreased. Although the mechanism of why the MMP-2 level at 6 h was slightly higher than that at 24 h with C+P treatment without temperature control is not clear, it suggests that the most vigorous action of MMP-2 was at the early stage and might be the best time-point for C+P targeted therapy. Similar levels of MMP-2 (0.7 vs. 1.8 at 6 h, ##p<0.01; 1.8 vs. 2.0 at 24 h, Fig. 4A) and MMP-9 (1.2 vs. 2.1 at 24 h, #p<0.05 Fig. 4B) reduction were also found in C+P treatment with temperature control groups.
Fig. 4.
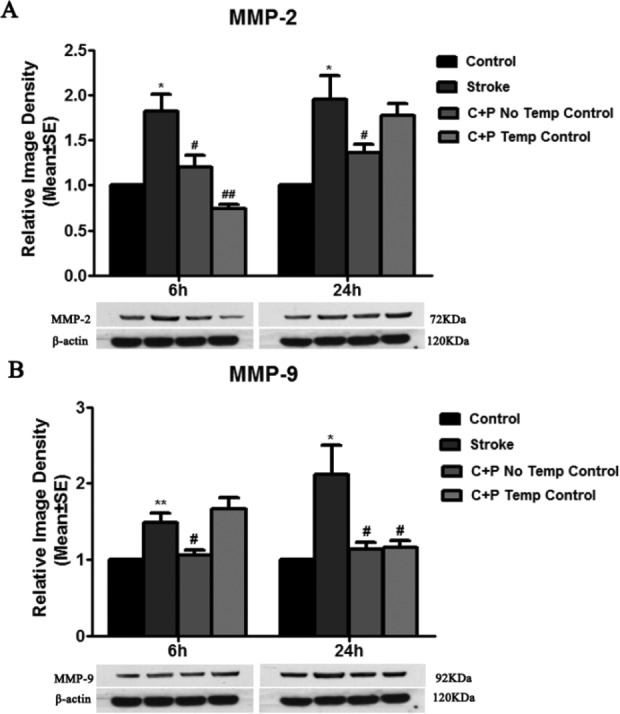
C+P downregulated MMP-2 and MMP-9 expression. (A) MMP-2 protein expression was significantly decreased by C+P with or without temperature control as compared with stroke groups without treatment at 6 h after reperfusion. A decrease in MMP-2 expression was slightly seen at 24 h by C+P. (B) MMP-9 protein expression was significantly increased after stroke and was reversed by C+P without temperature control at 6 h (#p<0.05) and 24 h (#p<0.05) after reperfusion. C+P with temperature control at 37°C decreased MMP-9 expression only at 24 h (#p<0.05). Representative immunoblots are presented.
Similarly, AQP-4 and AQP-9 levels were largely increased in ischemic rats at 6 h (**p<0.01), suggesting decreased BBB integrity. Levels of both AQP-4 (0.6 vs. 1.3 at 6 h, ##p<0.01; 0.7 vs. 0.9 at 24 h, Fig. 5A) and AQP-9 (1.0 vs. 1.6 at 6 h, ##p<0.01; 1.2 vs. 2.0 at 24 h, #p<0.01, Fig. 5B) were significantly decreased by C+P without temperature control groups. Similar levels of AQP-4 (0.3 vs. 1.3 at 6 h, ##p<0.01; 0.7 vs. 0.9 at 24 h, Fig. 5A) and AQP-9 (1.4 vs. 1.6 at 6 h; 1.1 vs. 2.0 at 24 h, #p<0.05 Fig. 5B) reduction were also found in C+P treatment with temperature control groups.
Fig. 5.
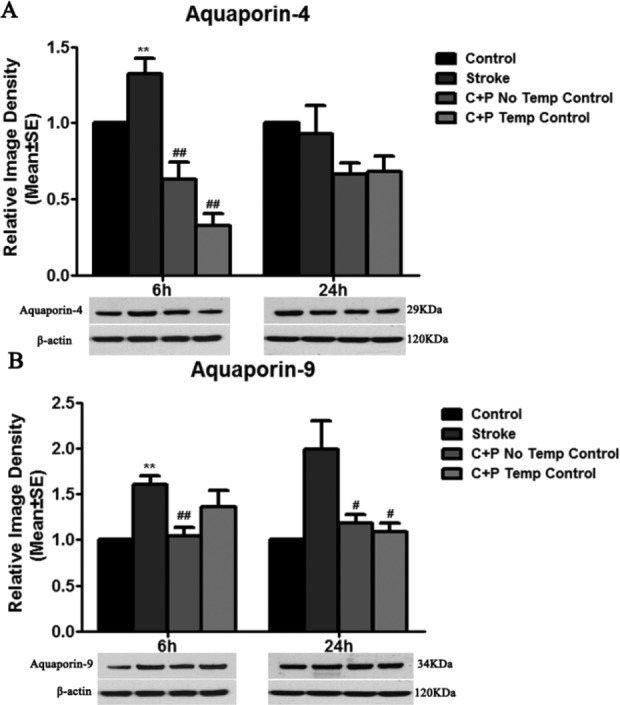
Stroke upregulated AQP-4 and AQP-9 expression. (A) AQP-4 protein expression was significantly decreased by C+P with or without temperature control as compared with stroke groups without treatment at 6 h (significantly, ##p<0.01) and 24 h (slightly) after reperfusion. (B) AQP-9 protein expression was significantly increased after stroke and reduced by C+P without temperature control at 6 h (##p<0.01) and 24 h (#p<0.05) after reperfusion. Similarly, a decrease in AQP-9 expression after stroke was induced by C+P with temperature control at 6 (slightly) and 24 h (significantly, #p<0.05). Representative immunoblots are presented.
Basal Lamina and Tight Junction Protein Expression
To test whether C+P treatment after stroke might influence BBB integrity, the expressions of claudin-1, claudin-5, occludin, zonula occludens (ZO)-1, and laminin were assessed. Compared with the sham-operated group referenced as 1, ischemia and reperfusion decreased protein expression of laminin, claudin-1, claudin-5, occluding, and ZO-1 (Fig. 6). Levels of claudin-1 (1.2 vs. 0.8 at 6 h; 1.1 vs. 0.9 at 24 h, Fig. 6A), claudin-5 (0.7 vs. 0.3 at 6 h, ##p<0.01; 1.0 vs. 0.6 at 24 h, #p<0.05, Fig. 6B), occludin (1.0 vs. 0.9 at 6 h; 1.0 vs. 0.6 at 24 h, #p<0.05, Fig. 6C), ZO-1 (0.9 vs. 0.7 at 6 h; 0.9 vs. 0.6 at 24 h, #p<0.05, Fig. 6D) and laminin (1.1 vs. 0.7 at 6 h; 1.1 vs. 0.3 at 24 h, #p<0.05, Fig. 6E) were significantly increased by C+P, most likely without temperature control.
Fig. 6.

(A) Claudin-1 protein expression was slightly increased with C+P as compared with stroke groups without treatment at 6 and 24 h after reperfusion. (B) A significant decrease in claudin-5 protein expression was seen after stroke, and this decrease was reversed by C+P± temperature control at 6 (##p<0.01) and 24 h (#p<0.05). Occludin protein expression (C) and ZO-1 protein expression (D) were increased by C+P without temperature control but not with temperature control as compared with stroke groups without treatment at 6 (slightly) and 24 h (significantly, #p<0.05) after reperfusion. (E) Laminin protein expression was increased by C+P with or without temperature control compared to stroke groups at 6 (slightly) and 24 h (significantly, #p<0.05) after reperfusion. Representative immunoblots are presented.
Discussion
In this study, we found that C+P in hypothermic or normothermic conditions can reduce infarct volume, improve functional neurological deficits, decrease brain edema, and attenuate BBB breakdown after cerebral ischemia in the rat. In addition, gap formations within the endothelial cell layer of the BBB were induced in the ischemic brain, and TJ proteins (including claudin-1, claudin-5, occluding, and ZO-1) were significantly increased after C+P treatment, along with induced expression levels of laminin and BBB integral proteins (including MMP-2, MMP-9, AQP-4, and AQP-9). Our study illustrates the influence of C+P on the brain’s microvascular system, with the results suggesting that C+P induces neuroprotection by improving microvascular integrity and reinforcement of the BL.
C+P, classical members of the phenothiazine drugs, have been widely used as neuroleptics since the 1950s. These two drugs, usually used in combination, are two of the oldest and most-studied drugs from the phenothiazine class. Because of their high lipophilicity, C+P can easily pass through the BBB to exert their depressive metabolic effects. We have previously reported an “artificial hibernation” effect of alcohol on the brain for suppressing metabolism that leads to neuroprotection following the onset of ischemic stroke. However, in comparison to C+P, there are many societal implications and side effects associated with alcohol which may limit its clinical translation. In addition to their clinical use for their antipsychotic and sedative effects, the present experimental studies along with previous studies support a new application of C+P as neuroprotectants in stroke for their ability to induce an “artificial hibernation” status.
Phenothiazines, specifically chlorpromazine and promethazine, have shown promise as drugs that could be repurposed to treat victims suffering from ischemic stroke. They hold the capacity of suppressing free radicals, reducing the uptake of Ca2+ and K+ leakage by plasma membrane, inhibiting the swelling of cells, and preventing the release of acidic hydrolyses. It has been demonstrated that phenothiazines protect the membrane by inhibiting phospholipase activation20. In addition, phenothiazines could slow down metabolism by uncoupling oxidative phosphorylation via inhibition of glucose oxidation20. The ability to decrease glucose utilization and uptake suggests that the target of these drugs involves the transport pathway. The mechanism is still debated, but it has been shown that these drugs have a marginal effect as antagonists on dopaminergic receptors (D2 dopamine receptors) and mediate the reduced uptake of glucose. Because of their marginal effect, it was also hypothesized that these drugs act on Ca2+ channels21. It has been reported that phenothiazines could inhibit apoptosis, reducing activation of caspases and cytochrome c release from mitochondria, which are most likely a result of protecting the mitochondria membrane from reactive oxygen species (ROS) and lipases that try to disrupt it22. When treated with promethazine and chlorpromazine, free radical-induced lipid peroxidation was significantly reduced in rats20. In addition, once phenothiazines were administered, cerebral blood flow was markedly reduced, and oxygen consumption was consequently reduced as well23.
TJs participate in forming the BBB and are composed of occludin (one of the transmembrane proteins), ZO-1 (an accessory cytoplasmic protein), and claudins (the key integral proteins), all of which play important roles in regulating BBB permeability and function24. Shi et al thought that blood occludin could be a clinically viable biomarker for evaluating BBB damage during ischemia/reperfusion in patients, which put forward the clinical significance of occludin on the BBB25. The disruption of occludin led to functional changes of the TJs in rats26,27 and was associated with hypoxia-induced vascular leakage28. The protective effects of occludin on BBB integrity, however, are not sufficiently understood. It was reported in an in vitro study that the activation of hypoxia-inducible factor-1α (HIF-1α) could lead to lower protein levels of occludin, resulting in an increase in BBB permeability29,30. HIF-1α regulates many kinds of protein expression in response to various pathophysiological conditions induced by hypoxia31, which may potentially explain the effect of occludin on BBB damage. In addition, by using lipopolysaccharide-induced dysregulation of the TJ, Qin et al. proposed another idea—activation of MMP-2 through augmenting the p38MAPK/JNK signaling pathways may be another way to cause occludin dysregulation in the BBB32.
Moreover, ZO-1 was found to be reduced after ischemic stroke, which was confirmed to be related to the functional changes in TJs33. The protective effects of ZO-1 on BBB integrity are various. It was reported that the EGFR-ERK1/2 pathway consists of key signaling proteins that regulate ZO-1 expression, which directly affects BBB permeability34. Recently, studies have also shown that Ras signaling induces the generation of ROS via activating the downstream effector mitogen-activated protein kinase (MEK)/extracellular signal regulated kinases (ERK), resulting in the alteration of TJs and BBB permeability. Jiang et al. reported that ZO-1 could be induced in brain endothelial cells via the Ras signaling pathway, which suggests another form of regulation for ZO-135. Furthermore, through an in vivo experiment, Zhang et al. demonstrated that enhanced autophagy could cause ZO-1 reduction and result in increasing BBB permeability, providing new insights into targeting ZO-1 regulation8.
Claudin is another class of TJs found to be decreased in lesioned vessels with BBB breakdown in the ischemic brain. When claudin-536,37 or claudin-138 expression is increased, BBB permeability is decreased. It was reported that vascular endothelial cadherin can upregulate claudin expression by preventing the nuclear accumulation of the transcriptional regulator forkhead box O1 (FoxO1)39. Our previous study had also confirmed that FoxO1 is regulated by HIF-1α, which is induced by ischemia. The HIF-1α-FoxO1 pathway may be the main signaling mechanism that regulates claudin expression in maintaining BBB integrity. In addition, increased transforming growth factor-β (TGF-β) was shown to decrease the activity of claudin, which compromises the integrity of endothelial sheets40. Claudin can also be modified by cyclic adenosine monophosphate (cAMP) via PKA-induced phosphorylation41 and mediated at Thr207 by Rho/ Rho kinase (RhoK) signaling via a PKA-independent pathway, resulting in phosphorylation and redistribution of claudin and ultimately in increased BBB permeability42.
Laminin, on the other hand, acts as a major structural protein of the BL and is known to be involved with BBB pathology43,44. It was reported that laminin regulates the maturation and function of the BBB, maintains BBB integrity, and regulates ventricular size or development43. Laminin may alleviate vasogenic edema through promoting migration of astrocytes to damaged or newly generated vessels to repair BBB disruption and reconstruction of the endothelial barrier45.
Recent studies have demonstrated a rapidly marked increase in the expression of MMP-2/9 at the site of ischemia, and occludin proved to be a direct substrate of MMPs after ischemic stroke46. The formation of vasogenic edema has been linked to the activation of MMPs, which are associated with BBB degradation and increased permeability/leakage47,48. Several studies have shown a significant increase in ROS leakage during reperfusion with BBB disruption49. The elevation in ROS contributes to the destabilization of the BBB by upregulating ERK1/2, which activates the NF-κB transcription factor. Activated NF-κB in turn activates the transcription of the MMP-9 gene to result in BBB damage50,51. Meanwhile, our previous study had reported that limb ischemic preconditioning can attenuate BBB disruption by decreasing ERK1/2 phosphorylation, therefore C+P treatment has the potential to inhibit MMP-9 regulated by the ERK1/2- NF-κB pathway. In addition, Machida et al. found the effect of thrombin-PAR1-PKCθ/δ axis on MMP-9-induced BBB leakage52. The protein kinase C (PKC) family is associated with TJ and cytoskeletal reorganization, endothelial barrier dysfunction, apoptosis, oxidative stress, and MMP production. Current evidence reveals that PKC-α and PKC-β isoforms play pivotal roles in TNF-α-mediated endothelial barrier dysfunction via regulation of MMP-253. In this study, our data suggest that the use of C+P can suppress brain edema by inhibiting MMP-2 or MMP-9. Combined with our previous study finding that C+P exert neuroprotective effects after ischemia by decreasing protein expression of PKC, we hypothesize that PKC may be a key protein for C+P regulation of MMPs in brain edema. Furthermore, another study drew the conclusion that MMP-2/9 activities can lead to decreases in TJ (claudin-5 and occludin) protein levels through the p38MAPK/JNK signaling pathways32.
Recent studies have suggested aquaporins as another molecular mechanism for edema formation in the BBB10,11. It has been reported that AQP-4 plays a vital role in brain edema10, but some studies indicated that AQP-4 expression does not alter BBB integrity54. There is no BBB alteration in AQP-4 knockout mice, suggesting no relationship between AQP-4 and BBB function. In our study, our data were consistent with the notion that the decrease in AQP-4 expression induced cerebral edema, not by directly contributing to BBB breakdown, but via transcellular water channels. The use of C+P suppressed brain edema by inhibiting AQP-4 or AQP-9. These results suggest that increased AQP expression in brain tissues can generate and exacerbate edema by upregulation of the water channel. Cytotoxic edema occurs rapidly and can persist 24 h after MCA occlusion55. Oxygen-glucose deprivation disrupts the self-regulation of neurons and astrocytes, producing an ionic gradient that allows uncontrolled influx of water through AQP channels due to osmotic pressure56 and leading to an obvious increase in intracellular fluid volume. When edema occurs, astrocyte swelling is more obvious than neuronal swelling because of astrocyte potassium spatial buffering and glutamate uptake, which causes osmotic overload of brain cells and the subsequent influx of water. The overexpression of AQPs in astrocytic end-feet also contributes to cytotoxic edema-induced apoptosis57, which results in BBB damage and eventual hemorrhagic transformation after reperfusion58. Furthermore, both AQP-4 and AQP-9 upregulation has been shown to potentiate brain edema after ischemic stroke by allowing increased water transport between fluid compartments through the BBB.
The protective mechanisms of C+P on BBB integrity are poorly understood. It has been shown that the TJs’ integrity is associated with HIF-1α, which may potentially explain the improvement in BBB integrity with C+P. Many studies have shown HIF-1α to be upregulated in traumatic brain injury models, and our previous study also showed that inhibition of HIF-1α causes increased expression of both BBB BL and TJ proteins59. This inhibition produced the opposite effect in AQP-4 and MMP-9 expression, suggesting that HIF-1α is a key protein in the formation of brain edema. Recent studies have demonstrated that HIF-1α and its target genes may contribute to cell death, tissue destruction, and the formation of brain edema primarily in the acute phase after ischemic brain damage, suggesting that inhibition of HIF-1α is likely to be beneficial during the acute phase60. In addition, FoxO1 was reported to have functions regulating claudin-5 in endothelial cells, which might be crucial for maintaining brain microvascular endothelial cell integrity61. In agreement with this, an accumulation of FoxO1 transcriptional activity was observed in the brain, correlating with the enhancement of BBB leakage62. In addition, the activation of HIF-1α, which could lead to upregulation of many genes involved in the recruitment, adhesion, and activation of leukocytes, may create a feedback loop enhancing BBB vascular damage63,64. HIF-1α has also been reported to play a critical role in regulating the interaction of FoxO1 in the pathogenesis of BBB breakdown65. Therefore, we will further determine the potential mechanism underlying the HIF-1α-FoxO1 signaling pathway in C+P induced neuroprotection in our future study.
In conclusion, the present study demonstrated a pathway involving claudin-1/5, occludin, ZO-1, laminin, MMP-2, MMP-9, AQP-4, and AQP-9 in brain edema formation and BBB disruption after ischemic stroke, and its improvement with C+P. Elucidation of the pathway can provide insights into effective pharmacological target therapies. To our knowledge, this is the first study to show the effect of C+P on MMPs, AQPs, and TJs leading to edema formation. In addition, C+P readily cross the BBB and easily diffuse through the collateral circulation into ischemic regions before reperfusion is even established. The clinical potential as stroke therapeutics of this widely available FDA-approved drug is apparent. Future studies of C+P in a clinical setting may move us closer toward the development of an efficacious neuroprotective therapy.
Footnotes
Ethical Approval: The study was approved by the Institutional Animal Investigation Committee of the Capital Medical University.
Statement of Human and Animal Rights: All of the experimental procedures involving animals were conducted in accordance with the Institutional Animal Investigation Committee of the Capital Medical University.
Statement of Informed Consent: Statement of Informed Consent is not applicable for this article.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by National Nature Science Foundation of China (no. 81871838 and 81802231); Beijing Tongzhou District Financial Fund; and Science and Technology Plan of Beijing Tongzhou District (KJ2018CX006-02).
Reference
- 1. Horng S, Therattil A, Moyon S, Gordon A, Kim K, Argaw AT, Hara Y, Mariani JN, Sawai S, Flodby P, Crandall ED, Borok Z, Sofroniew MV, Chapouly C, John GR. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J Clin Invest. 2017;127(8):3136–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, Keep RF, Shi Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 2017;163–164:144–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garbuzova-Davis S, Haller E, Lin R, Borlongan CV. Intravenously transplanted human bone marrow endothelial progenitor cells engraft within brain capillaries, preserve mitochondrial morphology, and display pinocytotic activity toward blood-brain barrier repair in ischemic stroke rats. Stem Cells. 2017;35(5):1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu C, Shi F, Chen Z, Yan S, Ding X, Lou M. Severe blood-brain barrier disruption in cardioembolic stroke. Front Neurol. 2018;9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Geng X, Li F, Yip J, Peng C, Elmadhoun O, Shen J, Ji X, Ding Y. Neuroprotection by chlorpromazine and promethazine in severe transient and permanent ischemic stroke. Mol Neurobiol. 2017;54(10):8140–8150. [DOI] [PubMed] [Google Scholar]
- 6. Liu X, Su P, Meng S, Aschner M, Cao Y, Luo W, Zheng G, Liu M. Role of matrix metalloproteinase-2/9 (MMP2/9) in lead-induced changes in an in vitro blood-brain barrier model. Int J Biol Sci. 2017;13(11):1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perez-Hernandez M, Fernandez-Valle ME, Rubio-Araiz A, Vidal R, Gutierrez-Lopez MD, O’Shea E, Colado MI. 3,4-Methylenedioxymethamphetamine (MDMA, ecstasy) produces edema due to BBB disruption induced by MMP-9 activation in rat hippocampus. Neuropharmacology. 2017;118:157–166. [DOI] [PubMed] [Google Scholar]
- 8. Zhang S, An Q, Wang T, Gao S, Zhou G. Autophagy- and MMP-2/9-mediated reduction and redistribution of ZO-1 contribute to hyperglycemia-increased blood-brain barrier permeability during early reperfusion in stroke. Neuroscience. 2018;377:126–137. [DOI] [PubMed] [Google Scholar]
- 9. Li J, Li C, Yuan W, Wu J, Li J, Li Z, Zhao Y. Mild hypothermia alleviates brain oedema and blood-brain barrier disruption by attenuating tight junction and adherens junction breakdown in a swine model of cardiopulmonary resuscitation. Plos One. 2017;12(3):e0174596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leitao RA, Sereno J, Castelhano JM, Goncalves SI, Coelho-Santos V, Fontes-Ribeiro C, Castelo-Branco M, Silva AP. Aquaporin-4 as a new target against methamphetamine-induced brain alterations: focus on the neurogliovascular unit and motivational behavior. Mol Neurobiol. 2018;55(3):2056–2069. [DOI] [PubMed] [Google Scholar]
- 11. Liu H, Yang M, Qiu GP, Zhuo F, Yu WH, Sun SQ, Xiu Y. Aquaporin 9 in rat brain after severe traumatic brain injury. Arq Neuropsiquiatr. 2012;70(3):214–220. [DOI] [PubMed] [Google Scholar]
- 12. Peng C, Li WA, Fu P, Chakraborty T, Hussain M, Guthikonda M, Rafols JA, Ding Y. At low doses ethanol maintains blood-brain barrier (BBB) integrity after hypoxia and reoxygenation: a brain slice study. Neurol Res. 2013;35(8):790–797. [DOI] [PubMed] [Google Scholar]
- 13. Forreider B, Pozivilko D, Kawaji Q, Geng X, Ding Y. Hibernation-like neuroprotection in stroke by attenuating brain metabolic dysfunction. Prog Neurobiol. 2017;157:174–187. [DOI] [PubMed] [Google Scholar]
- 14. Dave KR, Christian SL, Perez-Pinzon MA, Drew KL. Neuroprotection: lessons from hibernators. Comp Biochem Physiol B Biochem Mol Biol. 2012;162(1–3):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang F, Luo Y, Ling F, Wu H, Chen J, Yan F, He Z, Goel G, Ji X, Ding Y. Comparison of neuroprotective effects in ischemic rats with different hypothermia procedures. Neurol Res. 2010;32(4):378–383. [DOI] [PubMed] [Google Scholar]
- 16. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. [DOI] [PubMed] [Google Scholar]
- 17. Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke. 1996;27(9):1616–1622; discussion 1623. [DOI] [PubMed] [Google Scholar]
- 18. Wang F, Wang Y, Geng X, Asmaro K, Peng C, Sullivan JM, Ding JY, Ji X, Ding Y. Neuroprotective effect of acute ethanol administration in a rat with transient cerebral ischemia. Stroke. 2012;43(1):205–210. [DOI] [PubMed] [Google Scholar]
- 19. Ren C, Li N, Wang B, Yang Y, Gao J, Li S, Ding Y, Jin K, Ji X. Limb ischemic perconditioning attenuates blood-brain barrier disruption by inhibiting activity of MMP-9 and occludin degradation after focal cerebral ischemia. Aging Dis. 2015;6(6):406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. MacMillan V. Effects of promethazine on the energy metabolism of normoxic and hypoxic rat brain. Stroke. 1982;13(4):464–469. [DOI] [PubMed] [Google Scholar]
- 21. Dwyer DS, Liu Y, Bradley RJ. Dopamine receptor antagonists modulate glucose uptake in rat pheochromocytoma (PC12) cells. Neurosci Lett. 1999;274(3):151–154. [DOI] [PubMed] [Google Scholar]
- 22. Narayanan MV, Zhang W, Stavrovskaya IG, Kristal BS, Friedlander RM. Promethazine: a novel application as a neuroprotectant that reduces ischemia-mediated injury by inhibiting mitochondrial dysfunction. Clin Neurosurg. 2004;51:102–107. [PubMed] [Google Scholar]
- 23. Berntman L, Carlsson C. Influence of “lytic cocktail” on blood flow and oxygen consumption in the rat brain. Acta Anaesthesiol Scand. 1978;22(5):515–518. [DOI] [PubMed] [Google Scholar]
- 24. Haseloff RF, Dithmer S, Winkler L, Wolburg H, Blasig IE. Transmembrane proteins of the tight junctions at the blood-brain barrier: structural and functional aspects. Semin Cell Dev Biol. 2015;38:16–25. [DOI] [PubMed] [Google Scholar]
- 25. Shi S, Qi Z, Ma Q, Pan R, Timmins GS, Zhao Y, Shi W, Zhang Y, Ji X, Liu KJ. Normobaric hyperoxia reduces blood occludin fragments in rats and patients with acute ischemic stroke. Stroke. 2017;48(10):2848–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim S, Kim GH. Roles of claudin-2, ZO-1 and occludin in leaky HK-2 cells. Plos One. 2017;12(12):e0189221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang GS, Tian Y, Huang JY, Tao RR, Liao MH, Lu YM, Ye WF, Wang R, Fukunaga K, Lou YJ, Han F. The gamma-secretase blocker DAPT reduces the permeability of the blood-brain barrier by decreasing the ubiquitination and degradation of occludin during permanent brain ischemia. CNS Neurosci Ther. 2013;19(1):53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bauer AT, Burgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 2010;30(4):837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cao Y, Ni C, Li Z, Li L, Liu Y, Wang C, Zhong Y, Cui D, Guo X. Isoflurane anesthesia results in reversible ultrastructure and occludin tight junction protein expression changes in hippocampal blood-brain barrier in aged rats. Neurosci Lett. 2015;587:51–56. [DOI] [PubMed] [Google Scholar]
- 30. Zhao J, Hao J, Fei X, Wang X, Hou Y, Deng C. Isoflurane inhibits occludin expression via up-regulation of hypoxia-inducible factor 1alpha. Brain Res. 2014;1562:1–10. [DOI] [PubMed] [Google Scholar]
- 31. Berlow RB, Dyson HJ, Wright PE. Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature. 2017;543(7645):447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qin LH, Huang W, Mo XA, Chen YL, Wu XH. LPS induces occludin dysregulation in cerebral microvascular endothelial cells via MAPK signaling and augmenting MMP-2 levels. Oxid Med Cell Longev. 2015;2015:120641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khoyetsyan A, Kacimi R, Tsakanova G, Boyajyan A, Arakelyan A, Yenari MA. Activated complement protein C5a does not affect brain-derived endothelial cell viability and zonula occludens-1 levels following oxygen-glucose deprivation. Brain Circ. 2017;3(1):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu W, Wang P, Shang C, Chen L, Cai H, Ma J, Yao Y, Shang X, Xue Y. Endophilin-1 regulates blood-brain barrier permeability by controlling ZO-1 and occludin expression via the EGFR-ERK1/2 pathway. Brain Res. 2014;1573:17–26. [DOI] [PubMed] [Google Scholar]
- 35. Jiang W, Huang W, Chen Y, Zou M, Peng D, Chen D. HIV-1 transactivator protein induces ZO-1 and neprilysin dysfunction in brain endothelial cells via the Ras signaling pathway. Oxid Med Cell Longev. 2017;2017:3160360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang L, Li Z, Zhang X, Wang S, Zhu C, Miao J, Chen L, Cui L, Qiao H. Protective effect of shikonin in experimental ischemic stroke: attenuated TLR4, p-p38MAPK, NF-kappaB, TNF-alpha and MMP-9 expression, up-regulated claudin-5 expression, ameliorated BBB permeability. Neurochem Res. 2014;39(1):97–106. [DOI] [PubMed] [Google Scholar]
- 37. Lv J, Hu W, Yang Z, Li T, Jiang S, Ma Z, Chen F, Yang Y. Focusing on claudin-5: a promising candidate in the regulation of BBB to treat ischemic stroke. Prog Neurobiol. 2018;161:79–96. [DOI] [PubMed] [Google Scholar]
- 38. Yadav BK, Shin BS. Single-nucleotide polymorphisms of tight junction component claudin-1 associated with leukoaraiosis. J Stroke Cerebrovasc Dis. 2015;24(7):1662–1670. [DOI] [PubMed] [Google Scholar]
- 39. Gavard J, Gutkind JS. Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Galpha12/13 and Galpha11/q. J Biol Chem. 2008;283(44):29888–29896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shimizu F, Sano Y, Tominaga O, Maeda T, Abe MA, Kanda T. Advanced glycation end-products disrupt the blood-brain barrier by stimulating the release of transforming growth factor-beta by pericytes and vascular endothelial growth factor and matrix metalloproteinase-2 by endothelial cells in vitro. Neurobiol Aging. 2013;34(7):1902–1912. [DOI] [PubMed] [Google Scholar]
- 41. Ishizaki T, Chiba H, Kojima T, Fujibe M, Soma T, Miyajima H, Nagasawa K, Wada I, Sawada N. Cyclic AMP induces phosphorylation of claudin-5 immunoprecipitates and expression of claudin-5 gene in blood-brain-barrier endothelial cells via protein kinase A-dependent and -independent pathways. Exp Cell Res. 2003;290(2):275–288. [DOI] [PubMed] [Google Scholar]
- 42. Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV. Inflammation and brain edema: new insights into the role of chemokines and their receptors. Acta Neurochir Suppl. 2006;96:444–450. [DOI] [PubMed] [Google Scholar]
- 43. Gautam J, Zhang X, Yao Y. The role of pericytic laminin in blood brain barrier integrity maintenance. Sci Rep. 2016;6:36450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim YJ, Kim JY, Ko AR, Kang TC. Over-expression of laminin correlates to recovery of vasogenic edema following status epilepticus. Neuroscience. 2014;275:146–161. [DOI] [PubMed] [Google Scholar]
- 46. Chaturvedi M, Kaczmarek L. Mmp-9 inhibition: a therapeutic strategy in ischemic stroke. Mol Neurobiol. 2014;49(1):563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barichello T, Generoso JS, Michelon CM, Simoes LR, Elias SG, Vuolo F, Comim CM, Dal-Pizzol F, Quevedo J. Inhibition of matrix metalloproteinases-2 and -9 prevents cognitive impairment induced by pneumococcal meningitis in Wistar rats. Exp Biol Med (Maywood). 2014;239(2):225–231. [DOI] [PubMed] [Google Scholar]
- 48. Verma A, Prasad KN, Nyati KK, Singh SK, Singh AK, Paliwal VK, Gupta RK. Association of MMP-2 and MMP-9 with clinical outcome of neurocysticercosis. Parasitology. 2011;138(11):1423–1428. [DOI] [PubMed] [Google Scholar]
- 49. Bertuglia S, Giusti A. Microvascular oxygenation and oxidative stress during postischemic reperfusion. PO2, ROS, and NO during reperfusion. Adv Exp Med Biol. 2005;566:23–29. [DOI] [PubMed] [Google Scholar]
- 50. Graf R. Letter of apology. Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. J Cereb Blood Flow Metab. 2005;25(3):291. [DOI] [PubMed] [Google Scholar]
- 51. Wang Y, Fan X, Tang T, Fan R, Zhang C, Huang Z, Peng W, Gan P, Xiong X, Huang W, Huang X. Rhein and rhubarb similarly protect the blood-brain barrier after experimental traumatic brain injury via gp91(phox) subunit of NADPH oxidase/ROS/ERK/MMP-9 signaling pathway. Sci Rep. 2016;6:37098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Machida T, Dohgu S, Takata F, Matsumoto J, Kimura I, Koga M, Nakamoto K, Yamauchi A, Kataoka Y. Role of thrombin-PAR1-PKCtheta/delta axis in brain pericytes in thrombin-induced MMP-9 production and blood-brain barrier dysfunction in vitro. Neuroscience. 2017;350:146–157. [DOI] [PubMed] [Google Scholar]
- 53. Abdullah Z, Bayraktutan U. Suppression of PKC-alpha attenuates TNF-alpha-evoked cerebral barrier breakdown via regulations of MMP-2 and plasminogen-plasmin system. Biochim Biophys Acta. 2016;1862(7):1354–1366. [DOI] [PubMed] [Google Scholar]
- 54. Saadoun S, Tait MJ, Reza A, Davies DC, Bell BA, Verkman AS, Papadopoulos MC. AQP4 gene deletion in mice does not alter blood-brain barrier integrity or brain morphology. Neuroscience. 2009;161(3):764–772. [DOI] [PubMed] [Google Scholar]
- 55. Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739(1–2):88–96. [DOI] [PubMed] [Google Scholar]
- 56. Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4(12):991–1001. [DOI] [PubMed] [Google Scholar]
- 57. Manley GT, Binder DK, Papadopoulos MC, Verkman AS. New insights into water transport and edema in the central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience. 2004;129(4):983–991. [DOI] [PubMed] [Google Scholar]
- 58. Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, Pedrono E, Abo-Ramadan U, Tatlisumak T. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. 2008;153(1):175–181. [DOI] [PubMed] [Google Scholar]
- 59. Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, Ding JY, Dornbos D, 3rd, Li X, Guthikonda M, Rossi NF, Ding Y. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114(1):92–101. [DOI] [PubMed] [Google Scholar]
- 60. Chen W, Jadhav V, Tang J, Zhang JH. HIF-1alpha inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol Dis. 2008;31(3):433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Beard RS, Jr, Haines RJ, Wu KY, Reynolds JJ, Davis SM, Elliott JE, Malinin NL, Chatterjee V, Cha BJ, Wu MH, Yuan SY. Non-muscle Mlck is required for beta-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1beta-mediated barrier dysfunction in brain endothelial cells. J Cell Sci. 2014;127(Pt 8):1840–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang P, Xue Y, Shang X, Liu Y. Diphtheria toxin mutant CRM197-mediated transcytosis across blood-brain barrier in vitro. Cell Mol Neurobiol 2010;30(5):717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Witt KA, Mark KS, Huber J, Davis TP. Hypoxia-inducible factor and nuclear factor kappa-B activation in blood-brain barrier endothelium under hypoxic/reoxygenation stress. J Neurochem 2005;92(1):203–214. [DOI] [PubMed] [Google Scholar]
- 64. Zhang Z, Yan J, Shi H. Role of Hypoxia Inducible Factor 1 in hyperglycemia-exacerbated blood-brain barrier disruption in ischemic stroke. Neurobiol Dis. 2016;95:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen X, Iliopoulos D, Zhang Q, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014;508(7494):103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]