

Bhatt et al. show that autophagy ablation was synthetically lethal during Lkb1-deficient lung tumorigenesis in both tumor initiation and tumor growth.
Keywords: autophagy, LKB1, non-small cell lung cancer, lipid metabolism, energy metabolism
Abstract
Loss of tumor suppressor liver kinase B1 (LKB1) promotes cancer cell proliferation but also leads to decreased metabolic plasticity in dealing with energy crises. Autophagy is a protective process involving self-cannibalization to maintain cellular energy homeostasis during nutrient deprivation. We developed a mouse model for Lkb1-deficient lung cancer with conditional deletion of essential autophagy gene Atg7 to test whether autophagy compensates for LKB1 loss for tumor cells to survive energy crises. We found that autophagy ablation was synthetically lethal during Lkb1-deficient lung tumorigenesis in both tumor initiation and tumor growth. We further found that autophagy deficiency causes defective intracellular recycling, which limits amino acids to support mitochondrial energy production in starved cancer cells and causes autophagy-deficient cells to be more dependent on fatty acid oxidation (FAO) for energy production, leading to reduced lipid reserve and energy crisis. Our findings strongly suggest that autophagy inhibition could be a strategy for treating LKB1-deficient lung tumors.
Lung cancer remains the leading cause of cancer mortality worldwide, and non-small cell lung cancer (NSCLC) accounts for >85% of these cases (Siegel et al. 2015). Driver mutations are molecular alterations in tumors that play a significant role in tumor progression and growth. Over the past decade, the discovery of a number of driver mutations underlying NSCLC has led to significant advances in prognostication and treatment of these cancers (Bozic et al. 2010). However, patients harboring mutations in the tumor suppressor gene liver kinase B1 (LKB1) and the oncogene KRAS, two of the most common mutations in NSCLC, develop more aggressive lung tumors, show a high frequency of metastasis, and have limited treatment options (Ji et al. 2007; Matsumoto et al. 2007; Imielinski et al. 2012). Recent studies have found that LKB1 mutation causes KRAS mutant lung cancer patients to be resistant to immunotherapy (Skoulidis et al. 2018). Thus, new therapies for this subtype of lung cancer are urgently needed.
LKB1 signaling negatively regulates tumor growth and metastasis through direct phosphorylation and activation of the central metabolic sensor AMPK, which governs glucose and lipid metabolism in response to alterations in nutrients and intracellular energy levels (Shackelford and Shaw 2009; Shackelford 2013). Under energy stress, LKB1 activates AMPK, which in turn inhibits fatty acid (FA) synthesis through inhibitory phosphorylation of acetyl-CoA carboxylase (ACC) and sterol regulatory element-binding proteins (SREBP) and prevents protein synthesis through inactivation of mTORC1 signaling, leading to a restriction of cell growth and proliferation (Shackelford and Shaw 2009). Loss of LKB1 facilitates tumor growth and metastasis under energetically unfavorable conditions by causing disorganization of cell polarity, increasing glucose and glutamine uptake, and up-regulating de novo FA synthesis (Shaw et al. 2005; Faubert et al. 2014; Svensson et al. 2016). Loss of LKB1 also increases the production of reactive oxygen species (ROS) in tumors, leading to the transdifferentiation of adenocarcinoma to squamous cell carcinoma (Ji et al. 2007; Xu et al. 2014; Li et al. 2015). Moreover, oncogenic cooperation between the loss of LKB1 and activation of KRAS promotes tumorigenesis through the induction of the one-carbon pathway coupled to the generation of S-adenosylmethionine (Kottakis et al. 2016). Taken together, loss of LKB1 reprograms cancer cell metabolism to efficiently generate energy and biomass components for uncontrolled cell proliferation in order to expand and disseminate. However, such alterations in turn cause tumor cells to have less plasticity in response to an energy crisis, creating a metabolic vulnerability (Jeon et al. 2012; Parker et al. 2017). Therefore, targeting metabolic vulnerabilities is a valuable therapeutic approach to treat LKB1-deficient lung cancer. Indeed, LKB1-deficient NSCLC is sensitive to the metabolic-based drug phenformin, which is a mitochondrial inhibitor (Shackelford et al. 2013).
Cancer cells not only alter metabolism to promote macromolecular biosynthesis and maintain redox and energy homeostasis but also up-regulate nutrient-scavenging pathways, including autophagy, to provide metabolic substrates as fuel for their altered metabolism (Vander Heiden and DeBerardinis 2017). The catabolic process of autophagy captures proteins and organelles and then degrades and recycles them to provide metabolic substrates, a function that is critical when extracellular nutrients are limited. Autophagy also eliminates damaged proteins and organelles to maintain their quality and homeostasis (White 2012). Ras activation up-regulates basal autophagy and causes cancer cells to become “addicted” to autophagy during metabolic stress and tumorigenesis (Guo et al. 2011; Lock et al. 2011; Yang et al. 2011).
The support of tumor growth through the up-regulation of autophagy has been demonstrated in different types of tumors using genetically engineered mouse models (GEMMs) with distinct mechanisms (White et al. 2015; Amaravadi et al. 2016; Guo and White 2016; Sousa et al. 2016; Yang et al. 2018). In GEMMs for pancreatic ductal adenocarcinoma (PDAC), acute autophagy ablation suppresses PDAC progression through tumor cell-intrinsic as well as host effects (Yang et al. 2018). Host autophagy promotes tumor growth via maintaining circulating arginine (Poillet-Perez et al. 2018). Using GEMMs for NSCLC with or without p53, we demonstrated that autophagy promotes BrafV600E- and KrasG12D-induced lung tumor growth and prevents the conversion of adenocarcinoma to benign oncocytoma via maintaining functional mitochondria, energy homeostasis, redox balance, nucleotide pools, and amino acids (Guo et al. 2013a,b, 2016; Strohecker and White 2014). Acute systemic Atg7 deletion in mice with pre-existing NSCLC blocks tumor growth and promotes tumor cell death before destruction of normal tissues (Karsli-Uzunbas et al. 2014), indicating that there is a therapeutic window to treat lung cancer by autophagy inhibition.
The underlying mechanism of autophagy in supporting lung tumor growth is likely through the removal of damaged mitochondria for tumor cells to maintain energy homeostasis and reduce ROS production (Guo et al. 2011, 2013a). On the other hand, autophagy also recycles intracellular metabolites for cancer cells against nutrient deprivation. Pulse-chase studies with isotope-labeled nutrients reveal that autophagy deficiency causes defective mitochondrial substrate supply during starvation in Ras-driven lung cancer cells, which is associated with increased ROS, lower energy charge, and a dramatic drop in total nucleotide pools (Guo et al. 2016). Autophagy also plays unique roles in tumorigenesis via the tumor microenvironment. Stroma-associated pancreatic stellate cells are critical for PDAC metabolism through the secretion of nonessential amino acids, which is dependent on pancreatic satellite cell autophagy and stimulated by cancer cells (Sousa et al. 2016).
Cancer cells use a variety of nutrient sources to support mitochondrial tricarboxylic acid (TCA) cycle metabolism to meet high metabolic demand (Vander Heiden and DeBerardinis 2017). In addition to glucose, glutamine, and lactate (Hui et al. 2017), lipids are another major fuel for TCA cycle metabolism (Beloribi-Djefaflia et al. 2016). Although changes in lipid metabolism in tumors were commonly disregarded in the past, they are now well recognized. Lipids support cell proliferation through energy production via FA oxidation (FAO) and biomass buildup and as critical signaling molecules (Huang and Freter 2015; Beloribi-Djefaflia et al. 2016). The ability to up-regulate FA synthesis is a property of cancer cells that maintains the above functions and store energy as lipid droplets (LDs) (Shackelford and Shaw 2009; Merino Salvador et al. 2017; Wen et al. 2017). Additionally, hypoxia and acidosis promote the uptake of lipoproteins for LD storage in cancer cells, relieving the requirement for de novo synthesis (Bensaad et al. 2014; Menard et al. 2016). Systemic inhibition of ACC to prevent de novo FA synthesis is sufficient to block tumor growth in GEMMs of Lkb1-deficient NSCLC (Svensson et al. 2016). Colon cancer cells increase the uptake of FAs to survive nutrient starvation. Inhibition of autophagy attenuates the ability of colon cancer cells to use FAs and blocks the growth-promoting effect of adipocytes (Wen et al. 2017). Preventing lipid and FA uptake by an anti-CD36 neutralizing antibody impairs metastasis in human oral carcinomas (Pascual et al. 2017).
Here, using GEMMs for Lkb1-deficient NSCLC with conditional autophagy ablation, we found that Atg7 deficiency prevented the ability of activated Kras and deficient Lkb1 to initiate tumorigenesis and reduced the tumor growth. To further address the underlying mechanism, we generated tumor-derived cell lines (TDCLs) from Atg7+/+ or Atg7−/− tumors and found that LDs in Atg7−/−;KrasG12D/+;Lkb1flox/flox (KL) tumors and TDCLs were significantly lower than those in Atg7+/+ KL tumors and cells in starvation. We also found that autophagy deficiency caused cancer cells to be more sensitive to starvation-induced cell death due to the depletion of TCA cycle-related intermediates, leading to impaired energy production and nucleotide pool depletion. Palmitate supplementation was able to rescue the starvation survival of autophagy-deficient cells. In addition, inhibition of FAO accelerated starvation-induced Atg7-null cell death. Taken together, autophagy may sustain KL tumor growth by recycling substrates and sustaining free FA levels to maintain functional mitochondrial metabolism and by preventing excessive FAO to resist energy crises. Our study suggests that autophagy inhibition or a combination of autophagy inhibition and lipid metabolism blockade could be a potential therapeutic strategy for the treatment of LKB1-deficient lung cancer.
Results
Loss of Lkb1 causes Kras-driven lung tumors to escape adenoviral-Cre-mediated Atg7 deletion
Loss of LKB1 promotes cell growth but also results in broad defects in metabolic control in response to nutrient deprivation and other types of metabolic stress (Jeon et al. 2012; Parker et al. 2017). To test the hypothesis that autophagy is required to compensate for LKB1 loss-induced decrease in metabolic plasticity for tumor growth, KL mice were crossed with Atg7flox/flox mice possessing conditional deficiency in Atg7 (Komatsu et al. 2005) to generate a cohort that was either Atg7+/+ or Atg7flox/flox. Mice genotypes were confirmed by PCR for Lkb1, Kras, and Atg7 (Supplemental Fig. S1A). Initiation of tumorigenesis by Kras activation and Lkb1 deletion with and without Atg7 deletion was achieved by an intranasal delivery of Adenoviral-Cre to the mice. The delivery generates mice bearing Atg7+/+ or Atg7−/− KL lung tumors. The effects of the loss of Lkb1 and Atg7 on protein expression in tumor cells were examined by immunohistochemistry (IHC). Lkb1 protein expression was completely absent in KL tumor cells (Supplemental Fig. S1B). Unexpectedly, Atg7 protein expression was not homogeneously deleted in KL tumor cells at a later time point (8 wk after Adeno-Cre) (Supplemental Fig. S1B). In contrast, in Lkb1 intact Kras-driven tumors, Atg7 was fully deleted (Supplemental Fig. S1C). We further assessed the autophagy status by examining autophagy substrate p62 and microtubule-associated protein light chain 3 (LC3) in KL tumors. In comparison with Atg7+/+ tumors, there was an accumulation of p62 and LC3 in Atg7−/− KL tumors at 4 wk after infection, indicating the blockage of autophagy (Supplemental Fig. S1B). However, at a later time point (8 wk after infection), the accumulation of p62 and LC3 in most “Atg7−/−” tumors disappeared (Supplemental Fig. S1B). Consistent with the IHC of Atg7 in “Atg7−/−” tumors, we observed one in five “Atg7−/−” KL tumors showing increased Atg5–Atg12 conjugation and decreased LC3-I accumulation by Western blot (Supplemental Fig. S1D), indicating autophagy activation. Due to an incomplete autophagy ablation in “Atg7−/−” tumors, deletion of Atg7 only slightly extended mouse survival compared with mice bearing Atg7+/+ tumors (P = 0.05) (Supplemental Fig. S1E). The incomplete deletion of Atg7 in tumors may be due to the inability of transient expression of Adenoviral-Cre to effectively delete all floxed DNA sequences, resulting in heterogeneous growth of Atg7−/− and Atg7flox/flox wild-type KL tumors. Alternatively, loss of Lkb1 may select against autophagy-deficient tumor growth, causing an outgrowth of Atg7flox/flox wild-type tumors, which would suggest that autophagy is required for KL tumorigenesis.
Autophagy is required for KL tumor initiation and further tumor progression
The use of lentiviruses to deliver Cre (Lenti-Cre) is an alternative to induce lung tumors (DuPage et al. 2009). The advantage of Lenti-Cre is that lentiviruses will integrate into the genome of infected cells, allowing for further modification of the tumors by simultaneously introducing Cre recombinase, which can lead to higher efficiency in deleting target genes. To further test our hypothesis that autophagy compensates for LKB1 loss to sustain KL tumorigenesis, Lenti-Cre was intranasally delivered into Atg7+/+ or Atg7flox/flox KL GEMMs, and tumorigenesis was monitored from tumor initiation to tumor progression. Prior to 10 wk after Lenti-Cre infection, there was no significant difference in gross lung pathology as well as wet lung weight between mice bearing Atg7+/+ and Atg7−/− tumors. After 10 wk of tumor induction, wet lung weight gradually increased in mice bearing Atg7+/+ tumors, indicative of tumor growth, but was maintained in mice bearing Atg7−/− tumors (Fig. 1A,B). Quantification of tumor number in scanned lung sections shows that Atg7 ablation significantly reduced the tumor frequency (Fig. 1C,D). The difference between tumor burden in mice bearing Atg7+/+ and Atg7−/− tumors was apparent by examination of gross wet lung weight and tumor burden in scanned lung sections. In stark contrast to Atg7+/+ mice with significantly increased wet lung weight and tumor burden after tumor induction, the wet lung weight and tumor burden of Atg7−/− mice remained unchanged at the indicated times and were significantly lower than those of Atg7+/+ mice (Fig. 1B,C,E; Supplemental Fig. S2A). We further monitored KL tumorigenesis by IHC for the deficiency of Lkb1 expression in tumors and confirmed the inhibited growth of Atg7−/− tumors (Fig. 1F; Supplemental Fig. S2B). Moreover, Lkb1 deletion was confirmed by the Western blot of KL tumors (Fig. 1I). Most importantly, the extent of tumor growth inhibition by Atg7 deficiency in mice bearing KL tumors was much more dramatic than that in mice bearing KrasG12D/+;p53−/− (KP) tumors induced by Lenti-Cre. The extent of tumor growth inhibition was assessed via scanned lung sections, tumor number, and tumor burden at 14 and 18 wk after Lenti-Cre infection (Fig. 1C–E). In contrast to KP tumors where Atg7 deletion extended mouse survival by only 2 wk (Guo et al. 2013a), KL tumor reduction due to Atg7 loss significantly prolonged mouse survival, extending it by >2 mo (Fig. 1G). Mice bearing Atg7−/− KL tumors died from a combination of adenocarcinoma, squamous cell carcinoma, and inflammation based on tumor histology examined at 29 wk after Lenti-Cre infection (Supplemental Fig. S3A,B). Thus, autophagy inhibition could be particularly more effective in treating LKB1-defective KRAS-driven NSCLC than P53-deficient KRAS-driven NSCLC.
Figure 1.

Autophagy is required for Lkb1-deficient Kras mutant lung tumor initiation and progression. (A) Representative gross lung pathology at the indicated times. (B) Graph of wet lung weight at the indicated times. (C) Representative histology of scanned lung sections at the indicated times after Lenti-Cre infection. (D,E) Quantification of tumor number (D) and tumor burden (E) from C. (F) Representative IHC of Lkb1 at the indicated times. (G) Kaplan-Meier survival curve of mice bearing KL tumors that were intranasally infected with Lenti-Cre. P < 0.0001, log-rank test. (H) Representative IHC of Atg7, p62, and LC3 shows autophagy ablation in Atg7−/− tumors (red arrows indicate autophagosome in Atg7+/+ tumors) at 10 wk after Lenti-Cre infection. (I) Western blot of Lkb1, Atg7, Atg5-Atg12, p62, LC3, Tom20, and β-actin of KL lung tumors at 18 wk after Lenti-Cre infection. (J) Representative hematoxylin and eosin (H&E) staining of lung tumors shows increased necrotic cells (yellow arrows) in Atg7−/− tumors at the indicated times. (K) Representative IHC of Tom20 shows accumulation of mitochondria in Atg7-deficient tumors at 18 wk after Lenti-Cre infection. (L) Cytochrome c oxidase activity in tumors at 18 wk after Lenti-Cre infection. For all of the graphs, the error bar indicates ±SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. See also Supplemental Figures S2 and S3.
Autophagy ablation was confirmed by IHC for Atg7 expression and accumulation of autophagy substrates p62 and LC3 (Fig. 1H, Supplemental Fig. S2C–E). Autophagy was functionally blocked in KL tumors with Atg7 deletion, as indicated by the accumulation of p62 and LC3-I and loss of Atg5–Atg12 conjugation compared with normal lung (WT1) and Atg7+/+ KL tumors (Fig. 1I). However, at 22 wk after tumor induction, we observed that some KL tumor cells showed an escape of Lenti-Cre-mediated Atg7 deletion through examination of IHC for Atg7 (Supplemental Fig. S2C,D). Reduced tumor frequency and tumor growth in Atg7−/− mice correlated with the increased presence of necrotic cells in Atg7−/− tumors (Fig. 1J, Supplemental Fig. S3A) and increased accumulation of respiration-defective mitochondria examined by IHC and Western blot for Tom 20 (Fig. 1I,K) and cytochrome c oxidase assay (Fig. 1L). Taken together, autophagy deficiency alters the ability of activated Kras and Lkb1 deficiency to initiate lung tumorigenesis and diminishes further tumor growth.
Autophagy deficiency reduces residual AMPK activity in Kras-driven lung tumors
A recent study from the Shaw group (Eichner et al. 2018) found that AMPK activity is required for Kras-driven lung tumor growth. We therefore examined the AMPK activity in both KP and KL tumors by IHC of AMPK substrate phospho-ACC (Ser79) (pACCS79). Similar to the finding of Eichner et al. (2018), pACCS79 was detected in both KP and KL tumors with intact autophagy, indicating activation of AMPK (Fig. 2A,B). Although LKB1 is the major activating kinase of AMPK, other upstream kinases such as calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2) might promote AMPK activity upon Lkb1 loss to support tumorigenesis (Hardie and Alessi 2013; Eichner et al. 2018). We further found that AMPK activity was lower in both Atg7-deficient KP and KL tumors as examined by IHC of pACCS79, suggesting that autophagy might sustain AMPK activity for Kras-driven lung tumorigenesis.
Figure 2.

Autophagy deficiency impairs oncogenic signaling. (A,B) Representative IHC of pACCS79 in KP and KL tumors at 18 wk after Lenti-Cre infection at high magnification (A) and low magnification (B). (C–E) Representative IHC of pS6, pERK, and Ki67 at the indicated times. (F) Quantification of pS6, pERK, and Ki67 from C–E. (G) Representative IHC of active caspase-3 at the indicated times. (H) Quantification of active caspase-3 from G. (I) Representative images of positive β-galactosidase staining at 18 wk after Lenti-Cre infection. (J) Comparison of metabolites between Atg7+/+ and Atg7−/− KL tumors obtained by liquid chromatography/mass spectrometry (LC-MS). (K) Levels of metabolites that show a significant difference between Atg7+/+ and Atg7−/− KL tumors. For all of the graphs, the error bar indicates ±SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. See also Supplemental Figure S3.
Autophagy deficiency impairs RAS-induced oncogenic signaling and increases apoptosis
To determine the course of reduced tumor burden by autophagy ablation, the RAS downstream oncogenic signaling was assessed. The levels of phospho-S6 (pS6) and phospho-ERK1/2 (pERK) were significantly lower in Atg7−/− tumors compared with Atg7+/+ tumors (Fig. 2C,D,F; Supplemental Fig. S3C,D). We further examined tumor proliferation (Ki67) and apoptosis (active caspase-3) and found that tumors with Atg7 deletion had significantly fewer proliferating cells compared with those with intact Atg7 (Fig. 2E,F; Supplemental Fig. S3E). However, apoptosis was significantly increased in Atg7−/− tumors compared with Atg7+/+ tumors (Fig. 2G,H).
Reduced proliferation in tumors is often associated with the activation of senescence (Muñoz-Espín and Serrano 2014); therefore, the activity of senescence-associated β-galactosidase of Atg7 wild-type and deficient tumors was assessed at 18 wk after infection. We found that Atg7 deficiency increased the level of β-galactosidase activity in KL tumors (Fig. 2I). Thus, the impaired tumorigenesis by Atg7 deletion was attributed to increased apoptosis and senescence as well as ablation of proliferation and oncogenic signaling.
Autophagy is required to maintain KL tumor energy homeostasis
Loss of LKB1 causes less metabolic plasticity in response to energy crises. To broadly assess the metabolic consequences of Atg7 deficiency in tumors, unbiased metabolite profiling of KL tumors at 18 wk after infection was performed by liquid chromatography/mass spectrometry (LC-MS). We identified 110 metabolites, eight of which were down-regulated and 22 of which were up-regulated in Atg7−/− tumors compared with Atg7+/+ tumors (Fig. 2J,K). Altered metabolites included TCA cycle intermediates, amino acids, and those involved in nucleotide metabolism, transsulfuration, and catabolites of amino acids, vitamins, and glucose. Most interestingly, ATP was strikingly down-regulated in Atg7−/− tumors. Meanwhile, AMP was increased in Atg7−/− tumors compared with Atg7+/+ tumors. Similarly, we observed an increase in GMP, UMP, and CMP in autophagy-deficient tumors, indicators of lower energy levels (Fig. 2K). These observations suggest that autophagy is required to support KL tumor energy production.
Atg7-deficient TDCLs are sensitive to metabolic stress
To address the mechanism by which autophagy supports KL tumor growth, Atg7+/+ and Atg7−/− TDCLs were generated from Atg7 wild-type or Atg7-deficient KL lung tumors induced by Adenoviral-Cre. We found that 11 of 15 Atg7−/− TDCLs displayed accumulation of p62 and LC3-I and reduced Atg5–Atg12 conjugation—characteristics of autophagy defect (Fig. 3A; Supplemental Fig. S4A). However, four of 15 “Atg7−/−” TDCLs showed undeleted Atg7 and active autophagy, confirmed by Western blot and PCR (Supplemental Fig. S4A,B), which was consistent with our observation in KL tumors that Atg7 was incompletely deleted via Adenoviral-Cre infection (Supplemental Fig. S1B,D). We further confirmed that these “Atg7−/−” (Atgflox/flox) TDCLs were indeed still responding to Adenoviral-Cre mediated Atg7 deletion in vitro (Supplemental Fig. S4C), and acute Atg7 deletion inhibited KL cell proliferation (Supplemental Fig. S4D).
Figure 3.

Autophagy-deficient KL TDCLs are sensitive to metabolic challenge. (A) Western blot of Atg7, p62, LC3, and β-actin in KL TDCLs. (B) Cell proliferation of KL TDCLs in nutrient-rich RPMI medium analyzed using IncuCyte live-cell analysis system. (C,D) Clonogenic survival assay of TDCLs following Hank's balanced salt solution (HBSS; no glucose) starvation (C) or glucose (Gluc) or glutamine (Gln) deprivation (D). See also Supplemental Figure S4.
Upon characterization of TDCLs, we assessed the consequence of autophagy deficiency on cell proliferation in nutrient-rich medium and found no difference between Atg7 wild-type or Atg7-deficient cells (Fig. 3B), which could be due to in vitro cell culture selection and adaptation during the generating procedure. Since autophagy is activated during metabolic stress, we tested the hypothesis that autophagy sustains LKB1-deficient cell survival under metabolic stress by starving cells with Hank's balanced salt solution (HBSS; no glucose) and evaluated the cellular clonogenic survival. In contrast to TDCLs with intact Atg7, starvation of Atg7-deficient TDCLs inhibited the clonogenic survival (Fig. 3C; Supplemental Fig. S4E). Furthermore, we challenged Atg7+/+ and Atg7−/− TDCLs with other metabolic stresses, including deprivation of glucose and glutamine individually. Different from KP TDCLs, which were reported previously to be sensitive to glutamine deprivation (Guo et al. 2013a), KL TDCLs were much more sensitive to glucose deprivation (Fig. 3D), consistent with the previous report that loss of LKB1 increases glycolysis (Faubert et al. 2014). Despite this, deletion of Atg7 further accelerated the KL cell death induced by glucose or glutamine deprivation (Fig. 3D).
We further knocked down Atg5 in Atg7+/+ KL TDCLs using siRNA and found that knockdown of Atg5 significantly reduced cell proliferation and increased the sensitivity of KL cells to HBSS (no glucose) starvation-induced cell death (Supplemental Fig. S4F,G). Thus, KL TDCLs require autophagy to survive metabolic challenges.
Autophagy mediates substrate recycling for TCA metabolism to maintain energy homeostasis in starvation
Loss of autophagy as a metabolic input would be expected to also influence metabolite concentrations. However, we did not observe concentration changes of metabolites between Atg7+/+ and Atg7−/− TDCLs in nutrient-rich conditions (Fig. 4A; Supplemental Fig. S5A), which could be offset by the increased consumption from medium. Indeed, we observed that Atg7-deficient TDCLs increased the consumption of glucose, glutamine, serine, threonine, proline, and methionine from medium by assessing the levels of medium metabolites (Fig. 4B; Supplemental Fig. S5B). However, during HBSS starvation, the levels of the key TCA cycle intermediates and majority of amino acids were significantly lower in Atg7−/− TDCLs compared with Atg7+/+ TDCLs (Fig. 4A; Supplemental Fig. S5A), demonstrating that autophagy mediates intracellular substrate recycling in starvation.
Figure 4.

Autophagy mediates substrate recycling for TCA metabolism to maintain energy homeostasis in starvation. (A) Levels of TCA cycle intermediates and derivatives in nutrient-rich RPMI and after 1 h of HBSS starvation. (B) Glucose, glutamine, and proline consumption of TDCLs in nutrient-rich RPMI conditions. Each bar represents the average of four clones from Atg7+/+ or Atg7−/− TDCLs. (C,D) Clonogenic survival assay of Atg7+/+ and Atg7−/− TDCLs in HBSS and HBSS supplemented with 2 g/L glucose, 1 mM sodium pyruvate, 2 mM lactate, 2 mM glutamine, 1 mM membrane-permeable α-ketoglutarate (α-KG) analog (dimethyl-α-KG), or 2 mM proline. (E) Schematic of carbon contribution to TCA cycle. (F) Basal oxygen consumption rate (OCR) and relative OCR of TDCLs after 1 h of treatment with RPMI, HBSS, or HBSS supplemented with glutamine (Q). (G) The levels of adenosine phosphates (ATP, ADP, and AMP) in Atg7+/+ and Atg7−/− cells in RPMI, HBSS, or HBSS supplemented with glutamine after 1 h of treatment. For all of the graphs, the error bar indicates ±SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. See also Supplemental Figure S5.
To further determine which nutrients are essential for Atg7−/− TDCLs to survive starvation, we supplemented HBSS with individual amino acids, glucose, membrane-permeable α-ketoglutarate (α-KG) analog (dimethyl-α-KG), lactate, or pyruvate and assessed clonogenic survival. We found that supplementation of dimethyl-α-KG, glutamine, proline, glucose, pyruvate, and lactate, which can contribute carbons to the TCA cycle to maintain mitochondrial metabolism, rescued the survival of Atg7-null cells (Fig. 4C–E; Supplemental Fig. S4E). Consistent with impaired substrate supply during HBSS starvation (Fig. 4A), Atg7−/− TDCLs showed significantly reduced mitochondrial oxygen consumption rates (OCRs) compared with Atg7+/+ cells in starvation. The decreased OCR in Atg7−/− cells in HBSS was fully rescued by supplementation with glutamine (Fig. 4F). Glutamine supplementation also rescued Atg5 knockdown cells in starvation (Supplemental Fig. S4H). Thus, autophagy is required to supply substrates for maintaining mitochondrial metabolism in KL TDCLs.
The primary source of ATP in mammals is TCA cycle-driven oxidative phosphorylation. Decreased TCA cycle metabolites and OCR in starving autophagy-deficient cells (Fig. 4A,F) prompted us to hypothesize that autophagy makes a large contribution to maintain the energy homeostasis in starvation. As expected, ATP levels were not influenced by autophagy deficiency in nutrient-rich conditions but were significantly decreased in Atg7−/− TDCLs in starvation compared with cells that have intact autophagy (Fig. 4G). In association with decreased ATP levels, AMP levels were significantly increased in starved Atg7−/− TDCLs. Glutamine supplementation restored ATP and reduced AMP levels in starved Atg7−/− TDCLs (Fig. 4G). These findings suggest that substrates recycled from autophagy are required to maintain energy homeostasis of Lkb1-deficient tumor cells in starvation.
Atg7 prevents nucleotide catabolism in starvation
Our previous study demonstrated that autophagy deficiency causes profound nucleotide depletion and cell death in KP TDCLs during starvation, driven by nucleotide catabolism (Guo et al. 2016). Therefore, we examined whether autophagy is also required to maintain a nucleotide pool for KL cells to survive starvation. In addition to the reduced ATP levels (Fig. 4G), we found that there was a substantial accumulation of both purine and pyrimidine bases (inosine, hypoxanthine, xanthine, and uracil) in starved Atg7−/− KL cells (Supplemental Fig. S6A,B), indicating increased nucleotide catabolism by autophagy ablation. To understand the functional consequence of autophagy in sustaining nucleotide levels, we supplemented the KL TDCLs with nucleosides in starvation and found that adenosine, guanosine, and uridine were sufficient for the rescue of Atg7−/− cells (Supplemental Fig. S6C). Thus, in starvation, prevention of nucleotide pool degradation by autophagy is a common feature in both KP and KL lung cancer cells.
Autophagy deficiency alters the lipid metabolism of Lkb1-deficient tumor cells
The intracellular storage and utilization of LDs are critical to maintain cellular energy homeostasis. During nutrient deprivation, LDs are hydrolyzed into FAs via cytosolic lipolysis and autophagy-mediated lipophagy for FAO (Singh et al. 2009; Rambold et al. 2015; Zechner et al. 2017). LKB1/AMPK signaling regulates lipid metabolism to maintain energy homeostasis under energy stress (Shackelford and Shaw 2009; Faubert et al. 2014). To understand the role and underlying mechanism by which autophagy modulates lipid metabolism for KL tumorigenesis, we first examined the LDs in KL tumors by Oil Red O staining. Unlike KP lung tumors (Guo et al. 2013a), the LDs in Atg7−/− KL tumors were significantly lower than those in Atg7+/+ KL tumors (Fig. 5A). As in the primary tumors, there were fewer LDs in Atg7-deficient cells than Atg7 wild-type KL cells in starvation (Fig. 5B,C). However, more LDs were observed in Atg7−/− KP TDCLs than in Atg7+/+ KP cells (Fig. 5B,C; Guo et al. 2013a). Thus, the role of autophagy in lipid metabolism could be oncogenic driver-dependent.
Figure 5.

Autophagy deficiency alters lipid metabolism. (A,B) Representative Oil Red O staining of KL tumors (A) and KL and KP TDCLs in RPMI and HBSS starvation (B). (C, left) Quantification of Oil Red O staining of TDCLs from ImageJ. (Right) Quantification of Oil Red O staining normalized to RPMI to show significant reduction of LDs in Atg7−/− KL TDCLs in starvation. (D) Comparison of lipids between Atg7+/+ and Atg7−/− cells in nutrient-rich conditions obtained by LC-MS. (E) [U13C6] glucose tracer labeling to FAs of TDCLs in steady state. (F) Cell proliferation rate in the absence or presence of 50 µM FA synthase inhibitor fasnall in a nutrient-rich RPMI condition. For all of the graphs, the error bar indicates ±SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. See also Supplemental Figure S7.
To further confirm that autophagy modulates lipid metabolism in Lkb1-deficient lung cancer cells, we performed lipidomics in KL TDCLs by LC-MS and found that of 139 examined lipids, the levels of 114 lipids showed significant differences between Atg7+/+ and Atg7−/− cells, with 68 increasing and 46 decreasing by autophagy ablation in nutrient-rich conditions (Fig. 5D). We also found that of 97 examined saponified FAs, 75 saponified FAs showed significantly higher levels in Atg7−/− TDCLs compared with Atg7+/+ TDCLs in nutrient-rich conditions.
Synthesis of FAs is an essential cellular process that converts nutrients into metabolic intermediates for membrane biosynthesis, energy storage, and the generation of signaling molecules (Mashima et al. 2009; Beloribi-Djefaflia et al. 2016). Glucose contributes carbon for FA synthesis (Fig. 4E). To understand the mechanism by which autophagy modulates lipid metabolism and study the specific pathway of FA synthesis in which autophagy could be involved, we labeled Atg7+/+ or Atg7−/− TDCLs with [U13C6] glucose in steady state and examined the carbon contribution to FAs. As shown in Figure 5E and Supplemental Figure S7A, the short chain FAs that mainly come from de novo synthesis showed higher percentages of 13C labeling in Atg7−/− cells than that in Atg7+/+ cells. However, the long chain FA 13C-labeling pattern displayed a clear bimodal distribution. The heavy-labeled fractions come from de novo synthesis, while the light-labeled fractions come from elongating shorter unlabeled FAs. The Atg7−/− cells showed a much higher percentage of de novo synthesized long chain polyunsaturated FAs (Fig. 5E; Supplemental Fig. S7A). Thus, autophagy deficiency increases de novo FA synthesis and reduces FA elongation. Taken together, our observations demonstrate that autophagy deficiency alters KL tumor cell lipid metabolism.
Highly proliferating cancer cells need continuous de novo FA synthesis to support metabolism (Mashima et al. 2009; Beloribi-Djefaflia et al. 2016). Increased de novo FA synthesis by autophagy deficiency could be a metabolic bypass for cells to compensate for defective lipophagy, which could also be a vulnerable metabolic target in autophagy-deficient cells. To test this hypothesis, we treated Atg7+/+ or Atg7−/− KL TDCLs with a selective FA synthase inhibitor, fasnall, in nutrient-rich conditions and monitored cell proliferation. As expected, inhibition of FA synthesis by fasnall prevented cell proliferation and inhibited clonogenic survival. Moreover, autophagy depletion further accelerated cell death with fasnall treatment (Fig. 5F; Supplemental Fig. S7B). This result indicates that a combination of inhibiting autophagy and FA synthesis can be used as a therapeutic strategy for LKB1-deficient lung cancer.
Autophagy is required to maintain free FA levels for KL cell starvation survival
Defective intracellular recycling during starvation due to autophagy deficiency limits substrate supply to the TCA cycle in both KL (Fig. 4A; Supplemental Fig. S5A) and KP (Guo et al. 2016) cells. In response to energy crises, in addition to lactate, glucose, and glutamine, FAs also serve as substrates through β-oxidation to maintain energy homeostasis. To examine whether autophagy is required to maintain the proper levels of FAs in KL cells during starvation, the levels of saponified FAs in KL cells after 4 h of HBSS starvation were measured by LC-MS (Fig. 6A,B). Following starvation, the relative levels of saponified FAs were increased in both Atg7+/+ and Atg7−/− TDCLs (normalized to 0 h of starvation). Of the 95 saponified FAs examined, 26 FAs showed significant differences between Atg7+/+ and Atg7−/− KL cells, with 25 decreasing and one increasing due to autophagy ablation (Fig. 6A,B), indicating that autophagy is required to maintain the FA levels of KL cells during starvation.
Figure 6.
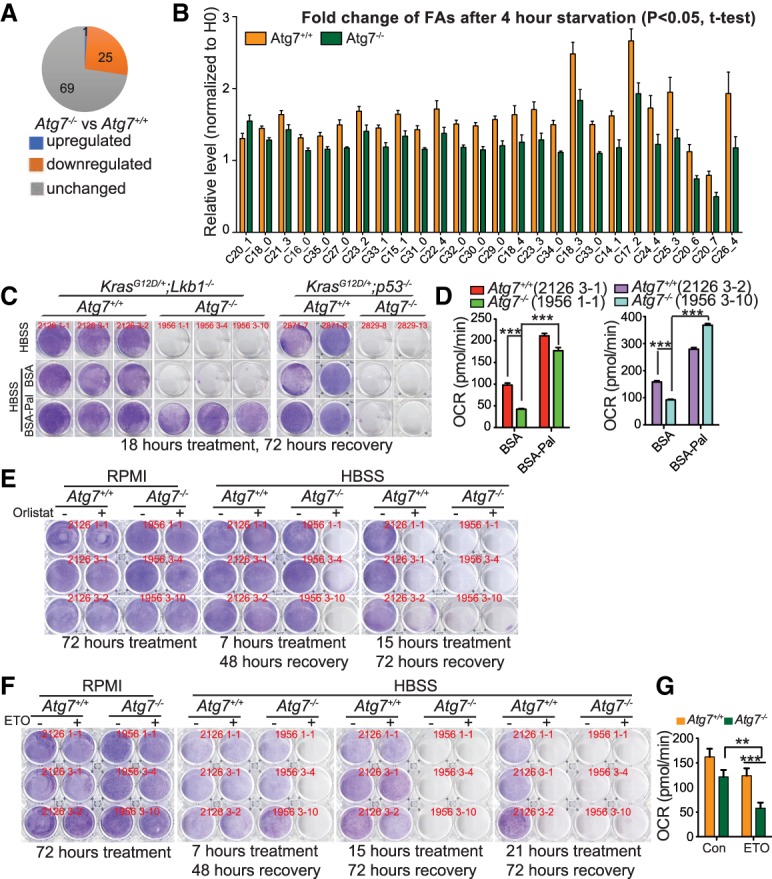
Autophagy deficiency leads to excessive FAO of KL cells in starvation. (A) Comparison of saponified FAs between Atg7+/+ and Atg7−/− cells in HBSS starvation obtained by LC-MS. (B) Saponified FAs with significant change between Atg7+/+ and Atg7−/− cells in HBSS starvation. Each bar represents the average of four clones from Atg7+/+ or Atg7−/− TDCLs. (C) Clonogenic survival assay of Atg7+/+ and Atg7−/− cells in HBSS supplemented with 20 µM bovine serum albumin (BSA) or BSA-conjugated palmitate (BSA-Pal). (D) Basal OCR of TDCLs after 1 h of HBSS starvation supplemented with 20 µM BSA or BSA-Pal. (E) Clonogenic survival assay of TDCLs in HBSS starvation supplemented with vehicle control or 50 µM lipase inhibitor orlistat. (F) Clonogenic survival assay of Atg7+/+ and Atg7−/− KL cells in HBSS or HBSS supplemented with vehicle control or etomoxir (ETO). (G) OCR of TDCLs in the absence or presence of ETO in starvation. Each bar represents the average of four clones from Atg7+/+ or Atg7−/− TDCLs. For all of the graphs, the error bar indicates ±SEM. (**) P < 0.01; (***) P < 0.001.
To elucidate the functional consequence of autophagy-mediated FA maintenance, we supplemented HBSS with bovine serum albumin (BSA)-conjugated palmitate (BSA-Pal) and found that the addition of BSA-Pal rescued Atg7−/− KL TDCLs in starvation. However, BSA-Pal supplementation failed to rescue starvation-induced autophagy-deficient KP cell death (Fig. 6C; Supplemental Fig. S4E). Additionally, BSA-Pal supplementation rescued the decreased OCR of Atg7−/− KL cells in HBSS starvation (Fig. 6D). Lipophagy is abolished in Atg7-deficient cells (Singh et al. 2009), which could promote lipase-mediated lipolysis to compensate for autophagy loss and maintain free FA levels in order for these cells to survive starvation. To test this possibility, we treated Atg7+/+ and Atg7−/− TDCLs with a lipase inhibitor, orlistat, in normal and starvation conditions and examined their clonogenic survival. Orlistat treatment did not impact cell proliferation in nutrient-rich conditions but accelerated starvation-induced Atg7−/− cell death (Fig. 6E). Taken together, autophagy maintains FA levels to ensure energy production through FAO for Lkb1-deficient cells to survive starvation.
Autophagy deficiency augments the intrinsic propensity of KL tumor cells to FAO dependency in starvation
To further address the mechanism by which autophagy deficiency causes reduced LD storage and inhibits tumorigenesis, we tested the hypothesis that in the absence of autophagy during starvation, defective intracellular recycling limits amino acids and glycolytic intermediates to support mitochondrial energy production (Fig. 4A; Supplemental Fig. S5A). This could cause autophagy-deficient cells to rely more on FAO for energy production. As expected, in nutrient-rich conditions, FAO inhibitor etomoxir (ETO) did not impact cell proliferation in both Atg7+/+ and Atg7−/− KL TDCLs (Fig. 6F; Supplemental Fig. S7C). However, ETO treatment accelerated starvation-induced Atg7−/− cell death (Fig. 6F). We further examined the OCR of KL TDCLs in response to ETO treatment during starvation. KL TDCLs were briefly starved for 1 h and then treated with ETO, and OCR was subsequently measured. OCR of Atg7-null cells was dramatically ablated with ETO treatment compared with Atg7+/+ cells (Fig. 6G). Thus, autophagy may sustain KL tumor growth by preventing excessive FAO to avoid an energy crisis.
Autophagy supports LKB1 mutant human lung cancer cells to survive starvation
To probe the significance of autophagy in human lung cancer cell lines with LKB1 mutation, the functional consequence of siRNA knockdown of ATG7 was assessed in KRAS mutant cells with wild-type LKB1 (H441) or mutant LKB1 (H23, HCC44, and H1355). ATG7 expression was suppressed by targeted siRNA, leading to the reduced LC3-II and increased LC3-I and p62 protein levels, indicative of autophagy blockage (Fig. 7A). Compared with LKB1 wild-type H441 cells, LKB1 mutant cells (H23, HCC44, and H1355) were much more sensitive to HBSS starvation-induced cell death (Fig. 7B), supporting the concept that loss of LKB1 leads to less plasticity in response to nutrient deprivation (Jeon et al. 2012; Parker et al. 2017). Furthermore, ATG7 knockdown not only inhibited cell proliferation of LKB1 mutant cells in nutrient-rich conditions (Fig. 7C) but also prevented clonogenic survival of LKB1 mutant cells in starvation but has no effect on LKB1 wild-type cells (Fig. 7D). Supplementation of glutamine or BSA-Pal in HBSS rescued the survival of ATG7 knockdown LKB1 mutant cells in starvation (Fig. 7E). Meanwhile, ATG7 knockdown decreased the OCR of HCC44 and H1355 cells in starvation, which was rescued by BSA-Pal supplementation (Fig. 7F). Knockdown of ATG5 in LKB1 mutant HCC44 and H1355 cells also reduced cell proliferation and increased sensitivity to HBSS starvation-induced cell death; supplementation of glutamine or BSA-Pal in HBSS rescued the survival of ATG5 knockdown HCC44 and H1355 cells in starvation (Supplemental Fig. S7D–F). Taken together, our results demonstrate that KRAS-driven human lung cancer cells with mutant LKB1 rely on autophagy to maintain mitochondrial metabolism to survive energy crises.
Figure 7.

KRAS-driven human lung cancer cells with mutant LKB1 require autophagy to support proliferation and survive starvation. (A) Western blot of ATG7, LC3, p62, and β-actin. (B) Clonogenic survival assay of human lung cancer cells in HBSS starvation. (C) Relative cell proliferation of human lung cancer cells without or with ATG7 knockdown in nutrient-rich RPMI medium. (D) Clonogenic survival assay of human lung cancer cells without or with ATG7 knockdown in HBSS starvation. (E) Clonogenic survival assay of LKB1 mutant human lung cancer cells without or with ATG7 knockdown in HBSS starvation supplemented with 2 mM glutamine or 20 µM BSA or BSA-Pal. (F) Basal OCR and relative OCR of LKB1 mutant human lung cancer cells without or with ATG7 knockdown after 1 h of treatment with RPMI or HBSS supplemented with 20 µM BSA or BSA-Pal. (G) Model of autophagy in supporting LKB1-deficient lung tumor growth. For all of the graphs, the error bar indicates ±SEM. (*) P < 0.05; (***) P < 0.001. See also Supplemental Figure S7.
Discussion
Intranasal delivery of both Adenoviral-Cre and Lenti-Cre can successfully induce Kras-driven NSCLC in GEMMs (DuPage et al. 2009; Gierut et al. 2014; Platt et al. 2014). In this study, Adenoviral-Cre and Lenti-Cre were used to induce Lkb1-deficient lung tumorigenesis separately. Unexpectedly, Atg7 was not homogeneously deleted in KL tumors with Adenoviral-Cre infection, leading to a mildly extended survival advantage with Atg7 ablation compared with mice bearing wild-type tumors. Li et al. (2015) reported that the deletion of Atg5 in KL tumors with Adenoviral-Cre intranasal infection did not impact tumor growth, which could be a result of incomplete Atg5 deletion. However, by using Lenti-Cre infection in KL mice, we found that the efficiency of Atg7 deletion from the KL tumor cells was dramatically increased. As a consequence of Atg7 deletion, tumor frequency and tumor burden were significantly reduced in mice bearing Atg7−/− tumors compared with mice bearing Atg7+/+ tumors. Our studies demonstrate that autophagy is essential for Lkb1-deficient lung tumorigenesis, which is consistent with our previous work (Guo et al. 2011, 2013b, 2016; Guo and White 2013) and supports the concept of inhibiting autophagy to metabolically compromise the growth and survival of Ras-driven cancers.
What is new here is that autophagy ablation is more effective in inhibiting KL tumorigenesis than in suppressing KP tumor growth. We found that autophagy ablation was synthetically lethal during KL tumorigenesis and dramatically impaired both the frequency of tumor initiation and tumor growth. However, autophagy deficiency does not affect KP tumor initiation (Guo et al. 2013a). Therefore, the role of autophagy in tumor initiation could be oncogenic driver-dependent. Regarding the consequences of autophagy ablation on tumor growth, few KL tumors with autophagy defects eventually formed, which is correlated with the decreased oncogenic signaling, increased apoptosis and senescence, and prolonged mouse survival. In mice bearing KP tumors, the loss of Atg7 extended mouse survival by just 2 wk (Guo et al. 2013a). However, Atg7 deficiency extended KL mouse life span by >2 mo (Fig. 1G). Loss of Lkb1 promotes transdifferentiation of adenocarcinoma to squamous cell carcinoma at the late stage of tumor progression in KL GEMMs (Ji et al. 2007; Han et al. 2014). We found that mice bearing Atg7−/− KL tumors died from a combination of adenocarcinoma, squamous cell carcinoma, and inflammation (Supplemental Fig. S3B). Therefore, the role of autophagy in regulating KL lung tumor differentiation needs further investigation. LKB1 loss promotes tumorigenesis but also leads to a lack of metabolic plasticity in response to an energy crisis (Parker et al. 2017), which is also confirmed in human cancer cells in response to starvation (Fig. 7B; Supplemental Fig. S7F). Therefore, LKB1-deficient cancer cells are driven to activate alternative metabolic pathways to prevent cell death. Our findings demonstrate that autophagy could be one of the alternatives, and autophagy inhibition could particularly be effective in the treatment of LKB1-defective KRAS-driven NSCLC that failed to respond to other treatments, including immunotherapy (Siegel et al. 2015; Skoulidis et al. 2018). In addition, a previous study demonstrated that Atg7 regulates the p53-dependent cell cycle and cell death pathway to survive metabolic stress (Lee et al. 2012). This could explain the different antitumor effects of Atg7 deletion in KL and KP tumors, which need further investigation. However, knockdown of Atg5 in KL cells inhibited cell proliferation and accelerated cell death in starvation, demonstrating that impaired tumorigenesis by Atg7 ablation is mainly through autophagy-dependent function.
AMPK activity is required for Kras-driven lung tumor growth (Eichner et al. 2018). Reduced AMPK activity in autophagy-deficient KP and KL tumors might contribute to the impaired tumorigenesis. However, the mechanism that promotes AMPK activation upon loss of Lkb1 and how autophagy deficiency prevents this processing need further investigation.
Autophagy degrades and recycles macromolecules to sustain cellular and tissue homeostasis in starvation. Different from a previous study showing that acute knockdown of Atg5 by siRNA attenuates glucose uptake (Roy et al. 2017), we found that glucose consumption is increased in Atg7−/− TDCLs in nutrient-rich conditions (Fig. 4B), which might be used to compensate for defective intracellular recycling for proper cell proliferation. A possible reason for these different phenotypes could be that Atg7−/− TDCLs went through in vitro cell culture selection and adaption during the generation process. The present study further supports our previous concept that autophagy sustains the level of mitochondrial TCA cycle intermediates and derivatives for energy homeostasis and prevents nucleotide catabolism during starvation in Kras-driven lung cancer cells (Guo et al. 2016). Different from p53 deletion, the loss of LKB1 significantly up-regulates de novo FA synthesis (Faubert et al. 2014; Svensson et al. 2016). FAO has been reported to play an important role in KL tumor mitochondrial energy production and tumor growth (Svensson et al. 2016). Here, we demonstrate that autophagy plays a prominent role in regulating lipid metabolism for Lkb1-deficient lung tumorigenesis. The possible mechanism is that in nutrient-rich conditions, both autophagy-intact and autophagy-deficient KL tumors are able to maintain energy homeostasis for cell proliferation. In response to nutrient deprivation, autophagy-mediated intracellular recycling is essential for supporting mitochondrial energy production and maintaining a LD reserve in Lkb1-deficient tumor cells. Without autophagy, however, tumor cells increase their reliance on FAO for energy homeostasis to compensate for defective recycling of TCA cycle intermediates and derivatives, which depletes lipid stores, likely causing an energy crisis (Fig. 7G). Additionally, we found that autophagy deficiency increases de novo FA synthesis and inhibits FA elongation, suggesting that autophagy ablation rewires the lipid metabolism, which could be a metabolic bypass for KL tumors to survive autophagy inhibition. In fact, we found that inhibiting lipolysis by orlistat or inhibition of FAO by ETO accelerated starvation-induced Atg7-null cell death. Therefore, a combination of autophagy inhibition and lipid metabolism interruption could be an effective therapy for LKB1-deficient RAS-driven lung cancer.
We also validated our findings in KRAS-driven human lung cancer cells with or without LKB1 mutations. Compared with LKB1 wild-type cells, LKB1 mutant cells are sensitive to starvation-induced cell death, which is aggravated by ATG7 or ATG5 knockdown, validating our findings in the KL mouse model and TDCLs. These results strongly demonstrate that autophagy is indispensable for the survival of LKB1-deficient KRAS mutant lung cancer cells during energy crises.
Materials and methods
Mice
Animal studies followed protocols approved by the Institutional Animal Care and Use Committee. KrasG12D/+; Atg7+/+ and KrasG12D/+;Atg7flox/flox mice generated in our previous study (Guo et al. 2013a) were crossed with Lkb1flox/flox mice (Jackson Laboratory) to generate Atg7+/+ and Atg7flox/flox KL mice. Mice were infected intranasally with Adenoviral-Cre (University of Iowa Viral Vector Core) at 4 × 107 plaque-forming units (pfu) per mouse or Lenti-Cre (University of Iowa Viral Vector Core) at 1 × 106 pfu per mouse at 6–8 wk of age to induce lung tumors. Atg7+/+ and Atg7flox/flox KP mice were infected intranasally with Lenti-Cre at 1 × 106 pfu per mouse at 6–8 wk of age to induce lung tumors.
Tumor burden quantification
Hematoxylin and eosin (H&E)-stained lung specimens were imaged at Rutgers Cancer Institute of New Jersey Biomedical Informatics shared resource using an Olympus VS120 whole-slide scanner (Olympus Corporation of the Americas) at 20× magnification. The image analysis protocol was custom-developed on the Visiopharm image analysis platform (Visiopharm A/S) to identify tissue area and compute tumor burden based on semiautomatically detected tumors. Tumor masks and whole-tissue masks were computed from low-resolution image maps, which were extracted from whole-slide images. Tumor and whole-tissue masks were created for each slide. The segmentation masks were used for generation of ratios of tumor burden.
Histology and IHC
Five mice from each group (Atg7+/+ or Atg7−/−) were sacrificed via cervical dislocation at 6, 10, 14, 18, and 22 wk after Lenti-Cre infection. Paraffin-embedded sections and frozen sections were prepared as described previously (Guo et al. 2013a). For IHC, paraffin-embedded sections were stained with antibodies against Atg7 (Sigma, A2856), LC3 (Nano Tools, LC3-5F10), p62 (Enzo Life Sciences, PW9860-0100), Ki67 (Abcam, ab-15580), pACCS79 (Cell Signaling, 3661S), pS6 (Cell Signaling, 4858S), p42/44 MAPK (p-ERK) (Cell Signaling, 4376), cleaved caspase3 (Cell Signaling, 9661S), Tom20 (Abcam, ab186735), and Lkb1 (Santa Cruz Biotechnology, sc-32245).
Cytochrome c oxidase staining
A modified Nadi reaction was conducted as follows: Freshly frozen sections were stained in incubating solution (75 mg of sucrose in 7.5 mL of water, 0.2 M phosphate buffer at pH 7.6, 5 mg of 3′ diaminobenzidine tetrachloride, 10 mg of cytochrome c, 20 µg of catalase) for 1 h at room temperature. Catalase (C-10), cytochrome C (C-2506), and DAB (D-5637) were purchased from Sigma. Slides were washed three times in deionized water and then dehydrated in ascending alcohols (50%, 70%, 80%, 95% twice, and then 100% twice) prior to mounting and imaging.
Senescence-associated β-galactosidase assessment
The β-galactosidase staining was performed per the manufacturer's recommendations as described previously (Guo et al. 2013a) (Cell Signaling, 9860). Tissue sections were visualized and imaged under a 20× objective using a Zeiss Primo Vert microscope with a Lumenera Infinity camera.
Reagents
[U13C6] glucose was purchased from Cambridge Isotope Laboratories. Dextrose (D-Glucose) was purchased from Fisher Scientific. Glutamine, pyruvate, lactate, dimethyl-α-KG, proline, uridine, adenosine, guanosine, inosine, BSA, palmitate, ETO, fasnall, and orlistat were purchased from Sigma-Aldrich. RPMI-1640, RPMI-1640 without glucose, and RPMI-1640 without glutamine media were purchased from Invitrogen. Negative control siRNA (51-01-14-03) and ATG7 siRNA (194439024) were purchased from IDT DNA. Atg5 siRNA (mouse; J-064838-05-0005, J-064838-06-0005, and J-064838-07-0005) were purchased from Dharmacon, Inc., and mixed to make a SMARTpool. ATG5 SMARTpool siRNA (human; 1-004374-04-0005) was purchased from Dharmacon, Inc. HBSS (no glucose) was configured according to the formula (Thermo Fisher, 14025092), but no glucose was added.
Cell culture
Atg7+/+ and Atg7−/− KL TDCLs were generated from Atg7+/+ KL or Atg7−/− KL lung tumors at 10 wk after tumor induction by Adenoviral-Cre. Atg7+/+ and Atg7−/− KP TDCLs were generated previously (Guo et al. 2013a). TDCLs were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 0.075% sodium bicarbonate at 38.5°C with 8.5% CO2.
LKB1 wild-type (H441) and LKB1 mutant (H23, HCC44, and H1355) human lung cancer cell lines with KRAS mutation were generously provided by the Pine laboratory at Rutgers Cancer Institute of New Jersey. The cells were cultured in RPMI-1640 medium containing 10% FBS, 1% penicillin/streptomycin, 0.075% sodium bicarbonate, and 2 mM glutamine at 37°C with 5% CO2.
Cell lines were routinely confirmed to be free of mycoplasma using the MycoAlert mycoplasma detection kit (Lonza).
Adenoviral-Cre-mediated deletion of Atg7 in Atg7flox/flox KL TDCLs
Atg7+/+ or Atg7flox/flox KL TDCLs were seeded in 10-cm dishes in normal growth medium. The following day, when cells were at 70%–80% confluence, fresh medium with Adenoviral-Cre (2.5 × 108 pfu) was supplemented and incubated for 24 h. The cells were then seeded in 12-well plates for cell proliferation and clonogenic survival assay in HBSS starvation or HBSS supplemented with 2 mM glutamine or 20 µM BSA or BSA-Pal. Cell lysates were collected for Western blot to confirm Atg7 deletion at 48 h after infection.
Using siRNA to knock down ATG5 and ATG7 in KL TDCLs or human lung cancer cell lines
Cells were seeded in six-well plates in normal growth medium. The following day, when cells were at 70%–80% confluence, RPMI was removed and replaced with fresh medium with control or targeted siRNAs (200 pmol siRNA for human ATG7; 500 pmol SMARTpool siRNA for mouse Atg5 or human ATG5) in Lipofectamine 3000 (Invitrogen, L3000-015) and opti-MEM and then incubated for 48 h. The cells were then seeded in 12-well plates for cell proliferation and clonogenic survival assay in HBSS starvation or HBSS supplemented with 2 mM glutamine or 20 µM BSA or BSA-Pal. Cell lysates were collected for Western blot to confirm protein knockdown at 48 h after transfection.
Cell proliferation assay
For IncuCyte measurement, cells were seeded at 4 × 104 cells per well in 12-well plates in nutrient-rich RPMI condition. Cell proliferation assay was performed for 4 d using IncuCyte live-cell analysis system and analyzed using IncuCyte Zoom.
For manual counting, cells were trypsinized off culture dishes and then counted in triplicates using a Vi-Cell counter.
Clonogenic survival assay
Starvation clonogenic survival assay was performed as described previously (Guo et al. 2011, 2013a) and stained with 1× Giemsa (Sigma-Aldrich).
Assessment of OCR
OCR of TDCLs was measured using a Seahorse Biosciences extracellular flux analyzer (XF24) as described previously (Guo et al. 2011, 2016). Briefly, cells were seeded at 2.5 × 104 to 3 × 104 cells in the XF24 plates overnight prior to XF assay. Cells were preincubated in RPMI, HBSS, and HBSS supplemented with 2 mM glutamine for 1 h, and basal OCRs were measured subsequently. Relative OCR (percentage) was normalized to the RPMI treatment.
For palmitate-induced OCR, cells were preincubated in HBSS supplemented with 20 µM BSA or BSA-Pal for 1 h; subsequently, a readout of basal OCR was measured.
For FAO assay in starvation, KL TDCLs were briefly starved in HBSS for 1 h and then treated with vehicle control or 50 µM ETO; a readout of basal OCR was subsequently measured upon treatment.
Glucose and amino acid consumption assay
TDCLs were plated at 0.5 × 106 cells per 6-cm dish. The following day, fresh RPMI medium was replaced, and medium was collected at 0, 12, and 24 h. The levels of glucose and amino acids in medium were measured using LC-MS. Cell number was measured at each time point for normalization.
Metabolomic analyses by LC-MS
Methanol:acetonitrile:water (40:40:20) with 0.5% formic acid solution followed by neutralization with 15% ammonium bicarbonate was used to extract metabolites from cells and tumors for LC-MS analysis, as described previously (Guo et al. 2016; Hui et al. 2017). Extracting saponified FAs from cells was performed as described previously (Guo et al. 2013a).
For de novo FAs synthesis analysis, TDCLs were cultured in 6-cm dishes in RPMI containing 2 g/L [U13C6] glucose for 3 d and assessed in triplicate. Fresh medium was changed every day to maintain nutrient-rich conditions. Saponified FAs were extracted as described above for LC-MS analysis after 3 d of labeling.
LC-MS analysis of the extracted metabolites, FAs, and lipids was performed on a Q Exactive Plus hybrid quadrupole orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to hydrophilic interaction chromatography (HILIC).
Detailed methods of metabolomic and lipidomic analysis by LC-MS are in the Supplemental Material.
Western blotting
Tumor samples were snap-frozen, ground using Cryomill in liquid nitrogen at 25 Hz for 2 min, and lysed in Tris lysis buffer. Protein concentration was assessed using the Bio-Rad BCA reagent. The following antibodies were used for Western blots: Atg7 (Sigma Aldrich, A2856), Lkb1 (Santa Cruz Biotechnology, sc-32245), LC3 (Novus Biologicals, NB600-1384), Atg5 (Abcam, ab108327), p62 (American Research Products, 03-GP62-C), TOM20 (Abcam, ab186735), and β-actin (Sigma, A1978).
PCR for mouse genotyping and TDCL characterization
For mouse genotyping, mouse tails (∼3 mm) were incubated with 200 µL 50 mM NaOH and heated for 45 min, and 100 µL of 1 M Tris-HCl was added, vortexed, and centrifuged at 12,000 rpm for 10 min to extract genomic DNA. Subsequently, the extracted genomic DNA was used for PCR.
For TDCL characterization, TDCL pellets were incubated in extraction buffer (Sigma Aldrich, XNAT-1000RXN) and heated for 30 min at 60°C; subsequently, neutralization buffer was added to stop the reaction. The extracted genomic DNA was then used for PCR.
Oil Red O staining and quantification
For the staining of lung tissues, fixed frozen tissue sections were air-dried and stained with prewarmed Oil red O solution (0.5 g of Oil Red O in 100 mL of propylene glycol) for 8–10 min in a 60°C oven, differentiated in 85% propylene glycol solution for 1 min, and rinsed twice with distilled water. Slides were then lightly stained with alum hematoxylin for 1 min, rinsed with distilled water, and mounted for microscopy.
For the staining of cells, TDCLs were plated on autoclaved glass coverslips in 24-well plates. The following day, cells were rinsed twice with PBS and treated with fresh RPMI or HBSS for 4 h. The cells were then rinsed twice with PBS and fixed with 10% formalin and 60% isopropanol. Prewarmed Oil Red O solution was then added to the cells for staining as described above.
A minimum of three high-magnification (100×) images was used for quantification of LDs. Briefly, each image was run through color deconvolution on ImageJ-Fiji software (National Institutes of Health), and a fixed threshold was adjusted between 70 and 200 for all images. Quantitative data were exported from ImageJ-generated histograms into Microsoft Excel software for further analysis and presentation. An average of Oil Red O percent area to the total area was calculated and normalized to cell number.
Statistics
Data were expressed as the mean ± SEM. Statistical analyses were carried out with Graphpad Prism version 5.0 or Microsoft Excel. Statistical significance was calculated by two-way ANOVA with Bonferroni post-test for the time course study of tumor number and tumor burden, and five mice from each genotype at each time point were used. Significance in the Kaplan-Meier analyses to determine and compare the progression-free survival was calculated using the log-rank test. Significance of metabolites, lipids, saponified FAs, OCR, and cell proliferation in drug treatments between autophagy wild-type and autophagy-deficient cells was determined by a paired two-tailed Student's t-test. For quantification of IHC for Ki67, caspase3, pS6, and p-ERK, six representative images of entire lung lobes were obtained at each time point for each genotype. A minimum of 150 cells from each image was scored. Statistical significance was calculated by a paired two-tailed Student's t-test. A P-value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We are grateful to Dr. Eileen White for her advice during the preparation of the manuscript; Dr. Sharon Pine for providing the human lung cancer cell lines; Dr. Chen Liu for providing consultation on tumor pathology; Sadia Nawshin, Taham Khan, Ellie Cheung, Sajid Fahumy, and Nuha Syed in the Guo laboratory for helping with mouse genotyping; and Jennifer Hostettler for assistance with editing. This work was supported by American Cancer Society pilot grant IRG-15-168-01 to J.Y.G., the Lung Cancer Research Foundation to J.Y.G., National Institutes of Health (NIH) grant K22 CA190521 to J.Y.G., NIH P30 CA072720 to Rutgers Cancer Institute of New Jersey (Metabolomics Shared Resource, Biomedical Informatics shared resource, and Biospecimen Repository Service Shared Resource at Rutgers Cancer Institute of New Jersey), and New Jersey Commission on Cancer Research DFHS18PPC021 to K.K.
Author contributions: J.Y.G. conceived the study and supervised the research. J.Y.G. and V.B. designed the experiments and interpreted data. V.B. performed most of the experiments and analysis. K.K. and W.K. performed some of the cell culture experiments. K.K. and A.L. performed the IHC and H&E staining. Z.S.H. maintained the mouse colony. X.S. performed metabolomics processing. J.Y.G. and V.B. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.320481.118.
References
- Amaravadi R, Kimmelman AC, White E. 2016. Recent insights into the function of autophagy in cancer. Genes Dev 30: 1913–1930. 10.1101/gad.287524.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloribi-Djefaflia S, Vasseur S, Guillaumond F. 2016. Lipid metabolic reprogramming in cancer cells. Oncogenesis 5: e189 10.1038/oncsis.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, et al. 2014. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9: 349–365. 10.1016/j.celrep.2014.08.056 [DOI] [PubMed] [Google Scholar]
- Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, Karchin R, Kinzler KW, Vogelstein B, Nowak MA. 2010. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci 107: 18545–18550. 10.1073/pnas.1010978107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, Jacks T. 2009. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4: 1064–1072. 10.1038/nprot.2009.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, Shokhirev MN, Leblanc M, Vera LI, Hutchins A, et al. 2018. Genetic analysis reveals AMPK is required to support tumor growth in murine Kras-dependent lung cancer models. Cell Metab 10.1016/j.cmet.2018.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, et al. 2014. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc Natl Acad Sci 111: 2554–2559. 10.1073/pnas.1312570111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierut JJ, Jacks TE, Haigis KM. 2014. In vivo delivery of lenti-Cre or adeno-Cre into mice using intranasal instillation. Cold Spring Harb Protoc 2014: 307–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, White E. 2013. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy 9: 1636–1638. 10.4161/auto.26123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, White E. 2016. Autophagy, metabolism, and cancer. Cold Spring Harb Symp Quant Biol 81: 73–78. 10.1101/sqb.2016.81.030981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25: 460–470. 10.1101/gad.2016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. 2013a. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27: 1447–1461. 10.1101/gad.219642.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Xia B, White E. 2013b. Autophagy-mediated tumor promotion. Cell 155: 1216–1219. 10.1016/j.cell.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD, White E. 2016. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 30: 1704–1717. 10.1101/gad.283416.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Li F, Fang Z, Gao Y, Li F, Fang R, Yao S, Sun Y, Li L, Zhang W, et al. 2014. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun 5: 3261 10.1038/ncomms4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Alessi DR. 2013. LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol 11: 36 10.1186/1741-7007-11-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Freter C. 2015. Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci 16: 924–949. 10.3390/ijms16010924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. 2017. Glucose feeds the TCA cycle via circulating lactate. Nature 551: 115–118. 10.1038/nature24057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. 2012. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150: 1107–1120. 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Chandel NS, Hay N. 2012. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485: 661–665. 10.1038/nature11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al. 2007. LKB1 modulates lung cancer differentiation and metastasis. Nature 448: 807–810. 10.1038/nature06030 [DOI] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. 2014. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 4: 914–927. 10.1158/2159-8290.CD-14-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169: 425–434. 10.1083/jcb.200412022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T, et al. 2016. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539: 390–395. 10.1038/nature20132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T. 2012. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 336: 225–228. 10.1126/science.1218395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Han X, Li F, Wang R, Wang H, Gao Y, Wang X, Fang Z, Zhang W, Yao S, et al. 2015. LKB1 inactivation elicits a redox imbalance to modulate non-small cell lung cancer plasticity and therapeutic response. Cancer Cell 27: 698–711. 10.1016/j.ccell.2015.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. 2011. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 22: 165–178. 10.1091/mbc.e10-06-0500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashima T, Seimiya H, Tsuruo T. 2009. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer 100: 1369–1372. 10.1038/sj.bjc.6605007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, Suzuki K, Nakamoto M, Shimizu E, Minna JD, et al. 2007. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 26: 5911–5918. 10.1038/sj.onc.1210418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard JA, Christianson HC, Kucharzewska P, Bourseau-Guilmain E, Svensson KJ, Lindqvist E, Indira Chandran V, Kjellen L, Welinder C, Bengzon J, et al. 2016. Metastasis stimulation by hypoxia and acidosis-induced extracellular lipid uptake is mediated by proteoglycan-dependent endocytosis. Cancer Res 76: 4828–4840. 10.1158/0008-5472.CAN-15-2831 [DOI] [PubMed] [Google Scholar]
- Merino Salvador M, Gomez de Cedron M, Merino Rubio J, Falagan Martinez S, Sanchez Martinez R, Casado E, Ramirez de Molina A, Sereno M. 2017. Lipid metabolism and lung cancer. Crit Rev Oncol Hematol 112: 31–40. 10.1016/j.critrevonc.2017.02.001 [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Serrano M. 2014. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15: 482–496. 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- Parker SJ, Svensson RU, Divakaruni AS, Lefebvre AE, Murphy AN, Shaw RJ, Metallo CM. 2017. LKB1 promotes metabolic flexibility in response to energy stress. Metab Eng 43: 208–217. 10.1016/j.ymben.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, et al. 2017. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541: 41–45. 10.1038/nature20791 [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. 2014. CRISPR–Cas9 knockin mice for genome editing and cancer modeling. Cell 159: 440–455. 10.1016/j.cell.2014.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W, et al. 2018. Autophagy maintains tumour growth through circulating arginine. Nature 563: 569–573. 10.1038/s41586-018-0697-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, Lippincott-Schwartz J. 2015. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32: 678–692. 10.1016/j.devcel.2015.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Leidal AM, Ye J, Ronen SM, Debnath J. 2017. Autophagy-dependent shuttling of TBC1D5 controls plasma membrane translocation of GLUT1 and glucose uptake. Mol Cell 67: 84–95.e5. 10.1016/j.molcel.2017.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB. 2013. Unravelling the connection between metabolism and tumorigenesis through studies of the liver kinase B1 tumour suppressor. J Carcinog 12: 16 10.4103/1477-3163.116323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. 2009. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 9: 563–575. 10.1038/nrc2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, et al. 2013. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23: 143–158. 10.1016/j.ccr.2012.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. 2005. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310: 1642–1646. 10.1126/science.1120781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. 2015. Cancer statistics, 2015. CA Cancer J Clin 65: 5–29. 10.3322/caac.21254 [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. 2009. Autophagy regulates lipid metabolism. Nature 458: 1131–1135. 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. 2018. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 8: 822–835. 10.1158/2159-8290.CD-18-0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. 2016. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536: 479–483. 10.1038/nature19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker AM, White E. 2014. Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov 4: 766–772. 10.1158/2159-8290.CD-14-0196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, et al. 2016. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med 22: 1108–1119. 10.1038/nm.4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, DeBerardinis RJ. 2017. Understanding the intersections between metabolism and cancer biology. Cell 168: 657–669. 10.1016/j.cell.2016.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YA, Xing X, Harris JW, Zaytseva YY, Mitov MI, Napier DL, Weiss HL, Mark Evers B, Gao T. 2017. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis 8: e2593 10.1038/cddis.2017.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. 2012. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12: 401–410. 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, Mehnert JM, Chan CS. 2015. Autophagy, metabolism, and cancer. Clin Cancer Res 21: 5037–5046. 10.1158/1078-0432.CCR-15-0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, Herter-Sprie GS, Akbay EA, Tchaicha JH, Altabef A, et al. 2014. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 25: 590–604. 10.1016/j.ccr.2014.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25: 717–729. 10.1101/gad.2016111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, et al. 2018. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov 8: 276–287. 10.1158/2159-8290.CD-17-0952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Madeo F, Kratky D. 2017. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol 18: 671–684. 10.1038/nrm.2017.76 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.