

Abstract
Neurofibromatosis type 1 (NF1) is a rare autosomal dominant disease linked to mutations of the Nf1 gene. Patients with NF1 commonly experience severe pain. Studies on mice with Nf1 haploinsufficiency have been instructive in identifying sensitization of ion channels as a possible cause underlying the heightened pain suffered by patients with NF1. However, behavioral assessments of Nf1+/−mice have led to uncertain conclusions about the potential causal role of Nf1 in pain. We used the clustered regularly interspaced short palindromic repeats (CRISPR)-associated 9 (CRISPR/Cas9) genome editing system to create and mechanistically characterize a novel rat model of NF1-related pain. Targeted intrathecal delivery of guide RNA/Cas9 nuclease plasmid in combination with a cationic polymer was used to generate allele-specific C-terminal truncation of neurofibromin, the protein encoded by the Nf1 gene. Rats with truncation of neurofibromin, showed increases in voltage-gated calcium (specifically N-type or CaV2.2) and voltage-gated sodium (particularly tetrodotoxin-sensitive) currents in dorsal root ganglion neurons. These gains-of-function resulted in increased nociceptor excitability and behavioral hyperalgesia. The cytosolic regulatory protein collapsin response mediator protein 2 (CRMP2) regulates activity of these channels, and also binds to the targeted C-terminus of neurofibromin in a tripartite complex, suggesting a possible mechanism underlying NF1 pain. Prevention of CRMP2 phosphorylation with (S)-lacosamide resulted in normalization of channel current densities, excitability, as well as of hyperalgesia following CRISPR/ Cas9 truncation of neurofibromin. These studies reveal the protein partners that drive NF1 pain and suggest that CRMP2 is a key target for therapeutic intervention.
Keywords: Neurofibromatosis type 1, CaV2.2, NaV1.7, CRMP2, CRISPR/Cas9, (S)-Lacosamide
1. Introduction
Neurofibromatosis type 1 (NF1) is a rare genetic disease characterized by tumors in the nervous system, neurological disorders and by chronic idiopathic pain.12,16 The prevalence of pain in patients with NF1 is unknown but quality of life-based questionnaires in Australia, Brazil, France, Italy, the United Kingdom, and the United States consistently identify both the intensity and quality of pain as having a major impact on patients with NF1.1,11,21,22,30,54,63,64
The mechanisms that promote pain in patients with NF1 are unclear. Sensitization of small-diameter nociceptive sensory neurons43 has been suggested to explain the pain reported by patients with NF1. Consistent with this possibility, small-diameter capsaicin-sensitive sensory neurons isolated from mice with a heterozygous truncation of the Nf1 gene, coding for the protein neurofibromin, were shown to have augmented excitability compared to neurons from wildtype mice.59,60 Sensory neurons from Nf1+/−mice, exhibit increased N-type voltage-gated Ca2+ (CaV2.2) currents.17,58 Furthermore, peak current densities for both tetrodotoxin-sensitive (TTX-S) and -resistant (TTX-R) sodium currents were significantly larger in Nf1+/−sensory neurons.59 Stimulus-evoked release of the neuropeptides including substance P and calcitonin gene-related peptide was also significantly greater from sensory neurons isolated from Nf1+/−mice.26 While these findings suggested a causal link between neurofibromin expression and pain, behavioral studies with Nf1+/−mice reported both increased and decreased pain sensitivity in male mice, without changes in sensitivity to acute nociceptive stimuli, or in models of inflammatory, or neuropathic pain.50 By contrast, female Nf1+/−mice were reported to have increased hyperalgesia.35 Collectively, the contribution of NF1 to expression of pain behaviors remained uncertain.
As altered expression of α subunits of CaV2.217 had not been observed in Nf1+/−mice, we examined neurofibromin-dependent pathways that might regulate CaV2.2 activity. We previously identified the axonal growth/specification collapsin response mediator protein 2 (CRMP2) as a protein that binds directly with CaV2.2 leading increased Ca2+ current density and increased transmitter release in sensory neurons.7 Collapsin response mediator protein 22, however, also interacts with the C-terminal domain of neurofibromin33,51 so that loss of neurofibromin increases CRMP2 phosphorylation,51 which in turn, increases its association with CaV2.2.4,46 In addition, CRMP2 was reported to control the TTX-S Na+ voltage-gated sodium channel NaV1.7,19 a major determinant of nociception,32 whose activity is increased in NF1.59
Over 1000 pathogenic allelic variants have been reported in the Nf1 gene.37,62 Since 80% of patients with NF1 express a truncated neurofibromin,20 we hypothesized that modifying the Nf1 gene rather than deleting 1 allele would recapitulate a pain phenotype allowing mechanistic investigation of pain in patients with NF1. On this basis, we evaluated (1) whether using a novel CRISPR/Cas9 strategy targeting Nf1 in sensory neurons could lead to ion channel dysregulation and hyperalgesia and (2) whether normalization of ion channel dysregulation could also normalize pain behaviors. Our data show that disruption of the interaction between CRMP2 and neurofibromin induced by CRISPR/Cas9 truncation of the Nf1 gene produces increased CaV2.2 and NaV1.7 currents along with behavioral hyperalgesia. Intrathecal targeted editing of Nf1 thereby provides direct evidence of neurofibromin involvement in pain. Further, we report a therapeutic strategy to normalize ion channel dysregulation and related pain by the targeting of the CRMP2/neurofibromin interaction via suppression of CRMP2 phosphorylation using (S)-lacosamide.
2. Methods
2.1. Animals
Pathogen-free, adult male and female Sprague-Dawley rats (225–250 g; Harlan Laboratories, Indianapolis, IN) were housed in temperature (23 ± 3˚C) and light (12-hour light/12-hour dark cycle; lights on 07:00–19:00) controlled rooms with standard rodent chow and water available ad libitum. The Institutional Animal Care and Use Committee of the College of Medicine at the University of Arizona approved all experiments. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Pain. Animals were randomly assigned to treatment or control groups for the behavioral experiments. Animals were initially housed 3 per cage but individually housed after the intrathecal cannulation on a 12-hour light–dark cycle with food and water ad libitum. All behavioral experiments were performed by experimenters who were blinded to the experimental groups and treatments.
2.2. Materials
All chemicals, unless noted, were purchased from Sigma (St. Louis, MO). Validated antibodies were purchased as follows: anti-CRMP2 polyclonal antibody (Sigma-Aldrich Cat# C2993 Research Resource Identifier (RRID):AB_1078573), CRMP2 pSer522 (ECM Biosciences Cat# CP2191, RRID:AB_2094486), anti-CaV2.2 polyclonal antibody (Origene Technologies, Inc, Rockville, MD),,46,47 anti-NaV1.7 (Cat# 75–103; NeuroMab, Davis, CA),18,19 anti-neurofibromin C-terminal (Abcam Cat# ab17963, RRID:AB_444142 or Cat# sc-67; Santa Cruz Bio-technology, Dallas, TX),20 anti-neurofibromin N-terminal (Cat# sc-68; Santa Cruz Biotechnology)20 and βIII-Tubulin (Cat# G712A; Promega, Madison, WI). (S)-N-benzyl 2-acetamido-3-methoxypropionamide ((S)-lacosamide; ((S)-LCM) was synthesized as described previously.9 CRMP2-S522D-DsRed, a plasmid encoding a constitutively phosphorylated form of CRMP2 has been described previously.18
2.3. Single guide RNA design for Nf1 gene targeting
Our strategy was to target the neurofibromin (Nf1) gene using a single guide RNA (sgRNA) designed to target the exon 39 to result in the expression of a truncated neurofibromin protein, as reported in 80% of patients with NF1. We identified and extracted the sequence of the Nf1 gene from the genomic sequence of chromosome 10 from Sprague-Dawley rats (AC_000078.1). To design the gRNA sequence, we screened for potential off-targets using the gRNA design tool (http://crispr.mit.edu).27 The sgRNA selected for targeting Nf1 exon 39 was located on the reverse strand: GGCAGTAACCCTTTGTCGTT, score 86 (Fig. 1A).
Figure 1.

Editing of Nf1 gene in rat dorsal root ganglia (DRG) neurons leads to an increase in calcium currents via voltage-gated N-type calcium (CaV2.2) channels. Exon/intron organization of rat Nf1 gene. Sequence of the single guide RNA (gRNA) against exon 39 is shown. Blue line, protospacer adjacent motif (PAM) sequence. The gRNA (sequence underlined in green) pairs with its DNA target followed by a 59NGG sequence. Cas9 catalyzes double stranded cleavage on the genomic DNA 3 bp before PAM sequence. Nucleotide positions indicated are based on the DNA sequence on the Nf1 gene. kbp: kilo base pair. (B) Representative micrographs of DRG sensory neurons transfected with either pSpCas9(BB)-2A-green fluorescent protein (GFP) (control sgRNA) or pSpCas9(BB)-2A-GFP-Nf1 sgRNA. Green fluorescent protein fluorescence identifies transfected neurons. In this experiment, neuron without GFP has robust expression of neurofibromin, whereas the adjacent neuron with GFP fluorescence (circled) demonstrates significantly decreased neurofibromin expression (marked by an arrow). (C) Representative family of current traces is illustrated for neurons transfected with control or Nf1 sgRNA. (D) Summary of current density (pA/pF) vs membrane potential curves from sensory neurons transfected with control or Nf1 sgRNA. (E) Peak current density, at −10 mV, for the indicated conditions (n = 15 cells for control sgRNA and n = 16 cells for Nf1 sgRNA). N-type calcium currents were pharmacologically isolated with toxins and blockers against the other channel subtypes (see Methods). Asterisks indicate significance compared with control sgRNA transfected cells (*P < 0.05, Student’s t test). (F) Boltzmann fits for activation for DRG treated as indicated. Values for V1/2, the voltage of half-activation, and slope factors (k) were not different between the 2 conditions. Error bars represent mean ± SEM.
2.3.1. Cloning of sgRNAs
We used the pSpCas9(BB)-2A-green fluorescent protein (GFP) plasmid (PX458, Cat#48138; Addgene, Cambridge, MA)52 that allows for simultaneous expression of the Cas9 enzyme with the gRNA and GFP to control for transfection efficiency. The plasmid was cut using BbsI restriction enzyme according to manufac-turer’s instructions (Cat#FD1014; Thermo Fisher Scientific, Waltham, MA). The digested plasmid was extracted from a 1% agarose gel (Cat#K0691; Thermo Fisher Scientific). Oligonucleotides were designed according to Ref. 52 for the cloning of exon 39 targeting gRNA (Nf1 forward: 5′-CACCGGCAGT AACCCTTTGTCGTT-3′; Nf1 reverse: 5′-AAACAACGACAAA GGGTTACTGCC-3′) and were obtained from Eurofins. A 10-µM solution of forward and reverse oligonucleotides was annealed in a thermocycler using the following protocol: 37˚C for 30 minutes, 95˚C for 5 minutes, with a cooling to 25˚C at a rate of 5˚C/min. Then, 100 ng of the digested pSpCas9(BB)-2A-GFP plasmid was set up for ligation with 50 nM of the annealed oligonucleotides at 22˚C for 5 minutes using Rapid DNA ligation Kit (Cat#K1422; Thermo Fisher Scientific). The ligation products were transformed into Escherichia coli DH5α competent bacteria (Cat#C2987; New England Biolabs, Ipswich, MA) according to manufacturer’s instructions. The integrity of all plasmids was checked by Sanger sequencing (Eurofins). A 100% efficiency was observed for the insertion of gRNA sequences into the pSpCas9 (BB)-2A-GFP plasmid. Plasmids were purified from DH5α E. coli using the NucleoBond Xtra Maxi kit (Cat# 740414; Macherey-Nagel, Düren, Germany).
2.4. Culturing and transfection of rat primary dorsal root ganglia neurons
Rat dorsal root ganglia (DRG) neurons were isolated from 225 to 250 Sprague-Dawley rats and then transfected using previously developed procedures.2 In brief, removing dorsal skin and muscle and cutting the vertebral bone processes parallel to the dissection stage exposed DRG. Dorsal root ganglia were then collected, trimmed at their roots, and digested in 3 mL bicarbonate-free, serum-free, sterile DMEM (Cat# 11965; Thermo Fisher Scientific) solution containing neutral protease (3.125 mg/mL, Cat#LS02104; Worthington, Lakewood, NJ) and collagenase Type I (5 mg/mL, Cat# LS004194; Worthington) and incubated for 45 minutes at 37˚C under gentile agitation. Dissociated DRG neurons (~1.5 × 106) were then gently centrifuged to collect cells and washed with DRG media DMEM containing 1% penicillin/streptomycin sulfate from 10,000 µg/mL stock, 30 ng.ml−1 nerve growth factor, and 10% fetal bovine serum (Hyclone). Collected cells were re-suspended in Nucleofector transfection reagent containing plasmids or siRNA at the working concentrations listed above. Then, cells were subjected to electroporation protocol O-003 in an Amaxa Biosystem (Lonza, Basel, Switzerland) and plated onto poly-D-lysine—and laminin-coated glass 12-or 15-mm cover-slips. Transfection efficiencies were routinely between 20% and 30% with about ~10% cell death. Small-diameter neurons were selected to target Aδ-and c-fiber nociceptive neurons. For rat DRG culture small cells were considered to be ~ <30 µm. All cultures were used 48 hours after transfection.
2.5. Whole-cell patch recordings of N-type Ca2+, Na+, and K+ currents in acutely dissociated dorsal root ganglia neurons
Recordings were obtained from acutely dissociated DRG neurons as described. To isolate calcium currents, Na+ and K+ currents were blocked with 500 nM TTX (Alomone Labs, Jerusalem, Israel) and 30 mM tetraethylammonium chloride (Sigma). Extracellular recording solution (at ~310 mOsm) consisted of the following (in millimolar): 128 N-methyl-D-glucamine, 10 BaCl2, 5 KCl, 2 MgCl2 10 tetraethylammonium chloride, 10 Na-HEPES, 10 D-glucose, pH at 7.4; with 1 µM TTX, and 10 µM nifedipine. The intracellular recording solution (at ~310 mOsm) consisted of the following (in millimolar): 150 CsCl2, 10 HEPES, 5 Mg-ATP, 5 BAPTA, pH at 7.4. To further isolate the contributions of N-type (CaV2.2) channels, we used the following subunit-selective blockers (all purchased from Alomone Labs): Nifedipine (10 µM, L-type); ω-agatoxin GIVA (200 nM, P/Q-type) 39; SNX-482 (200 nM, R-type)49; and 3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide (TTA-P2, 1 µM, T-type).8 Activation of ICa was measured by using a holding voltage of −90 mV with voltage steps 200 milliseconds in duration applied at 5-second intervals in +10 mV increments from −70 to +60 mV. Current density was calculated as peak ICa/cell capacitance. Steady-state inactivation of ICa was determined by applying an 800-millisecond conditioning prepulse (−100 to −20 mV in +10 mV increments) after which the voltage was stepped to −20 mV for 200 milliseconds; a 15-second interval separated each acquisition.
Whole-cell voltage clamp recordings were performed at room temperature (RT) using an EPC 10 Amplifier-HEKA as previously described.19 The internal solution for voltage clamp sodium current recordings contained (in millimolar): 140 CsF, 1.1Cs-EGTA, 10 NaCl, and 15 HEPES (pH 7.3, 290-310 mOsm/L) and external solution contained (in millimolar): 140 NaCl, 3 KCl, 30 tetraethylammonium chloride, 1 CaCl2, 0.5 CdCl2, 1 MgCl2, 10 D-glucose, and 10 HEPES (pH 7.3, 310–315 mosM/L).
Dorsal root ganglia neurons were subjected to current-density (I-V) and fast-inactivation voltage protocols as previously described.18,24 In the I-V protocol, cells were held at a −80 mV holding potential before depolarization by 20-millisecond voltage steps from −70 to +60 mV in 5-mV increments. This allowed for collection of current density data to analyze activation of sodium channels as a function of current vs voltage and also peak current density, which was typically observed near ~0 to 10 mV and normalized to cell capacitance (pF). In the fast-inactivation protocol, cells were held at a −80 mV holding potential prior to hyperpolarizing and repolarizing pulses for 500 milliseconds between −120 to −10 mV in 5 mV increments. This step conditioned various percentages of channels into fast-inactivated states so that a 0-mV test pulse for 20 milliseconds could reveal relative fast inactivation normalized to maximum current. Dorsal root ganglia were subjected to current-density (I-V) protocol and H-infinity (pre-pulse inactivation protocol). To estimate TTX-R contributions, I-V protocol was run after 10 minutes incubation with 500 nM TTX. Following holding at −100 mV, 200-millisecond voltage steps from −70 to +60 mV in 5-mV increments allowed for analysis of peak currents.
To isolate the potassium current (IK), neurons were superfused with a Ringer solution wherein NaCl was substituted with equimolar N-methyl-glucamine chloride and was composed of (in millimolar) 140 N-methyl-glucamine chloride, 5 KCl, 2 CaCl2,1 MgCl2, 10 HEPES, and 10 glucose, pH adjusted to 7.4 with KOH. Recording pipettes were filled with the following solutions (in millimolar): 140 KCl, 5 MgCl2, 4 ATP, 0.3 GTP, 2.5 CaCl2, 5 EGTA, and 10 HEPES, adjusted pH at 7.3 with KOH. The membrane was held at −60 mV. Activation of IK was determined by voltage steps 300 milliseconds in duration, which were applied at 5-s intervals in +10-mV increments from −80 to +60 mV. Steady-state inactivation of IK was measured by applying a 15-s conditioning prepulse (−100 to +20 mV in +20-mV increments) after which the voltage was stepped to +60 mV for 200 milliseconds; a 20-second interval separated each acquisition. The fast inactivating IKA was isolated by subtraction of the currents obtained for a conditioning prepulse to −40 mV from those obtained for a prepulse to −100 mV. Initially, a 4-s prepulse to −100 mV was applied followed by voltage steps of 500 milliseconds that ranged from −80 to +40 mV in +20-mV increments at 15-s intervals. This was followed by an identical voltage protocol using a prepulse to −40 mV. IA was obtained by digital subtraction of these current traces. Inactivation of IKA was determined by using a series of 4-second prepulses that ranged from −100 to −40 mV (+10-mV increments) that were immediately followed by a 200-millisecond step to −40 mV.
Fire-polished recording pipettes and 2 to 5 mV resistances were used for all recordings. Whole-cell recordings were obtained using a HEKA EPC-10 USB (HEKA Instruments Inc, Bellmore, NY); data were acquired using a Patchmaster (HEKA) and analyzed with a Fitmaster (HEKA). Capacitive artifacts were fully compensated, and series resistance was compensated by ~70%. Recordings made from cells with greater than a 5-MΩ shift in series resistance compensation error were excluded from analysis. All experiments were performed at RT (~23˚C).
2.6. Whole-cell patch recording of spontaneous and evoked action potential in acutely dissociated dorsal root ganglia neurons
For current-clamp recording, the internal solution contained (in millimolar) 137 KCl, 10 NaCl, 1 MgCl2, 1 EGTA, and 10 HEPES adjusted to pH 7.3 with KOH. The external solution contained (in millimolar) 154 NaCl, 5.6 KCl, 2 CaCl2, 1 MgCl2, 10 Glucose, and 8 HEPES adjusted to pH 7.4 with NaOH. The DRG neurons with a resting potential more hyperpolarized than −40 m V, stable baseline recordings, and evoked spikes that overshot 0 mV were used for experiments and analysis. Two protocols were used to access DRG excitability: first, a series of current steps that were 500-millisecond in duration and of 10-pA steps from −50 pA were used to determine the rheobase; second, neurons were held at resting potentials and injected with a 1-s ramp of depolarizing current that had a final amplitude of 1000 pA. The DRG neurons were then held at 0 pA to record spontaneous action potential for 5 minutes. Bridge balance was compensated to above 60% for the recorded DRG neurons. All recordings were made at RT.
2.7. Calcium imaging
Dorsal root ganglia neurons were loaded at 37˚C with 3 µM Fura-2AM (Cat#F-1221; Life Technologies, stock solution prepared at 1 mM in DMSO, 0.02% pluronic acid, Cat#P-3000 MP; Life Technologies) for 30 minutes (λex 340, 380 nm/λemi 512 nm) to follow changes in intracellular calcium ([Ca2+]c) in Tyrode solution (at ~310 mOsm) containing 119 mM NaCl, 2.5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 25 mM HEPES, pH 7.4, and 30 mM glucose. Incubation with (S)-LCM (10 µM) was done during the loading of the cells with Fura-2AM and the drugs were also added to the excitatory solution. All calcium-imaging experiments were done at RT (~23˚C). Baseline was acquired for 1 minute followed by stimulation (15 seconds) with an excitatory solution (at ~310 mOsm) composed of 32 mM NaCl, 90 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 25 mM HEPES, pH 7.4, and 30 mM glucose. Fluorescence imaging was performed with an inverted microscope, Nikon Eclipse Ti-U (Nikon Instruments Inc, Melville, NY), using objective Nikon Fluor 43 and a Photometrics cooled CCD camera CoolSNAP ES2 (Roper Scientific, Tucson, AZ) controlled by NIS Elements software (version 4.20, Nikon Instruments). The excitation light was delivered by a Lambda-LS system (Sutter Instruments). The excitation filters (340 ± 5 nm and 380 ± 7 nm) were controlled by a Lambda 10 to 2 optical filter change (Sutter Instruments). Fluorescence was recorded through a 505-nm dichroic mirror at 535 ± 25 nm. To minimize photobleaching and phototoxicity, the images were taken every 10 seconds during the time-course of the experiment using the minimal exposure time that provided acceptable image quality. The changes in [Ca2+]c were monitored by following the ratio of F340/F380, calculated after subtracting the background from both channels.
2.8. Immunohistofluorescence and epifluorescence imaging
L5 DRG were dissected from adult rats and then fixed using 4% paraformaldehyde for 4 hours at RT. Dorsal root ganglia were next transferred into a 30% sucrose solution and left at 4˚C until sinking of the tissues could be observed (~3 days). Tissues were cut at 10-µm thickness using Bright OTF 5000 Microtome Cryostat (Hacker Instruments and Industries, Inc, Winnsboro, SC) and fixed onto gelatin coated glass slides and kept at −20˚C until use. Prior to antibody staining, slides were dried at RT for 30 minutes and incubated with phosphate-buffered saline (PBS) containing 200 mM NH4Cl for 30 minutes. Next, the slides were incubated with PBS containing 3% sodium deoxycholate for 30 minutes at RT; these 2 PBS incubations were performed to reduce the background fluorescence of the tissue. Dorsal root ganglia slices were permeabilized and saturated using PBS containing 3% BSA, 0.3% triton X-100 solution for 30 minutes at RT, and then N-or C-terminal epitope neurofibromin antibodies were added overnight. The slices were then washed 3X in PBS and incubated with PBS containing 3% BSA, 0.3% triton X-100 containing secondary antibodies (Alexa 488 goat anti-rabbit or Alexa 594 goat anti-mouse secondary antibodies [Life Technologies]) for at least 3 hours at RT. After 3 washes (PBS, 10 minutes, RT), either DAPI was used to stain the nuclei of cells. Slides were mounted and stored at 4˚C until analysis. Immunofluorescent micrographs were acquired on an Olympus BX51 microscope with a Hamamatsu C8484 digital camera using a 20X UplanS Apo 0.75 numerical aperture objective. The freeware image analysis program ImageJ (http://rsb.info.nih.gov/ij/) was used to generate merged images. To quantitatively evaluate Nf1 editing, every fifth DRG section was selected from the consecutive serial sections (7–10 sections for each DRG), and in each selected section, the intensity of neurofibromin in all neuronal cells was determined. For a given experiment, all images were taken using identical acquisition parameters by individuals blinded to the treatment groups.
2.9. Implantation of intrathecal catheter
For intrathecal drug administration, rats were chronically implanted with catheters as described by Yaksh and Rudy.67 Rats were anesthetized with halothane and placed in a stereotactic head holder. The occipital muscles were separated from their occipital insertion and retracted caudally to expose the cisternal membrane at the base of the skull. Polyethylene tubing was passed caudally from the cisterna magna to the level of the lumbar enlargement. Animals were allowed to recover and were examined for evidence of neurologic injury. Animals with evidence of neuromuscular deficits were excluded.
2.10. In vivo transfection of CRISPR plasmids
For in vivo transfection, the plasmids pSpCas9(BB)-2A-GFP-Nf1-gRNA or pSpCas9(BB)-2A-GFP-Nf1-control-gRNA were diluted to 0.5 µg/µL in 5% sterile glucose solution. Then, Turbofect in vivo transfection reagent (Cat#R0541; Thermo Fisher Scientific) was added following manufacturer’s instructions and as published previously.44 Finally, 20 µL of the plasmid complexes were injected intrathecally in Sprague-Dawley rats.
2.11. Genomic DNA purification and sequencing of off-target effects
Genomic DNA purified from DRG and spinal cord tissues using Quick-gDNA MiniPrep Kit (Cat# 11–317AC; Genesee, San Diego, CA). PCR was performed using 5X HOT FIREPol Evagreen (Cat# 08–24-00020; Solis Biodyne, Tartu, Estonia) according to the manufacturer’s instructions. Specific primer sequences used are given in Supplementary Figure 1 (available online at http://links.lww.com/PAIN/A446). PCR efficiency was tested by running the samples on a 1% agarose gel containing SYBR Safe DNA Gel Stain (Cat# S33102; Thermo Fisher Scientific). Samples were sent for Sanger sequencing and obtained sequences were analyzed by alignment with the target regions within the Rattus norvegicus genome. As a control for off-target effects of our CRISPR/Cas9 editing strategy, we sequenced 11 potential off-target binding sites predicted using COSMID (crispr.bme.gatech. edu).10 We sequenced 11 genomic regions, in 16 tissues, DRG (L4, L5, and L6) and dorsal horn of the lumbar region of the spinal cord (n = 4 controls and n = 4 CRISPR Nf1 per tissue). We found only 1 occurrence of an off-target alteration among the 176 sites screened (Supplementary Figure 1, available online at http://links.lww.com/PAIN/A446); this occurred in a noncoding region of the DNA.
2.12. Measurement of thermal withdrawal latency
The method of Hargreaves et al.25 was used. Rats were acclimated within Plexiglas enclosures on a clear glass plate maintained at 30˚C. A radiant heat source (high-intensity projector lamp) was focused onto the plantar surface of the hind paw. When the paw was withdrawn, a motion detector halted the stimulus and a timer. A maximal cutoff of 33.5 seconds was used to prevent tissue damage.
2.13. Testing of allodynia
The assessment of tactile allodynia (ie, a decreased threshold to paw withdrawal after probing with normally innocuous mechanical stimuli) consisted of testing the withdrawal threshold of the paw in response to probing with a series of calibrated fine (von Frey) filaments. Each filament was applied perpendicularly to the plantar surface of the paw of rats held in suspended wire mesh cages. Withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (the “up and down” method), and data were analyzed with the nonparametric method of Dixon, as described by Chaplan et al.6 and expressed as the mean withdrawal threshold.
2.14. Assessment of mechanical hyperalgesia–Randall Selitto test (paw pressure test)
The assessment of mechanical hyperalgesia consisted of measuring the withdrawal threshold of the paw both ipsilateral and contralateral to the site of nerve injury or inflammation in response to continuously-increased pressure (noxious stimuli) using Randall Selitto method.53 Animals were exposed to the testing apparatus and the experimenter for 1 day prior to testing for 3 times. Paw withdrawal threshold was assessed using the Randall–Selitto nociceptive paw withdrawal flexion reflex test with an Ugo-Basile algesymeter (Stoelting, Chicago, IL), which applies a linearly increasing mechanical force to the plantar surface of the rat’s hind paw. A sharp withdrawal of the paw or vocalization is the end point. Testing was performed in alternating fashion on left and right hind paws for 3 times. Reported paw withdrawal thresholds are the mean of 3 tests. Paw withdrawal thresholds (in grams) are calculated according to the weight applied: 0 weight = 10× of the reading; 1 weight = 20× of the reading; 2 weights = 30× of the reading. Cutoff at 500 g. Compared to the baseline, an increase/decrease of the paw withdrawal threshold indicates the analgesic/hyperalgesic effects of the treatments.
2.15. Elevated plus maze
The elevated plus maze (EPM) consists of 4 elevated (50 cm) arms (50 cm long and 10 cm wide) with 2 opposing arms containing 30-cm high opaque walls. Elevated plus maze testing occurred in a quiet testing room with ambient lighting at ~500 lux. On day of testing, rats were allowed to acclimate to the testing room for 20 minutes. Each rat was placed in a closed arm, facing the enter platform and cage mates started in the same closed arm. Each rat was allowed 5 minutes to explore the EPM and then returned to its home cage. Between animals the EPM was cleaned thoroughly with Versa-Clean (Thermo Fisher Scientific). Elevated plus maze performance was recorded using an overhead video camera (MHD Sport 2.0 WiFi Action Camera; Walmart.com) for later quantification. Open and closed arm entries were defined as the front 2 paws entering the arm, and open arm time began the moment the front paws entered the open arm and ended upon exit. An anxiety index was calculated. The index combines EPM parameters into 1 unified ratio with values ranging from 0 to 1, with a higher value indicating increased anxiety.28 The following equation was used for calculation of the anxiety index:
2.16. Generation of spinal cord lysates
Spinal cord lysates prepared from adult Sprague-Dawley rats were generated by homogenization and sonication in RIPA buffer (50 mM Tris-HCl, pH 7.4, 50 mM NaCl, 2 mM MgCl2, 1% [vol/vol] NP40, 0.5% [mass/vol] sodium deoxycholate, 0.1% [mass/vol] SDS) as described previously.24 Protease inhibitors (Cat# B14002; Biotools, Houston, TX), phosphatase inhibitors (Cat# B15002; Biotools), and benzonase (Cat#71206; Millipore, Billerica, MA). Protein concentrations were determined using the BCA protein assay (Cat# PI23225; Thermo Fisher Scientific).
2.17. Immunoblot preparation and analysis
Indicated samples were loaded on 4% to 20% Novex gels (Cat# EC60285BOX; Thermo Fisher Scientific). Proteins were transferred for 1 hour at 120 V using TGS (25 mM Tris pH = 8.5, 192 mM glycine, 0.1% (mass/vol) SDS), 20% (vol/vol) methanol as transfer buffer to polyvinylidene difluoride membranes 0.45 µm (Cat# IPVH00010; Millipore), preactivated in pure methanol. After transfer, the membranes were blocked at RT for 1 hour with TBST (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20), 5% (mass/vol) nonfat dry milk, then incubated separately in indicated primary antibodies in TBST, 5% (mass/vol) BSA, overnight at 4˚C. Following incubation in horseradish peroxidase-conjugated secondary antibodies from Jackson immunoresearch, blots were revealed by enhanced luminescence (WBKLS0500; Millipore) before exposure to photographic film. Films were scanned, digitized, and quantified using Un-Scan-It gel version 6.1 scanning software by Silk Scientific Inc.
2.18. Confocal imaging
Immunocytochemistry was performed on DRG that were trans-fected as described above. Briefly, cells were fixed using 4% paraformaldehyde, 4% sucrose for 20 minutes at RT. Permeabilization was achieved by 30 minutes incubation in PBS, 0.1% Triton X-100, following which nonspecific binding sites were saturated by PBS containing 3% BSA for 30 minutes. Cell staining was performed with primary antibodies in PBS with 3% BSA overnight at 4˚C. Cells were then washed 3 times in PBS and incubated with PBS containing 3% BSA and secondary antibodies (Alexa 488 goat anti-mouse and Alexa 594 goat anti-rabbit from Life Technologies) for 1 hour at RT. Immunofluorescent micrographs were acquired on a Nikon C1si scanning confocal microscope using CFI Plan APO VC 360 oil immersion objective with 1.4 numerical aperture. Camera gain and other relevant settings were kept constant. Colocalization analysis preformed with NiS Elements-Nikon (version 4.20, Nikon Instruments). Membrane-to-cytosol ratio was determined by defining regions of interest on the cytosol and the membrane of each cell using ImageJ. Total fluorescence was normalized to the area analyzed and before calculating the ratios.
2.19. Statistical analyses
All data were first tested for a Gaussian distribution using a Shapiro-Wilk test (Graphpad Prism 7 Software). The statistical significance of differences between means was determined by either Student’s t test, parametric analysis of variance followed by post hoc comparisons (Tukey) using GraphPad Prism 7 Software. All behavioral data were analyzed by nonparametric 2-way analysis of variance (post hoc: Student-Neuman–Kuels) in FlashCalc (Dr. Michael H. Ossipov, University of Arizona, Tucson, AZ). Differences were considered to be significant if P ≤0.05. All data were plotted in GraphPad Prism 7. No outlier data were removed.
3. Results
3.1. Design of a CRISPR/Cas9 strategy for Nf1 gene editing
We designed a clustered regularly interspaced short palindromic repeats (CRISPR) associated protein-9 nuclease (Cas9) approach to induce a double stranded break in exon 39 of the rat Nf1 gene (Fig. 1A). The primary reasons for selecting this region of the gene, which would lead to deletion of the C-terminus, were (1) almost 80% of patients with NF1 express a C-terminally truncated neurofibromin20 and (2) more than 1000 pathogenic variants (insertions, microdeletions, copy number alterations) have been reported in the Nf1 gene.37,62 Given this wide mutational spectrum and the complexity of genotype-phenotype associations, we selected the C-terminus as it is a region recurrently altered in NF1. Non-end joining repair mechanisms are error prone and introduce mutations during DNA repair leading to early stop codons and expression of a truncated protein. Sensory neurons transfectedwith CRISPR/Cas9 plasmid with a sgRNA for Nf1 exhibited a loss of neurofibromin expression as revealed with a C-terminus specific antibody (Fig. 1B), demonstrating the efficiency of Nf1 gene editing in rat sensory neurons.
3.2. N-type Ca2+ channel current densities are increased after CRISPR/Cas9 editing of Nf1
We, and others, previously reported an increase in voltage-gated N-type Ca2+ (CaV2.2) channel current densities in neurons from Nf1+/−mice,17,58 placing CaV2.2 as the major contributor to whole cell Ca2+ currents in NF1. In addition, Nicol and colleagues reported that there were no significant differences in L-, P/Q-, T-and R-type currents between Nf1+/−and wildtype mice.17 Therefore, we asked if CRISPR/Cas9-mediated truncation of Nf1 could recapitulate the dysregulated CaV2.2 activity. Sensory neurons transfected with a CRISPR/Cas9 plasmid containing the Nf1 sgRNA exhibited increased CaV2.2 current densities compared to sensory neurons transfected with a plasmid with no sgRNA (Fig. 1C–E). CaV2.2 activation properties were unchanged by truncation of Nf1 (Fig. 1F). Thus, truncating neurofibromin elicits CaV2.2 dysregulation.
3.3. Tetrodotoxin-sensitive Na+ channel current densities are augmented after CRISPR/Cas9 editing of Nf1
Na+ peak current densities are increased in sensory neurons from Nf1+/−mice.59 Thus, we asked if CRISPR/Cas9 editing of Nf1 would recapitulate the dysregulated Na+ currents. Compared to sensory neurons transfected with a plasmid with a control sgRNA, sensory neurons transfected with a CRISPR/ Cas9 plasmid containing Nf1 sgRNA demonstrated increases in total (Fig. 2A–C) and TTX-S (Fig. 2D–F), but not TTX-R (Fig. 2G–I), voltage-gated Na+ channel current densities upon their isolation with a combined electrophysiology and pharmacological55 protocol. Because the DRG currents recorded were large (in some cases over 10 nA), even partially compensated series resistance could result in substantial voltage errors precluding accurate determination of changes in peak current densities, therefore we also plotted the data as normalized to conductance. Sodium conductance (G) was calculated using the net current and reversal potential recorded from each cell. The conductance-voltage relation was fit with the Boltzmann relation, and the conductance for each neuron was then normalized to the maximal value of G (Gmax) obtained from the fit. The G/Gmax-voltage relation is summarized in (Supplementary Figure 1, available online at http://links.lww.com/PAIN/A446). The Boltzmann fitting parameters, V0.5 and k, for the peak and steady-state measurements are shown in Table 1. There was a significant (P< 0.05) leftward shift in activation to more hyperpolarizing voltages (11 mV shift for total Na+ currents; 2.4 mV for TTX-S currents; and 6.9 mV for TTX-R currents) in neurons transfected with a CRISPR/Cas9 plasmid containing the Nf1 sgRNA compared to those with control sgRNA, suggesting that more channels are likely to be open at potentials between −10 and −40 mV. The slope k of activation was also significantly faster for total Na+ and TTX-R currents in neurons transfected with a CRISPR/Cas9 plasmid containing the Nf1 sgRNA compared to those with control sgRNA. No other changes were observed in biophysical properties of the recorded channels (Table 1) or on persistent sodium currents (Fig. 3).
Figure 2.

CRISPR/Cas9 Nf1 gene editing increases Na1 currents in sensory neurons. Representative family of total (A), tetrodotoxin-sensitive (TTX-S) (D), and TTX-resistant (TTX-R) (G) Na+ current traces are illustrated for sensory neurons transfected with pSpCas9(BB)-2A-green fluorescent protein (GFP) plasmid containing either control or Nf1 sgRNAs. Current versus voltage relationships (B, E, and H) and current densities at −20 mV (C, F, and I) for the indicated conditions. Numbers of cells are indicated in parentheses. Asterisks denote statistical significance compared with control sgRNA-transfected cells (P < 0.05, Student’s t test). n.s., not-significant; P > 0.05; unpaired Student’s t test.
TABLE 1.
Boltzmann parameters for the activation and inactivation of tetrodotoxin-sensitive (TTX-S) and resistant (TTX-R) INa in control and Nf1 sgRNA-transfected dorsal root ganglion neurons.
| V0.5 (mV) | k | |
|---|---|---|
| TTX-S | ||
| INa activation | ||
| Control (n = 9) | −27.2 ± 0.7 | 4.9 ± 0.3 |
| Nf1 sgRNA (n = 12) | −29.6 ± 0.3 | 4.3 ± 0.3 |
| Nf1 sgRNA 1 (S)-LCM (n = 6) | −32.6 ± 0.8 | 5.0 ± 0.5 |
| INa inactivation | ||
| Control (n = 13) | −63.9 ± 3.1 | 7.8 ± 1.6 |
| Nf1 sgRNA (n = 16) | −51.4 ± 6.9 | 17.3 ± 1.8* |
| Nf1 sgRNA 1 (S)-LCM (n = 12) | −64.7 ± 2.3 | 14.7 ± 1.2* |
| TTX-R | ||
| INa activation | ||
| Control (n = 13) | −20.1 ± 0.7 | 7.8 ± 0.3 |
| Nf1 sgRNA (n = 12) | −27.0 ± 0.3* | 5.8 ± 0.3* |
| Nf1 sgRNA 1 (S)-LCM (n = 7) | −21.6 ± 0.8 | 6.8 ± 0.5 |
| INa inactivation | ||
| Control (n = 9) | −30.0 ± 2.5 | 5.6 ± 1.6 |
| Nf1 sgRNA (n = 8) | −36.4 ± 5.2 | 8.3 ± 1.8 |
| Nf1 sgRNA 1 (S)-LCM (n = 6) | −34.9 ± 2.3 | 6.4 ± 1.2 |
p < 0.05 vs control (Student’s t-test).
Figure 3.

CRISPR/Cas9 Nf1 gene editing does not alter persistent Na1 currents. (A) Representative family of total Na+ current traces are illustrated for sensory neurons transfected with pSpCas9(BB)-2A-green fluorescent protein (GFP) plasmid containing either control sgRNA or Nf1 sgRNA. The currents were evoked by a series of 200-millisecond voltage pulses that ranged from −120 to +10 mV. The values for the persistent INa were obtained 100 milliseconds after the onset of the prepulse (noted by the vertical dotted line). The peak currents have been truncated for clarity of the persistent INa. Boxed regions illustrate an expanded region of the current traces. (B) Summary for the voltage dependence of the persistent INa measured as the percent of the maximum total transient current for cells transfected with control sgRNA or Nf1 sgRNA. (C) Representative family of Na+ current traces remaining after exposure to TTX are illustrated for sensory neurons transfected with control sgRNA or Nf1 sgRNA. (D) Summary for the voltage dependence of the tetrodotoxinR persistent INa measured as the percent of the maximum total transient current for cells transfected with control sgRNA or Nf1 sgRNA. No significant differences were noted between persistent currents in any conditions (n = 10 cells for control sgRNA and n = 14 cells for Nf1 sgRNA; P > 0.05; unpaired Student’s t test).
3.4. CRISPR/Cas9 editing of Nf1 increases sensory neuron excitability
As voltage-gated sodium channels contribute to neuron excitability, a phenomenon that is directly linked to transmission of a painful stimuli, we next investigated excitability properties of sensory neurons following truncation of Nf1. Consistent with the increase in total and TTX-S Na+ currents in neurofibromin-truncated neurons, CRISPR/Cas9-mediated truncation of Nf1 in sensory neurons resulted in increased action potential (AP) firing (3.5 ± 0.8 for control sgRNA [n = 12] vs 9.6 ± 2.0 for Nf1 sgRNA[n = 13]) (Fig. 4A–C); decreased rheobase (349.2 ± 57.0 for control sgRNA [n = 12] vs 179.2 ± 40.5 for Nf1 sgRNA [n = 13]), the current required to initiate an action potential (Fig. 4D–F); but no change in resting membrane potential (256.3 ± 1.8 for control sgRNA [n = 12] vs −52.4 ± 2.0 for Nf1 sgRNA [n = 13]) (Fig. 4G); AP spike height (72.4 ± 4.1 for control sgRNA [n = 12] vs 74.7 ± 2.0 for Nf1 sgRNA [n = 13]) (Fig. 4H); and AP half width (7.1 ± 1.1 for control sgRNA [n = 12] vs 7.4 ± 1.4 for Nf1 sgRNA [n = 13]) (Fig. 4I). Spontaneous activity was not observed in any of the Nf1-edited DRG (n = 13; data not shown). Together, these observations recapitulate the ionic dysregulation observed in sensory neurons of Nf1+/−mice.
Figure 4.

CRISPR/Cas9 Nf1 gene editing increases excitability in sensory neurons. (A) Representative recordings in response to a step of depolarizing current evoked action potentials (APs) in sensory neurons transfected with plasmids harboring control (A) or Nf1 sgRNAs (B). (C) Summary of the number of APs in the indicated conditions (n = 7 cells for control sgRNA and n = 11 cells for Nf1 sgRNA). Representative recordings in response to various steps of depolarizing current to measure rheobase in sensory neurons transfected with plasmids harboring control (D) or Nf1 sgRNAs (E). Rheobase is the current required for eliciting the first AP. (F) Summary of the measured rheobase in indicated conditions (n = 10 each). Summary of the resting membrane potential (in millivolts, mV) (G), AP spike height (in mV) (H), and AP half width (in milliseconds, ms) (I) in the indicated conditions. Asterisks indicate significance compared with control sgRNA transfected cells (*P < 0.05, Student’s t test). n.s., not-significant; P > 0.05; unpaired Student’s t test. At least 11 to 13 cells were recorded from for the parameters shown in (G-I).
3.5. Potassium currents are not affected by CRISPR/Cas9 editing of Nf1
Next, experiments were performed to determine if potassium current (IK) could account for the augmented excitability observed in sensory neurons expressing truncated neurofibromin compared with unedited neurofibromin. In these recordings from rat sensory neurons, 2 distinct phenotypes of IK were observed. One exhibited rapid activation with little time-dependent inactivation (Fig. 5A), whereas the other type showed rapid activation with more rapid inactivation kinetics (Fig. 5G). Both phenotypes of IK were observed in each edited condition. The current density-voltage relations for the peak (Fig. 5B) and steady-state (Fig. 5C) IKs in control and Nf1-edited neurons were nearly identical. The current values were transformed to G and plotted as described above. The G/Gmax-voltage relation is summarized in Figure 5D, E and indicates there is little difference in either the peak or steady-state values between the 2 groups. The Boltzmann fitting parameters, V0.5 and k, for the peak and steady-state measure-ments were also not different (data not shown). Similar to IK activation, there were no significant differences in the properties of steady-state inactivation for IK between neurons of the 2 edited conditions (Fig. 5F).
Figure 5.

CRISPR/Cas9 Nf1 gene editing does not alter K+ currents. Representative IKs (A) currents recorded from a Nf1 edited sensory neuron that exhibited rapid activation with little time-dependent inactivation. The peak (B) and steady-state (C) IKs in sensory neurons transfected with pSpCas9(BB)-2A-green fluorescent protein (GFP) plasmid containing either control sgRNA or Nf1 sgRNA are not different. The steady-state values were measured at the end of the voltage step. The conductance–voltage relations for the peak (D) and the steady-state (E) measurements. The points in (D) have been fitted by the Boltzmann relation and are shown as the continuous lines. The values in each panel of (D) and (E) represent the mean ± SEM obtained from 11 control sgRNA and 14 Nf1 sgRNA. (F) The steady-state inactivation of IKs in neurons from 11 control sgRNA and 14 Nf1 sgRNA. The steady-state inactivation voltage protocol consisted of a step to 160 mV after prepulses to −100 mV. Currents were normalized to the maximal value of G obtained for the −100 mV prepulse. The data points have been fitted by the Boltzmann relation and are shown as a continuous line. Representative IKA (G) currents recorded from a Nf1 edited sensory neuron. The peak (H) IKA in sensory neurons transfected with pSpCas9(BB)-2A-GFP plasmid containing either control sgRNA or Nf1 sgRNA are not different. The conductance–voltage relations for the peak (I). The points in (I) have been fitted by the Boltzmann relation and are shown as the continuous lines. (J) The steady-state inactivation of IK in neurons from 10 control sgRNA and 11 Nf1 sgRNA. The steady-state inactivation voltage protocol consisted of a step to 160 mV after prepulses to −100 mV. Currents were normalized to the maximal value of G obtained for the −100 mV prepulse. The data points have been fitted by the Boltzmann relation and are shown as a continuous line.
The rapidly inactivating type of IK known as IA was isolated by subtracting the currents obtained at a holding potential of −100 mV from those obtained at −40 mV (see Methods); the results for a representative wild-type neuron are shown in Figure 5G. All isolated IA traces, regardless of the edited condition, had a peak current that rapidly decayed over time and then reached a stable plateau (Fig. 5H). When the currents were normalized to cell capacitance, the pA/pF-voltage relations were not different (Fig. 5H). The G/Gmax-voltage relations are summarized in Figure 5I and demonstrate the similarities in voltage dependence for both activation and inactivation of IA in these 2 conditions. The relations for G/Gmax-voltage were fitted by the Boltzmann relation and the values for V0.5 and k, for the peak and steady-state measurements were not different (data not shown), for the activation of IA in the 2 genotypes were not different. Therefore, differences in IK or IA do not account for the enhanced excitability in Nf1-edited neurons.
3.6. Targeted intrathecal delivery of Nf1 sgRNA achieves neurofibromin editing in the spinal cord
Having demonstrated that CRISPR/Cas9 editing of Nf1 results in increased CaV2.2 and NaV1.7 current densities and an increase in excitability in sensory neurons, we next asked if this strategy would produce effects in vivo. We selected rats rather than mice as a model here for technical considerations as well as for data consistency because (1) repeated injections of the CRISPR plasmid were needed to achieve efficient editing (data now shown); lumbar puncture can be performed in mice, but the intrathecal catheterization method in rats is more reliable and with less chance of producing variable damage to the spinal nerves during repeated punctures; and (2) behavioral assays are generally more reliable in rats because of less spontaneous movement of rats compared with mice. Thus, lumbar intrathecal catheters were placed in rats to permit repeated spinal drug delivery of sgRNAs. Following 3 intrathecal (i.t.) injections of control or Nf1 sgRNAs, given once per day for 3 days, rats were monitored for various behaviors (see below) and sacrificed 7 days following the last i.t. injection. CRISPR/Cas9 plasmid containing Nf1 sgRNA given i.t. resulted in on-target editing of the Nf1 gene (Supplementary Figure 2, available online at http://links.lww.com/PAIN/A447) and in a 58.6 ± 14.0% (n = 146 neurons from 3 slices) loss of C-terminal neurofibromin immunoreactivity in L5 lumbar DRG (Fig. 6A–C) while N-terminal neurofibromin immunoreactivity was unchanged compared to control plasmid (Fig. 6A–C).
Figure 6.
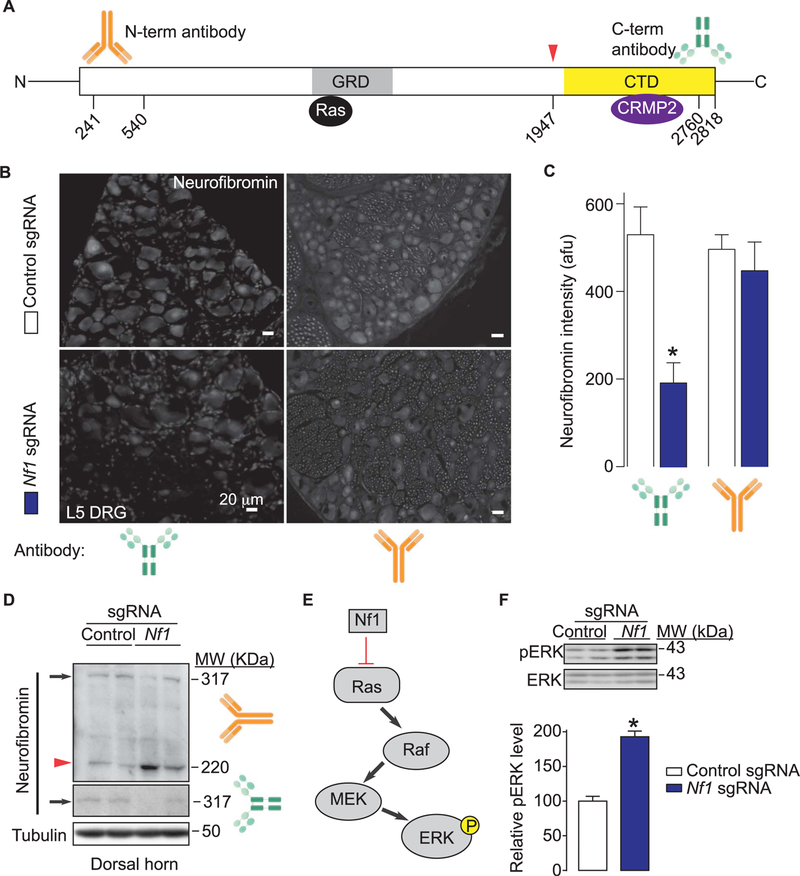
In vivo CRISPR/Cas9 Nf1 gene editing does not alter expression and function of neurofibromin. (A) Domain scheme of human neurofibromin protein with the catalytic Ras GTPase-activating protein (GAP)–related domain (GRD) and C-terminus domain (CTD) indicated. The location of the epitopes (amino acid numbers from human neurofibromin [Accession#: NP_000,258.1; GeneID4763]) used for raising antibodies against the N-terminus (orange antibody cartoon) or C-terminus (green antibody cartoon) are indicated. Red arrowhead and associated amino acid number denote the site of putative editing (ie, truncation) of neurofibromin. CRMP2 has been reported to bind to the CTD amino acids 2260 to 2818 of neurofibromin.51 (B) Micrographs of a 10-μm section of adult dorsal root ganglia (DRG) from animals having received intrathecal injection of either Nf1 sgRNA containing or control sgRNA plasmid (day 10) immunostained with neurofibromin antibodies as indicated. Scale bars: 20 μm. (C) Summary of intensity of neurofibromin staining (in arbitrary fluorescence units, afu) in DRG slices from rats injected intrathecally with plasmids harboring control (white bars) or Nf1 (red bars) sgRNAs (n = 8–12 slices from at least 3 independent rats in each condition). Asterisk indicates significance compared with control sgRNA-transfected cells (*P < 0.05, Student’s t test). (D) Western blots of neurofibromin from spinal cord dorsal horn lysates with the N-(top) and C-terminus (bottom) antibodies. β-tubulin is shown as loading control. Arrow represents full-length neurofibromin while the arrowhead denotes edited neurofibromin protein. Two replicates for each condition are shown. (E) Schematic representation of the Ras-ERK pathway. Unabated Ras activation leads to increased ERK phosphorylation. (F) Representative Western blot (top) and summary histograms (bottom) of ERK and phospho-ERK (p-ERK) from spinal cords of rats injected intrathecally with plasmids harboring control or Nf1 sgRNAs (n = 4 each; *P < 0.05, Student’s t test). Two replicates for each condition are shown. Error bars represent mean ± SEM.
Because neurofibromin is also abundantly expressed in the rodent spinal cord,14 it is conceivable that the effects of Nf1 sgRNA plasmid injection intrathecally could have editing effects not only on DRG neurons but also on second order sensory neurons in the spinal cord. To address this possibility, we performed immunoblotting of dorsal horn of the spinal cord from rats injected with control or Nf1 sgRNAs. Spinal cord lysates from Nf1 sgRNA-injected rats showed the presence of full-length as well as a C-terminally truncated neurofibromin protein in comparison to the predominantly full-length band observed in control-sgRNA injected samples (Fig. 6D; top). Immunoblotting with the N-terminal neurofibromin antibody revealed the presence of full-length protein that was reduced in samples from Nf1 sgRNA-injected rats compared to those from control-sgRNA– injected rats (Fig. 6D; bottom).
Neurofibromin is a negative regulator of the Ras signal transduction pathway13 (Fig. 6E). Loss or alteration of neurofibromin is thus expected to result in unopposed stimulation of the Ras signal transduction pathway leading to activation of the Raf/ MEK/ERK pathways. To test this, we evaluated phosphorylation of ERK, a downstream target of Ras signaling, in the dorsal horn spinal cord as this is a major termination zone of nociceptive primary afferents. We detected an increase in the levels of active ERK in dorsal horn spinal cord from Nf1 sgRNA-injected rats compared to those from control-sgRNA–injected rats (Fig. 6F). Taken together, these results show that (1) Nf1 gene editing leads to expression of a C-terminally truncated neurofibromin in the DRG and spinal cord and (2) ERK activity is increased despite preservation of neurofibromin’s GTPase-activating protein (GAP)–related domain, suggesting that ERK activation is independent of Ras in NF1, as reported previously.34
3.7. Nf1 gene editing in vivo elicits modality specific responses in rats
CRISPR/Cas9 editing of Nf1 resulted in increased CaV2.2 and NaV1.7 current densities and an increase in excitability of sensory neurons, suggesting that sensitization of nociceptive sensory neurons may contribute, in part, to the pain states observed in patients with NF1. To address if truncation of Nf1 in sensory neurons and spinal cord could alter pain behaviors, rats spinally injected with control or Nf1 sgRNA plasmids were evaluated for their ability to respond to noxious thermal and mechanical stimuli and to normally innocuous tactile stimuli. Rats injected i.t. with a Nf1 sgRNA plasmid demonstrated a time-dependent decrease in withdrawal latency to a thermal stimulus (Fig. 7A and B), indicating that truncation of neurofibromin results in development of thermal hyperalgesia. However, thresholds to mechanical probing with von Frey filaments were not changed between the conditions in either males or females (Fig. 7C and D). While a generalized enhancement in excitability of C-fibers would be expected to enhance mechanical pain from C-fibers, stimulation by normally innocuous von Frey hairs may depend more on activation of A-fiber mechanoreceptors. Therefore, we used Randall-Sellito stimulation to test for possible changes in threshold to mechanical pain. Male rats did not show any changes in their noxious mechanical thresholds irrespective of neurofibromin editing (Fig. 7E). By contrast, female rats injected with Nf1 sgRNA plasmid demonstrated a small but significant reduction in their nociceptive thresholds (~27% reduction) compared to control-edited rats (Fig. 7F).
Figure 7.
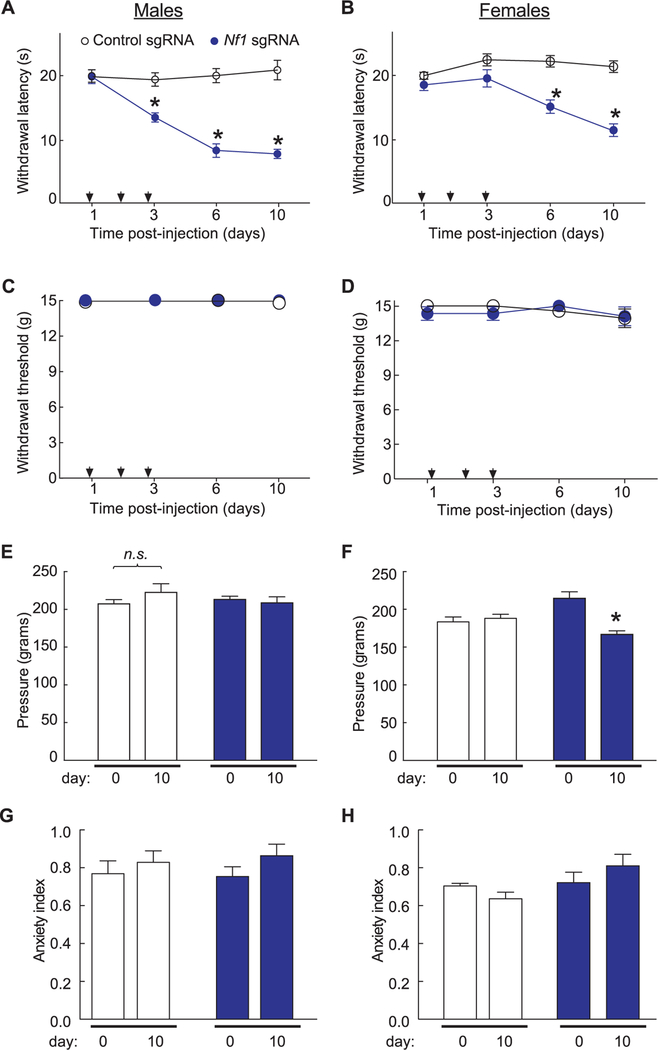
Comparison of targeted intrathecal editing of Nf1 in vivo reveals sex-specific and stimulus-intensity specific responses. For these experiments, rats received 1 injection (arrows) per day for 3 days of the indicated sgRNA plasmids. Both male (A) and female (B) rats injected with Nf1 sgRNA show a behavioral deficit in response to the radiant heat in the Hargreaves test in comparison to rats injected with control sgRNA (n = 6–8 males each and n = 13–14 females each;*P < 0.05, 1-way analysis of variance [ANOVA]). Male (C) and female (D) rats injected with Nf1 sgRNA show normal responses to von Frey hairs applied using the up-down method in comparison to rats injected with control sgRNA (n = 5–7 males each and n = 13–14 females each). (E) Male rats show normal responses to the Randall-Selitto apparatus applied to the paw indicating no change in mechanical withdrawal thresholds in either sgRNA condition (n = 6–8 male rats each; P > 0.05, 1-way ANOVA). (F) Female rats injected with Nf1 sgRNA show a significant increase in withdrawal threshold in response to the Randall-Selitto test at 10 days after injection compared to rats injected with control sgRNA (n = 19–20 female rats each; *P < 0.05, 2-way ANOVA). Both left (L) and right (R) paws were tested. The anxiety index, an integrated measure of times and entries into the arms of the elevated plus maze, was not different between male (G) and female (H) rats (n = 6–8 each) at baseline (ie, prior to any injections) and 10 days post-injection of control or Nf1 sgRNAs. Error bars represent mean ± SEM.
Taken together, these results show that Nf1 gene editing leads to expression of a C-terminally truncated neurofibromin, to increased Ca2+ and Na+ currents, and to neuronal excitability, culminating in enhanced nociceptive responses to noxious but not innocuous stimulation. In addition, Nf1 gene editing failed to alter anxiety-like behaviors as both male and female rats showed normal response in the EPM test (Fig. 7G and H).
3.8. (S)-Lacosamide normalizes the phenotype of CRISPR/ Cas9 editing of Nf1 in vitro and in vivo
A strong convergence of signaling pathways for neurofibromin and CRMP2 was suggested by findings that neurofibromin directly binds to CRMP2, as well as, indirectly modulates the kinases that phosphorylate CRMP2.51 We recently showed a direct positive effect of CRMP2 on calcium channel trafficking in sensory neurons.3 Thus, CRMP2 interaction with neurofibromin may be a novel mechanism for regulating phosphorylated CRMP2 levels in sensory neurons and, in turn, directing Ca2+ channel trafficking and activity (Fig. 8A). Consistent with the demonstration that loss of neurofibromin results in increased CRMP2 phosphorylation by cyclin dependent kinase 5 (Cdk5),51 a post-translational modification that increases CaV2.2 activity,4 we observed that editing of Nf1 results in an upregulation of Cdk5-phosphorylated levels of CRMP2 (Fig. 8B). We previously characterized (S)-lacosamide ((S)-LCM) as a small molecule inhibiting both CRMP2 phosphorylation by Cdk546 and CaV2.2 activity in sensory neurons.42 Specificity of (S)-LCM for CRMP2 phosphorylation was deduced from the following observations: (1) (S)-LCM did not inhibit Cdk5-mediated phosphorylation of a mutant CRMP2 engineered to be unable to bind this small molecule46; (2) neurite outgrowth—a canonical CRMP2-mediated function, which is normally blocked by (S)-LCM, was not affected in cells expressing a (S)-LCM binding-pocket mutant CRMP246; (3) calcium influx recorded from sensory neurons transfected with CRMP2-S522D, a construct that mimics constitutive Cdk5-mediated phosphorylation of CRMP2,4 were unaffected by (S)-LCM (Supplementary Figure 3, available online at http://links.lww.com/PAIN/A448); and (4) sodium influx recorded from sensory neurons transfected with CRMP2-S522D were unaffected by (S)-LCM (Supplementary Figure 3, available online at http://links.lww.com/PAIN/A448); the latter 2 results showing lack of effects on calcium and sodium influx imply that the functional effects of (S)-LCM are entirely due to inhibition of CRMP2 phosphorylation by Cdk5.
Figure 8.

(S)-LCM normalizes Ca21 and Na1 currents after CRISPR/Cas9 Nf1 editing in rats. (A) Schematic showing how convergent signaling involving CRMP2 and neurofibromin controls channel activity. Our previous work identified a direct binding between CaV2.2 and CRMP2 leading to a CRMP2-mediated increase in Ca2+ current density and increased transmitter release in sensory neurons. In addition, CRMP2 was reported to bind to neurofibromin. Loss of neurofibromin increases CRMP2 phosphorylation, which in turn, can increase its association with CaV2.2 and NaV1.7. The direct association of neurofibromin with non-phosphorylated CRMP2 may protect it from phosphorylation. Here, we propose to control the activity of CaV2.2 via an enantiomer of a clinically available small molecule ((S)-LCM) that interferes with the neurofibromin-CRMP2-channel interactions. (B) Representative Western blot (top) and summary histograms (bottom) of Cdk5-phophorylated (ie, p5222) and total CRMP2 from spinal cords of rats isolated 10 days after intrathecal injection with plasmids harboring control or Nf1 sgRNAs (n = 4 each; *P < 0.05, Student’s t test). (C) Peak CaV2.2 current density, at −10 mV, in dorsal root ganglia (DRG) neurons transfected by Nf1 sgRNA containing plasmid and treated with either 10 µM (S)-LCM or DMSO 0.04%. Line shows peak CaV.2.2 current level in DRG neurons transfected with the empty plasmid. (D) Peak current density for Total, tetrodotoxin-sensitive (TTX-S) or TTX-resistant (TTX-R) VGSC in DRG neurons transfected by Nf1 sgRNA containing plasmid and treated with either 10 µM (S)-LCM or DMSO 0.04%. Line shows peak current level in DRG neurons transfected with the empty plasmid. Representative micrographs (E) of and summary (F) of the relative membrane localization of CaV2.2 and NaV1.7 (compared to cytosol) in DRG neurons transfected by Nf1 sgRNA containing plasmid and treated with either 10 µM (S)-LCM (n = 11) or DMSO 0.04% (n = 12). Asterisks indicate significance compared with control sgRNA transfected cells (*P < 0.05, Student’s t test).
We hypothesized that restricting CRMP2 phosphorylation with (S)-LCM would reverse ion channel dysregulation observed following Nf1 gene editing. Increased Ca2+ currents (Fig. 8C) as well as increased total and TTX-S Na+ currents (Fig. 8D) recorded in Nf1 edited DRG neurons were normalized to wild type levels by incubation with (S)-LCM. (S)-LCM did not affect the biophysical properties of the channels, suggesting that the changes in current densities were likely due to alterations in the number of available Ca2+ (CaV2.2) and Na+ (NaV1.7) channels at the cell membrane. Confocal imaging revealed a decreased surface localization (Fig. 8E) of these channels following (S)-LCM treatment (Fig. 8F), thus confirming this hypothesis.
Excitability properties observed following CRISPR/Cas9-mediated truncation of Nf1 in sensory neurons were normalized by (S)-LCM, including a suppression in number of action potentials (Fig. 9A–C), an increase in rheobase (Fig. 9E–F), and no change in resting membrane potential (Fig. 9G), AP spike height (Fig. 9H), or AP half width (Fig. 9I). Finally, we tested whether (S)-LCM could reverse the thermal hyperalgesia induced by Nf1 gene editing in vivo. Oral administration of (S)-LCM reversed the decreased paw withdrawal latency in both male and female rats (Fig. 9J–K). These results identify CRMP2 phosphorylation by Cdk5 as a pathologic event underlying the increased pain sensitivity in NF1 and demonstrate the potential of using (S)-LCM as a therapeutic strategy.
Figure 9.
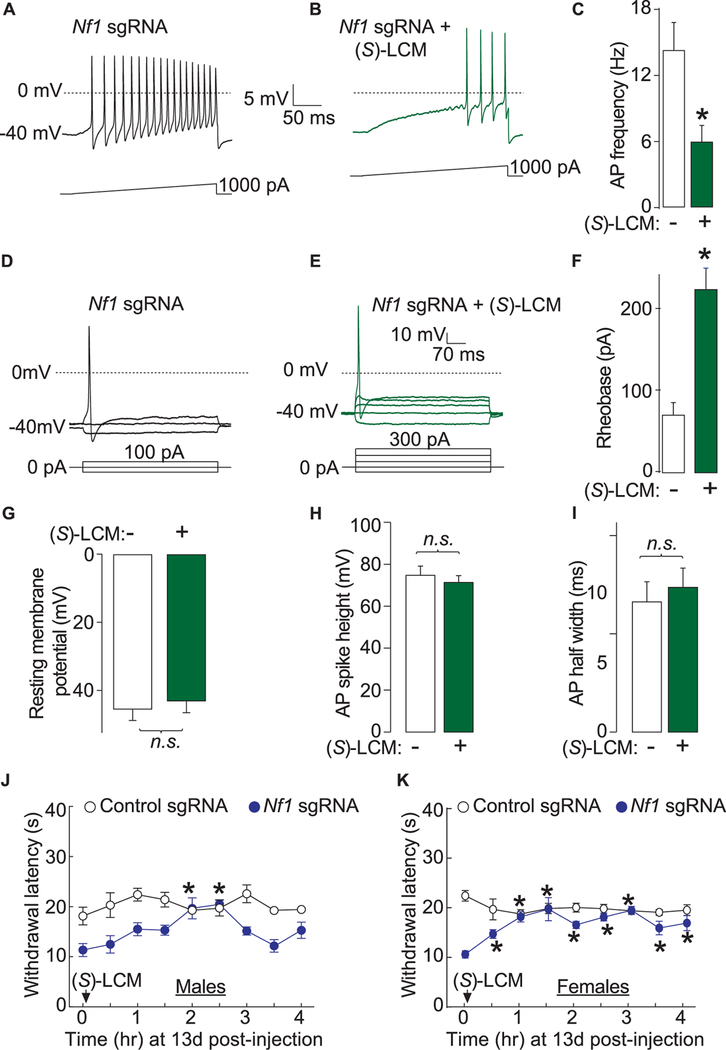
(S)-LCM reverses excitability and thermal hyperalgesia after CRISPR/Cas9 Nf1 editing in rats. Representative recordings in response to a step of depolarizing current evoked action potentials (APs) in sensory neurons transfected with neurons transfected by Nf1 sgRNA containing plasmid and treated with either DMSO 0.04% (A) or 10 μM (S)-LCM (B). (C) Summary of the number of APs in the indicated conditions (n = 11 cells for Nf1 sgRNA 1 DMSO 0.04% and n = 12 cells for Nf1 sgRNA +10 μM (S)-LCM). Representative recordings in response to various steps of depolarizing current to measure rheobase in sensory neurons transfected with neurons transfected by Nf1 sgRNA containing plasmid and treated with either DMSO 0.04% (D) or 10 μM (S)-LCM (E). Rheobase is the current required for eliciting the first AP. (F) Summary of the measured rheobase in indicated conditions (n = 11 each). Summary of the resting membrane potential (in millivolts, mV) (G), AP spike height (in mV) (H), and AP half width (in milliseconds, ms) (I) in the indicated conditions. Asterisks indicate significance compared with control sgRNA transfected cells (*P < 0.05, Student’s t test). n.s., not-significant; P > 0.05; unpaired Student’s t test. At least 11 to 14 cells were recorded from for the parameters shown in (G-I). (G) Thermal hyperalgesia (paw withdrawal latency) of male (J) and female (K) rats, 13 days after in vivo transfection with control or Nf1 sgRNA containing plasmids. Male rats (n = 6–8) were orally administered 30 mg/kg of (S)-LCM and their paw withdrawal latency was followed for 4 hours. *P < 0.05 vs control sgRNA, Student’s t test. Error bars represent mean ± SEM.
4. Discussion
The present study provides a direct link between neurofibromin expression and pain. Targeted truncation of neurofibromin, achieved by acute CRISPR/Cas9 editing of the Nf1 gene in adult rats, resulted in increases of Ca2+ (CaV2.2) and Na+ (TTX-S, mostly NaV1.7) currents confirming previous observations of channel dysregulation observed in sensory neurons from Nf1 haploinsufficient mice.17,59,60 These gains-of-function resulted in increased nociceptor excitability. Importantly, Nf1 editing resulted in thermal hyperalgesia in vivo in both male and female rats. Using this new rat model of NF1, we identify CRMP2 as a central protein contributing, in consort with neurofibromin, to CaV2.2 and NaV1.7 ion channels dysregulation. The concordance in normalization of ion channel dysregulation by a CRMP2-directed strategy and of hyperalgesia supports the translational targeting of CRMP2 to curb NF1-related pain.
While Nf1+/−mice have provided a basis for mechanistic investigation of disorders related to NF1, studies using these mice to understand the relationship of neurofibromin to pain have yielded contradictory results preventing definitive conclusions. Notably, no alteration of their baseline thermal hyperalgesia was observed. Also, these mice do not develop peripheral nerve sheath tumors or other characteristic symptoms of human NF1,29 suggesting that they do not fully recapitulate the disease. For this reason, we focused on mechanistic modeling of pain, an important symptom of patients with NF1. To this end, a recent computational functional genomics–based study identified neurofibromin as a pain chronification gene.57 Following CRISPR/ Cas9-mediated editing of Nf1 in vivo, rats developed thermal hyperalgesia indicating an increased sensitivity to a noxious stimulus. We did not, however, observe hypersensitivity to an innocuous mechanical stimulus in rats with Nf1 truncation. Touch-evoked allodynia is believed to be mediated by normally innocuous stimulation of large diameter (ie, Aβ) touch fibers in the presence of ongoing C-fiber input as would be present in the setting of injury and associated with central sensitization.65 The loss of neurofibromin was induced in these rats in the absence of injury and therefore this result is not surprising. The observed thermal hyperalgesia is consistent with increased nociceptor excitability. These findings also highlight the difference between neurofibromin truncation and single allelic loss of Nf1 gene in the heterozygous Nf1+/−mice, where heat hyperalgesia was not observed.50 Nf1+/−mice rely on global heterozygous truncation of 1 allele of the Nf1 gene rather than the expression of a truncated neurofibromin in the DRG/spinal dorsal horn. However, the different outcomes might be related to species differences. Female, but not male, rats with Nf1 editing had lowered nociceptive thresholds in the paw pressure Randall-Selitto test. While the reason for this gender-specific difference is not currently known, our data are in line with previous findings which reported that female Nf1+/−mice exhibit a subtle enhanced mechanical sensitivity to lower force Von Frey filaments.61 This observed enhancement in mechanical hyperalgesia is consistent with a generalized enhancement in excitability of C-fibers.
Importantly, truncation of neurofibromin in the brain recapitulates cognitive deficits that lead to alterations in memory formation56 and emotional processing,40 features noted in patients with NF1. In Nf1+/−mice, glutamate and GABA neurotransmission were also reportedly elevated together withça deficit in long-term potentiation in the amygdala.40 Pain sensation is believed to involve multidimensional processing in a distributed network of cortical, thalamic, and emotional brain centers48 including the amygdala.41 Together, these alterations could impair the emotional processing of a pain sensation thus explaining the conflicting pain behavior observed in this mouse model. The advantage of the spinal CRISPR/Cas9 approach used here is that it allows us to model a potential mechanism relevant to a symptom of patients with NF1, rather than attempting to capture the full spectrum of the disease itself. Importantly, anxiety-related behaviors in male and female rats were not affected by Nf1 editing, demonstrating that our reductionist approach of symptom-specific modeling of a multifaceted disease permits mechanistic studies into pain and NF1.
CRISPR/Cas9 is a powerful technique for genome editing.52 It involves directing the nuclease Cas9, via a guide RNA, to bind to a site in a gene (here, exon 39 for Nf1) to catalyze a double stranded break in the DNA. This break is repaired by a non-end-joining endogenous DNA repair mechanism, an error prone event, leading to random insertion/truncation events, which here lead to the appearance of an early stop codon and expression of a C-terminally truncated neurofibromin. Whether this editing resulted in a single copy of the Nf1 gene (ie, a hypomorphic allele) which models 80% of patients with inherited NF1 mutations20 that have a single full-length copy is not known and is therefore a potential limitation of our model. The editing guide RNA was targeted to exon 39 of Nf1 to avoid the Ras GAP domain of NF1. Inactivating mutations of the GAP domain in neurofibromin result in tumorigenesis, including malignant peripheral nerve sheath tumors. The tumors impinging on nerves in patients with NF1 have been associated with pain.12 As CRISPR/Cas9-mediated editing of Nf1 in vivo resulted in development of thermal and mechanical hyperalgesia, these findings argue against a role for the GAP domain in pain. The increased ERK phosphorylation in Nf1-edited spinal cord lysates is further evidence of a Ras-independent function of neurofibromin. Since Ras is over-activated following loss of neurofibromin in NF1, considerable effort has been geared to achieving inhibition of Ras signaling pathway for management of NF1 symptoms.69 Our approach identifies a new domain within neurofibromin’s C-terminus that is especially linked to pain in NF1 and can be targeted independently of Ras. It was recently reported that neurofibromin is a direct binding partner of G protein bg subunits and that its truncation prevents the opioid-receptor–induced activation of Ras.66 Taken together, this suggests that although the GAP domain of neurofibromin is not involved in nociceptive signal facilitation, it might be responsible for failure of pain management in patients with NF1.
In the course of studying mechanism(s) related to ion channel dysregulation induced by loss of neurofibromin, we discovered a novel regulatory pathway controlled by CRMP2, a protein that interacts with the C-terminal domain of neurofibromin.51 Recent studies from our group and others have established CRMP2 as a bona fide target for pain with CRMP2-derived peptides2,24 and adenoviruses23 as well CRMP2-modifying small molecules42 being anti-nociceptive. Furthermore, CRMP2 was found to interact with CaV2.2 to positively regulate its activity.3 CRMP2 was also described to positively regulate NaV1.7.18,19 This TTX-S Na+ channel controls sensory neuron excitability and mutations in this channel are linked to pain syndromes in humans.15 The fact that loss of neurofibromin resulted in increased peak current densities for these channels echoes with their regulation by the neurofibromin interacting protein: CRMP2. Our studies do not address which of the channels is more important. Collectively, these findings triangulate to suggest that loss of CRMP2 regulation by neurofibromin to be pathological event underlying the hyperalgesic phenotype of CRISPR/Cas9 Nf1 edited rats. While the lowered threshold for thermal stimulation is consistent with an enhanced excitability observed in Nf1-edited rats, the molecular basis for this remains unknown. One possibility, not tested here, could be upregulation of the heat sensor transient receptor potential cation channel V1.5 An upregulation of NaV1.7, as reported here, could alternatively explain the thermal hyperalgesia as a recent study demonstrated the necessity of NaV1.7 for heat nociception.38,68
Here, we report a novel strategy converging on the targeting of CRMP2: inhibiting CRMP2 phosphorylation by Cdk5 (an event regulated by neurofibromin) with the small molecule (S)-LCM. Blocking phosphorylation of CRMP2 reversed ion channel dysregulation, increased excitability to normal levels, and normalized pain behaviors elicited by Nf1 editing to control levels in vitro and in vivo. From a translational point of view, the small molecule (S)-LCM is an inactive enantiomer of an FDA-approved drug Vimpat©. (S)-LCM inhibits CRMP2 phosphorylation by Cdk5,46 which is required for CRMP2’s coupling to CaV2.24,46 and NaV1.7.18 Specificity of (S)-LCM for CRMP2 phosphorylation can be inferred from the lack of inhibition of calcium and sodium influx in cells expressing a constitutively phosphorylated version of CRMP2. That the anti-epileptic enantiomer (R)-LCM is unable to inhibit Cdk5-mediated CRMP2 phosphorylation or calcium influx42 also supports the contention that (S)-LCM’s actions are via inhibition of CRMP2 phosphorylation. In addition, we have previously reported that cephalic and extracephalic cutaneous allodynia induced in rats by activation of dural nociceptors with a cocktail of inflammatory mediators, was inhibited (peak effect observed at 2 hours) by oral administration of (S)-LCM.45 Notably, spinal administration of (S)-LCM did not result in motor deficits or sedation.42 (S)-LCM is 100% orally available and pharmacological studies have determined its half-life in serum to be ~3 hours.31 Our studies did not determine the site of action of oral (S)-LCM, which could reflect mechanisms within the brain, in the periphery or both. However, as spinal administration of (S)-LCM reversed postsurgical and neuropathic pain in preclinical animal models,42 the site of action is likely within the spinal cord. (S)-LCM would be expected to show a similar safety profile as Vimpat and to be a promising small molecule to be evaluated in human clinical trials for pain related to NF1.
Modeling pain of NF1 through facile CRISPR/Cas9 editing of the Nf1 gene can be added to the extensive toolbox for scientists as an alternative model for symptom-specific modeling of NF1. The parallel characterization of ion channel dysregulation and excitability related to pain behavior in Nf1 edited neurons identifies the molecular basis for increased pain sensitivity reported by patients with NF1.36,64 Our work identifies a novel CRMP2-related pathologic event responsible for increased pain sensitivity in NF1. Targeting CRMP2 may represent a new therapeutic approach for pain management in patients with NF1. Future studies could target editing of recurrent mutations found in human patients in a precision medicine style approach to interrogate the link to pain.
Supplementary Material
Acknowledgements
The authors thank Dr. Erik T. Dustrude (Indiana University School of Medicine) for helpful comments on the manuscript. This work was supported by a National Institutes of Health award (1R01NS098772 and 1R01DA042852-01); a Neurofibromatosis New Investigator Award from the Department of Defense Congressionally Directed Military Medical Research and Development Program (NF1000099); and a Children’s Tumor Foundation NF1 Synodos award to R. Khanna. A. Moutal was supported by a Young Investigator’s Award from the Children’s Tumor Foundation.
Appendix A. Supplemental Digital Content
Supplemental Digital Content associated with this article can be found online at http://links.lww.com/PAIN/A446, http://links.lww.com/PAIN/A447, and http://links.lww.com/PAIN/A448.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Conflict of interest statement
The authors have no conflict of interest to declare.
References
- [1].Bicudo NP, de Menezes Neto BF, da Silva de Avo LR, Germano CM, Melo DG Quality of life in adults with neurofibromatosis 1 in Brazil. J Genet Couns 2016;25:1063–74. [DOI] [PubMed] [Google Scholar]
- [2].Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, Fehrenbacher JC, Fitz SD, Khanna M, Park CK, Schmutzler BS, Cheon BM, Due MR, Brustovetsky T, Ashpole NM, Hudmon A, Meroueh SO, Hingtgen CM, Brustovetsky N, Ji RR, Hurley JH, Jin X, Shekhar A, Xu XM, Oxford GS, Vasko MR, White FA, Khanna R. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2)(1) channel complex. Nat Med 2011;17: 822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem 2009;284:31375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brittain JM, Wang Y, Eruvwetere O, Khanna R. Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett 2012;586:3813–18. [DOI] [PubMed] [Google Scholar]
- [5].Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997;389:816–24. [DOI] [PubMed] [Google Scholar]
- [6].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994; 53:55–63. [DOI] [PubMed] [Google Scholar]
- [7].Chi XX, Schmutzler BS, Brittain JM, Hingtgen CM, Nicol GD, Khanna R. Regulation of N-type voltage-gated calcium (CaV2.2) channels and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci 2009;23:4351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic-Todorovic V, Todorovic SM. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol 2011;80:900–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Choi D, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-Benzyl-2-acetamidopropionamide derivatives. J Med Chem 1996;39: 1907–16. [DOI] [PubMed] [Google Scholar]
- [10].Cradick TJ, Qiu P, Lee CM, Fine EJ, Bao G. COSMID. A web-based tool for identifying and validating CRISPR/cas off-target sites. Mol Ther Nucleic Acids 2014;3:e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Crawford HA, Barton B, Wilson MJ, Berman Y, McKelvey-Martin VJ, Morrison PJ, North KN. The impact of neurofibromatosis type 1 on the Health and wellbeing of Australian adults. J Genet Couns 2015;24:931–44. [DOI] [PubMed] [Google Scholar]
- [12].Creange A, Zeller J, Rostaing-Rigattieri S, Brugieres P, Degos JD, Revuz J, Wolkenstein P. Neurological complications of neurofibromatosis type 1 in adulthood. Brain 1999;122:473–81. [DOI] [PubMed] [Google Scholar]
- [13].Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, Parada LF, Mody I, Silva AJ. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008;135:549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron 1992;8:415–28. [DOI] [PubMed] [Google Scholar]
- [15].Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 2013;14:49–62. [DOI] [PubMed] [Google Scholar]
- [16].Drouet A, Wolkenstein P, Lefaucheur JP, Pinson S, Combemale P, Gherardi RK, Brugieres P, Salama J, Ehre P, Decq P, Creange A. Neurofibromatosis 1-associated neuropathies: a reappraisal. Brain 2004; 127:1993–2009. [DOI] [PubMed] [Google Scholar]
- [17].Duan JH, Hodgdon KE, Hingtgen CM, Nicol GD. N-type calcium current, Cav2.2, is enhanced in small-diameter sensory neurons isolated from Nf11/-mice. Neuroscience 2014;270:192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dustrude ET, Moutal A, Yang X, Wang Y, Khanna M, Khanna R. Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proc Natl Acad Sci U S A 2016;113:E8443–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dustrude ET, Wilson SM, Ju W, Xiao Y, Khanna R. CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J Biol Chem 2013; 288:24316–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Esposito T, Piluso G, Saracino D, Uccello R, Schettino C, Dato C, Capaldo G, Giugliano T, Varriale B, Paolisso G, Di Iorio G, Melone MA. A novel diagnostic method to detect truncated neurofibromin in neurofibromatosis 1. J Neurochem 2015;135:1123–8. [DOI] [PubMed] [Google Scholar]
- [21].Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol 2007;6:340–51. [DOI] [PubMed] [Google Scholar]
- [22].Ferner RE, Thomas M, Mercer G, Williams V, Leschziner GD, Afridi SK, Golding JF. Evaluation of quality of life in adults with neurofibromatosis 1 (NF1) using the Impact of NF1 on Quality of Life (INF1-QOL) questionnaire. Health Qual Life Outcomes 2017;15:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fischer G, Pan B, Vilceanu D, Hogan QH, Yu H. Sustained relief of neuropathic pain by AAV-targeted expression of CBD3 peptide in rat dorsal root ganglion. Gene Ther 2014;21:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Francois-Moutal L, Wang Y, Moutal A, Cottier KE, Melemedjian OK, Yang X, Wang Y, Ju W, Largent-Milnes TM, Khanna M, Vanderah TW, Khanna R. A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. PAIN 2015;156:1247–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. PAIN 1988;32:77–88. [DOI] [PubMed] [Google Scholar]
- [26].Hingtgen CM, Roy SL, Clapp DW. Stimulus-evoked release of neuropeptides is enhanced in sensory neurons from mice with a heterozygous mutation of the Nf1 gene. Neuroscience 2006;137: 637–45. [DOI] [PubMed] [Google Scholar]
- [27].Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 2013;31:827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Huynh TN, Krigbaum AM, Hanna JJ, Conrad CD. Sex differences and phase of light cycle modify chronic stress effects on anxiety and depressive-like behavior. Behav Brain Res 2011;222:212–22. [DOI] [PubMed] [Google Scholar]
- [29].Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet 1994;7:353–61. [DOI] [PubMed] [Google Scholar]
- [30].Kodra Y, Giustini S, Divona L, Porciello R, Calvieri S, Wolkenstein P, Taruscio D. Health-related quality of life in patients with neurofibromatosis type 1. A survey of 129 Italian patients. Dermatology 2009;218:215–20. [DOI] [PubMed] [Google Scholar]
- [31].Koo TS, Kim SJ, Ha DJ, Baek M, Moon H. Pharmacokinetics, brain distribution, and plasma protein binding of the antiepileptic drug lacosamide in rats. Arch Pharmacal Res 2011;34:2059–64. [DOI] [PubMed] [Google Scholar]
- [32].Lee JH, Park CK, Chen G, Han Q, Xie RG, Liu T, Ji RR, Lee SY. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell 2014;157:1393–404. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [33].Lin YL, Hsueh YP. Neurofibromin interacts with CRMP-2 and CRMP-4 in rat brain. Biochem Biophys Res Commun 2008;369:747–52. [DOI] [PubMed] [Google Scholar]
- [34].Mangues R, Corral T, Lu S, Symmans WF, Liu L, Pellicer A. NF1 inactivation cooperates with N-ras in in vivo lymphogenesis activating Erk by a mechanism independent of its Ras-GTPase accelerating activity. Oncogene 1998;17:1705–16. [DOI] [PubMed] [Google Scholar]
- [35].Marquez de PB, Hammond DL. Sex dependent enhancement of pain responses in a mouse model of neurofibromatosis Washington, DC: Proceedings of the Society for Neuroscience, 2011. [Google Scholar]
- [36].Martin S, Gillespie A, Wolters PL, Widemann BC. Experiences of families with a child, adolescent, or young adult with neurofibromatosis type 1 and plexiform neurofibroma evaluated for clinical trials participation at the National Cancer Institute. Contemp Clin Trials 2011;32:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, Paepe AD. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat 2000;15:541–55. [DOI] [PubMed] [Google Scholar]
- [38].Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, Malcangio M, Wood JN. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun 2012;3:791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature 1992;355:827–9. [DOI] [PubMed] [Google Scholar]
- [40].Molosh AI, Johnson PL, Spence JP, Arendt D, Federici LM, Bernabe C, Janasik SP, Segu ZM, Khanna R, Goswami C, Zhu W, Park SJ, Li L, Mechref YS, Clapp DW, Shekhar A. Social learning and amygdala disruptions in Nf1 mice are rescued by blocking p21-activated kinase. Nat Neurosci 2014;17:1583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Morland RH, Novejarque A, Spicer C, Pheby T, Rice AS. Enhanced c-Fos expression in the central amygdala correlates with increased thigmotaxis in rats with peripheral nerve injury. Eur J pain 2016;20:1140–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moutal A, Chew LA, Yang X, Wang Y, Yeon SK, Telemi E, Meroueh S, Park KD, Shrinivasan R, Gilbraith KB, Qu C, Xie JY, Patwardhan A, Vanderah TW, Khanna M, Porreca F, Khanna R. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. PAIN 2016;157:1448–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Moutal A, Dustrude ET, Khanna R. Sensitization of ion channels contributes to central and peripheral dysfunction in neurofibromatosis type 1. Mol Neurobiol 2017;54:3342–9. [DOI] [PubMed] [Google Scholar]
- [44].Moutal A, Dustrude ET, Largent-Milnes TM, Vanderah TW, Khanna M, Khanna R. Blocking CRMP2 SUMOylation reverses neuropathic pain. Mol Psychiatry 2017. [epub ahead of print]. [DOI] [PMC free article] [PubMed]
- [45].Moutal A, Eyde N, Telemi E, Park KD, Xie JY, Dodick DW, Porreca F, Khanna R. Efficacy of (S)-Lacosamide in preclinical models of cephalic pain. Pain Rep 2016;1:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moutal A, Francois-Moutal L, Perez-Miller S, Cottier K, Chew LA, Yeon SK, Dai J, Park KD, Khanna M, Khanna R. (S)-Lacosamide binding to collapsin response mediator protein 2 (CRMP2) regulates CaV2.2 activity by subverting its phosphorylation by Cdk5. Mol Neurobiol 2016;53: 1959–76. [DOI] [PubMed] [Google Scholar]
- [47].Moutal A, Li W, Wang Y, Ju W, Luo S, Cai S, Francois-Moutal L, Perez-Miller S, Hu J, Dustrude ET, Vanderah TW, Gokhale V, Khanna M, Khanna R. Homology-guided mutational analysis reveals the functional requirements for antinociceptive specificity of collapsin response mediator protein 2-derived peptides. Br J Pharmacol 2017. [epub ahead of print]. [DOI] [PMC free article] [PubMed]
- [48].Navratilova E, Morimura K, Xie JY, Atcherley CW, Ossipov MH, Porreca F. Positive emotions and brain reward circuits in chronic pain. J Comp Neurol 2016;524:1646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Newcomb R, Szoke B, Palma A, Wang G, Chen X, Hopkins W, Cong R, Miller J, Urge L, Tarczy-Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J, Miljanich G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry 1998;37:15353–62. [DOI] [PubMed] [Google Scholar]
- [50].O’Brien DE, Brenner DS, Gutmann DH, Gereau RW IV. Assessment of pain and itch behavior in a mouse model of neurofibromatosis type 1. J Pain 2013;14:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Patrakitkomjorn S, Kobayashi D, Morikawa T, Wilson MM, Tsubota N, Irie A, Ozawa T, Aoki M, Arimura N, Kaibuchi K, Saya H, Araki N. Neurofibromatosis type 1 (NF1) tumor suppressor, neurofibromin, regulates the neuronal differentiation of PC12 cells via its associating protein, CRMP-2. J Biol Chem 2008;283:9399–413. [DOI] [PubMed] [Google Scholar]
- [52].Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther 1957;111:409–19. [PubMed] [Google Scholar]
- [54].Riklin E, Talaei-Kheoi M, Merker VL, Sheridan MR, Jordan JT, Plotkin SR, Vranceanu AM. First report of factors associated with satisfaction in patients with neurofibromatosis. Am J Med Genet A 2017;173:671–7. [DOI] [PubMed] [Google Scholar]
- [55].Roy ML, Narahashi T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci 1992;12:2104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Silva AJ, Frankland PW, Marowitz Z, Friedman E, Laszlo GS, Cioffi D, Jacks T, Bourtchuladze R. A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nat Genet 1997;15: 281–4. [DOI] [PubMed] [Google Scholar]
- [57].Ultsch A, Kringel D, Kalso E, Mogil JS, Lotsch J. A data science approach to candidate gene selection of pain regarded as a process of learning and neural plasticity. PAIN 2016;157:2747–57. [DOI] [PubMed] [Google Scholar]
- [58].Wang Y, Brittain JM, Wilson SM, Hingtgen CM, Khanna R. Altered calcium currents and axonal growth in Nf1 haploinsufficient mice. Translational Neurosci 2010;1:106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang Y, Duan JH, Hingtgen CM, Nicol GD. Augmented sodium currents contribute to enhanced excitability of small diameter capsaicin-sensitive Nf11/-mouse sensory neurons. J Neurophysiol 2010;103:2085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang Y, Nicol GD, Clapp DW, Hingtgen CM. Sensory neurons from Nf1 haploinsufficient mice exhibit increased excitability. J Neurophysiol 2005; 94:3670–6. [DOI] [PubMed] [Google Scholar]
- [61].White S, Marquez de Prado B, Russo AF, Hammond DL. Heat hyperalgesia and mechanical hypersensitivity induced by calcitonin gene-related peptide in a mouse model of neurofibromatosis. PLoS One 2014;9:e106767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, Legius E, Callens T, Beiglbock H, Maertens O, Messiaen L. Spectrum of single-and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer 2006;45: 265–76. [DOI] [PubMed] [Google Scholar]
- [63].Wolkenstein P, Zeller J, Revuz J, Ecosse E, Leplege A. Quality-of-life impairment in neurofibromatosis type 1: a cross-sectional study of 128 cases. Arch Dermatol 2001;137:1421–5. [DOI] [PubMed] [Google Scholar]
- [64].Wolters PL, Burns KM, Martin S, Baldwin A, Dombi E, Toledo-Tamula MA, Dudley WN, Gillespie A, Widemann BC. Pain interference in youth with neurofibromatosis type 1 and plexiform neurofibromas and relation to disease severity, social-emotional functioning, and quality of life. Am J Med Genet Part A 2015;167A:2103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. PAIN 2011;152(3 suppl):S2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Xie K, Colgan LA, Dao MT, Muntean BS, Sutton LP, Orlandi C, Boye SL, Boye SE, Shih CC, Li Y, Xu B, Smith RG, Yasuda R, Martemyanov KA. NF1 is a direct G protein effector essential for opioid signaling to ras in the striatum. Curr Biol 2016;26:2992–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav 1976;17:1031–6. [DOI] [PubMed] [Google Scholar]
- [68].Yang L, Dong F, Yang Q, Yang PF, Wu R, Wu QF, Wu D, Li CL, Zhong YQ, Lu YJ, Cheng X, Xu FQ, Chen L, Bao L, Zhang X. FGF13 selectively regulates heat nociception by interacting with Nav1.7. Neuron 2017;93: 806–21.e9. [DOI] [PubMed] [Google Scholar]
- [69].Yap YS, McPherson JR, Ong CK, Rozen SG, Teh BT, Lee AS, Callen DF. The NF1 gene revisited-from bench to bedside. Oncotarget 2014;5: 5873–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.