

Abstract
Scleromyxoedema is a rare generalized cutaneous mucinosis, which in absence of thyroid disease, occurs almost invariably in patients with monoclonal gammopathies. A 54-year-old female patient presented with complaint of tightening of skin on the extremities, abdomen, forehead, gradually progressive since 1 year, episodes of generalized tonic–clonic convulsions, and acute psychosis since 5 days. Cutaneous examination revealed nonpitting edema over the face and sclerodermoid changes over extremities. Laboratory investigations showed presence of M-band on serum-protein electrophoresis and monoclonal spike of IgG lambda component on immunofixation. Magnetic resonance imaging of the brain showed periventricular subcortical lacunar infarcts. Skin biopsy with mucin staining was suggestive of scleromyxoedema. All other investigations were normal. Bone marrow biopsy showed a mild focal increase in plasma cells. The cutaneous, serological, and electrophoretic findings as well as the clinical profile of the patient were consistent with the diagnosis of monoclonal gammopathy of undetermined significance associated with scleromyxoedema. This case is presented because of its rare occurrence.
Keywords: Monoclonal gammopathy of undetermined significance, multiple myeloma, paraproteinemia, scleromyxoedema
Introduction
Scleromyxoedema, a progressive cutaneous mucinosis usually associated with a systemic involvement and paraproteinemia,[1] is an important cutaneous diagnostic marker of monoclonal gammopathy of undetermined significance (MGUS), which is a plasma cell dyscrasia spectrum disorder.
It has a chronic, disabling course because it is often associated with a systemic involvement of internal organs.[2] Here, we report a case of scleromyxoedema in association with plasma cell dyscrasia, presenting with concurrent central nervous system involvement.
Case Report
A 54-year-old female patient was referred to the dermatology outpatient department with a history of tightness of skin, involving the face, abdomen, and the extremities, which was gradually progressive in nature, which was persistent since 1 year and exacerbated for the past 1 month. It was asymptomatic in nature, with complaints of occasional pruritus and slight restriction of movement of small joints of hands. Patient also had an episode of generalized tonic–clonic convulsions followed by behavioral changes, unsteady gait, and altered sensorium since 5 days, which resolved temporarily after symptomatic treatment by the physician, only to recur after a gap of 3 days, and was subsequently resolved by symptomatic treatment without further recurrence. There was no history of Raynaud's phenomenon, breathlessness, or dysphagia or any other medical comorbidities. Blood pressure, electrocardiogram, random blood sugar, and serum electrolytes were within normal limits, and there was no family history of hematological malignancy or similar cutaneous or systemic complaints.
Cutaneous examination revealed sclerodermoid changes over face, extremities and abdomen, as evident by deepening of the furrows over forehead, infiltration over ears. [Figure 1], multiple grouped papular lesions over the lower back [Figure 2], taut shiny skin over the shins, and a prominence of follicular ostia over forearms [Figure 3].
Figure 1.
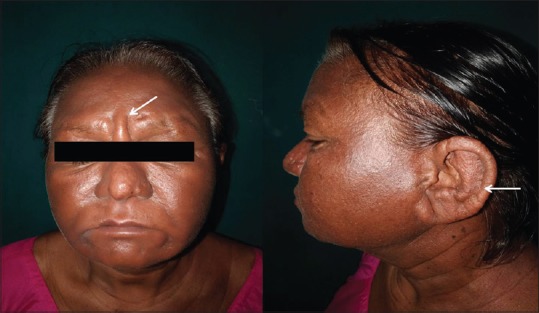
Sclerodermoid changes over the face with deepening of furrows over the forehead. Diffuse thickening of the skin. There is diffuse infiltration over the entire helix and earlobe. Few papulonodular lesions can be appreciated over the helix
Figure 2.
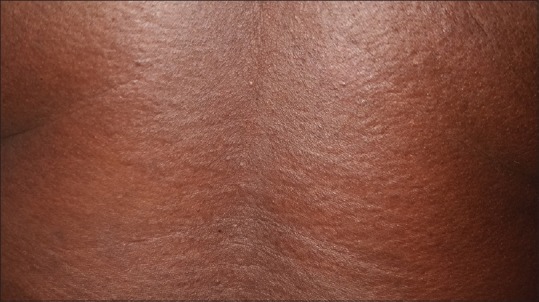
On close inspection of the back, multiple closely grouped papular lesions can be appreciated over the back, giving a rough, cobblestone-like texture to the skin
Figure 3.
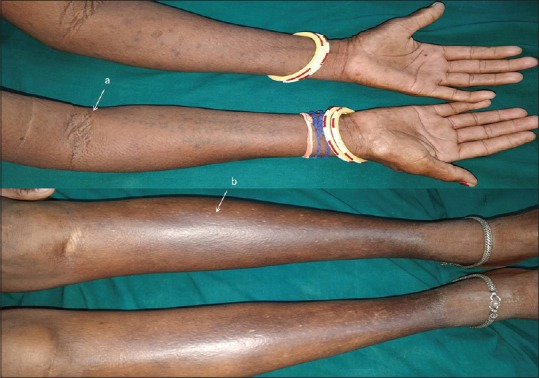
There is a prominence of follicular ostia visible near the cubital fossae (a). Sclerodermoid skin changes over the shins and (b) forearms
The cutaneous features were suggestive of a clinical diagnosis of scleromyxoedema which was confirmed by histopathological examination. Hematoxylin and eosin staining of the skin biopsy specimen showed widely separated dermal collagen fibers and fibroblastic proliferation [Figure 4]. Colloidal iron special staining showed an increased mucin deposition between the dermal collagen fibers [Figure 5], which further clinched the diagnosis. The presence of scleromyxoedema prompted a series of investigations to rule out the multiple systemic associations which have been known to be associated with this rare dermatological condition, with a special emphasis on hematological and neurological workup. Baseline liver and renal function tests, thyroid profile, serum antinuclear antibody profile were within normal limits. Hematological investigations showed mild anemia, a peripheral smear examination showing presence of toxic granules in the neutrophils, and an increase in the circulating plasma cells. Bone marrow biopsy was suggestive of a trilineage hematopoesis and a focal increase in plasma cells (14%). Serum protein electrophoresis studies revealed hypoalbuminemia (serum albumin, 2.82 g/dL) presence of M-band (M protein, 0.91 g/Dl), and raised serum B2 microglobulin levels [Supplementary Figure 1 (366.7KB, tif) ]. Immunofixation studies showed a monoclonal spike of IgG lambda component, with levels suggestive of monoclonal gammopathy of undetermined significance. Urine test for Bence–Jones proteins was found to be negative.
Figure 4.
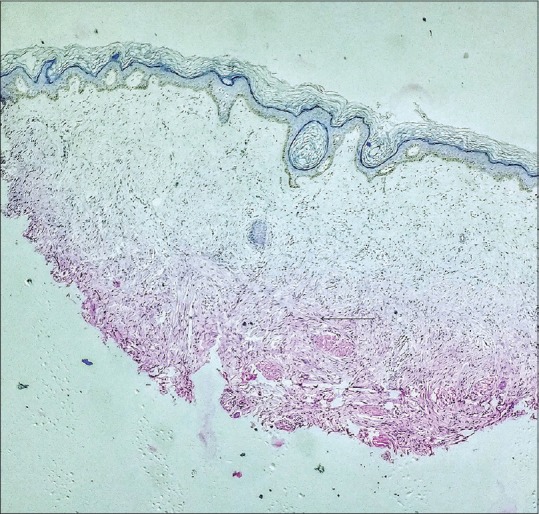
Histopathological examination reveals excessive fibroblastic proliferation and dermal fibrosis (H and E, ×4)
Figure 5.
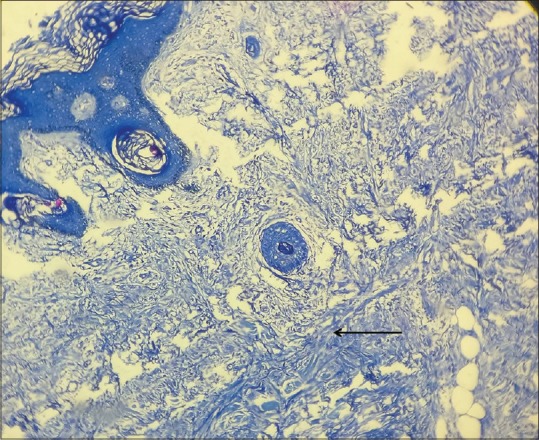
Special staining with colloidal iron showed amorphous blue-colored mucin deposition throughout the dermis, which in correlation with the histopathological picture is diagnostic for scleromyxoedema. Counter stain used: Hematoxylin (Colloidal iron stain: ×10)
Report suggestive of monoclonal gammopathy of undetermined significance as reported by a pathologist
Magnetic resonance imaging of the brain showed subcortical periventricular lacunar infarcts. Contrast-enhanced computed tomography scan did not reveal any major vessel blocks.
The patient was referred to a hemato-oncosurgeon and was started initially on prednisolone and cyclophosphamide. However, due to a lack of clinical response and worsened anemia and unchanged M-protein levels on repeat electrophoresis, patient was started on injection bortezumib 2 mg subcutaneously, oral cyclophosphamide 300 mg, and dexamethasone 40 mg every week for 8 cycles. The patient was followed up after 8 weeks with an improvement in anemia, with minimal improvement of skin condition.
Thus, the dermatological diagnosis of scleromyxoedema along with the hematological, electrophoretic, and bone marrow findings suggestive of monoclonal gammopathy of undetermined significance highlight the importance of scleromyxedema as a cutaneous surrogate of paraproteinemia.
Discussion
Scleromyxoedema is classified as a generalized papular and sclerodermoid variant of lichen myxedematosus. Diagnosis of scleromyxedema should fulfill the following criteria: (1) generalized papular and sclerodermoid eruption; (2) mucin deposition, fibroblast proliferation, and fibrosis; (3) monoclonal gammopathy; and (4) the absence of thyroid disease.[3] The etiology of the disorder remains an enigma; the precise mechanisms whereby increased fibroblast activity results in mucin deposition remain to be defined.[4] To date, there is no completely satisfactory therapeutic approach to scleromyxoedema.
Patients with scleromyxoedema have variable multisystem manifestations commonly involving the gastrointestinal tract, musculoskeletal, pulmonary, cardiovascular, renal, and central nervous systems, leading to significant morbidity and mortality. Prominent symptoms include dysphagia, proximal muscle weakness, and dyspnea on exertion; less common, but important findings, include central nervous system involvement in the form of encephalopathy, seizures, coma, and psychosis.
In our patient, prominent extracutaneous involvement occurred in the form of an acute organic brain syndrome presenting with seizures and acute psychosis, which abated after symptomatic treatment upon admission with complete normalisation of mental status; only to recur after a gap of 3 days. Such an involvement of the central nervous system is an unusual and rarely reported manifestation of scleromyxoedema. It has been proposed that an increased blood viscosity with impaired microcirculation along with an increase in serum factor VIII, and von Willebrand factor caused by paraproteinemia may result in encephalopathy-like symptoms.[5] In our case, the central nervous system involvement may be attributed to this mechanism, however, it could not be investigated further owing to the relatively short duration of the symptoms. Further, a cerebrospinal fluid examination was desirable, but could not be conducted as the patient gave a negative consent. This remains a limitation of this case report.
The immunofixation studies showed a monoclonal spike of predominantly IgG type in the lambda region behind the point of origin with its level suggestive of monoclonal gammopathy of undetermined significance of non-IgM type. This is the most well-characterized subtype of monoclonal gammopathy of undetermined significance, in which the M-component isotype can be IgG (69%), IgA (11%), or biclonal (3%); IgD and IgE are rare.[6] The cumulative incidence of full progression to multiple myeloma at 10 years is 7%, 13%, and 18%, respectively for IgG, A, and M types.[7] Further, the rate of progression of all non-IgM monoclonal gammopathy of undetermined significance to multiple myeloma is approximately 1% per year.[8]
For a dermatologist, the diagnostic importance of scleromyxedema lies in the fact that is a rare disorder which is commonly associated with paraproteinemia. Hence, the presence of scleromyxoid changes should prompt a clinician to thoroughly workup for plasma cell dyscrasias for early diagnosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understand that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Koronowska SK, Osmola-Mańkowska A, Jakubowicz O, Zaba R. Scleromyxedema: A rare disorder and its treatment difficulties. Postepy Dermatol Alergol. 2013;30:122–6. doi: 10.5114/pdia.2013.34165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serdar ZA, Altunay IK, Yasar SP, Erfan GT, Gunes P. Generalized papular and sclerodermoid eruption: Scleromyxedema. Indian J Dermatol Venereol Leprol. 2010;76:592. doi: 10.4103/0378-6323.69096. [DOI] [PubMed] [Google Scholar]
- 3.Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273–81. doi: 10.1067/mjd.2001.111630. [DOI] [PubMed] [Google Scholar]
- 4.Allam M, Ghozzi M. Scleromyxedema: A case report and review of the literature. Case Rep Dermatol. 2013;5:168–75. doi: 10.1159/000353178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savran Y, Akarsu S. Dermato-neuro syndrome in a case of scleromyxedema. Eur J Rheumatol. 2015;2:160–2. doi: 10.5152/eurjrheum.2015.0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354:1362–9. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 7.Rajkumar SV, Dispenzieri A, Kyle RA. Monoclonal gammopathy of undetermined significance, waldenström macroglobulinemia, AL amyloidosis, and related plasma cell disorders: Diagnosis and treatment. Mayo Clin Proc. 2006;81:693–703. doi: 10.4065/81.5.693. [DOI] [PubMed] [Google Scholar]
- 8.Zingone A, Kuehl WM. Pathogenesis of monoclonal gammopathy of undetermined significance and progression to multiple myeloma. Semin Hematol. 2011;48:4–12. doi: 10.1053/j.seminhematol.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Report suggestive of monoclonal gammopathy of undetermined significance as reported by a pathologist