

The transmembrane E3 ubiquitin ligases ZNRF3 and RNF43 antagonize Wnt signaling by promoting degradation of frizzled receptors. Here, Basham et al. demonstrate that adrenocortical-specific loss of ZNRF3, but not RNF43, results in adrenal hyperplasia that depends on Porcupine-mediated Wnt ligand secretion.
Keywords: Wnt signaling, adrenal zonation, mouse models, organ maintenance, proliferation
Abstract
Spatiotemporal control of Wnt signaling is essential for the development and homeostasis of many tissues. The transmembrane E3 ubiquitin ligases ZNRF3 (zinc and ring finger 3) and RNF43 (ring finger protein 43) antagonize Wnt signaling by promoting degradation of frizzled receptors. ZNRF3 and RNF43 are frequently inactivated in human cancer, but the molecular and therapeutic implications remain unclear. Here, we demonstrate that adrenocortical-specific loss of ZNRF3, but not RNF43, results in adrenal hyperplasia that depends on Porcupine-mediated Wnt ligand secretion. Furthermore, we discovered a Wnt/β-catenin signaling gradient in the adrenal cortex that is disrupted upon loss of ZNRF3. Unlike β-catenin gain-of-function models, which induce high Wnt/β-catenin activation and expansion of the peripheral cortex, ZNRF3 loss triggers activation of moderate-level Wnt/β-catenin signaling that drives proliferative expansion of only the histologically and functionally distinct inner cortex. Genetically reducing β-catenin dosage significantly reverses the ZNRF3-deficient phenotype. Thus, homeostatic maintenance of the adrenal cortex is dependent on varying levels of Wnt/β-catenin activation, which is regulated by ZNRF3.
The Wnt pathway is an essential regulator of development and homeostasis in many tissues (MacDonald et al. 2009; Nusse and Clevers 2017). Signaling is initiated by secreted Wnt ligands, which bind frizzled (FZD) membrane receptors on cells within close proximity (Farin et al. 2016) to trigger a canonical (β-catenin-dependent or Wnt/β-catenin) or noncanonical (β-catenin-independent) Wnt response. As aberrant Wnt signaling is associated with many diseases, including cancer (Nusse and Clevers 2017), the strength of Wnt signaling must be tightly regulated in vivo.
ZNRF3 (zinc and ring finger 3) and its homolog, RNF43 (ring finger protein 43), are newly identified negative regulators of Wnt signaling (Fearon and Spence 2012). Znrf3 and Rnf43 were first discovered by gene expression profiling as β-catenin target genes (Hao et al. 2012) that are also enriched in LGR5+ intestinal stem cells (Koo et al. 2012). Once induced, ZNRF3 and RNF43 function as transmembrane E3 ubiquitin ligases to induce endocytosis of FZD receptors from the cell surface (Hao et al. 2012; Koo et al. 2012). Integral components of this signaling module also include R-spondin (RSPO) proteins (de Lau et al. 2012), which are secreted factors that regulate the activity of ZNRF3/RNF43. Specifically, RSPOs promote clearance of ZNRF3/RNF43 from the membrane (Hao et al. 2012) through both LGR-dependent and LGR-independent mechanisms (Lebensohn and Rohatgi 2018; Szenker-Ravi et al. 2018). Consequently, RSPO proteins neutralize the negative effects of ZNRF3/RNF43 on Wnt signaling.
Alterations in the RSPO–ZNRF3/RNF43 module have been frequently identified in human cancer (Hao et al. 2016). These primarily include loss-of-function (LOF) events in ZNRF3/RNF43 caused by homozygous deletion or truncation mutation (Ong et al. 2012; Chan-On et al. 2013; Nord et al. 2013; Ryland et al. 2013; Assie et al. 2014; Giannakis et al. 2014; Robinson et al. 2015; Witkiewicz et al. 2015; Zheng et al. 2016) and gain-of-function (GOF) events in RSPOs caused by translocations (Seshagiri et al. 2012; Robinson et al. 2015). Unlike many of the previously identified Wnt pathway mutations in human cancer that alter the intracellular trafficking and stability of β-catenin (Kinzler et al. 1991; Nishisho et al. 1991; Morin et al. 1997; Rubinfeld et al. 1997; Satoh et al. 2000; Nusse and Clevers 2017), perturbations in RSPO–ZNRF3/RNF43 act upstream to control Wnt receptor availability. Thus, the loss of ZNRF3/RNF43 or gain of RSPOs has the potential to enhance both β-catenin-dependent and β-catenin-independent signaling, and the precise molecular consequences remain unclear.
To examine the mechanisms downstream from RSPO–ZNRF3/RNF43, we used the adrenal cortex as a model tissue. The function of the adrenal cortex is to produce steroid hormones that are essential for life and regulate key biological processes (Walczak and Hammer 2015). In order to achieve both a rapid and precise response, the adrenal cortex uses hormonal feed-forward–feedback systems that function in the context of histologically distinct adrenocortical zones (Xing et al. 2015). These layers, the outer zona glomerulosa (zG), intermediate zona fasciculata (zF), and inner zona reticularis (zR), produce mineralocorticoids, glucocorticoids, and androgens, respectively. Like other more prototypical epithelial tissue models, including the intestinal crypt (Clevers 2013) and skin epidermis (Gonzales and Fuchs 2017), cells of the adrenal cortex are continually renewed throughout life. This process is governed predominantly by centripetal migration and differentiation, where multipotent progenitor cells in the surrounding mesenchymal capsule and outer zG give rise to concentric layers of differentiated cortex (King et al. 2009; Freedman et al. 2013; Wood et al. 2013). Accordingly, the adrenocortical homeostatic unit is an elegant model of progenitor cell dynamics and cell fate conversion.
Previously, Wnt/β-catenin signaling has been shown to be essential for normal adrenal gland formation and homeostasis (Kim et al. 2008). High Wnt/β-catenin signaling is constrained to the outer cortex (Walczak et al. 2014), where it promotes zG differentiation and subsequent mineralocorticoid production (Berthon et al. 2014). However, as cells of the zG are centripetally displaced, Wnt/β-catenin signaling is inhibited to allow conversion into zF cells (Drelon et al. 2016). Spatial restriction of Wnt/β-catenin activation to the outermost cortex is established in part by capsular RSPO3, which promotes Wnt/β-catenin signaling and is required for zG cell fate (Vidal et al. 2016) as well as active PKA signaling in the zF, which represses Wnt/β-catenin signaling (Drelon et al. 2016). Notably, genetic mouse models of constitutive β-catenin activation, where such restriction is lost, display expanded zG differentiation and mineralocorticoid excess with concomitant loss of the zF (Berthon et al. 2010). These observations suggest that the level of Wnt/β-catenin signaling in the adrenal cortex must be tightly regulated to maintain functional zonation and proper steroid hormone production.
Here, we investigated the potential role of ZNRF3 and RNF43 in homeostatic regulation of the adrenal cortex. We show that loss of ZNRF3, but not RNF43, leads to severe adrenocortical hyperplasia. Through ablation of porcupine (PORCN), a key enzyme required for Wnt ligand secretion, we demonstrate that the effects of ZNRF3 loss are highly dependent on Wnt signaling. Furthermore, using high-resolution single-cell techniques, we discovered a Wnt/β-catenin signaling gradient in the adrenal cortex. We show that loss of ZNRF3 increases Wnt/β-catenin activity specifically in the inner cortex, resulting in disruption of this gradient. Moreover, genetically reducing the dosage of β-catenin significantly rescues the ZNRF3-deficient phenotype. These results suggest that adrenocortical cells are highly sensitive to varying levels of Wnt/β-catenin activation and that ZNRF3 plays an essential role in the adrenal cortex to maintain a Wnt/β-catenin signaling gradient.
Results
ZNRF3 is expressed throughout the adrenal cortex, beneath the RSPO-producing capsule
We sought to generate a conditional knockout (cKO) mouse model to study the molecular mechanisms downstream from RSPO–ZNRF3/RNF43 signaling. In order to design an appropriate targeting strategy, we first assessed the expression and localization of Rspos, Rnf43, and Znrf3 in the normal adrenal glands of 6-wk-old mice. Consistent with previous studies (Vidal et al. 2016), we found that Rspo3 from the outer adrenal capsule was the main source of RSPOs (Supplemental Fig. S1). Next, we measured the expression and localization of Rnf43 and Znrf3, which we hypothesized would be enriched in the zG, since they have been characterized previously as Wnt/β-catenin target genes (Hao et al. 2012). Indeed, Rnf43 was predominately expressed at low levels in the zG (Fig. 1A). In contrast, Znrf3 was more highly expressed and displayed a wider expression pattern that included the zG as well as the zF (Fig. 1B). This broad range of Znrf3 expression corresponded to a 6.1-fold and 3.5-fold higher level of Znrf3 compared with Rnf43 in female and male mice, respectively (Fig. 1C,D). Thus, ZNRF3 is the dominant homolog expressed in the adrenal gland. Furthermore, since RSPO3 is predicted to neutralize ZNRF3 (Hao et al. 2012), these observations suggest a gradient of increasing ZNRF3 activity toward the inner cortex.
Figure 1.
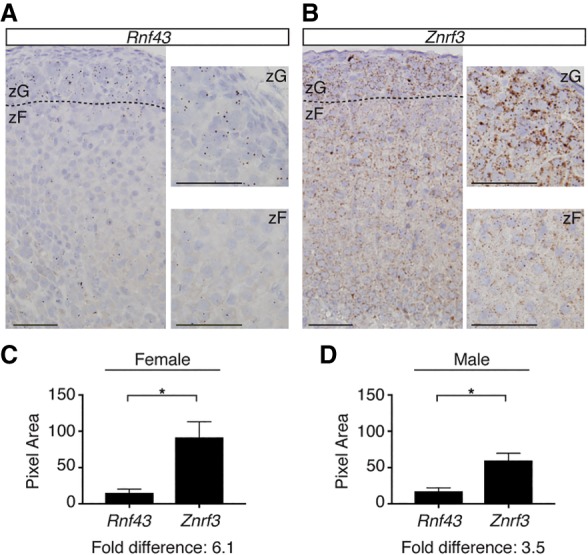
ZNRF3 is expressed throughout the adrenal cortex, beneath the RSPO-producing capsule. (A,B) Rnf43 is expressed predominately in the zG (A), while Znrf3 is expressed throughout the cortex (B). The dashed line marks the histological zG/zF boundary. Bars, 50 µm. Representative images of single-molecule in situ hybridizations (ISHs) from 6-wk-old female mice are shown. (C,D) Quantification of ISHs based on pixel area. Statistical analysis was performed using two-tailed Student's t-test. (*) P < 0.05.
Loss of ZNRF3 during adrenal development triggers hyperplasia due to proliferative expansion of the zF
Based on the wide expression pattern of Znrf3, we chose Steroidogenic factor 1 (SF1)-Cre, in which Cre is expressed in all cells of the adrenal cortex (Bingham et al. 2006), to generate a cKO mouse model. We crossed SF1-Cre mice with Znrf3- and/or Rnf43-floxed mice, which contain loxP sites flanking the RING domain of each gene (Koo et al. 2012). This approach resulted in single-cKO mice lacking functional ZNRF3 or RNF43 in all adrenocortical cells as well as double-knockout mice to assess potential compensation. At 6 wk of age, we observed a marked increase in the size of the adrenal glands in Znrf3 cKO, but not Rnf43 cKO, mice (Fig. 2A; Supplemental Fig. S2A). Loss of ZNRF3 specifically resulted in an average 8.4-fold (female) (Fig. 2B) and 7.7-fold (male) (Supplemental Fig. S2B) increase in normalized adrenal weight. This significant enlargement in adrenal size was accompanied by an increase in cellularity (Fig. 2C; Supplemental Fig. S2C) and loss of normal adrenocortical architecture, as visualized by immunohistochemistry (IHC) for the vascular marker CD31 (Fig. 2D). These results support a critical role for ZNRF3 in regulating adrenal growth and indicate that Rnf43 does not compensate for Znrf3 loss in the adrenal cortex (Supplemental Fig. S3).
Figure 2.
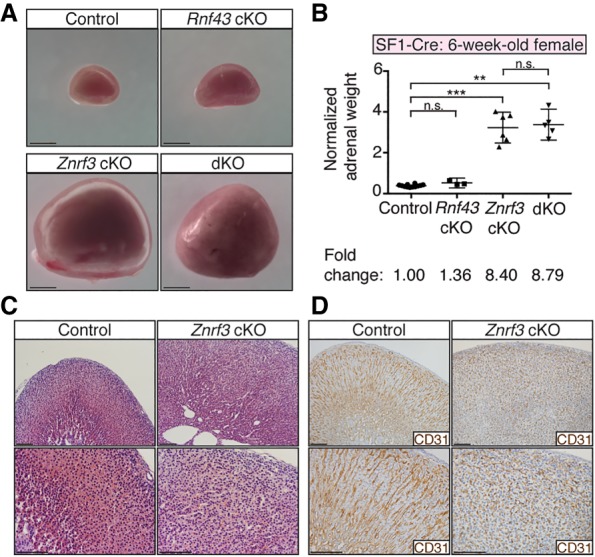
Loss of ZNRF3 induces rapid adrenal growth. (A) Whole adrenals from Znrf3 cKO and Rnf43;Znrf3 double-knockout (dKO) mice are significantly larger in size at 6 wk compared with control or Rnf43 cKO mice. Bars, 1 mm. (B) Normalized adrenal weights shown as mean and 95% confidence interval (CI). Statistical analysis was performed using Welch's one-way ANOVA followed by Games-Howell post hoc test. (**) P < 0.01; (***) P < 0.001. (C,D) Loss of ZNRF3 disrupts normal adrenocortical architecture, as shown by H&E (C) and IHC (D) for the vascular marker CD31. Bars, 100 µm. All data shown are from female mice.
To define the histological changes caused by ZNRF3 deficiency, we next evalulated the expression of distinct adrenal markers. Znrf3 cKO and double-knockout mice displayed an increase in SF1-positive adrenocortical cells and disruption of the innermost tyrosine hydroxylase (TH)-expressing medulla (Fig. 3A; Supplemental Fig. S4A). Immunofluorescence (IF) staining for the zG marker aldosterone synthase (AS; CYP11B2) and the zF marker steroid 11β-hydroxylase (CYP11B1) revealed that the expanded cell population expressed CYP11B1 but not CYP11B2, suggesting a zF phenotype (Fig. 3B; Supplemental Fig. S4B). To rule out potential involvement of the X zone, a transient region of the mouse adrenal cortex that forms adjacent to the medulla and ultimately regresses (Xing et al. 2017), we stained for 20α-hydroxysteroid dehydrogenase (20α-HSD). In contrast to control and Rnf43 cKO mice, we were unable to detect 20α-HSD-positive cells in Znrf3 cKO and double-knockout mice (Fig. 3C; Supplemental Fig. S4C), indicating that the increase in SF1-positive adrenocortical cells observed with ZNRF3 loss was not due to either expansion or failed regression of the X zone. Since we could not readily visualize the X zone by IHC, we also performed quantitative PCR on whole adrenals for Pik3c2g and Akr1c18 (the gene encoding 20α-HSD). As opposed to 6-wk-old male mice that undergo X-zone regression at puberty, we detected Pik3c2g and Akr1c18 in 6-wk-old female control and Znrf3 cKO mice (Supplemental Fig. S5), suggesting that X-zone cells were still present in Znrf3 cKOs but had become highly dispersed, similar to the medulla. Finally, we performed IHC for Ki67 to measure cell proliferation and assessed EdU incorporation to more specifically detect cells in S phase. We observed a significant increase in both Ki67-positive and EdU-positive cells within the zF of Znrf3 cKO mice (Fig. 3D–G; Supplemental Fig. S4D,E). Taken together, these results suggest that loss of ZNRF3 leads to hyperplastic growth of the zF.
Figure 3.
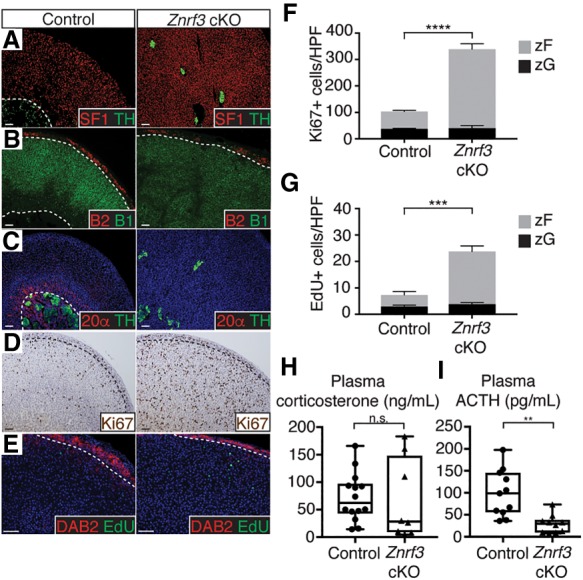
Loss of ZNRF3 results in proliferative expansion of the zF. (A–C) ZNRF3 loss expands the zF and disrupts organization of the inner adrenal medulla. Dashed lines mark histological cortical/medullary and zG/zF boundaries. SF1 (cortex), TH (medulla), B2 (zG), B1 (zF), and 20α-HSD (X zone) were used as markers. (D,E) ZNRF3 loss significantly increases proliferation in the zF based on IHC for Ki67 (D) and EdU (E) incorporation. DAB2 marks the zG. (F,G) Quantification of Ki67 and EdU based on the number of positive cells per high-power field (HPF) within the histological zG or zF. Results are shown as mean ± SEM with three biological replicates per genotype. Statistical analysis was performed using two-way ANOVA followed by Tukey's post hoc test. (***) P < 0.001; (****) P < 0.0001. Reported statistical results represent comparison between zF compartments (gray). No significant difference was observed between zG compartments (black). (H,I) Znrf3 cKO mice maintain normal plasma corticosterone concentrations (H) concomitant with decreased adrenocorticotropic hormone (ACTH) (I). Statistical analysis was performed using two-tailed Welch's t-test. (**) P < 0.01. All data shown are from female mice. Bars, 50 µm.
Since the zF normally functions to synthesize glucocorticoids, we next evaluated hormone production in control and Znrf3 cKO mice. We observed no significant difference in plasma concentrations of corticosterone (Fig. 3H), which is the main glucocorticoid produced in rodents. However, glucocorticoid production is dynamically regulated by adrenocorticotropic hormone (ACTH) from the anterior pituitary as part of the hypothalamic–pituitary–adrenal (HPA) axis. This classic endocrine system is predicated on the integration of feed-forward activation and negative feedback regulation. We found that plasma ACTH was suppressed in Znrf3 cKO mice compared with controls (Fig. 3I), consistent with an increase in the number of steroidogenic zF cells and intact negative feedback. These results suggest that loss of ZNRF3 leads to functional expansion of the zF and that the observed increase in proliferation is not ACTH-dependent.
ZNRF3 deficiency in the adrenal glands of adult mice disrupts homeostasis
Our SF1-Cre-driven cKO mouse model strongly implicated a critical role for ZNRF3 in regulating growth of the zF. However, SF1-Cre is expressed as early as embryonic day 10.5 (E10.5) in the developing embryo (Bingham et al. 2006), and a developmental analysis of Znrf3 cKO mice revealed expansion of the adrenal cortex as early as postnatal day 0 (P0) (Supplemental Fig. S6). Therefore, whether ZNRF3 functions primarily during embryonic and postnatal adrenal development or also during homeostatic maintenance of the adult cortex (after 6 wk of age) remained unclear. Moreover, SF1-Cre is also expressed in the ventromedial hypothalamic nucleus, anterior pituitary, spleen, and gonads (Bingham et al. 2006). Given that ACTH-producing corticotroph cells within the anterior pituitary directly regulate adrenal function, we confirmed that there were no significant histological changes in the pituitary of Znrf3 cKO mice compared with controls (Supplemental Fig. S7A). These results are consistent with SF1-Cre activity being restricted to gonadotrophs rather than corticotrophs in the anterior pituitary (Supplemental Fig. S7B; Bingham et al. 2006). However, we still could not completely exclude the possibility that ZNRF3 loss in other tissues affected adrenocortical growth. To clarify the function of ZNRF3 in the adrenal gland, we generated a cKO mouse model driven by AS-Cre (Freedman et al. 2013). AS (CYP11B2) is an essential steroidogenic enzyme required for mineralocorticoid production (i.e., aldosterone) and thus is expressed only in the adrenal cortex, where it is restricted to the zG (Giroud et al. 1956). However, since the adrenal cortex is maintained by a continual process of centripetal migration and differentiation, AS-expressing cells in the zG replenish the inner cortex over time (Freedman et al. 2013). Notably, as confirmed by lineage tracing (Fig. 4A), the majority of the zF is derived from AS-expressing cells by 12 wk of age in female mice (Freedman et al. 2013; Dumontet et al. 2018).
Figure 4.
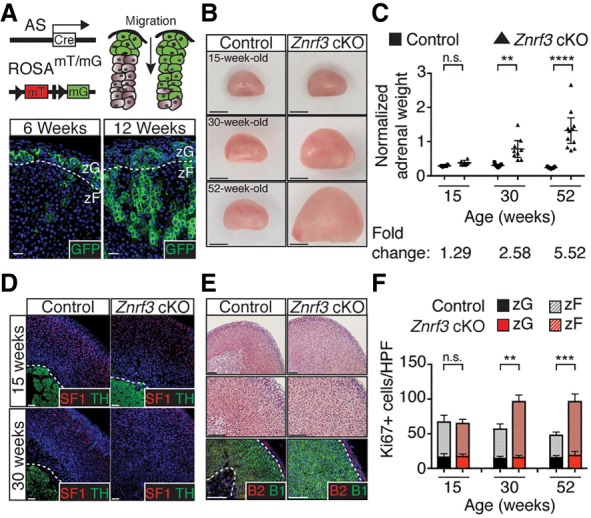
AS-Cre recapitulates SF1-Cre-driven ZNRF3 loss, supporting an adrenal-specific defect in the zF. (A) AS-Cre expression begins in the zG, which gives rise to the zF. Bars, 20 µm. (B) Adrenal glands from AS-Cre-driven Znrf3 cKO mice are significantly larger in size by 30 wk compared with controls. Bars, 1 mm. (C) Normalized adrenal weights are shown as mean and 95% CI. Statistical analysis was performed using two-tailed Welch's t-test. (**) P < 0.01; (****) P < 0.0001. (D,E) AS-Cre-driven loss of ZNRF3 recapitulates the phenotype observed with SF1-Cre, including progressive disruption of the innermost TH-expressing medulla by SF1-positive (D) and CYP11B1-positive (E) zF cells. Bars, 100 µm. (F) AS-Cre-driven loss of ZNRF3 significantly increases proliferation. Quantification of Ki67 based on the number of positive cells per high-power field (HPF) within the histological zG or zF. Results are shown as mean ± SEM with at least three biological replicates per genotype. Statistical analysis was performed using two-way ANOVA followed by Tukey's post hoc test. (**) P < 0.01; (***) P < 0.001. Reported statistical results represent comparison between zF compartments (dashed line). No significant difference was observed between zG compartments (solid line). Dashed lines mark histological cortical/medullary and zG/zF boundaries. All data shown are from female mice.
Using AS-Cre, we generated Znrf3 cKO mice and assessed the size of the adrenal gland beginning at 6 wk of age (data not shown). We first observed a significant increase (2.58-fold) in normalized adrenal weight at 30 wk of age, which progressed to an average 5.52-fold increase by 52 wk (Fig. 4B,C). Consistent with our SF1-Cre-driven Znrf3 cKO mouse model, AS-Cre-driven loss of ZNRF3 resulted in an expanded zF and disrupted adrenocortical organization (Fig. 4D,E; Supplemental Fig. S8), concomitant with increased proliferation in the zF (Fig. 4F). These results demonstrate that adrenal-specific loss of ZNRF3 is sufficient to induce adrenocortical hyperplasia. Moreover, since the phenotype is not observed until after the zF is fully derived from the AS-expressing cell lineage, these results also support a key role for ZNRF3 in homeostatic maintenance of the zF.
Loss of ZNRF3 acts through a Wnt-dependent mechanism
Previous studies have implicated ZNRF3 as a critical Wnt inhibitor that acts by reducing the level of FZD receptors on the cell surface (Hao et al. 2012, 2016; Koo et al. 2012). Based on these observations, we next genetically inactivated Porcn to test whether the hyperplastic adrenal phenotype seen in ZNRF3-deficient mice was Wnt-dependent. PORCN is an enzyme that posttranslationally modifies all Wnt ligands before their secretion (Lum and Clevers 2012; Rios-Esteves and Resh 2013). As a result, loss of PORCN acts upstream in the Wnt pathway to block ligand availability (Barrott et al. 2011). We used SF1-Cre to generate conditional Porcn;Znrf3 double-knockout mice and assessed adrenal size and morphology at 6 wk of age. Loss of PORCN alone had little effect on the cortex (Fig. 5A,B; Supplemental Fig. S9), likely due to ligand secretion being maintained in SF1-negative capsular cells. However, Porcn;Znrf3 double-knockout mice displayed significantly reduced adrenal weight compared with Znrf3 cKOs (Fig. 5A). Moreover, Porcn;Znrf3 double-knockout mice had relatively normal adrenocortical architecture (Fig. 5B), including a more consolidated medullary compartment and the presence of radial cords within the zF. Thus, loss of ZNRF3 promotes adrenal hyperplasia via Wnt signaling.
Figure 5.
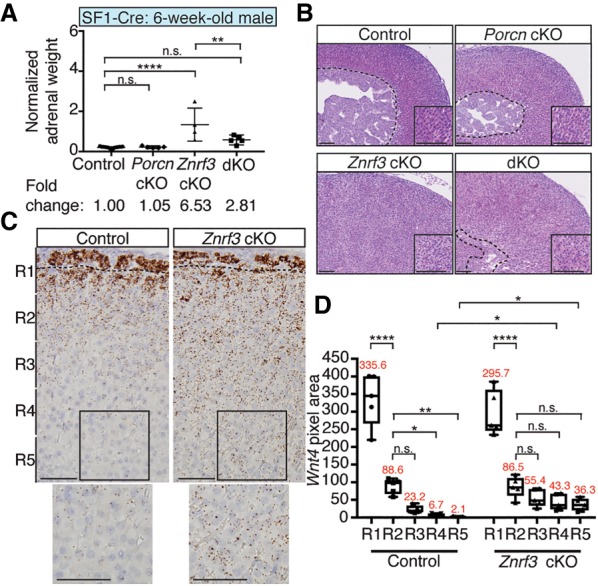
Manifestation of the ZNRF3-deficient phenotype requires Wnt ligand secretion. (A,B) Loss of PORCN significantly rescues the increase in adrenal weight (A) and disrupted adrenocortical architecture (B) observed with ZNRF3 loss. (dKO) Double knockout. Normalized adrenal weight is shown as mean and 95% CI. Statistical analysis was performed using one-way ANOVA followed by Tukey's post hoc test. (**) P < 0.01; (****) P < 0.0001. Representative H&Es from male cKOs are shown. The dashed line marks the histological cortical/medullary boundary. Insets show zF. Bars, 100 µm. (C) Wnt4, which is expressed along a gradient in the normal adrenal cortex, is significantly increased in the inner cortex of Znrf3 cKOs. Single-molecule ISHs were performed in 6-wk-old females. The dashed line marks the histological zG/zF boundary. Bars, 50 µm. (D) Quantification of Wnt4 ISH data based on pixel area within five equal-sized regions (R1–R5) extending from the outer capsule. Results are shown as mean ± SEM with five biological replicates per genotype. The mean pixel area for each region is noted in red. Statistical analysis was performed using one-way ANOVA followed by Tukey's post hoc test. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001.
ZNRF3 loss disrupts a Wnt ligand gradient in the adrenal cortex
Next, to identify candidate Wnt ligands that may contribute to the ZNRF3-deficient phenotype, we performed single-molecule in situ hybridizations (ISHs) for Wnt ligands in adrenals from 6-wk-old cKO mice. Given the short range across which Wnt ligands are thought to act (Boutros and Niehrs 2016; Farin et al. 2016), we were particularly interested in those expressed in the zF, where loss of ZNRF3 drives adrenocortical expansion. Here, we identified only two ligands, Wnt4 and Wnt5a, which were expressed in the zF of the normal adrenal cortex and induced in Znrf3 cKOs (Fig. 5C,D, Supplemental Fig. S10A,B).
WNT4 has been implicated previously as a driver of Wnt/β-catenin signaling in the adrenal cortex that serves to promote zG differentiation (Drelon et al. 2016; Vidal et al. 2016). Consistent with these studies, Wnt4 was highly expressed in the zG of control mice (Fig. 5C). However, we also observed a Wnt4 gradient that continued into the upper zF of the normal adrenal cortex (Fig. 5C,D). Znrf3 cKO mice showed comparable levels of high-intensity Wnt4 expression in the zG (Fig. 5C,D), consistent with the observed normal CYP11B2 levels in these mice. Unexpectedly, Znrf3 cKO mice lost the Wnt4 gradient in the zF and instead displayed moderate-level Wnt4 expression throughout the entire zF (Fig. 5C,D). These results were also observed in 52-wk-old AS cKO mice (Supplemental Fig. S10C,D). In addition, Wnt5a, which has not been characterized previously in the adrenal cortex, was restricted to the zG and upper zF of control mice (Supplemental Fig. S10A,B) but showed significantly increased expression throughout the zG and zF in ZNRF3-deficient mice (Supplemental Fig. S10A,B). Taken together, these results suggest that loss of ZNRF3 acts through a Wnt-dependent mechanism that may be mediated by an elevated and broader expression pattern of WNT4 and/or WNT5A.
Loss of ZNRF3 increases Wnt/β-catenin signaling in the zF to promote adrenal hyperplasia
Given the known role of WNT4 as a driver of Wnt/β-catenin signaling in the adrenal cortex (Drelon et al. 2016; Vidal et al. 2016), we next assessed β-catenin activation in Znrf3 cKO mice. We performed IHC for nonphospho (active) β-catenin and found that control mice displayed high-intensity active β-catenin staining only in the zG (Fig. 6A, top panel), similar to reported in vivo TCF/LEF reporter activity (Ferrer-Vaquer et al. 2010; Walczak et al. 2014). In addition, however, we observed a gradient of β-catenin activation in the inner cortex, with moderate signal in the upper zF and no signal in the innermost zF. This β-catenin pattern corresponded precisely to the Wnt4 gradient. High-intensity active β-catenin was similarly confined to the zG in Znrf3 cKO mice (Fig. 6A, bottom panel). However, consistent with the expansion of Wnt4 expression, moderate-intensity β-catenin staining was present throughout the zF. To confirm these observations, we performed single-molecule ISH for Axin2, a well-characterized Wnt/β-catenin target gene (Jho et al. 2002; Lustig et al. 2002). Mirroring activated β-catenin, we detected a gradient of Axin2 expression in the normal adrenal gland and sustained moderate-level Axin2 expression throughout the zF of Znrf3 cKOs (Fig. 6B,C; Supplemental Fig. S11). These results suggest that loss of ZNRF3 in the adrenal cortex results in a significant increase in Wnt/β-catenin activity within the zF that disrupts the normal signaling gradient.
Figure 6.
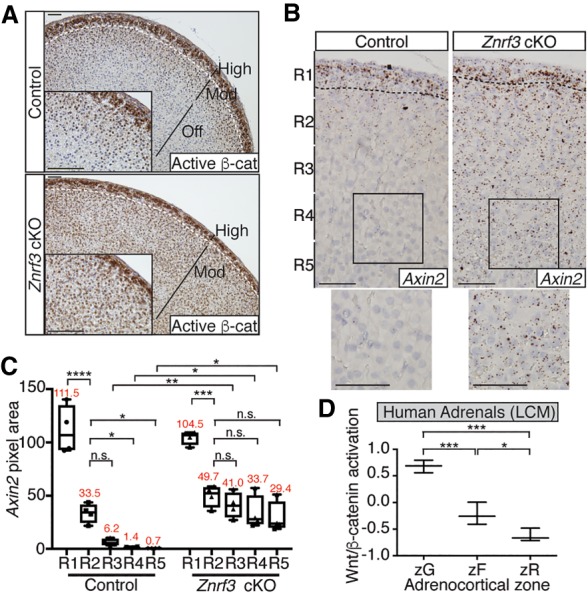
Loss of ZNRF3 increases moderate-level Wnt/β-catenin signaling to promote adrenal hyperplasia. (A–C) ZNRF3 loss increases activated β-catenin (A) and Axin2 (B,C) expression in the zF. Representative images from 6-wk-old females are shown. The dashed line marks the histological zG/zF boundary. Bars, 50 µm. Axin2 quantification based on pixel area within five equal-sized regions (R1–R5) extending from the outer capsule. Results are shown as mean ± SEM with four biological replicates per genotype. The mean pixel area for each region is noted in red. Statistical analysis was performed using one-way ANOVA followed by Tukey's post hoc test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. (D) Wnt/β-catenin activation follows a gradient in the normal human adrenal cortex. Each zone was isolated by laser capture microdissection (LCM). Statistical analysis was performed using one-way ANOVA followed by Tukey's post hoc test. (*) P < 0.05; (***) P < 0.001.
Previously, combined loss of ZNRF3 and RNF43 in the intestinal epithelium was reported to robustly and uniformly activate Wnt/β-catenin signaling (Koo et al. 2012). By comparison, ZNRF3 loss in the mouse adrenal cortex appeared to specifically increase moderate-level Wnt/β-catenin activity. To extend these observations, we assessed differential Wnt/β-catenin pathway activation in normal human adrenal tissue, where each zone was isolated by laser capture microdissection, and in human adrenocortical carcinomas (ACCs), where activating mutations in CTNNB1 (the gene encoding β-catenin) and LOF alterations in ZNRF3 are found in a mutually exclusive 16% and 20% of cases, respectively (Assie et al. 2014; Zheng et al. 2016). We developed a Wnt/β-catenin activation score based on the combined expression of five bona fide adrenocortical Wnt/β-catenin target genes: APCDD1, AXIN2, LEF1, LGR5, and NKD1 (Kim et al. 2008; Berthon et al. 2010; Walczak et al. 2014; Drelon et al. 2016). We then measured the relative level of Wnt/β-catenin activity in each zone of the normal human adrenal gland (Nishimoto et al. 2015) as well as in tumors from The Cancer Genome Atlas (TCGA) ACC project (Zheng et al. 2016). In the normal human adrenal, the zG showed the highest level of Wnt/β-catenin activation, as expected. However, in agreement with our observations of mice, the zF showed a lower level of Wnt/β-catenin activation compared with the zG that was significantly higher than the innermost zR (Fig. 6D). We validated these results by single-molecule ISH for AXIN2 in a normal human adrenal sample, where we visualized a gradual decrease in AXIN2 from the outer to the inner cortex (Supplemental Fig. S12). In human tumors, ACCs harboring CTNNB1-activating mutations had a significantly higher Wnt/β-catenin activation score compared with those without known Wnt pathway mutations (Fig. 7A). However, tumors with ZNRF3 alterations (either homozygous deletion or mutation) showed significantly lower levels of Wnt/β-catenin activation compared with CTNNB1-mutated tumors.
Figure 7.
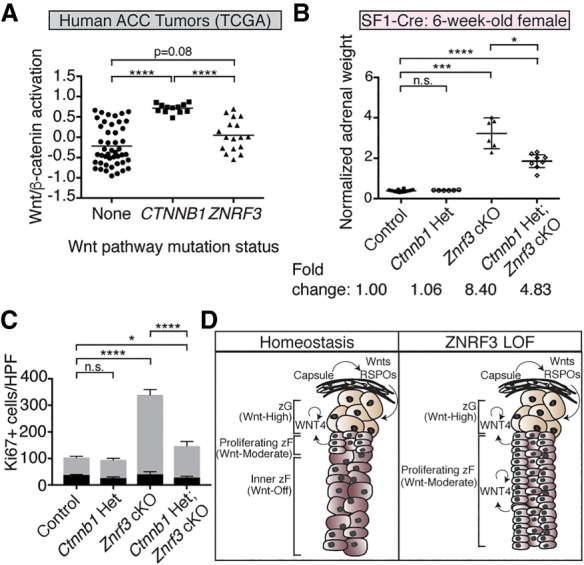
Reduced β-catenin dosage significantly reverses the ZNRF3-deficient phenotype. (A) Wnt/β-catenin activation in ZNRF3-altered human ACCs is significantly lower compared with CTNNB1-activated tumors. Statistical analysis was performed using Welch's one-way ANOVA followed by Games-Howell post hoc test. (****) P < 0.0001. (B,C) Loss of a single copy of Ctnnb1 in the context of ZNRF3 loss significantly rescues adrenal weight (B) and proliferation (C). Normalized adrenal weights are shown as mean and 95% CI. Statistical analysis was performed using Welch's one-way ANOVA followed by Games-Howell post hoc test. (*) P < 0.05; (***) P < 0.001; (****) P < 0.0001. Quantification of Ki67 based on the number of positive cells per high-power field (HPF) within the histological zG or zF. Results are shown as mean ± SEM with three biological replicates per genotype. Statistical analysis was performed using two-way ANOVA followed by Tukey's post hoc test. (*) P < 0.05; (****) P < 0.0001. Reported statistical results represent comparison between zF compartments (gray). No significant difference was observed between zG compartments (black). (D) Model for how varying levels of Wnt/β-catenin signaling regulate adrenal homeostasis. High Wnt/β-catenin signaling in the zG, which is driven in part by capsular-derived Wnts and RSPOs, helps promote zG differentiation. As zG cells migrate centripetally, moderate-level Wnt/β-catenin signaling supports proliferation and conversion into zF. Wnt/β-catenin signaling is ultimately shut off in the innermost cortex to facilitate proper homeostatic turnover. With loss of ZNRF3, moderate-level Wnt/β-catenin signaling is sustained throughout the inner cortex, resulting in increased proliferation and hyperplasia.
Depletion of β-catenin levels partially restores adrenal homeostasis after loss of ZNRF3
Taken together, our observations in both mouse and human models of ZNRF3 deficiency in the adrenal gland suggested that loss of ZNRF3 specifically increases moderate-level Wnt/β-catenin signaling. To test the functional significance of these results, we sought to generate a cKO mouse model lacking both Znrf3 and β-catenin (Ctnnb1). However, since SF1-Cre-driven loss of Ctnnb1 precludes adrenal development beyond E16.5 (Kim et al. 2008), we were unable to generate any Znrf3;Ctnnb1 double-knockout animals (data not shown). Instead, we assessed the role of β-catenin in our ZNRF3-deficient phenotype by inactivating a single copy of Ctnnb1. We found that adrenals from compound Ctnnb1 heterozygous:Znrf3 cKO mice were significantly smaller (Fig. 7B; Supplemental Fig. S13) and had significantly reduced proliferation (Fig. 7C) compared with Znrf3 cKOs. Specifically, Ctnnb1 heterozygosity resulted in an average 42.3% reduction in adrenal size. These results demonstrate that the phenotype of Znrf3 cKOs is at least in part β-catenin-dependent. Furthermore, these results suggest that adrenocortical cells are sensitive to varying levels of Wnt/β-catenin activity and that ZNRF3 acts in the adrenal cortex to maintain a Wnt/β-catenin gradient required for normal homeostasis.
Discussion
Using high-resolution single-molecule approaches, we discovered a Wnt/β-catenin signaling gradient in the adrenal cortex. Furthermore, ZNRF3 is required to maintain this gradient and adrenal homeostasis. Consistent with previous studies, high Wnt/β-catenin signaling is restricted to the zG (Kim et al. 2008; Walczak et al. 2014; Drelon et al. 2016). However, Wnt/β-catenin activity is not uniformly inhibited throughout the zF. Rather, signal strength gradually decreases as cells of the zG move centripetally and convert to zF cells. Genetic inactivation of ZNRF3 in the adrenal cortex leads to aberrant Wnt/β-catenin signaling specifically in the zF, which disrupts the normal Wnt/β-catenin gradient. We demonstrate the functional significance of these observations by genetically reducing the dosage of β-catenin, which significantly rescues the ZNRF3-deficient phenotype.
Although consistent with an expected increase in Wnt/β-catenin signaling, our Znrf3 cKO mouse model displays a fundamentally different phenotype than previous models of β-catenin GOF. Specifically, β-catenin GOF in the adrenal cortex expands the zG, resulting in an overproduction of aldosterone (Berthon et al. 2010), while ZNRF3 LOF expands the zF. These results suggest that the dosage of Wnt/β-catenin activity influences adrenocortical cell fate. High levels of Wnt/β-catenin, which are achieved with direct β-catenin stabilization, help drive proliferation as well as glomerulosa differentiation. β-Catenin has been shown to activate transcription of key zG genes, including AT1R and CYP11B2, in vitro (Berthon et al. 2014), and activating mutations in CTNNB1 are associated with increased glomerulosa function in both mice (Berthon et al. 2010) and humans (Åkerström et al. 2016). Conversely, inactivation of Wnt/β-catenin in the adrenal gland through deletion of either Ctnnb1 (Kim et al. 2008), Wnt4 (Heikkilä et al. 2002; Drelon et al. 2016), or Rspo3 (Vidal et al. 2016) leads to a reduction in CYP11B2 expression and impaired aldosterone production (when measured). These data indicate that high Wnt/β-catenin signaling is required for functional zG differentiation. In contrast, lower levels of Wnt/β-catenin activation, which are achieved with loss of ZNRF3, are not sufficient to drive zG identity and primarily promote proliferation. This is observed in the normal adrenal cortex, where the highest levels of proliferation occur at the zG/zF boundary (Kataoka et al. 1996) along the Wnt/β-catenin gradient, as well as in Znrf3 cKO mice, where proliferation and moderate levels of Wnt/β-catenin signaling are sustained. Taken together, these observations suggest that throughout the process of centripetal differentiation, varying levels of Wnt/β-catenin signaling have different functional consequences (Fig. 7D). High Wnt/β-catenin signaling is necessary in the outer cortex to drive zG differentiation, moderate Wnt/β-catenin signaling in the intermediate cortex allows for proliferation and zF cell fate conversion, and complete inhibition of Wnt/β-catenin signaling is important in the innermost cortex for proper homeostatic turnover.
At the molecular level, it remains unclear how ZNRF3 LOF in the adrenal cortex sustains only moderate levels of Wnt/β-catenin activation as compared with the high levels that might have been initially predicted based on previous studies in other tissues. One potential mechanism, which has been described recently in the adrenal cortex, is active repression of Wnt/β-catenin signaling by PKA. Specifically, PKA signaling in the zF has been shown to inhibit zG differentiation through WNT4 repression (Drelon et al. 2016). Thus, in our model, the competing effects of ZNRF3 loss (WNT4 activation) and PKA activation (WNT4 repression) in a zF cell may result in an overall moderate level of WNT4 ligand and subsequent Wnt/β-catenin signaling. In addition, the level of Wnt/β-catenin activation in response to ZNRF3 loss may be constrained by the physical distance between the capsule, where RSPOs (Vidal et al. 2016) and additional Wnt ligands (Drelon et al. 2015) are produced, and the zF, where the ZNRF3-deficient phenotype is manifest. Capsule-derived RSPOs and Wnts would likely have minimal impact on cells greater than one to two cell diameters away due to the short range over which they are thought to act (Kazanskaya et al. 2004; Rocha et al. 2015; Farin et al. 2016). Consequently, despite an increase in membrane receptor complexes, zF cells in Znrf3 cKO mice may not receive the Wnt ligands/agonists necessary for high Wnt/β-catenin signaling. Finally, the level of Wnt/β-catenin activation with ZNRF3 LOF may be dampened by cross-talk with β-catenin-independent signaling, including the planar cell polarity (Wnt/PCP) and calcium (Wnt/Ca2+) pathways. FZD proteins that are regulated by ZNRF3 and required for Wnt/β-catenin signaling are also integral to Wnt/PCP and Wnt/Ca2+ (Katoh and Katoh 2007; Hao et al. 2012). As a result, loss of ZNRF3 may concurrently activate Wnt/PCP or Wnt/Ca2+, which have been shown previously to inhibit Wnt/β-catenin signaling (Grumolato et al. 2010). Activation of Wnt/PCP or Wnt/Ca2+ would be Wnt ligand-dependent and could be mediated by WNT5A, which is ectopically expressed in Znrf3 cKO mice. However, noncanonical Wnt signaling and the role of WNT5A in the adrenal cortex have yet to be elucidated. Further studies are needed to test the functional impact of PKA, stromal RSPOs/Wnts, and noncanonical Wnt activation on ZNRF3 LOF. These studies will shed light on the molecular basis for the observed functional difference between β-catenin GOF (i.e., Wnt/β-catenin-high) and ZNRF3 LOF (i.e., Wnt/β-catenin-moderate).
Somewhat surprisingly, we observed a difference in the level and expression pattern of Rnf43 and Znrf3 in the adrenal cortex. Moreover, we did not observe functional redundancy or compensation between the homologs, which suggests that these proteins may be inherently distinct. These observations are particularly interesting given the differences between RNF43 and ZNRF3 revealed previously by their crystal structures (Zebisch et al. 2013). An unexpected and highly conserved dimerization mode specific to ZNRF3 was identified, which was speculated to mediate a yet unknown difference in their function and/or regulation. While we cannot conclude a definitive difference in function given the higher Znrf3 expression level, Rnf43 and Znrf3 are clearly differentially regulated in the adrenal cortex. Rnf43 expression is restricted to the Wnt/β-catenin-high zG, while Znrf3 is expressed at higher levels and in both the zG (Wnt/β-catenin-high) and zF (Wnt/β-catenin-moderate and Wnt/β-catenin-off). Notably, a similar paradigm has been observed in the liver, where Rnf43 is limited to the first few layers surrounding the central vein, while Znrf3 is expressed in all hepatocytes (Planas-Paz et al. 2016). These observations suggest a broader function for ZNRF3 beyond the progenitor cell niche. Furthermore, these results suggest that Rnf43 and Znrf3 are differentially regulated, which could be explained by varying thresholds of Wnt/β-catenin signaling activating different transcriptional targets or by additional pathways that may regulate Znrf3 expression.
In addition to progenitor cell dynamics, our studies have important implications for human cancers, where the RSPO–ZNRF3/RNF43 signaling module is frequently altered. ZNRF3 was identified recently as the most commonly altered gene in ACC, with an average 20% of cases harboring biallelic inactivation (Assie et al. 2014; Zheng et al. 2016). However, ZNRF3 is also recurrently deleted in osteoblastoma (Nord et al. 2013) and advanced prostate cancer (Robinson et al. 2015) and down-regulated in gastric adenocarcinoma (Zhou et al. 2013) and papillary thyroid cancer (Qiu et al. 2016). RNF43 mutations range from 4.0% to 18.9% in a variety of cancers, including colorectal adenocarcinoma (Giannakis et al. 2014), mucinous ovarian carcinoma (Ryland et al. 2013), pancreatic cancer (Witkiewicz et al. 2015), and cholangiocarcinoma (Ong et al. 2012; Chan-On et al. 2013), and RSPO fusions are frequently observed in advanced prostate cancer (Robinson et al. 2015) and colon cancer (Seshagiri et al. 2012). Importantly, these alterations are predicted to confer Wnt ligand sensitivity, which can be therapeutically targeted with newly developed PORCN inhibitors (Liu et al. 2013; Madan and Virshup 2015). Although our mouse model of ZNRF3 deficiency does not fully recapitulate human tumors, our studies demonstrate a critical role of Wnt ligands in mediating the effects of ZNRF3 loss. Notably, we observed a highly significant rescue with genetic ablation of Porcn, which supports further testing of PORCN inhibitors against ZNRF3-deficient human tumors, including ACCs. Since loss of Porcn did not completely rescue the ZNRF3-deficient phenotype, it is possible that ZNRF3 may have additional Wnt-independent functions. However, due to the use of SF1-Cre in this experiment, secretion of Wnt ligands from the outer mesenchymal capsule was maintained and could account for the incomplete rescue. Pharmacological PORCN inhibitors may be more effective at inhibiting Wnt activation in this model, since they will target Wnt production in both the capsular and cortical compartments. Thus, our results support further testing of upstream Wnt pathway inhibitors in RSPO–ZNRF3/RNF43-altered tumors.
In conclusion, our data demonstrate that ZNRF3 is a critical mediator of adrenal homeostasis. Through a series of in vivo genetic manipulations, we show that ZNRF3 specifically controls a Wnt/β-catenin signaling gradient, which is essential for maintaining cortical cell renewal and zonal differentiation.
Materials and methods
Mice
All experiments were carried out in accordance with protocols approved by the Auvergne Ethics Committee (Porcn) and the University Committee on Use and Care of Animals at the University of Michigan (all others). All mouse lines—SF1-Crehigh (Bingham et al. 2006), AS+/Cre (Freedman et al. 2013), Znrf3fx/+::Rnf43fx/+ (Koo et al. 2012), Ctnnb1fx/+ (Brault et al. 2001), Porcnfx/+ (Barrott et al. 2011), and ROSAmT/mG (Muzumdar et al. 2007)—have been described previously. Adrenals were cleaned of excess fat, weighed, and frozen in liquid nitrogen or fixed as described. Adrenal weights were compared based on the sum of both adrenal glands and normalized to body weight. We observed no significant difference in body weight between genotypes (Supplemental Fig. S14). SF1-Cre-driven control and Znrf3 cKO cohorts from all experiments performed at the University of Michigan were combined for analysis.
Single-molecule ISH
Adrenals were fixed in 10% normal buffered formalin (VWR) for 24 h at room temperature, paraffin-embedded, and cut into 5-µm sections. The RNAscope Brown detection kit (Advanced Cell Diagnostics) was used according to the manufacturer's instructions. Probes are listed in Supplemental Table S1. Images were acquired on an Olympus BX-51 microscope with an Olympus DP-70 camera. Automated image analysis was performed on 40× images using ImageJ (version 1.48) as described previously (Grabinski et al. 2015). A description of image analysis with examples is in Supplemental Figure S15. The average of three independent images per sample and at least three biological replicates per condition was analyzed.
Histology
Adrenals were fixed in 4% paraformaldehyde for 1 h at 4°C, paraffin-embedded, and cut into 5-µm sections. Antibody information, dilutions, and unmasking conditions are listed in Supplemental Table S1. For DAB-based detection, the ImmPRESS Excel staining kit (Vector Laboratories) was used according to the manufacturer's instructions and images were acquired on an Olympus BX-51 microscope with an Olympus DP-70 camera. For IF, unmasked slides were blocked for 1 h at room temperature followed by primary antibody incubation overnight at 4°C. Primary antibodies were detected with Dylight-conjugated secondary antibodies (1:800; Jackson ImmunoResearch) or HRP-polymer solution (Vector Laboratories) and Alexa fluor tyramide reagent (Thermo Fisher). Nonspecific staining was blocked using 10% normal goat serum (NGS) and 0.5% SDS in TBS (anti-CYP11B1-555), 2.5% horse serum, and 1% bovine serum albumin (BSA; detection with HRP polymer) or 5% NGS and 3% BSA in TBST. The M.O.M. kit (Vector Laboratories) was used for all primary mouse antibodies according to the manufacturer's instructions, and nuclei were counterstained with DAPI. Slides were mounted using ProLong Gold (Life Technologies) and imaged on a Zeiss ApoTome microscope with an AxioCam MRm camera. Quantification of Ki67 or EdU was performed on 40× images using ImageJ (version 1.48). The average of three independent images per sample and at least three biological replicates per condition was analyzed.
EdU
EdU (Invitrogen) was resuspended in 10 mg/mL sterile PBS, and mice were injected intraperitoneally 2 h prior to sacrifice (100 mg/kg). The Click-iT EdU Alexa fluor imaging kit (Invitrogen) was used according to the manufacturer's instructions.
Wnt/β-catenin score
Relative Wnt/β-catenin activation in each tumor from TCGA ACC was calculated using gene set variation analysis (GSVA) (Hänzelmann et al. 2013). A composite enrichment score was determined based on the “gsva” function and five selected bona fide Wnt/β-catenin target genes: APCDD1, AXIN2, LEF1, LGR5, and NKD1. Wnt/β-catenin activation was compared between tumors based on Wnt pathway mutation status. Two tumors with APC deletions and one tumor with an unknown Wnt mutation status were excluded. For normal human adrenals, laser capture microdissection microarray data were downloaded from Gene Expression Omnibus (.CEL files; GSE68889), and the R/Bioconductor “affy” package was used to read the data and perform RMA normalization. Expression values for probes 225016_at, 222695_s_at, 221558_a_at, 210393_at, and 229613_at (APCDD1, AXIN2, LEF1, LGR5, and NKD1, respectively) were used to calculate the Wnt/β-catenin activation score in each zone as described.
Hormone measurements
Blood samples were obtained between 9:00 a.m. and 10:00 a.m. by decapitation and within 30 sec of handling to minimize stress-induced ACTH secretion. Core trunk blood was collected using sodium heparin-coated evacuated tubes (Fisher Scientific) and centrifuged at 568g for 15 min at 4°C to obtain plasma. When >75 µL was obtained, samples were divided into two aliquots for ACTH and steroid measurement and stored at −80°C prior to analysis. If <75 µL was obtained, samples were assigned randomly for ACTH or steroid measurement. ACTH levels were determined by immunoassay (MD Bioproducts) in duplicate according to the manufacturer's instructions. Two independent samples with known concentrations of synthetic ACTH were included in each assay as internal controls. Plasma corticosterone concentrations were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as described in the Supplemental Material.
Statistics
Statistical analyses were performed using R and Graphpad Prism 7. For comparison of two groups, a two-tailed Student's t-test or two-tailed Welch's t-test (if normal distribution could not be assumed) was performed. For more than two groups, one-way ANOVA followed by Tukey's post hoc test or Welch's one-way ANOVA followed by Games-Howell post hoc test (if normal distribution could not be assumed) was performed. Two-way ANOVA followed by Tukey's post hoc test was used when analyzing two independent variables. Supplemental Table S2 contains detailed statistical information.
Supplementary Material
Acknowledgments
We are indebted to the late Keith Parker for providing the SF1-Cre allele, and Charlie Murtaugh (Salt Lake City, UT) for providing Porcn-floxed mice. We are also grateful to Typhanie Dumontet (Ann Arbor, MI), Sining Leng (Boston, MA), and Angela Andersen (Life Science Editors) for critically reading this manuscript; Leonard Cheung (Ann Arbor, MI) for assistance with pituitaries; and Bill Rainey (Ann Arbor, MI) for providing human adrenal tissue. This work was supported by National Institutes of Health (NIH) grants RO1-CA134606 and RO1-DK043140. S.R. and P.V. were supported by Worldwide Cancer Research grant 16-1052. K.J.B. was supported by the Heather Rose Kornick Research Fund, a NIH Postdoctoral Training Grant (T32-DK071212), and an American Cancer Society-Michigan Cancer Research Fund Postdoctoral Fellowship (PF-17-227-01-DDC).
Author contributions: G.D.H. and K.J.B. designed the project. K.J.B. carried out all experiments unless noted otherwise. S.R. and P.V. generated Porcn;Znrf3 double-knockout mice, A.F.T. performed LC-MS/MS, A.M.L. generated the Wnt/β-catenin score, C.Y.L. and R.N. provided expertise and reagents for ISHs, and M.R.R. performed IHC. C.E.G.-S. provided antibodies, and D.T.B., B.-K.K., and H.C. provided mouse strains. K.J.B. wrote the manuscript, and all authors provided editorial input.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.317412.118.
References
- Åkerström T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, Stalberg P, Robinson B, Alexander Iwen K, Dralle H, et al. 2016. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep 6: 19546 10.1038/srep19546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F, et al. 2014. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46: 607–612. 10.1038/ng.2953 [DOI] [PubMed] [Google Scholar]
- Barrott JJ, Cash GM, Smith AP, Barrow JR, Murtaugh LC. 2011. Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc Natl Acad Sci 108: 12752–12757. 10.1073/pnas.1006437108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, Taketo MM, Tissier F, Bertherat J, Lefrancois-Martinez AM, et al. 2010. Constitutive β-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet 19: 1561–1576. 10.1093/hmg/ddq029 [DOI] [PubMed] [Google Scholar]
- Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, Samson-Couterie B, Zennaro MC, Plouin PF, Skah S, et al. 2014. WNT/β-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet 23: 889–905. 10.1093/hmg/ddt484 [DOI] [PubMed] [Google Scholar]
- Bingham NC, Verma-Kurvari S, Parada LF, Parker KL. 2006. Development of a steroidogenic factor 1/Cre transgenic mouse line. Genesis 44: 419–424. 10.1002/dvg.20231 [DOI] [PubMed] [Google Scholar]
- Boutros M, Niehrs C. 2016. Sticking around: short-range activity of Wnt ligands. Dev Cell 36: 485–486. 10.1016/j.devcel.2016.02.018 [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. 2001. Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128: 1253–1264. [DOI] [PubMed] [Google Scholar]
- Chan-On W, Nairismagi ML, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, et al. 2013. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet 45: 1474–1478. 10.1038/ng.2806 [DOI] [PubMed] [Google Scholar]
- Clevers H. 2013. The intestinal crypt, a prototype stem cell compartment. Cell 154: 274–284. 10.1016/j.cell.2013.07.004 [DOI] [PubMed] [Google Scholar]
- de Lau WB, Snel B, Clevers HC. 2012. The R-spondin protein family. Genome Biol 13: 242 10.1186/gb-2012-13-3-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drelon C, Berthon A, Mathieu M, Martinez A, Val P. 2015. Adrenal cortex tissue homeostasis and zonation: a WNT perspective. Mol Cell Endocrinol 408: 156–164. 10.1016/j.mce.2014.12.014 [DOI] [PubMed] [Google Scholar]
- Drelon C, Berthon A, Sahut-Barnola I, Mathieu M, Dumontet T, Rodriguez S, Batisse-Lignier M, Tabbal H, Tauveron I, Lefrancois-Martinez AM, et al. 2016. PKA inhibits WNT signalling in adrenal cortex zonation and prevents malignant tumour development. Nat Commun 7: 12751 10.1038/ncomms12751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumontet T, Sahut-Barnola I, Septier A, Montanier N, Plotton I, Roucher-Boulez F, Ducros V, Lefrancois-Martinez AM, Pointud JC, Zubair M, et al. 2018. PKA signaling drives reticularis differentiation and sexually dimorphic adrenal cortex renewal. JCI Insight 3: 98394 10.1172/jci.insight.98394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DV, de Punder K, Angers S, Peters PJ, Maurice MM, et al. 2016. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 530: 340–343. 10.1038/nature16937 [DOI] [PubMed] [Google Scholar]
- Fearon ER, Spence JR. 2012. Cancer biology: a new RING to Wnt signaling. Curr Biol 22: R849–R851. 10.1016/j.cub.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, Hadjantonakis AK. 2010. A sensitive and bright single-cell resolution live imaging reporter of Wnt/β-catenin signaling in the mouse. BMC Dev Biol 10: 121 10.1186/1471-213X-10-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman BD, Kempna PB, Carlone DL, Shah MS, Guagliardo NA, Barrett PQ, Gomez-Sanchez CE, Majzoub JA, Breault DT. 2013. Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev Cell 26: 666–673. 10.1016/j.devcel.2013.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, Saksena G, Lawrence MS, Qian ZR, Nishihara R, et al. 2014. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 46: 1264–1266. 10.1038/ng.3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroud CJ, Stachenko J, Venning EH. 1956. Secretion of aldosterone by the zona glomerulosa of rat adrenal glands incubated in vitro. Proc Soc Exp Biol Med 92: 154–158. 10.3181/00379727-92-22416 [DOI] [PubMed] [Google Scholar]
- Gonzales KAU, Fuchs E. 2017. Skin and its regenerative powers: an alliance between stem cells and their niche. Dev Cell 43: 387–401. 10.1016/j.devcel.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabinski TM, Kneynsberg A, Manfredsson FP, Kanaan NM. 2015. A method for combining RNAscope in situ hybridization with immunohistochemistry in thick free-floating brain sections and primary neuronal cultures. PLoS One 10: e0120120 10.1371/journal.pone.0120120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumolato L, Liu G, Mong P, Mudbhary R, Biswas R, Arroyave R, Vijayakumar S, Economides AN, Aaronson SA. 2010. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev 24: 2517–2530. 10.1101/gad.1957710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, Guinney J. 2013. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14: 7 10.1186/1471-2105-14-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H, et al. 2012. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485: 195–200. 10.1038/nature11019 [DOI] [PubMed] [Google Scholar]
- Hao HX, Jiang X, Cong F. 2016. Control of Wnt receptor turnover by R-spondin–ZNRF3/RNF43 signaling module and its dysregulation in cancer. Cancers 8: E54 10.3390/cancers8060054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkilä M, Peltoketo H, Leppäluoto J, Ilves M, Vuolteenaho O, Vainio S. 2002. Wnt-4 deficiency alters mouse adrenal cortex function, reducing aldosterone production. Endocrinology 143: 4358–4365. 10.1210/en.2002-220275 [DOI] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. 2002. Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22: 1172–1183. 10.1128/MCB.22.4.1172-1183.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka Y, Ikehara Y, Hattori T. 1996. Cell proliferation and renewal of mouse adrenal cortex. J Anat 188 (Pt 2): 375–381. [PMC free article] [PubMed] [Google Scholar]
- Katoh M, Katoh M. 2007. WNT signaling pathway and stem cell signaling network. Clin Cancer Res 13: 4042–4045. 10.1158/1078-0432.CCR-06-2316 [DOI] [PubMed] [Google Scholar]
- Kazanskaya O, Glinka A, del Barco Barrantes I, Stannek P, Niehrs C, Wu W. 2004. R-Spondin2 is a secreted activator of Wnt/β-catenin signaling and is required for Xenopus myogenesis. Dev Cell 7: 525–534. 10.1016/j.devcel.2004.07.019 [DOI] [PubMed] [Google Scholar]
- Kim AC, Reuter AL, Zubair M, Else T, Serecky K, Bingham NC, Lavery GG, Parker KL, Hammer GD. 2008. Targeted disruption of β-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development 135: 2593–2602. 10.1242/dev.021493 [DOI] [PubMed] [Google Scholar]
- King P, Paul A, Laufer E. 2009. Shh signaling regulates adrenocortical development and identifies progenitors of steroidogenic lineages. Proc Natl Acad Sci 106: 21185–21190. 10.1073/pnas.0909471106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, et al. 1991. Identification of FAP locus genes from chromosome 5q21. Science 253: 661–665. 10.1126/science.1651562 [DOI] [PubMed] [Google Scholar]
- Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM, et al. 2012. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488: 665–669. 10.1038/nature11308 [DOI] [PubMed] [Google Scholar]
- Lebensohn AM, Rohatgi R. 2018. R-spondins can potentiate WNT signaling without LGRs. Elife 7: e33126 10.7554/eLife.33126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D, et al. 2013. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci 110: 20224–20229. 10.1073/pnas.1314239110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum L, Clevers H. 2012. Cell biology. The unusual case of Porcupine. Science 337: 922–923. 10.1126/science.1228179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, et al. 2002. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol 22: 1184–1193. 10.1128/MCB.22.4.1184-1193.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. 2009. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan B, Virshup DM. 2015. Targeting Wnts at the source—new mechanisms, new biomarkers, new drugs. Mol Cancer Ther 14: 1087–1094. 10.1158/1535-7163.MCT-14-1038 [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. 1997. Activation of β-catenin—Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 275: 1787–1790. 10.1126/science.275.5307.1787 [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. 2007. A global double-fluorescent Cre reporter mouse. Genesis 45: 593–605. 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, Liu CJ, Sanjanwala AR, Edwards MA, Gomez-Sanchez CE, et al. 2015. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci 112: E4591–E4599. 10.1073/pnas.1505529112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P. 1991. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 253: 665–669. 10.1126/science.1651563 [DOI] [PubMed] [Google Scholar]
- Nord KH, Nilsson J, Arbajian E, Vult von Steyern F, Brosjo O, Cleton-Jansen AM, Szuhai K, Hogendoorn PC. 2013. Recurrent chromosome 22 deletions in osteoblastoma affect inhibitors of the Wnt/β-catenin signaling pathway. PLoS One 8: e80725 10.1371/journal.pone.0080725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, Clevers H. 2017. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169: 985–999. 10.1016/j.cell.2017.05.016 [DOI] [PubMed] [Google Scholar]
- Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH, et al. 2012. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet 44: 690–693. 10.1038/ng.2273 [DOI] [PubMed] [Google Scholar]
- Planas-Paz L, Orsini V, Boulter L, Calabrese D, Pikiolek M, Nigsch F, Xie Y, Roma G, Donovan A, Marti P, et al. 2016. The RSPO–LGR4/5–ZNRF3/RNF43 module controls liver zonation and size. Nat Cell Biol 18: 467–479. 10.1038/ncb3337 [DOI] [PubMed] [Google Scholar]
- Qiu W, Yang Z, Fan Y, Zheng Q. 2016. ZNRF3 is downregulated in papillary thyroid carcinoma and suppresses the proliferation and invasion of papillary thyroid cancer cells. Tumour Biol 37: 12665–12672. 10.1007/s13277-016-5250-4 [DOI] [PubMed] [Google Scholar]
- Rios-Esteves J, Resh MD. 2013. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep 4: 1072–1081. 10.1016/j.celrep.2013.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. 2015. Integrative clinical genomics of advanced prostate cancer. Cell 161: 1215–1228. 10.1016/j.cell.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha AS, Vidal V, Mertz M, Kendall TJ, Charlet A, Okamoto H, Schedl A. 2015. The angiocrine factor Rspondin3 is a key determinant of liver zonation. Cell Rep 13: 1757–1764. 10.1016/j.celrep.2015.10.049 [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. 1997. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science 275: 1790–1792. 10.1126/science.275.5307.1790 [DOI] [PubMed] [Google Scholar]
- Ryland GL, Hunter SM, Doyle MA, Rowley SM, Christie M, Allan PE, Bowtell DD, Australian Ovarian Cancer Study Group, Gorringe KL, Campbell IG. 2013. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. J Pathol 229: 469–476. 10.1002/path.4134 [DOI] [PubMed] [Google Scholar]
- Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, et al. 2000. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 24: 245–250. 10.1038/73448 [DOI] [PubMed] [Google Scholar]
- Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, et al. 2012. Recurrent R-spondin fusions in colon cancer. Nature 488: 660–664. 10.1038/nature11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szenker-Ravi E, Altunoglu U, Leushacke M, Bosso-Lefevre C, Khatoo M, Thi Tran H, Naert T, Noelanders R, Hajamohideen A, Beneteau C, et al. 2018. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature 557: 564–569. 10.1038/s41586-018-0118-y [DOI] [PubMed] [Google Scholar]
- Vidal V, Sacco S, Rocha AS, da Silva F, Panzolini C, Dumontet T, Doan TM, Shan J, Rak-Raszewska A, Bird T, et al. 2016. The adrenal capsule is a signaling center controlling cell renewal and zonation through Rspo3. Genes Dev 30: 1389–1394. 10.1101/gad.277756.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak EM, Hammer GD. 2015. Regulation of the adrenocortical stem cell niche: implications for disease. Nat Rev Endocrinol 11: 14–28. 10.1038/nrendo.2014.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak EM, Kuick R, Finco I, Bohin N, Hrycaj SM, Wellik DM, Hammer GD. 2014. Wnt signaling inhibits adrenal steroidogenesis by cell-autonomous and non-cell-autonomous mechanisms. Mol Endocrinol 28: 1471–1486. 10.1210/me.2014-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. 2015. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6: 6744 10.1038/ncomms7744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Acharya A, Finco I, Swonger JM, Elston MJ, Tallquist MD, Hammer GD. 2013. Fetal adrenal capsular cells serve as progenitor cells for steroidogenic and stromal adrenocortical cell lineages in M. musculus. Development 140: 4522–4532. 10.1242/dev.092775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Lerario AM, Rainey W, Hammer GD. 2015. Development of adrenal cortex zonation. Endocrinol Metab Clin North Am 44: 243–274. 10.1016/j.ecl.2015.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Morohashi KI, Ingraham HA, Hammer GD. 2017. Timing of adrenal regression controlled by synergistic interaction between Sf1 SUMOylation and Dax1. Development 144: 3798–3807. 10.1242/dev.150516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebisch M, Xu Y, Krastev C, MacDonald BT, Chen M, Gilbert RJ, He X, Jones EY. 2013. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat Commun 4: 2787 10.1038/ncomms3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, et al. 2016. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 29: 723–736. 10.1016/j.ccell.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Lan J, Wang W, Shi Q, Lan Y, Cheng Z, Guan H. 2013. ZNRF3 acts as a tumour suppressor by the Wnt signalling pathway in human gastric adenocarcinoma. J Mol Histol 44: 555–563. 10.1007/s10735-013-9504-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.