

SUMMARY
B cell development is a highly regulated process that requires stepwise rearrangement of immunoglobulin genes to generate a functional B cell receptor (BCR). The polycomb group protein BMI1 is required for B cell development, but its function in developing B cells remains poorly defined. We demonstrate that BMI1 functions in a cell-autonomous manner at two stages during early B cell development. First, loss of BMI1 results in a differentiation block at the pro-B cell to pre-B cell transition due to the inability of BMI1-deficient cells to transcribe newly rearranged Igh genes. Accordingly, introduction of a pre-rearranged Igh allele partially restored B cell development in Bmi1−/− mice. In addition, BMI1 is required to prevent premature p53 signaling, and as a consequence, Bmi1−/− large pre-B cells fail to properly proliferate. Altogether, our results clarify the role of BMI1 in early B cell development and uncover an unexpected function of BMI1 during VDJ recombination.
Graphical Abstract
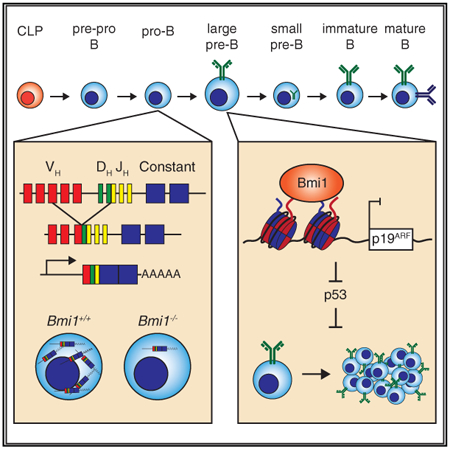
In Brief
Cantor et al. identify a cell-autonomous role for the polycomb group protein BMI1 in early B cell development. At the pro-B cell to pre-B cell transition, BMI1 promotes the expression of newly rearranged Igh genes in pro-B cells and subsequently prevents premature p53 activation and enables large pre-B cell proliferation.
INTRODUCTION
B cell development depends on successful gene rearrangements at the immunoglobulin (Ig) loci to ensure that mature B cells express a diverse repertoire of antibodies. During this process, a VDJH joint is assembled at the Ig heavy (Igh) chain locus in pro-B cells, which, if in the correct reading frame, leads to the expression of an Igμ protein. Igμ assembles with surrogate light chains to form a surface pre-B cell receptor (pre-BCR), which guides the pro-B cell to pre-B cell transition. Following signaling by the pre-BCR, pre-B cells undergo clonal expansion and subsequent rearrangement of the Ig light (IgL) chain loci. Successfully rearranged light chains assemble with an Igμ chain, forming a surface BCR, and drive the progression to immature B cells that will form the reservoir of long-lived mature B cells (Herzog et al., 2009).
The process of V(D)J recombination presents inherent risks to the organism because of the generation of double-strand breaks and subsequent non-homologous end joining of DNA fragments at the Ig loci. Thus, numerous mechanisms exist to ensure the proper generation of a clonal surface BCR on B cells while preventing deleterious events. These include the sequential rearrangement of Ig loci, with several checkpoints along the sequential development to assess rearrangement status, and the programming of developing B cells for either clonal expansion or apoptosis, depending on pre-BCR and BCR signaling cues (Melchers, 2015). While extensive work has elucidated many of these mechanisms, our understanding of the molecular pathways critical for B cell development remains fragmentary.
Polycomb group (PcG) proteins are a group of regulatory factors that form multimeric protein complexes and are critical for maintaining cell identity and cell proliferation by modifying chromatin structure and silencing genes (Sauvageau and Sauvageau, 2010). Polycomb repressive complex 1 (PRC1) and polycomb repressive complex 2 (PRC2) were the earliest complexes described, although more recent work has identified both alternative and novel polycomb complexes. The core components of PRC1 consist of one member of each CBX, HPH, PCGF, and RING1 protein family, which monoubiquitinate histone H2A on lysine 119 (H2AK119), whereas the core components of PRC2 are EED, Suz12, EZH1/2, and RBBP4/7, which methylate H3K27 (Di Croce and Helin, 2013; Simon and Kingston, 2013). PRC1 and PRC2 are thought to cooperate to regulate gene expression, because PRC2 deposition of H3K27me3 recruits PRC1 through its CBX family member (Blackledge et al., 2015). However, inactivation of core PRC2 factors in mammalian cells only partially affects PRC1 recruitment to its target loci and minimally changes global H2AK119ub levels, suggesting that PRC2-independent mechanisms exist for PRC1 recruitment (Tavares et al., 2012).
Through genetic studies in the mouse, it became apparent that PcG proteins are critical for B cell lymphopoiesis. EZH2 regulates distal VH gene usage during VH-DJH recombination and prevents IgL loci rearrangement in pro-B cells (Mandal et al., 2011; Su et al., 2003). The PRC1 component BMI1, also known as PCGF4, is required for normal lymphocyte development at least partly through the repression of the Ink4/Arf locus, which encodes the two tumor suppressor proteins, p16INK4A and p19ARF (Bruggeman et al., 2005; Oguro et al., 2010). In developing T cells, BMI1 prevents premature p19ARF-mediated stabilization of p53 to promote the proliferation and survival of progenitor T cells in response to pre-T cell receptor (TCR) signaling (Miyazaki et al., 2008). However, Bmi1−/− Ink4a/Arf−/− mice still exhibit defective B cell lymphopoiesis, suggesting that BMI1 functions partly through Ink4a/Arf-independent mechanisms during B cell development (Oguro et al., 2010).
We found that BMI1 is required for pro-B cells to differentiate into pre-B cells in a cell-autonomous manner. Loss of BMI1 results in the upregulation of p53 target genes in pro-B cells, precluding the expansion of these cells at the large pre-B stage. Furthermore, expression of rearranged Igh genes and production of the Igμ chain are impaired in Bmi1−/− pro-B cells. Accordingly, introduction of a pre-rearranged Igh allele partially restored B cell development in Bmi1−/− mice. Altogether, these results identify a critical role for BMI1 in B cell development through the regulation of rearranged Igh gene expression and expansion of pre-B cells.
RESULTS
BMI1 Is Required for the Pro-B Cell to Pre-B Cell Transition
Previous studies have identified BMI1 as essential for B cell development in the mouse (Oguro et al., 2010; van der Lugt et al., 1994). However, the mechanisms that BMI1 engages to promote B cell development remain unknown. To begin to dissect the function of BMI1 in progenitor B cells, we first assessed its expression levels throughout early B cell development using data acquired through the Immunological Genome Project (Heng et al., 2008; Painter etal., 2011). Bmi1 is highly expressed in pro-B cells and large pre-B cells and is downregulated as large pre-B cells transition into small pre-B cells (Figure S1A). The expression of Cdkn2a, which encodes the tumor suppressor proteins p16INK4A and p19ARF and is repressed by BMI1, inversely correlates with Bmi1 expression at the pro-B cell to pre-B cell transition (Figure S1A). This correlation disappears in mature B cells, likely pointing to a more critical role for a Bmi1-Cdkn2a axis at the pro-B cell to pre-B cell transition. The high expression of Bmi1 and the resulting repression of Cdkn2a early in B cell development are reminiscent of what has been observed in early T cell development, in which BMI1 represses p19ARF to prevent apoptosis in proliferating DN3 T cells (Miyazaki et al., 2008). Furthermore, the inverse correlation of Bmi1 and Cdkn2a levels is consistent with studies demonstrating a modest rescue of B cell development in Bmi1−/− Ink4a/Arf−/− mice (Miyazaki et al., 2008; Oguro et al., 2010).
To further probe BMI1’s function in early B cell development, we determined the frequency and numbers of early B cell progenitors in Bmi1+/+ and Bmi1−/− mice. The bone marrow of Bmi1−/− mice was devoid of B cells, correlating with the accumulation of pro-B cells and their failure to differentiate into pre-B cells (Figures 1A and 1B; Figure S1B). The differentiation block at the pro-B cell to pre-B cell transition resulted in drastically fewer mature B cells in the peripheral blood and spleen of Bmi1−/− mice and a corresponding increase in the frequency of CD11b+ myeloid cells (Figures 1C and 1D). Consistent with previous reports (van der Lugt et al., 1994), we observed a drastic reduction in the cellularity of various hematopoietic organs of Bmi1−/− mice as a consequence of the overall reduction in lymphocyte numbers (Figure S1C).
Figure 1. BMI1 Is Required for the Pro-B Cell to Pre-B Cell Transition.

(A) Representative fluorescence-activated cell sorting (FACS) plots of developing B cells in the bone marrow of indicated mice. Shown is the frequency of each cell population in the bone marrow of indicated animals. Pro-B: B220+ CD19+ IgM− IgD− ckit+ CD25−. Pre-B: B220+ CD19+ IgM− IgD− ckit+ CD25−. Immature: B220+ CD19+ IgM+ IgD−. Mature: B220+ CD19+ IgM+ IgD+.
(B) Frequency of indicated cell populations in the bone marrow of animals. Each point represents one animal; the line represents the mean. n = 6 for each genotype.
(C) Frequency of indicated cell type in the peripheral blood of indicated animals. Each point represents one animal; the line represents the mean. n = 6 for Bmi1+/+ animals; n = 5 for Bmi1−/− animals.
(D) Frequency of B220+ cells in the spleens of indicated mice. Each point represents one animal; the line represents the mean. n = 4 for Bmi1+/+ animals; n = 7 for Bmi1−/− animals.
***p < 0.001, **p < 0.01. BM, bone marrow; PB, peripheral blood.
See also Figure S1.
Reduced Igμ Chain Expression in Bmi1−/− Pro-B Cells
The pro-B cell to pre-B cell transition and subsequent expansion of large pre-B cells depend on the successful rearrangement of the Igh locus, the expression of Igm chain, the assembly of a pre-BCR on the surface of the cell, and signaling downstream of the pre-BCR. Failure in any of these steps will impair pro-B cell to pre-B cell differentiation (Herzog et al., 2009). We therefore determined whether BMI1 was required for the expression of Igμ chain in pro-B cells. By intracellular flow cytometry analysis, we observed a decrease in the frequency of pro-B cells expressing Igμ chain in Bmi1−/− mice to about half of the levels observed in Bmi1+/+ mice (Figure 2A). In addition, we observed a decrease in the median Igμ fluorescence intensity in Igμ-expressing pro-B cells from Bmi1−/− mice (Figure 2B).
Figure 2. BMI1 Is Required for the Expression of Rearranged Igh Genes in Pro-B Cells.

(A) Representative FACS plot and quantification of Igμ chain-positive pro-B cells in Bmi1+/+ and Bmi1−/− mice. Each point represents one animal; the line represents the mean. n = 4 for Bmi1+/+ animals; n = 5 for Bmi1−/− animals.
(B) Igμ MFI in Igμ-expressing pro-B cells. Each dot represents one animal; the line represents the mean. n = 4 for Bmi1+/+ animals; n = 5 for Bmi1−/− animals.
(C) Semiquantitative PCR analysis of genomic DNA from sorted Bmi1+/+ and Bmi1−/− pro-B cells assessing the frequency of Igh locus rearrangement using either proximal (VH7183) or distal (VHJ558) VH gene segments. DNA from DP T was used as a negative control.
(D) Frequency of proximal and distal VH segments used in individually sequenced VDJH coding joints from sorted pro-B cells. VDJH joints were amplified using a promiscuous forward primer (MsVHe) and a reverse primer specific to JH3. n = 37 VDJH sequences for Bmi1+/+ pro-B cells, and n = 48 VDJH sequences for Bmi1−/− pro-B cells sorted from 3 animals for each genotype.
(E) qRT-PCR analysis of mRNA expression of rearranged Igh genes. DP T were used as negative controls. Data are shown as a Tukey box-and-whisker plot; the center line represents the median. n ≥ 5 for each genotype.
***p < 0.001, **p < 0.01, *p < 0.05. MFI, median fluorescence intensity; DP T, double-positive thymocytes.
See also Figure S2.
BMI1 Is Dispensable for VDJH Recombination but Required for Rearranged Igh Gene Expression in Pro-B Cells
We next determined the basis for the reduction in Igμ chain expression in Bmi1−/− pro-B cells. This reduction could result from inefficient VDJH recombination and/or failure to express rearranged Igh genes. To assess whether BMI1 inactivation alters VDJH recombination, we determined the frequency of rearrangement for two VH gene families, VH7183 and VHJ558, in Bmi1+/+ and Bmi1−/− pro-B cells. The VH7183 gene family is the most proximal VH family and the VHJ558 family is the most distal VH family to DJH genes (Figure S2A). Because the PRC2 component EZH2 is required for distal VH gene usage (Su et al., 2003), we tested whether BMI1 similarly enabled distal VH gene usage. On genomic DNA isolated from pro-B cells, we performed semiquantitative PCR using degenerative forward primers that amplify either VH7183 genes or VHJ558 genes and a reverse primer specific to the JH3 gene. Bmi1−/− pro-B cells and their wild-type counterparts contained comparable levels of DNA that had recombined either VH7183 or VHJ558 genes with DJH genes (Figure 2C). In addition, we isolated and sequenced individual VDJH joints from Bmi1+/+ and Bmi1−/− pro-B cells using a promiscuous forward primer that amplifies various genes across all VH families. Again, we observed no difference in the ratio of proximal to distal VH gene usage between Bmi1+/+ and Bmi1−/− pro-B cells, supporting the conclusion that BMI1 is not required for VDJH recombination in pro-B cells (Figure 2D).
In contrast, when we assessed the expression levels of rearranged Igh genes, we observed a drastic reduction in the expression levels of all VH family genes tested in Bmi1−/− pro-B cells, consistent with a combined decreased frequency of Bmi1−/− pro-B cells expressing Igm and decreased Igμ protein levels in Igμ-expressing Bmi1−/− pro-B cells (Figure 2E). The decrease in rearranged Igh gene expression was not the result of a generic reduction in expression at the Igh locus, because we detected no difference in the expression of DH-Cu transcripts (Figure 2E). Altogether, these data support a model in which BMI1 is dispensable for VDJH recombination but is critical for expression of recently rearranged VDJH genes in pro-B cells.
BMI1 Is Not Required for Igh Germline Transcription or Igh Contraction
PRC1 has been shown to regulate high-order chromatin architecture (Simon and Kingston, 2013). For RAG complexes to access VH genes and initiate recombination, the surrounding chromatin needs to be accessible (Ebert et al., 2013; Fuxa et al., 2004). The transcription of Igh genes in germline configuration (germline transcription) has been hypothesized to be critical for RAG accessibility to these genes (Yancopoulos and Alt, 1985). We therefore first assessed whether BMI1 loss altered the ability of the germline Igh loci to be transcribed in pro-B cells as a surrogate assay for chromatin accessibility. However, we did not observe differences in germline transcription for either VH7183 or VHJ558 gene families between Bmi1+/+ and Bmi1−/− pro-B cells (Figure S2B). Because we observed that BMI1 modulates the expression of recombined Igh genes in pro-B cells, and given the impact of chromatin architecture on Igh recombination (Roldán et al., 2005), we next assessed the impact of BMI1 on high-order organization of the Igh loci. With 3D microscopy, we determined whether BMI1 is required for Igh contraction by using DNA-fluorescence in situ hybridization (FISH) probes against the 5′ Igh (distal) and 3′ Igh (proximal) regions. Consistent with our finding that Bmi1−/− pro-B cells efficiently use both proximal and distal VH gene families during VH-DJH joining, the contraction of the Igh locus in pro-B cells was not affected by the absence of BMI1 (Figure S2C).
A Pre-rearranged Igh Allele Restores B Cell Development in Bmi1−/− Mice
We postulated that if the process of VDJH recombination at the Igh locus affects the expression of rearranged Igh genes in Bmi1−/− pro-B cells and is responsible for the differentiation block of developing B cells in Bmi1−/− mice, the introduction of a pre-rearranged VDJh allele would rescue B cell development. To test this, we generated Bmi1+/+ and Bmi1−/− mice carrying a recombined VDJH element inserted into the endogenous JH locus (B1.8i) (Sonoda et al., 1997). Consistent with our hypothesis, introduction of the B1.8i allele largely restored B cell development to Bmi1−/− animals (Figures 3A–3C; Figure S3). The frequencies of immature, mature, and splenic B cells in Bmi1−/− B1.8+ mice were comparable to that of Bmi1+/+ mice (Figures 3A–3C). Total numbers of Bmi1−/− B1.8+ B cells, while still decreased compared to those of Bmi1+/+ animals, were drastically increased compared to those of Bmi1−/− animals (Figure S3). Moreover, when we assessed Igμ expression in pro-B cells, we found that the B1.8 allele increased the frequency of Bmi1−/− pro-B cells expressing Igμ and restored Igμ protein levels in Igμ-expressing Bmi1−/− pro-B cells (Figure 3D). Altogether, these data demonstrate that bypassing VDJh recombination enables Bmi1−/− pro-B cells to express Igμ and subsequently differentiate.
Figure 3. A pre-rearranged Igh Locus Restores B Cell Development to Bmi1−/− Mice.

(A) Representative FACS plot of developing B cells in the bone marrow of indicated mice. Shown is the frequency of each cell population in the bone marrow of indicated animals.
(B) Frequency of indicated cell type in the bone marrow of indicated mice. Data are shown as a Tukey box-and-whisker plot; the center line represents the median. n ≥ 5 for each genotype.
(C) Frequency of B220+ cells in the spleen of indicated mice. Each point represents one animal; the line represents the mean. n ≥ 5 for each genotype.
(D) Histogram showing the frequency of Igμ-positive pro-B cells from indicated animals. Igμ MFI in Igμ-expressing pro-B cells is indicated in the top right of each plot.
***p < 0.001, *p < 0.05. BM, bone marrow; MFI, median fluorescence intensity.
See also Figure S3.
BMI1 Loss Results in the Upregulation of p53 Signaling in Pro-B Cells
Although expression of a pre-rearranged Igh locus partially rescues B cell development in Bmi1−/− mice, Bmi1−/− B1.8i+ bone marrow still contains lower frequencies and numbers of pre-B cells compared to wild-type mice (Figure 3; Figure S3). We therefore sought to identify additional mechanisms engaged by BMI1 that control the pro-B cell to pre-B cell transition in developing B cells. We assessed the expression levels of the B cell transcription factors Pax5 and Ebf1, because BMI1 regulates their expression in early progenitors before lymphoid commitment (Oguro et al., 2010), but we found no difference in their expression between Bmi1−/− pro-B cells and their wild-type counterparts (Figure S4A). To uncover other altered pathways, we sorted pro-B cells from Bmi1+/+ and Bmi1−/− mice and performed genome-wide expression analysis. Using a false discovery rate (FDR) < 0.05 as a cutoff for significance, we identified 122 upregulated transcripts and 165 downregulated transcripts in Bmi1−/− pro-B cells compared to Bmi1+/+ pro-B cells (Table S1). Numerous p53 targets, including Bbc3, Pmaip1, Phlda3, and Cdkn1a, were found among the upregulated transcripts in Bmi1−/− pro-B cells (Figure 4A; Table S1). Consistent with this observation, gene ontology analysis of upregulated genes in Bmi1−/− pro-B cells showed enrichment for pathways associated with apoptosis, proliferation, DNAdamage, and p53 signaling (Figure S4B). Similarly, the p53 pathway was enriched in Bmi1−/− pro-B cells when analyzed by gene set enrichment analysis (GSEA) (Figure S4C). As expected, p19Arf expression was drastically upregulated in Bmi1−/− pro-B cells, demonstrating de-repression of the Ink4a/Arf locus, and we confirmed the upregulation of specific p53 target genes, including Bbc3, Pmaip1, Phlda3, and Cdkn1a, in Bmi1−/− pro-B cells from independent animals (Figure 4B). We did not detect changes in Trp53 transcript levels, suggesting BMI1-dependent post-transcriptional regulation of p53 activity (Figure 4B). Together with previous work demonstrating that deletion of p19Arf partially restores B cell development to Bmi1−/− mice (Akala et al., 2008; Miyazaki et al., 2008; Oguro et al., 2006) and our analysis of the expression of Bmi1 and Ckdn2a in progenitor B cells (Figure S1A), these findings are consistent with a role for BMI1 in preventing p53 signaling through the repression of the Ink4a/Arf locus in developing pro-B cells.
Figure 4. BMI1 Prevents the Upregulation of p53 Signaling and Maintains Large Pre-B Cell Cycling.

(A) RNA sequencing (RNA-seq) MA (log ratio and mean average) plot of Bmi1+/+ versus Bmi1−/− pro-B cells. Known targets of p53 are depicted in red.
(B) qRT-PCR expression analysis of indicated genes from sorted pro-B cells of indicated mice. Each point represents one animal; the line represents the mean. n = 4 for Bmi1+/+ animals; n = 5 for Bmi1−/− animals.
(C) Frequency of apoptotic (as indicated by annexin V positivity) pro-B cells and pre-B cells of indicated mice. Each point represents one animal; the line represents the mean. n = 5 for each genotype.
(D) Frequency of large pre-B cells present in the pre-B cell compartment of indicated mice. Each point represents one animal; the line represents the mean. n = 6 for each genotype.
(E) FACS plot of developing B cells in the bone marrow of indicated mice. Shown is the frequency of each cell population in the bone marrow of indicated animals. Representative of 3 independent experiments.
(F) Frequency of B220+ cells in the spleen and peripheral blood of indicated mice. Each point represents one animal; the line represents the mean. n = 3 for each genotype.
***p < 0.001, **p < 0.01, *p < 0.05. PB, peripheral blood.
p53 signaling generally results in either apoptosis or cell-cycle arrest (Kruse and Gu, 2009). In progenitor T cells, BMI1 prevents apoptosis through the inhibition of the p19ARF-p53 pathway (Miyazaki et al., 2008). Therefore, we first tested whether increased apoptosis was occurring in developing Bmi1−/− B cells. However, we did not observe differences in the frequency of annexin V-positive pro-B cells or pre-B cells in Bmi1−/− mice compared to wild-type counterparts (Figure 4C). Following successful VDJh recombination, signaling through the pre-BCR provides survival cues and promotes the proliferation of large pre-B cells. Following this proliferative burst, pre-B cells become quiescent, differentiate into small pre-B cells, and subsequently rearrange their IgL loci (Herzog et al., 2009). Therefore, we assessed whether the increased p53 signaling prevented the capacity of Bmi1−/− large pre-B cells to clonally expand. Large and small pre-B cells can be distinguished by gating on forward scatter within the pre-B cell population. Using this strategy, we observed that Bmi1−/− mice have a drastically lower proportion of large pre-B cells in their pre-B cell compartment (Figure 4D). Moreover, the frequency of proliferating cells, as assessed either by DNA content or by bromodeoxyuridine (BrdU) incorporation, in the pre-B cell compartment was decreased in Bmi1−/− animals (Figures S4D and S4E). We subsequently hypothesized that Bmi1−/− B1.8+ mice may still exhibit decreased pre-B cells numbers (Figure 3; Figure S3) due to their inability to clonally expand. Consistent with this hypothesis, there was a comparable reduction both in the proportion of large pre-B cells in the pre-B cell compartment and in the frequency of BrdU-positive pre-B cells between Bmi1−/− mice and Bmi1−/− B1.8+ mice (Figures S4F and S4G).
Finally, to demonstrate that BMI1 functions upstream of p53 to prevent B cell development, we generated a cohort of animals (p53F/F Bmi1+/+, p53F/F Bmi1+/+ Mx1-cre, p53F/F Bmi1−/− and p53F/F Bmi1−/− Mx1-cre, hereafter referred to as p53WT Bmi1+/+, p53CKO Bmi1+/+, p53WT Bmi1−/−, and p53CKO Bmi1−/−), in which p53 was acutely deleted following serial injections of poly(I:C). We found that acute p53 deletion partially restored the pro-B cell to pre-B cell transition and the frequency of mature B cells in the periphery in Bmi1−/− mice (Figures 4E and 4F). Altogether, these data demonstrate that aberrant p53 signaling caused by BMI1 loss prevents the clonal expansion of large pre-B cells.
BMI1 Is Required for the Pro-B Cell to Pre-B Cell Transition in a Cell-Autonomous Manner
It has previously been postulated that the microenvironment of Bmi1−/− mice is defective in supporting B cell lymphopoiesis (Oguro et al., 2010). To test whether the defects observed in Bmi1−/− mice reflected non-cell-autonomous effects of BMI1 on developing B cells, we generated animals in which Bmi1 is deleted selectively in pro-B cells and their progeny, using the Mb1-cre transgene (Hobeika et al., 2006). We confirmed efficient deletion of Bmi1 and the resulting increased expression of the BMI1-target gene p19Arf in pro-B cells sorted from Bmi1F/F Mb1-cre mice (Figure 5A). Bmi1F/F Mb1-cre mice presented with reduced total cell numbers in their bone marrow, spleen, and peripheral blood and a roughly 2-fold reduction in the frequency of B cells in the bone marrow (Figures S5A and S5B). Consistent with our observations in Bmi1−/− mice, cell-autonomous BMI1 loss impaired the pro-B cell to pre-B cell transition, as evidenced by the increased frequency of pro-B cells and the decreased frequency of pre-B cells, in the bone marrow of Bmi1F/F Mb1-cre mice (Figures 5B and 5C). In addition, we observed a reduction in the frequency of B cells in the peripheral tissues of Bmi1F/F Mb1-cre mice (Figure 5D). The fraction of pro-B cells expressing intracellular Igμ chain was reduced in Bmi1F/F Mb1-cre mice (Figure 5E), consistent with the decreased expression of rearranged Igh genes (Figure 5F). Again, we observed no difference in the expression of Pax5 or Ebf1 in Bmi1F/F Mb1-cre pro-B cells (Figure S5C), but we did observe higher levels of p53 target genes (Figure 5G). Finally, subsequent deletion of p53 alleviated the pro-B cell to pre-B cell differentiation block and largely restored the frequency of mature B cells in the periphery to Bmi1F/F Mb1-cre mice (Figures 5H and 5I; Figure S5D). Altogether, these data demonstrate that the defects in B cell differentiation observed in Bmi1−/− mice are largely recapitulated by cell-autonomous loss of BMI1.
Figure 5. BMI1 Regulates the Pro-B Cell to Pre-B Cell Transition in a Cell-Autonomous Manner.

(A) mRNA expression levels as determined by qRT-PCR of indicated genes from pro-B cells of indicated mice. Each point represents one animal; the line represents the mean. n = 3 for each genotype.
(B) Representative FACS plot of developing B cells in the bone marrow of indicated mice. Shown is the frequency of each cell population in the bone marrow of indicated animals.
(C) Frequency of developing B cells in the bone marrow of indicated mice. Each point represents one animal; the line represents the mean. n = 5 for Bmi1F/F animals; n = 6 for Bmi1F/F Mb1-cre animals.
(D) Frequency of B220+ cells in the spleen and peripheral blood of indicated mice. Each point represents one animal; the line represents the mean. n = 4 for Bmi1F/F animals; n = 6 for Bmi1F/F Mb1-cre animals.
(E) Frequency of Igμ-positive pro-B cells in indicated mice. Each point represents one animal; the line represents the mean. n = 5 for Bmi1F/F animals; n = 6 for Bmi1F/F Mb1-cre animals.
(F) qRT-PCR analysis of gene expression of rearranged Igh genes in indicated mice. Data are shown as a box-and-whisker plot, with whiskers showing minimum and maximum values; the center line represents the median. n = 6 for each genotype.
(G) qRT-PCR expression analysis of indicated genes from sorted pro-B cells from indicated mice. Data are shown as a box-and-whisker plot, with whiskers showing minimum and maximum values; the center line represents the median. n = 6 for each genotype.
(H) Ratio of the frequency of pro-B cells to pre-B cells in the bone marrow of indicated mice. Data represent mean ± SEM; n = 3 for each genotype.
(I) Frequency of B220+ cells in the spleen and peripheral blood of indicated mice. Data represent mean ± SEM; n = 3 for each genotype.
**p < 0.01, *p < 0.05. BM, bone marrow; PB, peripheral blood.
See also Figure S5.
DISCUSSION
Prior studies have shown that BMI1 is critical for developing lymphocytes; however, the molecular pathways BMI1 engages to promote B cell development have remained poorly defined, and whether the role of BMI1 in B cell development was cell intrinsic remained to be elucidated (Akala et al., 2008; Oguro et al., 2010; van der Lugt et al., 1994). Our analysis identified a cell-autonomous role for BMI1 at two stages during early B cell development. Specifically, BMI1 promotes the expression of rearranged Igh genes in pro-B cells and enables large pre-B cell proliferation following pre-BCR signaling through its inhibition of p53 signaling.
In adult stem cells, PRC1 directly binds to and compacts the Ink4a/Arf locus, thus repressing the expression of the cell-cycle inhibitors p16INK4A and p19ARF and maintaining stem cell self-renewal capacity (Jacobs et al., 1999; Lessard and Sauvageau, 2003; Molofsky et al., 2003; Park et al., 2003). In BMI1 null mice, the failure to develop mature lymphocytes is due to increased expression of p19ARF, but not increased expression of p16INK4A, as demonstrated by genetic studies in mice (Akala et al.,2008). In support of this observation, loss of BMI1 in developing thymocytes results in the upregulation of p19ARF and the premature stabilization of p53, preventing DN3 T cell proliferation downstream of pre-TCR signaling (Miyazaki et al., 2008). In agreement with these studies, we found that in developing B cells, the expression level of Bmi1 is high in proliferating pro-B cells and large pre-B cells and subsequently downregulated in quiescent small pre-B cells and immature B cells. Moreover, loss of BMI1 resulted in the increased expression of p53 target genes in pro-B cells and prevented large pre-B cell expansion following pre-BCR signaling. Our data indicating that genetic inactivation of p53 restored B cell development in Bmi1−/− mice, with previous studies showing a partial rescue of peripheral B cells in Bmi1−/− p19Arf−/− double-knockout mice, support a model whereby BMI1 repression of p19Arf is critical to prevent the stabilization of p53 and premature cell-cycle withdrawal of cycling pre-B lymphocytes.
Past studies had demonstrated that genetic inaction of the Ink4a/Arf locus only partially restores B cell development to BMI1 null mice (Oguro et al., 2010). These observations suggested that BMI1 functioned through unknown Ink4a/Arf-independent mechanisms to promote B cell development. It was initially postulated that this may be through non-cell-autonomous pathways, because transplantation studies suggested the bone marrow of Bmi1−/− mice was defective at supporting B cell development (Oguro et al., 2010). We found that in Bmi1−/− mice, developing B cells fail to progress through the pro-B cell to pre-B cell transition due to the failure of pro-B cells to express rearranged Igh genes. Accordingly, the introduction of a pre-rearranged Igh locus largely restored B cell development to BMI1 null mice. The pre-rearranged Igh allele did not restore the ability of Bmi1−/− large pre-B cells to clonally expand, highlighting the two distinct roles BMI1 plays during early B cell development. Finally, we recapitulated the B cell defects observed in Bmi1−/− mice using a pro-B cell-specific Cre transgene, indicating that cell-autonomous mechanisms of BMI1 in progenitor B cells are sufficient to explain the B cell phenotypes observed in BMI1 null mice.
The exact mechanism BMI1 engages to promote the expression of rearranged Igh genes remains unclear. The identification that PRC1 is recruited to sites of double-strand breaks (DSBs) and is required for their efficient repair in vitro suggests that a similar process may occur during VDJH recombination (Facchino et al., 2010; Ginjala et al., 2011; Ismail et al., 2010). Moreover, BMI1 associates with and recruits factors known to be critical for repair of VDJH joints, such as ATM, KU70, and 53BP1, to sites of DSBs (Facchino et al., 2010; Pan et al., 2011; Ui et al., 2015). However, our data demonstrate that BMI1 is not required for VDJHrecombination. In addition, we did not observe an increased frequency of γH2A.X foci at the Igh locus in Bmi1−/− pro-B cells (data not shown). These differences may stem from the fact that BMI1 is not absolutley required but instead increases the efficiency of DSB resolution, coupled with the higher-sensitivity assays used to detect DSBs in the absence of BMI1 in vitro (Facchino et al., 2010; Ismail et al., 2010; Pan et al., 2011). Alternatively, BMI1 may function downstream of DSB repair to enable to expression of recently damaged loci. Immediately following the generation of DSBs, BMI1 is required to prevent premature transcription of damaged genes through the deposition of H2AK119ub and the inhibition of both the FACT (facilitates chromatin transcription) histone chaperone complex and RNA polymerase II elongation (Sanchez et al., 2016; Ui et al., 2015). In the wake of this temporal transcriptional silencing, new uniquely marked histones, such as H3.3, are deposited. The deposition of H3.3 primes recently damaged chromatin and is required for transcription restart (Adam et al., 2013). Another study demonstrated that FACT-mediated histone exchange was required for transcription restart following UV damage (Dinant et al., 2013). However, no studies have demonstrated what happens to transcription restart at sites of DSBs if the initial transcriptional repression does not occur. It is therefore possible that BMI1-dependent transcriptional silencing following the generation of DSBs is required to permit the proper turnover of histones and subsequent re-expression of repaired genes, in this case the recently rearranged Igh genes.
BMI1 is overexpressed in several hematologic malignancies, and its upregulation predicts poor prognosis in pediatric B cell acute lymphoblastic leukemia (B-ALL), the most common cancer in children (Peng et al., 2017; Sahasrabuddhe, 2016). Work has shown that pre-BCR and downstream signaling promote the aggressiveness of certain subtypes of B-ALL and can be targeted to eliminate human and mouse B-ALL cells (Eswaran et al., 2015; Feldhahn et al., 2005; Geng et al., 2015; Rickert, 2013). It is therefore tempting to speculate that increased BMI1 expression promotes the inappropriate proliferation of B-ALL cells downstream of pre-BCR signaling in a process analogous to what we observed in normal developing B cells. If so, BMI1-dependent signaling might present a novel therapeutic approach to target B-ALL cell growth. Along these lines, a chemical inhibitor specific to BMI1 was identified and well tolerated in mice (Kreso et al., 2014). Moreover, the inhibitor has been shown to be effective in various cancer cells (Kreso et al., 2014; Yong et al., 2016).
Our study clarifies the function of BMI1 in early B cell development, revealing a critical role for BMI1 for the expression of rearranged Igh genes in pro-B cells and the clonal expansion of large pre-B cells, and adds to the expanding range of BMI1’s function in vivo.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gregory David (Gregory.David@nyulangone.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mx1-Cre, Bmi1F/+, Mb1-cre, and p53F/+ mice were purchased from The Jackson Laboratory and have been described (Hobeika et al., 2006; Kühn et al., 1995; Marino et al., 2000; Maynard et al., 2014). Bmi1+/− mice were a kind gift from Maarten van Lohuizen and have been described (van der Lugt et al., 1994). B1.8i mice were a kind gift from Klaus Rajewsky and have been described (Sonoda et al., 1997). Animals were housed in specific pathogen-free conditions at the Skirball Animal Facility. Aged matched controls were used for Bmi1 germline mutant mice whereas littermate controls were used for Bmi1F/F Mb1-cre mice. Female and male mice were used depending on availability. All mice were analyzed at 4-8 weeks of age. All animal experiments were done in accordance with the guidelines of the NYU School of Medicine Institutional Animal Care and Use Facility.
METHOD DETAILS
Mouse treatment
For induction of Cre-mediated deletion of loxP-flanked p53 alleles in the Bmi1 p53 Mx1-Cre animal cohort, poly(I:C) (InvivoGen) was administered by i.p. injection at 5μg kg−1 in PBS for 3 injections on alternating days at 3 weeks of age. Animals were analyzed two weeks following the last poly(I:C) injection.
Flow cytometry analysis and cell sorting
Single cell suspensions were derived from bone marrow (femur and tibia of both hind legs), spleen, thymus, and peripheral blood. Red blood cells were lysed with ACK lysis buffer. Cell counts were determined using a cell counter (Beckman Coulter) set to detect nuclei between 3.5uM and 10uM or manually with a hemocytometer. All cells were blocked with purified rat IgG (20ug ml−1) for 15 min on ice prior to antibody staining. The following antibodies were used for analysis (from Biolegend): anti-B220 (RA3-6B2), anti-CD19 (6D5), anti-CD4 (RM4-5), anti-CD8 (53-6.7), anti-CD11b (M1-70), anti-Gr1 (RB6-8C5), anti-TER119 (TER119), anti-CD25 (PC61), anti-IgM (RMM-1), anti-IgD (11-26c.2a), and anti-ckit (2B8). Cells were stained with primary antibodies for 30 min on ice. For intracellular staining of Igμ chain, cells were first stained for surface markers followed by fixation and permeabilization with BD Cytofix/Cytoperm for 30 min at room temperature. Cells were then incubated with an Igμ specific Fab fragment (Jackson ImmunoResearch) for 45 min on ice. PI (1ug ml−1), DAPI (500ng ml−1), 7aad (1ug ml−1), or Zombie fixable dye was added following staining for the exclusion of dead cells. Cells were identified based on the cell-surface phenotypes described in Table S2. Cells were analyzed on a LSRII (BD Biosciences) or sorted on an AriaI or AriaII (BD Biosciences). Data were analyzed with FlowJo software (FlowJo, LLC).
RNA-sequencing
RNA was isolated from FACS purified pro-B cells. RNA was harvested from 2 biological replicates using the RNeasy Micro Kit (QIAGEN). RNA quantification and quality was determined using an Aglient 1200 Bioanalyzer. Strand-specific libraries were prepared using the SMARTer low-input RNA kit. Libraries were sequenced on an Illumina HiSeq2500 using 50bp paired-end reads. Fastq files were aligned to mm9 using TopHat. Differential expression tests were done using the Cuffdiff module of Cufflinks against the Refseq annotation. We used an FDR < 0.05 as a cutoff for significance. Gene set enrichment analysis was performed using log2 fold change as metric for ranking genes and 1,000 permutations. Gene sets used in this study were identified from the Molecular Signatures Database (Curated v5.0 and Hallmarks v5.0).
Cell cycle analysis
For cell cycle analysis of pre-B cells, cells were stained for surface markers, fixed and permeabilized with BD Cytofix/Cytoperm for 30 min at room temperature, and incubated with DAPI (10μg ml−1) and RNaseA(50μg ml−1) for 30 min at room temperature. For BrdU incorporation analysis of pre-B cells, mice were i.p. injected with 1mg BrdU and sacrificed 2 hr later. Cells were stained for surface markers and analyzed for BrdU incorporation as described using the BD PharMingen BrdU Flow Kit. Apoptotic cells were detected using the Annexin V Apoptosis Detection Kit (Biolegend) according to the manufacturer’s instructions.
RT-qPCR analysis
RNA from sorted cell populations was isolated using the RNeasy Mini Kit (QIAGEN). Prior to reverse transcription, RNA was incubated with DNase I (Promega). RNA was subsequently converted into cDNA using either M-MLV reverse transcriptase (Promega) or Maxima reverse transcriptase (Thermo Scientific) according to the manufacturer’s instructions, using an equal mix of oligo dT and random hexamers as primers. Real-time quantitative PCR (RT-qPCR) was performed on a Bio-rad iCycler using Maxima SYBR Green master mix (Thermo Scientific) according to the manufacturer’s instructions. No-RT controls were included to ensure no genomic DNA contamination was present. All values were normalized to Hprt levels using the ΔCt method. RT-PCR primer pairs are shown in Table S3.
Semiquantitative PCR analysis of VDJH joints
Genomic DNA from sorted cell populations was isolated using the DNeasy Blood and Tissue kit (QIAGEN). Igh rearrangements were amplified as previously described (Fuxa et al., 2004). Briefly, serial dilutions of genomic DNA were amplified using a forward primer specific to either the DH, the VH7183 family, or the VHJ558 family and a reverse primer specific to JH3. Primers specific to Cμ were used as a loading control. PCRs were run in the linear range and products were run on a 1.25% agarose gel. Primer pairs are shown in Table S4.
Sub-clonal sequencing of VDJH joints
Genomic DNA from sorted pro-B cells was isolated using the DNeasy Blood and Tissue kit (QIAGEN). Igh rearrangements were amplified using a promiscuous forward primer (MsVHe) (5′-GGGAATTCGAGGTGCAGCTGCAGGAGTCTGG-3′) that binds to genes within all VH families and a reverse primer specific to JH3 (5′-GTCTAGATTCTCACAAGAGTCCGATAG ACCCTGG-3′). A high fidelity polymerase (Roche) was used to minimize point mutations during PCR amplification. The amplified product was run on a 1% agarose gel and the expected band size (~450bp) was excised and purified. The purified DNA product was inserted into the pGEM-T easy vector (Promega) according to the manufacturers instructions. Subcloned DNA was column purified (QIAGEN) and sequenced using T7 primer (Macrogen). VDJH sequences were analyzed using the National Center for Biotechnology Information’s IgBlast program.
DNA FISH
DNA FISH was performed with the probes BACs CT7-199M11 (3’ Igh) and CT7-526A21 (5’ Igh) labeled by nick translation. For each 22×22 mm coverslip, 1 μg of labeled BAC DNA, 10 μg of sheared salmon sperm DNA and 1 μg of Cot-1 (Life Technologies) were precipitated and resuspended in 16 μL of hybridization buffer (50% formamide/20% dextran sulfate/5X denhardt’s solution). DNA FISH was carried out as previously described (Chaumeil et al., 2008). Briefly, cells were adhered to poly-L-lysine coated coverslips for 10 min, fixed for 10 min at 22°C with 2% (wt/vol) paraformaldehyde/PBS and permeabilized for 10 min with 0.5% (vol/vol) Triton/ PBS. Coverslips were incubated with RNaseA (0.1 mg/ml in PBS, 30 min at 37°C), then re-permeabilized for 10 min at 0°C in 0.7% (vol/vol) Triton X-100 / 0.1 M HCl. Cells were then placed in 50% formamide/2XSSC for 1 hr at 22°C. Coverslips and resuspended probes were then placed at 75°C for 2 min, before probes were applied to coverslips, sealed onto slides with rubber cement, followed by incubation overnight at 37°C. Cells were then washed two times with 50%formamide/2xSSC at 42°C, two times with 2xSSC at 42°C and 1xSSC at 42°C, for 5 min each and were mounted in Prolong Gold (Life Technologies) containing 1.5 μg/ml DAPI.
Confocal microscopy and analysis
DNA FISH was imaged by confocal microscopy on a Leica SP5 AOBS system (Acousto-Optical Beam Splitter). Optical sections separated by 0.3 μm were collected and only cells with signals from both alleles were analyzed using ImageJ software (NIH). For distance measurements in DNA FISH, ImageJ was used to measure the center of mass of each focus and calculate intralocus distances.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using an unpaired Student’s two-tailed t test. Differences in the distribution of intralocus distance between the 5’ and 3’ Igh FISH probes was compared using the Kolmogorov-Smirnov test. All experiments were performed in the number of biological replicates mentioned as n in the figure legends or shown as individual points in figures. N represents individual sequences for pro-B cell subclonal VDJH joint sequencing, individual cells for DNA FISH assessing Igh contraction, and individual animals for all other experiments. A p value < 0.05 was considered statistically significant. All data were analyzed using GraphPad Prism software.
DATA AND SOFTWARE AVAILABILITY
RNA-sequencing data were deposited at GEO and is available under accession GEO: GSE119422.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-mouse B220 – APCCy7 (clone: RA3-6B2) | Biolegend | Cat#103223; RRID: AB_313006 |
| Rat anti-mouse CD19 – Pacific Blue (clone: 6D5) | Biolegend | Cat#115526; RRID: AB_493341 |
| Rat anti-mouse ckit – APC (clone: 2B8) | Biolegend | Cat#105805; RRID: AB_313214 |
| Rat anti-mouse CD25 – PE (clone: PC61) | Biolegend | Cat#102007; RRID: AB_312856 |
| Rat anti-mouse IgM – PECy7 (clone: RMM-1) | Biolegend | Cat#406513; RRID: AB_10640069 |
| Rat anti-mouse IgD – PerCP5.5 (clone: 11-26c.2a) | Biolegend | Cat#405709; RRID: AB_1575115 |
| Rat anti-mouse CD4 – FITC (clone: RM4-5) | Biolegend | |
| Rat anti-mouse CD8 – APCCy7 (clone: 53-6.7) | Biolegend | Cat#100713; RRID: AB_312752 |
| Rat anti-mouse B220 – Pacific Blue (clone: RA3-6B2) | Biolegend | Cat#103230; RRID: AB_492877 |
| Rat anti-mouse Gr1 – PE (clone: RB6-8C5) | Biolegend | Cat#108407; RRID: AB_313372 |
| Rat anti-mouse TER119 – PerCP (clone: TER119) | Biolegend | Cat#116225; RRID: AB_893637 |
| Rat anti-mouse CD11b – PECy7 (clone: M1-70) | Biolegend | Cat#101215; RRID: AB_312798 |
| AffiniPure Fab Fragment Goat anti-mouse IgM, μ chain specific – FITC | Jackson ImmunoResearch | Cat#115-097-020; RRID: AB_2338618 |
| IgG from rat serum | Sigma Aldrich | Cat#I4131; RRID: AB_1163627 |
| Bacterial and Virus Strains | ||
| NEB 5-alpha competent E. coli | New England Biolabs | Cat#C2987I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DAPI | Sigma Aldrich | Cat#32670; CAS#28718-90-3 |
| Critical Commercial Assays | ||
| FITC BrdU Flow Kit | BD Biosciences | Cat#559619; RRID: AB_2617060 |
| FITC Annexin V Apoptosis Detection Kit | Biolegend | Cat#640932 |
| Deposited Data | ||
| Pro-B cell RNA-sequencing data | This paper | GEO: GSE119422 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Bmi1+/−: FVB.129P2-Bmi1tm1Brn/MvlJ | Maarten van Lohuizen (van der Lugt et al., 1994) | JAX: 024584 |
| Mouse: B1.8+: B6.129P2(C)-Ightm2Cgn/J | Klaus Rajewsky (Sonoda et al., 1997) | JAX: 012642 |
| Mouse: p53F/+: B6.129P2-Trp53tm1Brn/J | The Jackson Laboratory | JAX: 008462 |
| Mouse: Mx1-cre: B6.Cg-Tg(Mx1-cre)1Cgn/J | The Jackson Laboratory | JAX: 003556 |
| Mouse: Bmi1F/+: Bmi1tm1.1Lees/J | The Jackson Laboratory | JAX: 028572 |
| Mouse: mb1-cre: B6.C(Cg)-Cd79atm1(cre)Reth/EhobJ | The Jackson Laboratory | JAX: 020505 |
| Oligonucleotides | ||
| mRNA expression RT-PCR primer pairs, see Table S3 | This paper | N/A |
| Primer pairs for analysis of VDJH joints, see Table S4 | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com |
| Prism | Graphpad | https://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| NCBI IgBlast | NIH | https://www.ncbi.nlm.nih.gov/igblast/ |
| Other | ||
| Poly(I:C) | InvivoGen | Cat#tlrl-picw |
| BD Cytofix/Cytoperm | BD Biosciences | Cat#554714 |
| Zoombie UV Fixable Viability Dye | Biolegend | Cat#423107 |
| Propidium Iodide Staining Solution | BD Biosciences | Cat#556463 |
| M-MLV Reverse Transcriptase | Promega | Cat#M1701 |
| Maxima Reverse Transcriptase | Thermo Scientific | Cat#EP0741 |
| Maxima SYBR Green Master Mix | Thermo Scientific | Cat#K0241 |
| Roche Expand High Fidelity PCR System | Roche | Cat#11732641001 |
| pGEM-T Easy Vector System | Promega | Cat#A1360 |
| RNeasy Micro Kit | QIAGEN | Cat#74004 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| DNAeasy Blood and Tissue Kit | QIAGEN | Cat#69504 |
| SMARTer low-input RNA kit | Clontech | Cat#634938 |
Highlights.
BMI1 is required at the pro-B cell to pre-B cell transition in a cell-autonomous manner
BMI1 promotes the expression of newly rearranged Igh genes in pro-B cells
BMI1 prevents premature p53 activation and enables large pre-B cell proliferation
ACKNOWLEDGMENTS
We are grateful to all members of the David laboratory for helpful discussions during the preparation of the manuscript. We wish to acknowledge the Skirball Institute of Biomolecular Medicine for hosting our laboratory following Hurricane Sandy. We thank the NYU Cytometry and Cell Sorting Core (supported in part by NIH/NCI support grant P30CA016087) for help with analysis and cell sorting. We thank the NYU Genome Technology Center (supported in part by NIH/NCI support grant P30CA016087) for help with RNA sequencing and bioinformatic analysis. This work was funded by the NIH (R01CA148639, R21CA155736, and R21CA206013 to G.D.), the Samuel Waxman Cancer Research Foundation (to G.D.), and a Feinberg NYU individual grant (to G.D.). D.J.C. was supported by a NIH MSTP training grant (T32GM007308), a predoctoral NIH/NCI training grant (T32CA009161), and a predoctoral NIH/NCI NRSA (F30CA203047).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and four tables and can be found with this article online at https://doi.Org/10.1016/j.celrep.2018.12.030.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adam S, Polo SE, and Almouzni G (2013). Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell 155, 94–106. [DOI] [PubMed] [Google Scholar]
- Akala OO, Park I-K, Qian D, Pihalja M, Becker MW, and Clarke MF (2008). Long-term haematopoietic reconstitution by Trp53−/− p16Ink4a−/− p19Arf−/− multipotent progenitors. Nature 453, 228–232. [DOI] [PubMed] [Google Scholar]
- Blackledge NP, Rose NR, and Klose RJ (2015). Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nat. Rev. Mol. Cell Biol 16, 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E, Hulsman D, Leung C, Arsenijevic Y, Marino S, and van Lohuizen M (2005). Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 79, 1438–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil J, Augui S, Chow JC, and Heard E (2008). Combined immunofluorescence, RNA fluorescent in situ hybridization, and DNA fluorescent in situ hybridization to study chromatin changes, transcriptional activity, nuclear organization, and X-chromosome inactivation. Methods Mol. Biol 463, 297–308. [DOI] [PubMed] [Google Scholar]
- Di Croce L, and Helin K (2013). Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol 20, 1147–1155. [DOI] [PubMed] [Google Scholar]
- Dinant C, Ampatziadis-Michailidis G, Lans H, Tresini M, Lagarou A, Grosbart M, Theil AF, van Cappellen WA, Kimura H, Bartek J, et al. (2013). Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol. Cell 51, 469–479. [DOI] [PubMed] [Google Scholar]
- Ebert A, Medvedovic J, Tagoh H, Schwickert TA, and Busslinger M (2013). Control of antigen receptor diversity through spatial regulation of V(D) J recombination. Cold Spring Harb. Symp. Quant. Biol 78, 11–21. [DOI] [PubMed] [Google Scholar]
- Eswaran J, Sinclair P, Heidenreich O, Irving J, Russell LJ, Hall A, Calado DP, Harrison CJ, and Vormoor J (2015). The pre-B-cell receptor checkpoint in acute lymphoblastic leukaemia. Leukemia 29, 1623–1631. [DOI] [PubMed] [Google Scholar]
- Facchino S, Abdouh M, Chatoo W, and Bernier G (2010). BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci 30, 10096–10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldhahn N, Klein F, Mooster JL, Hadweh P, Sprangers M, Wartenberg M, Bekhite MM, Hofmann WK, Herzog S, Jumaa H, et al. (2005). Mimicry of a constitutively active pre-B cell receptor in acute lymphoblastic leukemia cells. J. Exp. Med 201, 1837–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxa M, Skok J, Souabni A, Salvagiotto G, Roldan E, and Busslinger M (2004). Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. Genes Dev. 18, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H, Hurtz C, Lenz KB, Chen Z, Baumjohann D, Thompson S, Goloviznina NA, Chen WY, Huan J, LaTocha D, et al. (2015). Self-enforcing feedback activation between BCL6 and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell 27, 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginjala V, Nacerddine K, Kulkarni A, Oza J, Hill SJ, Yao M, Citterio E, van Lohuizen M, and Ganesan S (2011). BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol. Cell. Biol 31, 1972–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TS, and Painter MW; Immunological Genome Project Consortium (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Herzog S, Reth M, and Jumaa H (2009). Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat. Rev. Immunol 9, 195–205. [DOI] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, and Reth M (2006). Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. USA 103, 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail IH, Andrin C, McDonald D, and Hendzel MJ (2010). BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol 191, 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JJL, Kieboom K, Marino S, DePinho RA, and van Lohuizen M (1999). The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164–168. [DOI] [PubMed] [Google Scholar]
- Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W, Sydorenko N, et al. (2014). Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med 20, 29–36. [DOI] [PubMed] [Google Scholar]
- Kruse JP, and Gu W (2009). Modes of p53 regulation. Cell 137, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn R, Schwenk F, Aguet M, and Rajewsky K (1995). Inducible gene targeting in mice. Science 209, 1427–1429. [DOI] [PubMed] [Google Scholar]
- Lessard J, and Sauvageau G (2003). Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423, 255–260. [DOI] [PubMed] [Google Scholar]
- Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, Kee BL, Dinner AR, and Clark MR (2011). Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat. Immunol 12, 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Maynard MA, Ferretti R, Hilgendorf KI, Perret C, Whyte P, and Lees JA (2014). Bmi1 is required for tumorigenesis in a mouse model of intestinal cancer. Oncogene 33, 3742–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchers F (2015). Checkpoints that control B cell development. J. Clin. Invest 125, 2203–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Itoi M, Katoh Y, Guo Y, Kanno R, Katoh-Fukui Y, Honda H, Amagai T, van Lohuizen M, et al. (2008). Thymocyte proliferation induced by pre-T cell receptor signaling is maintained through polycomb gene product Bmi-1-mediated Cdkn2a repression. Immunity 28, 231–245. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, and Morrison SJ (2003). Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425, 962–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, and Nakauchi H (2006). Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J. Exp. Med 203, 2247–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H, Yuan J, Ichikawa H, Ikawa T, Yamazaki S, Kawamoto H, Nakauchi H, and Iwama A (2010). Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell 6, 279–286. [DOI] [PubMed] [Google Scholar]
- Painter MW, Davis S, Hardy RR, Mathis D, and Benoist C; Immunological Genome Project Consortium (2011). Transcriptomes of the B and T lineages compared by multiplatform microarray profiling. J. Immunol 186, 3047–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MR, Peng G, Hung WC, and Lin SY (2011). Monoubiquitination of H2AX protein regulates DNA damage response signaling. J. Biol. Chem 286, 28599–28607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, and Clarke MF (2003). Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423, 302–305. [DOI] [PubMed] [Google Scholar]
- Peng HX, Liu XD, Luo ZY, Zhang XH, Luo XQ, Chen X, Jiang H, and Xu L (2017). Upregulation of the proto-oncogene Bmi-1 predicts a poor prognosis in pediatric acute lymphoblastic leukemia. BMC Cancer 17, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert RC (2013). New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat. Rev. Immunol 13, 578–591. [DOI] [PubMed] [Google Scholar]
- Roldán E, Fuxa M, Chong W, Martinez D, Novatchkova M, Busslinger M, and Skok JA (2005). Locus ‘decontraction’ and centromeric recruitment contribute to allelic exclusion of the immunoglobulin heavy-chain gene. Nat. Immunol 6, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasrabuddhe AA (2016). BMI1: a biomarker of hematologic malignancies. Biomark. Cancer 8, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A, De Vivo A, Uprety N, Kim J, Stevens SM Jr., and Kee Y (2016). BMI1-UBR5 axis regulates transcriptional repression at damaged chromatin. Proc. Natl. Acad. Sci. USA 113, 11243–11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, and Sauvageau G (2010). Polycomb group proteins: multifaceted regulators of somatic stem cells and cancer. Cell Stem Cell 7, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, and Kingston RE (2013). Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 49, 808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Pewzner-Jung Y, Schwers S, Taki S, Jung S, Eilat D, and Rajewsky K (1997). B cell development under the condition of allelic inclusion. Immunity 6, 225–233. [DOI] [PubMed] [Google Scholar]
- Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, and Tarakhovsky A (2003). Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol 4, 124–131. [DOI] [PubMed] [Google Scholar]
- Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H, et al. (2012). RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ui A, Nagaura Y, and Yasui A (2015).Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol. Cell 58, 468–482. [DOI] [PubMed] [Google Scholar]
- van der Lugt NM, Domen J, Linders K, van Roon M, Robanus-Maandag E, te Riele H, van der Valk M, Deschamps J, Sofroniew M, van Lohuizen M, et al. (1994). Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 protooncogene. Genes Dev. 8, 757–769. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, and Alt FW (1985). Developmentally controlled and tissue-specific expression of unrearranged VH gene segments. Cell 40, 271–281. [DOI] [PubMed] [Google Scholar]
- Yong KJ, Basseres DS, Welner RS, Zhang WC, Yang H, Yan B, Alberich-Jorda M, Zhang J, de Figueiredo-Pontes LL, Battelli C, et al. (2016). Targeted BMI1 inhibition impairs tumor growth in lung adenocarcinomas with low CEBPα expression. Sci. Transl. Med 8, 350ra104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data were deposited at GEO and is available under accession GEO: GSE119422.