

Abstract
Chronic hepatitis B virus (HBV) infection represents a major health threat. Current FDA-approved drugs do not cure HBV. Targeting HBV core protein (Cp) provides an attractive approach toward HBV inhibition and possibly infection cure. We have previously identified and characterized a 5-amino-3-methylthiophene-2,4-dicarboxamide (ATDC) compound as a structurally novel hit for capsid assembly effectors (CAEs). We report herein hit validation through studies on absorption, distribution, metabolism and excretion (ADME) properties and pharmacokinetics (PK), and hit optimization via analogue synthesis aiming to probe the structure-activity relationship (SAR) and structure-property relationship (SPR). In the end, these medicinal chemistry efforts led to the identification of multiple analogues strongly binding to Cp, potently inhibiting HBV replication in nanomolar range without cytotoxicity, and exhibiting good oral bioavailability (F). Two of our analogues, 19o (EC50 = 0.11 μM, CC50 >100 μM, F = 25%) and 19k (EC50 = 0.31 μM, CC50 >100 μM, F = 46%), displayed overall lead profiles superior to reported CAEs 7–10 used in our studies.
Graphical abstract
Introduction
HBV chronically infects an estimated 250 million people worldwide and remains a major health threat [1]. Despite a successful vaccine, there are still around 1 million people newly infected each year. Chronic HBV infection is often associated with severe liver diseases which typically progress through fibrosis, cirrhosis and eventually develop into hepatocellular carcinoma (HCC) [2]. It is estimated that HBV-associated liver diseases result in approximately 660,000 deaths annually. Current FDA-approved HBV drugs include the immunomodulatory agent pegylated interferon alpha (IFN-α) [3] and direct acting nucleos(t)ide analogues (NAs) which target reverse transcriptase (RT) of the HBV P protein complex [4]. These NAs are based on all four endogenous nucleosides with distinct approaches with respect to mimicking the ribose moiety (Figure 1), including β-L-nucleosides [5–6] lamivudine (1, 3TC, analogue of dC) and telbivudine (2, LdT, analogue of T), the carbocyclic nucleoside entecavir (3, ETV, analogue of dG) [7], the acyclic nucleoside phosphonate (ANPs) [8] adefovir (4, ADV, analogue of dA) and two forms of tenofovir (TFV, analogue of dA): tenofovir disoproxil fumarate (5, TDF) and tenofovir alafenamide (6, TAF). While NAs are generally well tolerated [9] and largely effective [10–11] in suppressing viral load, they do not cure HBV infection presumably because they do not eliminate HBV covalently closed circular DNA (cccDNA) which is the main viral reservoir for persistent infection [12]. HBV cure calls for novel antiviral approaches to completely eliminate or functionally inactivate cccDNA [13–15]. Otherwise, clinical management of chronic HBV infection requires lifetime treatment with current drugs.
Figure 1.

FDA-approved NAs for treating chronic HBV infection. 1: lamivudine (3TC); 2: telbivudine (LdT); 3: entecavir (ETV); 4: adefovir (ADV); 5: tenofovir disoproxil fumarate (TDF); and 6: tenofovir alafenamide (TAF).
The HBV replication cycle [16] entails a critical step where Cp dimers are assembled into functional viral capsids which encapsulate both viral pregenomic RNA (pgRNA) and polymerase complex for active reverse transcription to generate genomic, partially double-stranded relaxed circular DNA (RC-DNA). Interestingly, RC-DNA-containing mature nucleocapsids are either enveloped then secreted as new infectious virions, or enter the nucleus to replenish the cccDNA pool, constituting the intracellular amplification pathway of cccDNA. Disrupting the proper assembly of capsid can therefore inhibit HBV by blocking the production of infectious viruses and depleting cccDNA replenishment, and potentially contribute to HBV cure [17]. Therefore, targeting HBV Cp represents an attractive approach for treating chronic HBV infection. A few chemotypes have been reported as potent CAEs (Figure 2) [14, 18–19]. Amongst these, the heteroaryldihydropyrimidine (HAP) [20–24] chemotype as represented by compound 7 and the sulfamoylbenzamide (SBA) chemotype [25–26] as represented by compound 8 are particularly well studied with analogues in clinical development [27–29]. Mechanistically, all known CAEs accelerate capsid assembly, albeit with different assembled products. The HAP series constitutes a class of its own by inducing misassembly to form aberrant nonfunctional capsid particles, whereas SBA and other chemotypes, such as phenylpropenamide (9) [30] and NZ-4 (10) [31], promote the formation of empty, morphologically normal capsids [32].
Figure 2.

Major CAE chemotypes reported. 7 (BAY-7690) represents the HAP chemotype; 8 (DVR-56) represents the SBA chemotype.
Recently we conducted a high-throughput screening (HTS) of commercial libraries in a thermal shift assay (TSA) that measures binding to HBV Cp and identified compound 11 as a strong binder (Figure 3) [33]. In the subsequent antiviral assay 11 inhibited HBV total DNA production (EC50 = 3.4 μM) with no cytotoxicity observed (CC50 >100 μM). Mechanistically, compound 11 promoted the formation of large Cp aggregates and prevented Cp from entering nucleus [33]. These results confirmed 11 as a valid CAE hit. We report herein the optimization of CAE hit 11 through analogue synthesis, SAR as well as ADME and PK studies.
Figure 3.

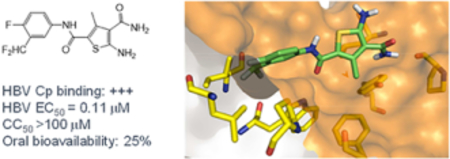
Our CAE hit. Compound 11 strongly bound to Cp, inhibited viral DNA production without cytotoxicity, and disrupted capsid assembly. 11 also demonstrated favorable ADME properties in vitro and decent oral bioavailability in vivo. These properties, combined with its low MW and high LE, render 11 a high quality lead.
Results and Discussion
In the present work, compound 11 was first evaluated in various in vitro assays for its physicochemical and ADME properties, including aqueous solubility and stability, plasma stability, plasma protein binding and microsomal stability. Overall, compound 11 exhibited favorable ADME properties (Figure 3) predicting good oral bioavailability. This was confirmed through in vivo PK studies in mice from which the oral bioavailability of 11 was determined to be 31%. In addition, compound 11 has relatively low molecular weight (MW) and a good ligand efficiency (LE), two highly desired attributes that bode well with lead optimization. Based on these, 11 was considered a high quality CAE lead.
Analogue design and synthesis.
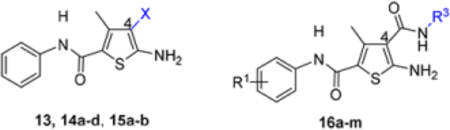
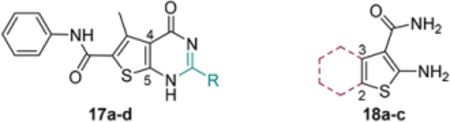
To optimize hit 11, analogues were designed and synthesized to explore the SAR around the central thiophene ring (Figure 4). Specifically, analogue synthesis includes subtype 12 which aims to study the SAR of the C5 amino group; subtypes 13–16 which concern the SAR around the C4 carboxamide moiety; and subtype 19 which probes the phenyl ring of the C2 carboxamide. In addition, 4,5-cyclized analogues (subtype 17) and 2,3cyclized analogues (subtype 18) were also synthesized (Figure 4) to explore the impact of molecular rigidity.
Figure 4.

SAR design for hit 11. Sites for structural variation include the C5 amino group (subtype 12), the C4 carboxamide moiety (subtypes 13—16), and the C2 carboxamide phenyl ring (subtype 19). In addition, 4,5-cyclized analogues (subtype 17) and 2,3-cyclized analogues (subtype 18) are also designed.
Synthetically, all these analogues can be easily accessed via different versions of the Gewald synthesis [34]. This synthesis features a three-component one-pot reaction to construct the thiophene ring with the desired substitution pattern (Scheme 1) [35–36].
Scheme 1.

a Analogue synthesis for hit 11
Biology and SAR.
All analogues were first evaluated in our recently developed TSA [33] which is not quantitative but does provide effective monitoring of HBV capsid assemble states. From this assay, hits with significant binding, as observed from the TSA curve (see Figure 5A for an example), were then tested in an antiviral assay which measures total intracellular HBV DNA production. Compounds with significant antiviral activity were further evaluated in a cytotoxicity assay.
Figure 5.

Characterization of compound 19h. (A) TSA curve (dotted line). Solid line depicts the curve of DMSO control; (B) dose-response antiviral testing; (C) TEM images of HBV capsid. HBV capsid aggregation observed in the presence of compound 19h as compared to the DMSO control.
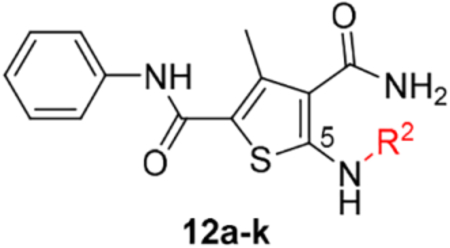
SAR around C-5 amino group.
This domain of SAR concerns mainly the effect of derivatizing the C-5 amino group. Toward this end, the amino group was converted into urea, aliphatic amides, aromatic amides and tetrazole. In the binding assay, the urea analogue (12a) and the tetrazole compound (12k) were completely inactive and aliphatic amides (12b-c) showed weak Cp binding (Table 1). By contrast, strong binding was preserved for all aromatic amides (12d-j), with the exception of compound 12h (Table 1). In addition, all aromatic amides (12d-j) remained potent in the antiviral assay with EC50 values (1.4–4.7 μM) comparable to that of the lead compound 11 (EC50 = 3.4 μM). These results suggest that acylating the C-5 amino group is not tolerated with an aliphatic acyl group, and tolerated or even moderately beneficial with an aromatic acyl group. Particularly interesting is compound 12e which exhibited improved antiviral potency (EC50 = 1.4 μM). However, this moderate potency improvement was conferred by the addition of a relatively large group, and hence may reflect a reduced ligand efficiency.
Table 1.
SAR around C-5 amino group.
| Compound | Structure | TSAa | EC50b (μM) | CC50c (μM) |
|---|---|---|---|---|
| 11 | 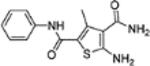 |
+++ | 3.4 ± 0.6 | >100 |
| 12a | 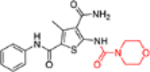 |
− | NTd | NT |
| 12b | 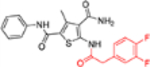 |
+ | ~10 | >100 |
| 12c | 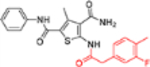 |
+ | >10 | >100 |
| 12d | 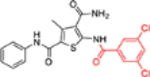 |
+++ | 2.7 ± 1.9 | 49.5 ± 0.1 |
| 12e | 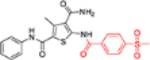 |
+++ | 1.4 ± 0.02 | >100 |
| 12f | 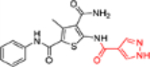 |
+++ | 4.7 ± 0.4 | >100 |
| 12g | 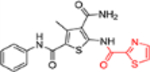 |
+++ | 4.1 ± 2.2 | >100 |
| 12h | 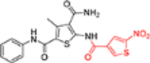 |
+ | 2.9 ± 0.7 | >100 |
| 12i | 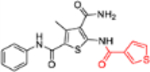 |
+++ | 2.1 ± 0.3 | >100 |
| 12j | 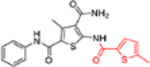 |
+++ | 3.5 ± 2.1 | >100 |
| 12k | 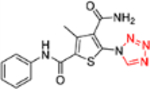 |
− | NT | NT |
Thermal shift assay that measures changes in the thermal stability of HBV Cp upon binding of a small molecule: “−” denotes no binding, “+” weak binding, “++” moderate binding and “+++” strong binding.
Concentration of a compound inhibiting HBV total DNA production by 50%, expressed as the mean ± standard deviation from at least two independent experiments.
Concentration of a compound causing 50% cytotoxicity, expressed as the mean ± standard deviation from at least two independent experiments.
NT = not tested
SAR around C-4 carboxamide.
In this SAR domain, the C-4 carboxamide group was first changed to three different bioisosteres: the thiocarboxamide, cyano and ester (Table 2). Strong binding to Cp was retained with the thiocarboxamide (13) and the esters (15a-b), though no antiviral activity was observed with either 13 or 15a at concentrations up to 10 μM, and 15b exhibited reduced potency (EC50 = 6.1 μM). In the meantime, replacing carboxamide with cyano also led to the loss of binding affinity to Cp (14a and 14d). The binding was restored by introducing substituents at the para and meta positions of the phenyl ring on the left (14b-c), with analogue 14c demonstrating slightly improved antiviral potency compared to lead 11. These results suggest that thiocarboxamide, cyano and ester groups at C-4 are either not tolerated or inferior to the carboxamide group. Further SAR in this domain then focused on the substitution of amide through the synthesis of analogues 16a-m (Table 2). When the amide is substituted with a phenyl ring, the resulting analogues all showed strong Cp binding (16a-f) with the single exception of compound 16g. Most of these analogues (16a-d) also inhibited HBV DNA production with potency similar to lead 11. Of particular interest is analogue 16f, which exhibited antiviral potency in the nanomolar range (EC50 = 0.68 μM), a major potency improvement over lead 11. However, phenyl substitution on the amide also resulted in moderate cytotoxicity (CC50 = 17–40 μM), rendering these analogues less attractive than lead 11. Interestingly, when a methylene group is inserted between the phenyl substituent and the amide, the resulting analogues (16h-i) were rendered completely inactive in the antiviral assay, suggesting that alkyl substitution may not be tolerated. This is confirmed through the synthesis of alkyl and cycloalkyl substituted analogues (16j-m) which showed no binding affinity to Cp and no antiviral activity at concentrations up to 10 μM. The only exception is 16m, which retained strong Cp binding without conferring antiviral activity. Based on all these results, unsubstituted carboxamide as seen in lead 11 is preferred over all other variants.
Table 2.
SAR around C-4 carboxamide.
| Compound | Structure | TSAa | EC50b (μM) | CC50c (μM) |
|---|---|---|---|---|
| 11 | 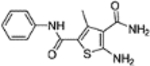 |
+++ | 3.4 ± 0.6 | >100 |
| 13 | 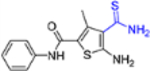 |
+++ | >10 | >100 |
| 14a | 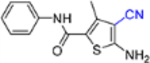 |
− | NTd | NT |
| 14b | 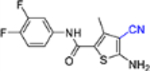 |
+++ | ~10 | >100 |
| 14c | 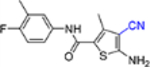 |
+++ | 2.0 ± 0.2 | 96 ± 10 |
| 14d | 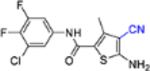 |
− | NT | NT |
| 15a | 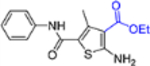 |
+++ | >10 | 76 ± 1.8 |
| 15b | 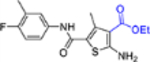 |
+++ | 6.1 ± 2.8 | >100 |
| 16a | 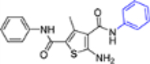 |
+++ | 3.2 ± 0.3 | >100 |
| 16b | 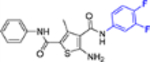 |
+++ | 2.4 ± 0.2 | 40 ± 5.1 |
| 16c | 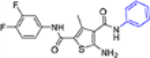 |
+++ | 3.4 ± 0.3 | 26 ± 2.7 |
| 16d | 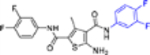 |
++ | 3.6 ± 1.6 | 17 ± 1.3 |
| 16e | 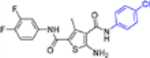 |
+++ | >10 | NT |
| 16f | 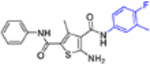 |
+++ | 0.68 ± 0.01 | 40 ± 2.6 |
| 16g | 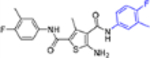 |
− | NT | NT |
| 16h | +++ | >10 | NT | |
| 16i | +++ | >10 | NT | |
| 16j | 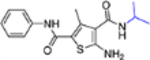 |
− | >10 | NT |
| 16k | 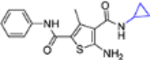 |
− | >10 | NT |
| 16l | 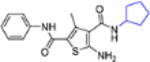 |
− | >10 | NT |
| 16m | 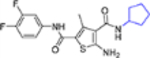 |
+++ | >10 | NT |
Thermal shift assay that measures changes in the thermal stability of HBV Cp upon binding of a small molecule: “−” denotes no binding, “+” weak binding, “++” moderate binding and “+++” strong binding.
Concentration of a compound inhibiting HBV total DNA production by 50%, expressed as the mean ± standard deviation from at least two independent experiments.
Concentration of a compound causing 50% cytotoxicity, expressed as the mean ± standard deviation from at least two independent experiments.
NT = not tested
SAR with C-4, C-5 cyclization and C-2, C-3 cyclization.
This part of the SAR mainly probes the impact of added rigidity to the chemotype. Since binding events typically result in entropy loss that needs to be compensated, rigidifying a molecule could potentially benefit target binding by reducing the required entropy compensation. Toward this end, a few C-4, C-5 cyclized analogues (17a-d) and C-2, C-3 cyclized analogues (18a-c) were synthesized (Table 3). Unfortunately, none of these analogues showed any binding affinity toward Cp (Table 3), suggesting that the attempted cyclization is not tolerated.
Table 3.
SAR with C-4, C-5-cyclized analogues and C-2, C-3-cyclized analogues.
| Compound | Structure | TSAa | EC50b (μM) | CC50c (μM) |
|---|---|---|---|---|
| 11 | 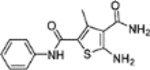 |
+++ | 3.4 ± 0.6 | >100 |
| 17a | 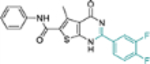 |
− | NTd | NT |
| 17b | 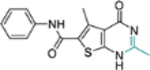 |
− | NT | NT |
| 17c | 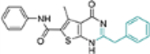 |
− | NT | NT |
| 17d | 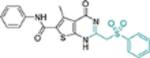 |
− | NT | NT |
| 18a | 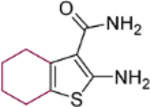 |
− | NT | NT |
| 18b | 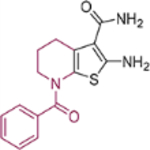 |
− | NT | NT |
| 18c | 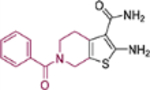 |
− | NT | NT |
Thermal shift assay that measures changes in the thermal stability of HBV Cp upon binding of a small molecule: “−” denotes no binding, “+” weak binding, “++” moderate binding and “+++” strong binding.
Concentration of a compound inhibiting HBV total DNA production by 50%, expressed as the mean ± standard deviation from at least two independent experiments.
Concentration of a compound causing 50% cytotoxicity, expressed as the mean ± standard deviation from at least two independent experiments.
NT = not tested
SAR around the phenyl ring of C-2 carboxamide.
This domain of SAR began with the synthesis of analogue 19a. With the addition of a flexible methylene group between the amide and the phenyl ring, strong Cp binding was preserved while antiviral potency dropped considerably (Table 4), suggesting that direct phenyl substitution is preferred at the C-2 carboxamide. The remaining SAR then focused on exploring the effects of substituents at the para and meta positions of the phenyl ring. Toward this end, compounds with a mono substituent at the para position (19b-f) were first synthesized. Among them, only the analogue with a para F group (19e) retained both strong Cp binding and antiviral potency when compared to lead 11. Cp binding was preserved also with para OMe substituted compound (19b), albeit with much reduced antiviral activity. None of the other para substituted analogues showed binding affinity toward Cp (Table 4). These SAR results suggest that mono substitution at the para position is generally undesired, with only the F group tolerated. The next series of compounds consists of analogues with a para F group and a meta substituent (19g-h, 19k-m and 19o). The addition of a meta substituent drastically improved the activity profile, as all analogues showed strong Cp binding, markedly improved antiviral potency (EC50 = 0.11–1.2 μM), and no cytotoxicity at concentrations up to 100 μM (Table 4). Interestingly, when the para F group is replaced with a Cl or Br group, the resulting analogues were much less potent (19i-j vs 19h, 19n vs 19m). In addition, when a second meta position is also substituted, the resulting tri-substituted analogue (19p) exhibited significantly reduced antiviral potency compared to the di-substituted analogues. These results indicate that a para F group in combination with one meta substituent on the phenyl ring confers the optimal antiviral potency.
Table 4.
SAR around C-2 carboxamide phenyl ring.
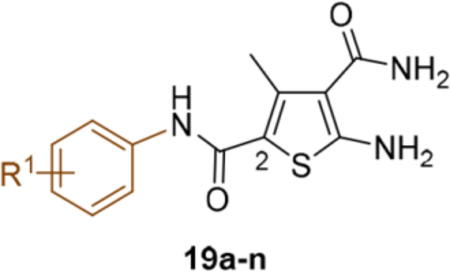
| Compound | Structure | TSAa | EC50b (μM) | CC50c (μM) |
|---|---|---|---|---|
| 11 | 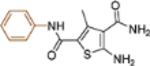 |
+++ | 3.4 ± 0.6 | >100 |
| 19a | 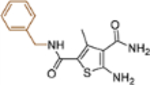 |
+++ | ~10 | NTd |
| 19b | 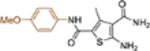 |
+++ | ~10 | NT |
| 19c | 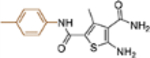 |
− | >10 | NT |
| 19d | 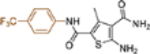 |
− | NT | NT |
| 19e | 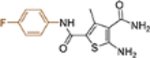 |
+++ | 3.0 ± 0.7 | >100 |
| 19f | 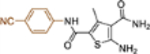 |
− | NT | NT |
| 19g | 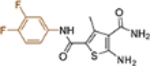 |
+++ | 0.76 ± 0.22 | >100 |
| 19h | 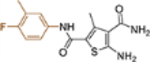 |
+++ | 0.21 ± 0.09 | >100 |
| 19i | 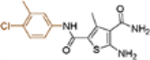 |
+++ | 1.7 ± 0.4 | >100 |
| 19j | 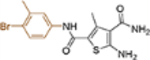 |
+++ | 7.1 ± 1.7 | 107 ± 7.2 |
| 19k | 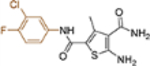 |
+++ | 0.31 ± 0.07 | >100 |
| 19l | 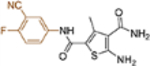 |
+++ | 0.47 ± 0.25 | >100 |
| 19m | 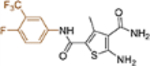 |
+++ | 1.2 ± 0.03 | >100 |
| 19n | 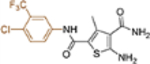 |
+++ | 4.6 ± 0.4 | 72 ± 6.4 |
| 19o | 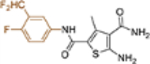 |
+++ | 0.11 ± 0.01 | >100 |
| 19p | 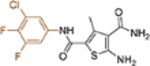 |
+++ | 4.6 ± 1.1 | 72 ± 4.1 |
Thermal shift assay that measures changes in the thermal stability of HBV Cp upon binding of a small molecule: “−” denotes no binding, “+” weak binding, “++” moderate binding and “+++” strong binding.
Concentration of a compound inhibiting HBV total DNA production by 50%, expressed as the mean ± standard deviation from at least two independent experiments.
Concentration of a compound causing 50% cytotoxicity, expressed as the mean ± standard deviation from at least two independent experiments.
NT = not tested
Overall, our SAR efforts led to the identification of five analogues with strong Cp binding, nanomolar antiviral potency and no cytotoxicity (Table 5). To benchmark our CAEs, we also resynthesized representative compounds (7-10) of all four known CAE chemotypes (Figure 2) and tested them in our assays. As shown in Table 5, AT-61 (9) and NZ-4 (10) were at least one order of magnitude less potent than our CAEs, while HAP analogue 7 and SBA analogue 8 exhibited antiviral potency largely comparable to that of our nanomolar CAEs (Table 5). In addition, significant cytotoxicity was observed with all but one control compounds (8-10, CC50 = 17–61 μM), whereas our CAEs of subtype 19 showed no cytotoxicity at concentrations up to 100 μM. Direct comparison between HAP analogue 7 and our nanomolar CAEs revealed that our best compound 19o was about four times as potent as 7.
Table 5.
Summary of nanomolar CAEs of the ATDC series
| Compound | TSA | EC50(μM) | CC50(μM) |
|---|---|---|---|
| 19g | +++ | 0.76 ± 0.22 | >100 |
| 19h | +++ | 0.21 ± 0.09 | >100 |
| 19k | +++ | 0.31 ± 0.07 | >100 |
| 19l | +++ | 0.47 ± 0.25 | >100 |
| 19o | +++ | 0.11 ± 0.01 | >100 |
| 7 | +++ | 0.41 ± 0.05 | >100 |
| 8 | +++ | 0.25 ± 0.07 | 17 ± 4.4 |
| 9 | +++ | 4.8 ± 0.6 | 61 ± 25 |
| 10 | +++ | 10 ± 0.5 | 29 ± 10 |
To confirm the mechanism of action our novel CAEs, a selected compound 19h was further investigated using TEM for its effect on capsid assembly. As shown in Figure 5C, comparing to the DMSO control, 19h promoted the aggregation of HBV capsid, an effect similar to that observed with hit molecule 11 [33]. The impact of 19h on capsid assembly is also reflected in the TSA curve (Figure 5A). The TSA was conducted under conditions to allow two denaturation peaks corresponding to the Cp dimer and the assembled capsid, respectively (DMSO control). However, in the presence of 19h, only the denaturation peak corresponding to fully assemble capsid was observed, corroborating the mechanism that 19h confers potent antiviral activity (Figure 5B) by promoting capsid assembly.
Physicochemical properties, in vitro ADME and in vivo PK.
To assess drug-like properties of our best CAE subtype 19, analogues with nanomolar antiviral activity (19g-h, 19k-l and 19o) were tested in various physicochemical and in vitro ADME studies (Table 6). First, aqueous stability and solubility were evaluated in Dulbecco’s Phosphate-Buffered Saline (DPBS). While all tested compounds were highly stable in DPBS, a few of them (19g, 19l and 19o) exhibited poor aqueous solubility. Second, all tested compounds were stable in both human and mouse plasma. Third, plasma protein binding was in a reasonable range for all compounds with higher binding observed in human plasma (80–94%) than in mouse plasma (50–90%). Fourth, all compounds exhibited excellent phase I and phase II microsomal metabolic stability, with the exception of 19h which demonstrated greater susceptibility to phase I NADPH-dependent metabolism particularly in mouse liver microsomes. Finally, two compounds were also tested in Caco-2 cell line for permeability, and the obtained high apparent permeability coefficient (Papp > 10×10−6 cm/s) suggested a good in vitro drug absorption with minimal efflux (Papp[B→A]/Papp[A→B] < 2). Taken together, except for the poor solubility of a few analogues, our potent CAEs possess favorable physicochemical and ADME properties.
Table 6.
Physicochemical, in vitro ADMEa, and oral bioavailability profile of selected analogues
| Compd | Aqueous Solubility (μM)a |
Plasma Stability t1/2(h) | Plasma Protein Binding (%)b |
Microsomal Stability CLintc (Phase I / Phase II) | Caco-2 Pappd (10−6 cm/s) | Fg (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Human | Mouse | Human | Mouse | Human | Mouse | Ae to Bf | B to A | |||
| 11 | 19 | >24 | >24 | 86 | 50 | 5.0/2.0 | 18/4.4 | − | 34 | |
| 19g | 11 | >24 | >24 | 86 | 80 | 3.4/< 0.1 | 25/5.2 | 24.8 | 27.6 | − |
| 19h | 31 | >24 | >24 | 91 | 82 | 14/<0.1 | 92/5.2 | 18.6 | 26.9 | 11 |
| 19k | 31 | >24 | >24 | 94 | 90 | 7.6/< 0.1 | 28/1.6 | − | − | 46 |
| 191 | 6.7 | >24 | >24 | 80 | 69 | 2.4/0.1 | 4.2/2.0 | − | − | − |
| 19o | 9.5 | >24 | >24 | 89 | 82 | 5.8/3.2 | 22/10 | − | − | 25 |
Aqueous stability of selected analogues were determined in Dulbecco’s Phosphate-Buffered Saline (DPBS). All analogues showed excellent stability with remaining percentage at 24 h > 90%.
Percent of fraction bound.
CLint: intrinsic clearance, μl/min/mg protein.
Papp: The apparent permeability coefficient.
A: apical side.
B: basolateral side.
F: Oral bioavailability in mice.
Based on the relatively high solubility and good cell permeability as well as the superb antiviral potency, analogue 19h was initially selected for in vivo PK studies. Unfortunately, the oral bioavailability was found to be quite low (F=11%), which likely reflects the poor metabolic stability of 19h. Upon further examination, the main structural difference between compound 19h and all other potent CAEs is the additional methyl group at the meta position of the phenyl ring (Figure 6). We hypothesized that this metabolically labile methyl group could be the culprit for low bioavailability. Therefore, we decided to replace this methyl group with a metabolically more stable -CF3 bioisostere (19m, Figure 6). Unfortunately, this replacement resulted in a six fold drop in antiviral potency (Table 4, 19m vs 19h), presumably due to the complete loss of the C-H bond which can hyperconjugate using its σ* orbital with a π system in the protein. Such a hyperconjugation forms the molecular basis for the well-known magic methyl effect [37]. To restore the ability for hyperconjugation, the -CF3 group was changed to -CHF2 group and the resulting analogue 19o showed not only substantially improved oral bioavailability (F = 25%), but also enhanced antiviral potency (EC50 = 0.11 μM, Figure 6). The improved bioavailability correlated well with increased phase I metabolic stability as the intrinsic clearance for 19o falls within the low to medium range of clearance according to literature[38] in both human and mouse liver microsomes (Table 6 and Figure 6).
Figure 6.

Lead optimization from 19h to 19o. The highlighted -CH3 group in 19h is a metabolic liability which likely accounts for the low F. Replacing the -CH3 group with a -CF3 group substantially reduced antiviral potency (19m), whereas a -CHF2 substituent enhanced antiviral activity while improving metabolic stability (19o).
Lastly, animal PK studies also included analogue 19k, which displayed good balance between antiviral potency (EC50 = 0.31 μM) and ADME profile, particularly solubility and microsomal stability. The oral bioavailability of 19k was determined to be 46% (Table 6), the highest among analogues tested. This observation further confirms that solubility and metabolic stability are key factors in achieving good oral bioavailability.
Molecular modeling.
To gain understanding on the binding mode of our new CAEs, docking analysis was performed using Glide XP (version 6.9) [39–40]. The predicted binding mode of compound 19o within HBV Cp suggests a key interaction between the carbonyl oxygen of benzamide group in 19o and W102. The halogenated benzamide group of 19o extends into a deep pocket lined by several hydrophobic residues; P25, L30, T33, W102 and I105 and makes CH-π interaction between the phenyl of benzamide group and P25 (Figure 7A). A similar interaction was observed and found to be critical for the reported SBA inhibitor of HBV Cp [41]. An overlay of ligand 19o and SBA analog (29) is shown in Figure 7C. The aminothiophene carboxamide within 19o was predicted to occupy another large hydrophobic sub pocket lined by several residues F23, F110, Y118 and F122. These residues are located at the dimer-dimer interface and are known to be critical for icosahedral capsid assembly [41–43]. The amine of carboxamide at 3 position of thiophene makes hydrogen bond interaction with backbone carbonyl of P138. The methyl group at 4 position of thiophene core makes hydrophobic interactions within the pocket with F23, Y118 and F122. The interactions associated with thiophene group of 19o within this large hydrophobic sub pocket was observed to be similar to that of SBA (Figure 7C). The bis(fluoranyl)pyrrolidine-2-carboxylic acid within HAP (30) occupies this sub pocket and makes interactions identical to the thiophene group of 19o (Figure 7D).
Figure 7.

Molecular modeling of 19o. (A) Predicted binding mode of 19o within the crystal structure of HBV Cp (PDB code: 5T2P). (B) Structures of SBA analogue (29) and HAP analogue (30) in reported co-crystal structures. (C) Structural overlay of predicted binding mode of 19o and 29 within the HBV capsid. (D) Structural overlay of predicted binding mode of 19o and 30 within the HBV capsid. Key residues are highlighted in yellow sticks. H-bond interactions are depicted as black dotted lines. Alkyl-π interactions is represented as double headed arrow in black.
Conclusion.
ATDC analogue 11, previously identified as a CAE hit, was confirmed as a CAE lead via further ADME and PK studies. Extensive SAR revealed that a para F substitution along with a meta substitution on the phenyl ring confers the best antiviral activity, with five analogues of subtype 19 exhibiting nanomolar antiviral activity and no cytotoxicity. Interestingly, analogue 19o was identified via a combination of SAR and SPR from a highly potent compound 19h. In the end, our SAR and SPR efforts identified two analogues, 19o (EC50 = 0.11 μM, CC50 >100 μM, F = 25%) and 19k (EC50 = 0.31 μM, CC50 >100 μM, F = 46%), with potent antiviral activity, no cytotoxicity and good oral bioavailability. Such lead profiles render 19o and 19k discernibly superior to the reported CAEs 7-10 used in our studies and excellent candidates for preclinical development.
Experimental
Chemistry
General Procedures.
All commercial chemicals were used as supplied unless otherwise indicated. Dry solvents were either purchased (toluene and dioxane, EtOH) or dispensed under argon from an anhydrous solvent system with two packed columns of neutral alumina or molecular sieves. Flash chromatography was performed on a Teledyne Combiflash RF-200 with RediSep columns (silica) and indicated mobile phase. All moisture sensitive reactions were performed under an inert atmosphere of ultra-pure argon with oven-dried glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer or Bruker 400 MHz spectrometer. Mass data were acquired on an Agilent TOF II TOS/MS spectrometer capable of ESI and APCI ion sources. Melting points were determined in open glass capillaries using a MEL-TEMP melting-point apparatus. Analysis of sample purity was performed on a Varian Prepstar SD-1 HPLC system with a Phenomenex Gemini, 5 micron C18 column (250mm x 4.6 mm). HPLC conditions: solvent A = H2O, solvent B = MeCN; flow rate = 1.0 mL/min; compounds were eluted with a gradient of 20% MeCN/H2O to 100% MeCN for 30 min. Purity was determined by total absorbance at 254 nm. All tested compounds have a purity ≥ 96%. Characterization data for final compounds other than the representative analogues are shown in Supplementary Data.
5-Amino-3-methyl-N2-phenylthiophene-2,4-dicarboxamide (11).
To a solution of 2cyanoacetamide (59 mg, 0.70 mmol) in anhydrous EtOH (3mL) was added acetoacetanilide (124 mg, 0.70 mmol) and sulfur (23 mg, 0.70 mmol), followed by the addition of morpholine (121 mg, 1.40 mmol) dropwise. The reaction mixture was stirred at 80°C until no starting material left as shown by TLC. Solvent was removed and the residue was purified by flash chromatography on silica gel using hexanes:EtOAc (1:2) to obtain compound 11 (170 mg, 89 %). Mp 234−235°C 1H NMR (600 MHz, DMSO- d6) δ 9.57 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.36 (m, 4H), 7.12 (m, 1H), 7.05 (s, 2H), 2.41 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.9, 162.0, 161.3, 140.2, 139.7, 128.9, 123.5, 120.6, 112.4, 112.0, 16.3; C13H14N3O2S [M+H]+ 276.0803, found 276.0801.
General procedure for the synthesis of compounds 12a-k.
To a solution of a carboxylic acid derivative (0.4 mmol) in anhydrous DMF (1mL) was added PyAOP (0.42 mmol) and DIEA (0.8 mmol). The resulting mixture was stirred at rt for 30 min before 5-amino-3-methyl-N2phenylthiophene-2,4-dicarboxamide (0.4 mmol) was added. This reaction mixture was stirred at rt overnight, and quenched by adding H2O (5 mL). The aqueous was extracted with EtOAc (4 × 15 mL, and the combined organics were washed with brine (2 × 40 mL) and dried over MgSO4. Solvent was removed and the residue was purified by flash chromatography on silica gel using hexanes: EtOAc (3:2) to obtain 12a-k.
3-Methyl-5-(morpholine-4-carboxamido)-N2-phenylthiophene-2,4-dicarboxamide (12a).
Yield 57 %; Mp 270−271°C; 1H NMR (600 MHz, DMSO- d6) δ 12.68 (s, 1H), 10.32 (s, 1H), 8.28 (s, 2H), 7.78 (d, J = 7.8 Hz, 1H), 7.45 (m, 2H), 7.22 (m, 2H), 3.41 (s, 4H), 2.82 (s, 4H), 2.62 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 167.9, 162.1, 161.4, 140.1, 139.7, 129.0, 123.6, 120.7, 112.4, 112.2, 63.7, 43.1, 16.3; HRMS (ESI+) calcd. for for C18H21N4O4S [M+H]+ 389.1282, found 389.1280.
5-Amino-4-carbamothioyl-N-(3,4-difluorophenyl)-3-methylthiophene-2-carboxamide (13) [44].
To a solution of 2-cyanoacetamide (0.3 mmol) in anhydrous EtOH (1 mL) was added 2cyanoethanethioamide (0.3 mmol), followed by the addition of morpholine (0.6 mmol) dropwise. The resulting mixture was stirred at 80 °C until no starting material left as shown by TLC. Solvent was removed and the residue was purified by flash chromatography on silica gel using hexanes:EtOAc (1:2) to give compound 13 (61%). Mp 175−176°C; 1H NMR (600 MHz, CD3OD) δ 7.64 (d, J = 7.8 Hz, 2H), 7.44 (t, J = 7.8 Hz, 2H), 7.23 (t, J = 7.2 Hz, 1H), 2.56 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 167.9, 162.0, 161.3, 140.1, 139.7, 128.9, 123.5, 120.6, 112.4, 112.0, 16.2; HRMS (ESI+) calcd. for C13H14N3OS2[M+H]+ 292.0575, found 292.0573.
General procedure for the synthesis of compounds 14a-d [45].
To a solution of malononitrile (0.7 mmol) in anhydrous EtOH (3mL) was added an acetoacetanilide derivative (0.7 mmol) and sulfur (0.7 mmol), followed by the addition of morpholine (1.4 mmol) dropwise. The resulting mixture was stirred at 80 °C until no starting material left as shown by TLC. Solvent was removed and the residue was purified by flash chromatography on silica gel using hexanes:EtOAc (1:2) to give compounds 14a-d.
5-Amino-4-cyano-N-(4-fluoro-3-methylphenyl)-3-methylthiophene-2-carboxamide(14c).
Yield 47 %; Mp 212−213°C; 1H NMR (600 MHz, CD3OD) δ 7.67 (d, J = 6.6 Hz, 1H), 7.62 (m, 1H), 7.27 (t, J = 9.0 Hz, 1H), 2.71 (s, 3H), 2.53 (s, 3H); 13C NMR (150 MHz, DMSO) δ 165.5, 160.8, 158.1, 156.6, 140.8, 135.3, 124.4, 123.4, 120.1, 115.9, 113.3, 88.4, 15.3, 14.8; HRMS (ESI+) calcd. for C14H13FN3OS [M+H]+ 290.0758, found 290.0760.
General procedure for the synthesis of compounds 15a-b [46–47].
To a solution of ethyl 2cyanoacetate (0.5 mmol) (0.5 mmol) in anhydrous EtOH (2.0 mL) was added was added acetoacetanilide (0.5 mmol) and sulfur (16 mg, 0.5 mmol), followed by the addition of morpholine (1.0 mmol) dropwise. The resulting mixture was stirred at 80 °C until no starting material left as shown by TLC. Solvent was removed and the residue was purified by flash chromatography on silica gel using hexanes:EtOAc (1:2) to give compounds 15a-b.
Ethyl 2-amino-4-methyl-5-(phenylcarbamoyl)thiophene-3-carboxylate (15a).
Yield 83 %; Mp 180−181°C; 1H NMR (600 MHz, DMSO-d6) δ 9.66 (s, 1H), 7.72 (s, 2H), 7.59 (d, J = 7.2 Hz, 2H), 7.30 (t, J = 7.8 Hz, 2H), 7.04 (t, J = 7.2 Hz, 1H), 4.23 (q, J = 6.6 Hz, 2H), 2.48 (s, 2H), 1.29 (t, J = 6.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 165.5, 165.4, 161.8, 140.7, 139.6, 129.0, 123.7, 120.5, 113.4, 105.9, 59.7, 17.0, 14.8; HRMS (ESI-) calcd. for C15H15N2O3S [M-H]− 303.0798, found 303.0802.
General procedure for the synthesis of compounds 16a-m.
To a solution of a cyanoacetamide derivative (0.4 mmol) in anhydrous EtOH (2mL) was added an acetoacetanilide derivative (0.4 mmol) and sulfur (0.4 mmol), followed by the addition of morpholine (0.8 mmol) dropwise. The resulting mixture was stirred at 80 °C until no starting material left as shown by TLC. Solvent was removed and the residue was purified by flash chromatography on silica gel using hexanes:EtOAc (1:2) to give compounds 16a-m. (55–83 %).
5-Amino-3-methyl-N2,N4-diphenylthiophene-2,4-dicarboxamide (16a).
Yield 65 %; 1H NMR (600 MHz, CD3OD) δ 7.61 (d, J = 7.8 Hz, 2H), 7.54 (d, J = 7.8 Hz, 2H), 7.34 (m, 4H), 7.11 (m, 2H), 2.45 (s, 3H); 13C NMR (100 MHz, DMSO-d6) 163.9, 161.8, 159.2, 140.7, 139.7, 139.6, 129.0, 128.9, 123.6, 120.7, 120.6, 120.2, 114.9, 112.3, 15.9; HRMS (ESI+) calcd. for C19H18N3O2S [M+H]+ 352.1116, found 352.1113.
General procedure for the synthesis of compounds 17a-d.
An aminoester, (1 mmol), and a nitrile (1 mmol) were placed in a 15 mL seal tube at 0 °C. To this was added saturated HCl solution in dioxane (2 mL) dropwise. The tube was carefully sealed and heated at 100 °C with stirring for 4–16 h before cooled to rt. The tube was then opened (Caution! Excessive pressure inside), and the reaction mixture was poured into water (25 mL). The precipitate formed was filtered and washed with a small amount of cold EtOH three times to afford compounds 17a-d as off-white solid (56–85%).
2-Benzyl-5-methyl-4-oxo-N-phenyl-1,4-dihydrothieno[2,3-d]pyrimidine-6-carboxamide (17c).
Yield 85 %; Mp 270−271°C; 1H NMR (600 MHz, DMSO-d6) δ 12.81 (s, 1H), 10.22 (s, 1H), 7.73 (d, J = 7.8 Hz, 2H), 7.41 (m, 6H), 7.33 (m, 1H), 7.19 (t, J = 7.2 Hz, 1H), 4.04 (s, 2H), 2.77 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 165.2, 161.1, 159.6, 159.1, 138.9, 136.8, 136.5, 129.3, 129.1, 129.0, 127.7, 127.4, 124.5, 121.7, 120.8, 40.6, 15.4; HRMS (ESI-) calcd. for C21H16N3O2S [M-H]− 374.0969, found 374.0972.
General procedure for the synthesis of compounds 18a-c.
Cyclohexanone (for 18a), 1benzoylpiperidin-3-one (for 18b) or 1-benzoylpiperidin-4-one (for 18c) (0.42 mmol), cyanoacetamide (34 mg, 0.40 mmol) and sulfur (16 mg, 0.50 mmol) were suspended in EtOH (1 mL). To this was added morpholine (70 mg, 0.80 mmol). The resulting mixture was refluxed gently with stirring for 4 h, and was allowed to cool to room temperature. Solvent was removed and the residue was purified by flash chromatography on silica gel using hexanes:EtOAc (1:2) to give compounds 18a-c as off white powder.
2-Amino-6-benzoyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxamide (18c).
Yield 77 %; Mp 221−222°C; 1H NMR (600 MHz, DMSO-d6) δ 7.56 (m, 5H), 7.06 (s, 2H), 6.68 (s, 2H), 4.60 (s, 2H), 3.54 (t, J = 6.6 Hz, 2H), 2.82 (m, 2H); 13C NMR (100 MHz, DMSO-d6) 169.7, 167.9, 160.2, 136.4, 130.2, 129.6, 128.9, 127.2, 112.2, 107.6, 40.6, 40.4, 26.2; HRMS (ESI-) calcd. for C15H14N3O2S [M-H]− 300.0801, found 300.0804.
General procedure for the synthesis of compounds 19a-p.
To a solution of 2-cyanoacetamide (0.70 mmol) in anhydrous EtOH (3mL) was added a substituted acetoacetanilide (0.70 mmol) and sulfur (23 mg, 0.70 mmol), followed by the addition of morpholine (121 mg, 1.40 mmol) dropwise. The resulting mixture was stirred at 80°C until no starting material left as shown by TLC. Solvent was removed and the residue was purified by flash chromatography on silica gel using hexanes:EtOAc (1:2) to give compounds 19a-p as off-white solid.
5-Amino-3-methyl-N2-(p-tolyl)thiophene-2,4-dicarboxamide (19c).
Yield 82 %; Mp 231232°C; 1H NMR (600 MHz, DMSO-d6) δ 9.48 (s, 1H), 7.55 (d, J = 8.4 Hz, 2H), 7.36 (s, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.03 (s, 2H), 2.56 (s, 3H), 2.32 (s, 3H); 13C NMR (100 MHz, DMSOd6) δ 168.0, 161.9, 161.3, 139.9, 137.2, 132.5, 129.3, 120.7, 112.3, 112.2, 20.9, 16.3; HRMS (ESI+) calcd. for C14H14N3O2S [M+H]+ 290.0958, found 290.0960
Biology
Reagents
Biologicals.
HepG2 (ATCC) cells were maintained in complete media [Dulbecco’s Modified Eagle Medium (DMEM), 10% fetal bovine serum (FBS)]. HepAD38 cells (ATCC) [48] were maintained in tet media [complete media plus 0.4 μg/ml tetracycline (tet) and 400 μg/ml G418 (Gibco)]. All cells were incubated at 37°C with 5% CO2.
Chemicals.
The Maybridge Hitfinder chemical library of compounds (version 6) was purchased from Maybridge.
Cp Purification
A gBlock Gene Fragment coding for the 149 amino acid assembly domain of HBV capsid protein with an added C-terminal cysteine (C150) [49–51] with NdeI and BamHI restriction sites was synthesized by Integrated DNA Technologies and cloned into the pET11a expression vector (Novagen). HBV C150 was expressed and purified as previously described [50–52] with minor modifications. The C150 expression plasmid was transformed into BL21 (DE3) E. coli, grown at 37°C to an OD600 of ~0.8, and induced for 3 h with 1 mM IPTG at 37°C. Cells were pelleted and resuspended in 50 mM Tris (pH 7.5), 1 mM EDTA, 20 mM 2-mercaptoethanol (2-ME), 1 mM PMSF, 150 μg/ml lysozyme, and 0.2 mg/ml DNase I. The suspension was incubated on ice for 30 min and lysed by sonication. Polyethylenimine (PEI) was added to a final concentration of 0.15% w/v to precipitate DNA, and the lysate was centrifuged at 16,000xg for 1 h. Ammonium sulfate was added to the supernatant to 40% saturation. The solution was gently stirred for 1 h, then centrifuged at 16,000xg for 1 h. The pellet was resuspended in Buffer A [100 mM Tris (pH 7.5), 100 mM NaCl, 10 mM 2-ME] to ~10 mg/ml, centrifuged at 16,000xg for 20 min, loaded onto a Buffer A-equilibrated HiLoad 26/60 Superdex 200 prep grade (GE Healthcare) column, and eluted at 2.5 ml/min. Fractions were pooled based on the chromatogram and SDS-PAGE, concentrated to ~5 mg/ml, and dialyzed into Buffer N [50 mM sodium bicarbonate (pH 9.6), 10 mM 2-ME]. Solid urea was added to 3 M and stirred for 1 h at 4°C. The solution was loaded onto a Buffer N-equilibrated HiLoad 26/60 Superdex 200 prep grade column and eluted at 2.5 ml/min. Fractions containing the C150 dimer (C1502) were pooled, concentrated, and stored at 80°C. Final protein concentration was determined spectrophotometrically using an extinction coefficient of 60,900 [51].
Thermal shift assay
The development and use of the thermal shift assay for drug discovery has been described previously [53–54], and optimization for Cp analysis has been described [33]. Briefly, in a final reaction volume of 20 μl, 10 μl of C1502 (15 μM) in Buffer N was mixed with 10 μl assembly buffer [100 mM HEPES (pH 7.5), 1 M NaCl] containing 2x Sypro Orange Protein Gel Stain (Life Technologies). Compounds were added at a final concentration of 20 μM, and reactions contained 1% DMSO. Samples were heated in a PikoReal Real-Time PCR System (Thermo Scientific) from 25°C to 95°C in steps of 1°C every 50 s. Melting curves were analyzed with PikoReal Software.
Antiviral assays
The dot blot assay was performed similarly to previously described [22–23, 26]. HepAD38 cells were seeded in 96-well plates (3 × 104 cells/well) in tet media. The following day, the cells were washed twice with PBS, and complete media containing 2-fold serial dilutions of test compounds was added to the wells with 1% final DMSO concentration. After 2 days, the media was removed, and fresh media containing compounds was added. After 2 additional days, cells were washed with PBS and lysed with 10 mM Tris (pH 7.5), 1mM EDTA, 100mM NaCl, 1% NP-40 at 37 °C for 30 min. 60 μl of lysate was added to 60 μl of 1 M NaOH, 1.5 M NaCl and incubated at RT for 5 min to denature DNA. 120 μl of 1 M Tris (pH 7.4), 2 M NaCl was added, and samples were transferred to positively charged nylon membranes (Roche) using a vacuum dot blot manifold. The wells were washed with 200 μl of 20x SSC (3M NaCl, 300 mM sodium citrate), and the DNA was UV crosslinked to the membranes. The membrane was subjected to Southern blot using a 500 base pair digoxigenin (DIG)-labeled HBV-specific probe synthesized from HepAD38 cells using 5’-GGCCTTTCTGTGTAAACAATACCTGAACC-3’ and 5’GTAATCGAGCTCCGGTGGTCTCCATGCGAC-3’ primers with the PCR DIG Probe Synthesis Kit (Roche), as described previously [55]. Membranes were incubated with CDP-Star (Roche), imaged by chemiluminescence, and quantified by densitometry. Values were plotted in GraphPad Prism 5 and analyzed with the log (inhibitor) vs. normalized response – variable slope equation.
Transmission electron microscopy
This was done as previously described [56]. Briefly, C150 (18 μM dimer) in buffer N was assembled in the presence of 1% DMSO or 10 μM Bay 38–7690 by addition of an equal volume of 100 mM HEPES (pH 7.5) with 1 M NaCl and incubation at room temperature (RT) for 1 h. Assemblies were absorbed to glow-discharged carbon-coated 200 mesh copper grids (Electron Microscopy Sciences), stained with 2% uranyl acetate, and imaged with a JEOL JEM-1400 transmission electron microscope.
Cytotoxicity Assays
HepG2 cells were plated in complete media and treated with compounds as in the antiviral screening above. At the end of treatment duration, cell viability was assessed with the Cell Proliferation Kit II (XTT) (Roche) according to the manufacturer’s instructions. Values were plotted in GraphPad Prism 5 and analyzed with the log (inhibitor) vs. normalized response ‒ variable slope equation.
Physicochemical properties and ADME assays
Aqueous stability assay.
The aqueous stability assay was performed in triplicate by incubating each selected compound (typically 1 μM final concentration) in DPBS at 37 °C. At 0, 1, 3, 6, and 24 h, aliquots of the incubation mixture were taken and quenched with 3 volumes of acetonitrile containing an appropriate internal standard. The samples were then vortexed and centrifuged at 14,000 rpm for 5 min. The supernatants were collected and analyzed by LC-MS/MS.
Aqueous solubility assay [57].
The aqueous solubility of each selected compound was determined in Dulbecco’s Phosphate-Buffered Saline (DPBS) under thermodynamic solubility conditions. Briefly, a saturated solution was made by adding DPBS to the solid compound. The mixture was shaken at 200 rpm for 72 h in a MaxQ 6000 orbital shaker at ambient temperature to allow equilibrium between the solid and dissolved compound. The suspension was then filtered through a 0.45 μm PVDF syringe filter and the filtrate was collected for analysis using LCMS/MS.
Plasma stability assay.
The plasma stability assay was performed in triplicate by incubating each selected compound (typically 1 μM final concentration) in normal mouse and human plasma at 37 °C. At 0, 1, 3, 6, and 24 h, aliquots of the plasma mixture were taken and quenched with 3 volumes of acetonitrile containing an appropriate internal standard. The samples were then vortexed and centrifuged at 14,000 rpm for 5 min. The supernatants were collected and analyzed by LC-MS/MS to determine the half-life time (t1/2).
Plasma protein binding assay.
The plasma protein binding assay was performed in duplicate using equilibrium dialysis in normal mouse and human plasma. Briefly, selected compounds were spiked into plasma at a concentration of 5 μM and dialyzed against DPBS for 5 h at 37 °C using a Rapid Equilibrium Dialysis 96-well plate [58] (Thermo Fisher Scientific). At the end of incubation, aliquots were taken from plasma and DPBS chambers and quenched with 3 volumes of acetonitrile containing an appropriate internal standard. The samples were then vortexed and centrifuged at 14,000 rpm for 5 min. The supernatants were collected and analyzed by LCMS/MS to determine the percent of compound bound to plasma.
Microsomal stability assay [59–60].
The in vitro microsomal stability assay was conducted in triplicate in mouse and human liver microsomal systems, which were supplemented with either nicotinamide adenine dinucleotide phosphate (NADPH) or uridine 5′-diphosphoglucuronic acid (UDPGA) as a cofactor for phase I or phase II metabolism, respectively. In a typical phase I microsomal incubation, a compound (typically 1 μM final concentration) was spiked into the reaction mixture containing 0.5 mg/mL of liver microsomal protein and 1 mM of NADPH in 0.1 M potassium phosphate buffer (pH 7.4) at 37 °C. In a typical phase II microsomal incubation (glucuronidation), a compound (typically 1 μM final concentration) was spiked into the reaction mixture containing 0.5 mg/mL of liver microsomal protein, 50 μg/mg protein of alamethicin, 1 mM of MgCl2 and 5 mM of UDPGA in 0.1 M potassium phosphate buffer (pH 7.4) at 37 °C.
At various time points during either incubation, 1 volume of reaction aliquot was taken and quenched with 3 volumes of acetonitrile containing an appropriate internal standard. The samples were then vortexed and centrifuged at 14,000 rpm for 5 min. The supernatants were collected and analyzed by LC-MS/MS to determine the in vitro metabolic half-life (t1/2) and intrinsic clearance (CLint). Verapamil and 7-hydroxycoumarin were used as a positive control for phase I and phase II metabolism, respectively.
Caco-2 Permeability Assay [61].
The human colon adenocarcinoma cell line Caco-2 was purchased from ATCC (American Type Culture Collection, Manassas, VA), and was cultured in a humidified atmosphere of 5 % CO2 at 37 °C. Caco-2 cells were cultured in DMEM with 10 % FBS, 1 % NEAA, 1% GlutaMax and 1 % penicillin and streptomycin solution. The culture medium was changed every other day during cell growth and differentiation. On achieving 80–90 % confluence, the cells were rinsed with pre-warm DPBS (pH 7.4) and split using trypsin. The cells were then seeded on membranes of Millipore Millicell-24 cell culture device. The transepithelial electrical resistance (TEER) were assessed to reflect membrane integrity using a Millicells® ERS-2 (Millipore, USA). The Caco-2 cell monolayers were used for transport experiments on day 21 post-seeding with TEER values >500 Ω·cm2.
Before the transport experiments, the cell monolayer was washed three times with HBSS. Then, the plates were incubated in fresh permeability assay buffer (HBSS containing 10 mM glucose and 20 mM HEPES, pH 7.4) at 37 °C. The experiments were conducted by spiking test compounds to either the apical (A, 0.4 mL) or basolateral side (B, 0.8 mL), while the receiving chamber contained the corresponding volume of pre-warmed permeability assay buffer. Every experiment was repeated in duplicate, and the plates were incubated in an orbital shaker at 37 °C, 50 rpm/min. To assess the drug transport, at the incubation time of 20, 40, 60 and 80 min, aliquot was removed and was immediately replenished with an equal volume of permeability assay buffer. The samples were then subjected to LC/MS/MS analysis. The apparent permeability coefficient was indicated by the absorption rate constant Papp. It was measured either in B to A or A to B direction using the equation of Papp = (dQ/dt)/(A × C0). dQ/dt is the rate at which the compound appears in the receiver chamber (nmol/s), A is the surface area of the filter membrane (0.7 cm2) and C0 is the initial concentration in the donor chamber (μM).
LC-MS/MS Bioanalysis.
Quantification and analysis of compounds in biological samples were carried out on an AB Sciex QTrap 5500 mass spectrometer coupled with an Agilent 1260 Infinity HPLC. The chromatographic separation of compounds was achieved using a Phenomenex Kinetex C18 column (50 × 2.1 mm, 2.6 μm), and MS/MS analysis was conducted using an ESI ion source with MRM detection at positive mode. The MS/MS detection parameters including declustering potential (DP), entrance potential (EP), collision energy (CE), and collision cell exit potential (CXP) were optimized for each compound.
Animal PK
The following mouse PK study was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Minnesota and the experiments were conducted in compliance with IACUC policies and internal animal welfare guidelines. Briefly, selected compounds were evaluated in male CD-1 mice (25–35 g) via two administration routes: IV and PO (n = 4 per route). Compounds were dissolved in Dulbecco’s phosphate-buffered saline (DPBS) containing 2-hydroxypropyl-β-cyclodextrin (2-HPBCD) in different percentages (up to 42%) depending upon solubility. Typical doses of 2 mg/kg (IV) and 10 mg/kg (PO) were used. Blood samples (20 μL/time point) were collected into EDTA-fortified tubes serially by saphenous vein puncture at various time points (up to 24 h and 8 time points). The samples were then centrifuged at 3000 rpm for 10 min at 4 °C to obtain plasma. Plasma concentrations were determined by LC-MS/MS, and the concentration-time data were analyzed using Phoenix WinNonlin (v7.0, Pharsight Corporation) with non-compartmental modeling. Oral bioavailability (F %) were determined by comparing the area under the curve (AUC) of the PO and IV administration routes: F (%) = (AUCpo/AUCiv)×(Doseiv/Dosepo).
Molecular modeling and docking analysis
Molecular modeling and docking was performed using the Schrodinger small molecule drug discovery suite 2015–4 [23]. The crystal structure of Hepatitis B virus core protein Y132A mutant in complex with sulfamoylbenzamide (29) was extracted from the protein data bank (PDB code: 5T2P) [25] as reported by Z Zhou et.al.[41] The above structure was analyzed using Maestro [55] (Schrodinger Inc.) and subjected to docking protocol which involves several steps including preparing protein of interest, grid generation, ligand preparation and docking. The crystal structure above was refined using protein preparation wizard [33, 62] (Schrodinger Inc.), in which missing hydrogen atoms and side chains were added and minimized using OPLS 2005 force field [63] to optimize hydrogen bonding network and converge the heavy atoms to an rmsd of 0.3 Å. The processed model indicates that sulfamoylbenzamide forms a key hydrogen bond interaction between the carbonyl oxygen of benzamide group and W102 of HBV core protein. The benzamide group extends in to a deep hydrophobic pocket lined by P25, L30, T33, W102 and I105. The piperidyl group of the ligand extends into the solvent front. The receptor grid generation tool in Maestro [33, 55, 62] (Schrodinger Inc.) was used to define binding pocket around the native ligand (29) to cover all the residues within 12 Å. Compounds were drawn using Maestro and subjected to Lig Prep [56] to generate conformers, possible protonation at pH of 7±3 that serves as an input for docking process. All the dockings were performed using Glide XP [39–40] (Glide, version 6.9) with the van der Waals radii of nonpolar atoms for each of the ligands were scaled by a factor of 0.8. The solutions were further refined by post docking and minimization under implicit solvent to account for protein flexibility.
Supplementary Material
Highlights.
Validation and optimization of an ATDC analogue as a CAE hit.
SAR led to six nanomolar CAEs from the micromolar hit.
SPR led to improved metabolic stability and oral bioavailability.
Molecular modeling study revealed a binding mode.
The best CAEs from this series are better than reported CAEs.
Acknowledgements
This research was supported by the National Institutes of Health (R01AI121315 to SGS and ZW).
Abbreviations
- HBV
hepatitis B virus
- Cp
core protein
- ATDC
5-amino-3-methylthiophene-2,4-dicarboxamide
- CAE
capsid assembly effector
- ADME
absorption, distribution, metabolism and excretion
- PK
pharmacokinetics
- SAR
structure-activity relationship
- SPR
structure-property relationship
- F
oral bioavailability
- HCC
hepatocellular carcinoma
- IFN-α
interferon alpha
- NA
nucleos(t)ide analogue
- RT
reverse transcriptase
- ANP
acyclic nucleoside phosphonate
- cccDNA
covalently closed circular DNA
- pgRNA
pregenomic RNA
- RC-DNA
relaxed circular DNA
- HAP
heteroaryldihydropyrimidine
- SBA
sulfamoylbenzamide
- HTS
high-throughput screening
- TSA
thermal shift assay
- MW
molecular weight
- LE
ligand efficiency
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supplementary data
Synthesis and characterization data of final compounds as well as intermediates 22 and 27. This material is available free of charge via the Internet at
References
- 1.Global Hepatitis Report, http://apps.who.int/iris/bitstream/10665/255016/1/9789241565455-eng.pdf?ua=1 (2017).
- 2.Chan SL; Wong VWS; Qin SK; Chan HLY, Infection and Cancer: The Case of Hepatitis B. J. Clin. Oncol 34 (2016) 83–90. [DOI] [PubMed] [Google Scholar]
- 3.Tian YJ; Chen WL; Ou JHJ, Effects of Interferon-alpha/beta on HBV Replication Determined by Viral Load. PLoS Pathog. 7 (2011) e1002159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang L; Pan J; Wu J; Hu J; Sun Q; Tang J, Anti-HBV Drugs: Progress, Unmet Needs, and New Hope. Viruses 7 (2015) 4960–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng YC, L-Nucleoside Analogues as a Novel Class of HBV Drugs. Antiviral Ther. 7 (2002) L83–L83. [Google Scholar]
- 6.Furman PA; Wilson JE; Reardon JE; Painter GR, The Effect of Absolute-Configuration on the Anti-Hiv and Anti-Hbv Activity of Nucleoside Analogs. Antiviral Chem. Chemother 6 (1995) 345–355. [Google Scholar]
- 7.Tang H; Griffin J; Innaimo S; Lehman-Mckeeman L; Llamoso C, The Discovery and Development of a Potent Antiviral Drug, Entecavir, for the Treatment of Chronic Hepatitis B. J. Clin. Transl. Hepatol 1 (2013) 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ying C; Holy A; Hockova D; De Clercq E; Neyts J, Novel Acyclic Nucleoside Phosphonate Analogues as Potent and Selective anti-HBV Agents. Antiviral Res. 62 (2004) A45–A45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lampertico P; Chan HLY; Janssen HLA; Strasser SI; Schindler R; Berg T, Review Article: Long-term Safety of Nucleoside and Nucleotide Analogues in HBV-monoinfected Patients. Aliment Pharm Ther 44 (2016) 16–34. [DOI] [PubMed] [Google Scholar]
- 10.Piano S; Tonon M; Romano A; Fasolato S; Morando F; Cavallin M; Rosi S; Stanco M; Gola E; Brocca A; Sticca A; Gatta A; Angeli P, The Impact of Nucleoside/Nucleotide Analogues on the Natural History of Patients with Hbv-Related Cirrhosis: A Prospective Comparative Study Towards Patients with Hcv-Related Cirrhosis in the Interferon and Ribavirin Era. Dig. Liver Dis 46 (2014) E141–E141. [Google Scholar]
- 11.Ma K; Wang JS; Han MF; Guo W; Huang JQ; Yang DF; Zhao XP; Song JX; Tian DY; Qi JY; Huang YC; Ning Q, Nucleoside Analogs Prevent Disease Progression in HBVRelated Acute-on-Chronic Liver Failure: Validation of the TPPM Model. Hepatology 58 (2013) 225a–225a. [DOI] [PubMed] [Google Scholar]
- 12.Nassal M, HBV cccDNA: Viral Persistence Reservoir and Key Obstacle for a Cure of Chronic Hepatitis B. Gut 64 (2015) 1972–1984. [DOI] [PubMed] [Google Scholar]
- 13.Tang L; Zhao Q; Wu S; Cheng J; Chang J; Guo JT, The Current Status and Future Directions of Hepatitis B Antiviral Drug Discovery. Expert Opin. Drug Discovery 12 (2017) 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei YM; Wang CT; Yan SF; Liu G, Past, Current, and Future Developments of Therapeutic Agents for Treatment of Chronic Hepatitis B Virus Infection. J. Med. Chem 60 (2017) 6461–6479. [DOI] [PubMed] [Google Scholar]
- 15.Durantel D; Zoulim F, New Antiviral Targets for Innovative Treatment Concepts for Hepatitis B Virus and Hepatitis Delta Virus. J. Hepatol 64 (2016) S117–S131. [DOI] [PubMed] [Google Scholar]
- 16.Tong S; Revill P, Overview of Hepatitis B Viral Replication and Genetic Variability. J. Hepatol 64 (2016) S4–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boucle S; Bassit L; Ehteshami M; Schinazi RF, Toward Elimination of Hepatitis B Virus Using Novel Drugs, Approaches, and Combined Modalities. Clin. Liver Dis 20 (2016) 737–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li XL; Zhou K; He HY; Zhou Q; Sun Y; Hou LJ; Shen L; Wang XF; Zhou YD; Gong Z; He SB; Jin HT; Gu ZX; Zhao SY; Zhang L; Sun CY; Zheng SS; Cheng Z; Zhu YD; Zhang MH; Li J; Chen SH, Design, Synthesis, and Evaluation of Tetrahydropyrrolo[1,2-c]pyrimidines as Capsid Assembly Inhibitors for HBV Treatment. Acs Med Chem Lett 8 (2017) 969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mani MN; Cole AG; Phelps JR; Abbott C; Ardzinski A; Bechard J; Burns R; Chiu T; Cuconati A; Dorsey BD; Evangelista E; Fu L; Guo HT; Harasym TO; Jarosz A; Kadhim S; Kultgen S; Kwak K; Lee AC; Li AH; Majeski S; McClintock K; Miller A; Pasetka CJ; Reid SP; Rijnbrand R; Shapiro A; Stever K; Tang S; Teng XW; Wang XH; Zhang H; Sofia J, Antiviral Characterization of a Next Generation Chemical Series of HBV Capsid Inhibitors in vitro and in vivo. Hepatology 66 (2017) 510a–510a.28437844 [Google Scholar]
- 20.Belloni L; Li L; Palumbo GA; Chirapu SR; Calvo L; Finn M; Zlotnick A; Levrero M, HAPs Hepatitis B Virus (HBV) Capsid Inhibitors Affect Core Protein Interaction with the Minichromosome and Target cccDNA Function. J. Hepatol 58 (2013) S153–S153. [Google Scholar]
- 21.Belloni L; Li LC; Palumbo GA; Chirapu SR; Calvo L; Finn M; Lopatin U; Zlotnick A; Levrero M, HAPs Hepatitis B Virus (HBV) Capsid Inhibitors Block Core Protein Interaction with the Viral Minichromosome and Host Cell Genes and Affect cccDNA Transcription and Stability. Hepatology 58 (2013) 277a–277a. [Google Scholar]
- 22.Qiu ZX; Lin XF; Zhang WX; Zhou MW; Guo L; Kocer B; Wu GL; Zhang ZS; Liu HX; Shi HG; Kou BY; Hu TS; Hu YM; Huang MW; Yan SF; Xu ZH; Zhou Z; Qin N; Wang YF; Ren S; Qiu HX; Zhang YX; Zhang Y; Wu XY; Sun K; Zhong S; Xie JX; Ottaviani G; Zhou Y; Zhu LN; Tian XJ; Shi LP; Shen F; Mao Y; Zhou X; Gao L; Young JAT; Wu JZ; Yang G; Mayweg AV; Shen HC; Tang GZ; Zhu W, Discovery and Pre-Clinical Characterization of Third-Generation 4-H Heteroaryldihydropyrimidine (HAP) Analogues as Hepatitis B Virus (HBV) Capsid Inhibitors. J. Med. Chem 60 (2017) 3352–3371. [DOI] [PubMed] [Google Scholar]
- 23.Qiu ZX; Lin XF; Zhou MW; Liu YF; Zhu W; Chen WM; Zhang WX; Guo L; Liu HX; Wu GL; Huang MW; Jiang M; Xu ZH; Zhou Z; Qin N; Ren S; Qiu HX; Zhong S; Zhang YX; Zhang Y; Wu XY; Shi LP; Shen F; Mao Y; Zhou X; Yang WG; Wu JZ; Yang G; Mayweg AV; Shen HC; Tang GZ, Design and Synthesis of Orally Bioavailable 4-Methyl Heteroaryldihydropyrimidine Based Hepatitis B Virus (HBV) Capsid Inhibitors. J. Med. Chem 59 (2016) 7651–7666. [DOI] [PubMed] [Google Scholar]
- 24.Weber O; Schlemmer KH; Hartmann E; Hagelschur I; Paessens A; Graef E; Deres K; Goldmann S; Niewoehner U; Stoltefuss J; Haebich D; Ruebsamen-Waigmann H; Wohlfeil S, Inhibition of Human Hepatitis B Virus (HBV) by a Novel Non-nucleosidic Compound in a Transgenic Mouse Model. Antiviral Res. 54 (2002) 69–78. [DOI] [PubMed] [Google Scholar]
- 25.Campagna MR; Liu F; Mao R; Mills C; Cai D; Guo F; Zhao X; Ye H; Cuconati A; Guo H; Chang J; Xu X; Block TM; Guo JT, Sulfamoylbenzamide Derivatives Inhibit the Assembly of Hepatitis B Virus Nucleocapsids. J. Virol 87 (2013) 6931–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sari O; Boucle S; Cox BD; Ozturk T; Russell OI; Bassit L; Amblard F; Schinazi RF, Synthesis of Sulfamoylbenzamide Derivatives as HBV Capsid Assembly Effector. Eur. J. Med. Chem 138 (2017) 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gane EJ; Schwabe C; Walker K; Flores L; Hartman GD; Klumpp K; Liaw S; Brown NA, Phase 1a Safety and Pharmacokinetics of NVR 3–778, a Potential First-In-Class HBV Core Inhibitor. Hepatology 60 (2014) 1279a–1279a. [Google Scholar]
- 28.Yuen MF; Kim DJ; Weilert F; Chan HLY; Lalezari JP; Hwang SG; Nguyen TT; Liaw S; Brown N; Klumpp K; Flores L; Hartman GD; Gane EJ, Phase 1b Efficacy and Safety of NVR 3–778, a First-In-Class HBV Core Inhibitor, in HBeAg-Positive Patients with Chronic HBV Infection. Hepatology 62 (2015) 1385a–1386a. [Google Scholar]
- 29.Wu GY; Liu B; Zhang YJ; Li J; Arzumanyan A; Clayton MM; Schinazi RF; Wang ZH; Goldmann S; Ren QY; Zhang FX; Feitelson MA, Preclinical Characterization of GLS4, an Inhibitor of Hepatitis B Virus Core Particle Assembly. Antimicrob. Agents Chemother. 57 (2013) 5344–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang PY; Naduthambi D; Mosley RT; Niu CR; Furman PA; Otto MJ; Sofia MJ, Phenylpropenamide derivatives: Anti-Hepatitis B Virus Activity of the Z Isomer, SAR and the Search for Novel Nnalogs. Bioorg. Med. Chem. Lett 21 (2011) 4642–4647. [DOI] [PubMed] [Google Scholar]
- 31.Yang L; Shi LP; Chen HJ; Tong XK; Wang GF; Zhang YM; Wang WL; Feng CL; He PL; Zhu FH; Hao YH; Wang BJ; Yang DL; Tang W; Nan FJ; Zuo JP, Isothiafludine, a Novel Non-nucleoside Compound, Inhibits Hepatitis B Virus Replication through Blocking Pregenomic RNA Encapsidation. Acta Pharmacol. Sin 35 (2014) 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katen SP; Chirapu SR; Finn MG; Zlotnick A, Trapping of Hepatitis B Virus Capsid Assembly Intermediates by Phenylpropenamide Assembly Accelerators. ACS Chem. Biol 5 (2010) 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huber AD; Pineda DL; Liu D; Boschert KN; Gres AT; Wolf JJ; Coonrod EM; Tang J; Laughlin TG; Yang Q; Puray-Chavez MN; Ji J; Singh K; Kirby KA; Wang Z; Sarafianos SG, Novel Hepatitis B Virus Capsid-Targeting Antiviral that Aggregates Core Particles and Inhibits Nuclear Entry of Viral Cores. ACS Infect. Dis. Submitted [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang YJ; Domling A, The Gewald Multicomponent Reaction. Mol. Diversity 15 (2011) 3–33. [DOI] [PubMed] [Google Scholar]
- 35.Sabnis RW; Rangnekar DW; Sonawane ND, 2-Aminothiophenes by the Gewald reaction. J. Heterocycl. Chem 36 (1999) 333–345. [Google Scholar]
- 36.Puterova Z; Krutosikova A; Vegh D, Gewald Reaction: Synthesis, Properties and Applications of Substituted 2-Aminothiophenes. Arkivoc (2010) 209–246. [Google Scholar]
- 37.Nishio M; Umezawa Y; Hirota M; Takeuchi Y, Tetrahedron Report Number-378 - the CH/Pi Interaction - Significance in Molecular Recognition. Tetrahedron 51 (1995) 8665–8701. [Google Scholar]
- 38.Kerns EH; Di L, Drug-like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization 2nd Edition ed.; Academic Press: 2016. [Google Scholar]
- 39.Release Schrödinger 2015–4: Glide version 6.9, Schrödinger, LLC, New York, NY, 2015. [Google Scholar]
- 40.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem 49 (2006) 6177–6196. [DOI] [PubMed] [Google Scholar]
- 41.Zhou Z; Hu TS; Zhou X; Wildum S; Garcia-Alcalde F; Xu ZH; Wu DZ; Mao Y; Tian XJ; Zhou Y; Shen F; Zhang ZS; Tang GZ; Najera I; Yang G; Shen HC; Young JAT; Qin N, Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Sci. Rep 7 (2017) doi: 10.1038/srep42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crowther RA; Kiselev NA; Bottcher B; Berriman JA; Borisova GP; Ose V; Pumpens P, Three Dimensional Structure of Hepatitis-B Virus Core Particles Determined by Electron Cryomicroscopy. Cell 77 (1994) 943–950. [DOI] [PubMed] [Google Scholar]
- 43.Wynne SA; Crowther RA; Leslie AGW, The Crystal Structure of the Human Hepatitis B Virus Capsid. Mol. Cell 3 (1999) 771–780. [DOI] [PubMed] [Google Scholar]
- 44.Gewald K; Schindler R, Cyclizations of Cyanothioacetamide in the Presence of Sulfur. J. Prakt. Chem 332 (1990) 223–228. [Google Scholar]
- 45.Mohareb RM; Elkholy YM; Sayed NIA, The Uses of Polyfunctionally Substituted Thiophenes in Heterocyclic Synthesis: Synthesis of Benzo[b]thiophene, Thieno[2,3-b] pyridine Derivatives. Phosphorus Sulfur Relat. Elem 106 (1995) 193–201. [Google Scholar]
- 46.Karci F; Karci F, Synthesis and Tautomeric Structures of Some Novel Thiophene-Based Bis-Heterocyclic Monoazo Dyes. J. Mol. Struct 1024 (2012) 117–122. [Google Scholar]
- 47.Bogolubsky AV; Ryabukhin SV; Plaskon AS; Stetsenko SV; Volochnyuk DM; Tolmachev AA, Dry HCl in Parallel Synthesis of Fused Pyrimidin-4-ones. J. Comb. Chem 10 (2008) 858–862. [DOI] [PubMed] [Google Scholar]
- 48.Ladner SK; Otto MJ; Barker CS; Zaifert K; Wang GH; Guo JT; Seeger C; King RW, Inducible Expression of Human Hepatitis B Virus (HBV) in Stably Transfected Hepatoblastoma Cells: a Novel System for Screening Potential Inhibitors of HBV Replication. Antimicrob. Agents Chemother. 41 (1997) 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bourne CR; Finn MG; Zlotnick A, Global Structural Changes in Hepatitis B Virus Capsids Induced by the Assembly Effector HAP1. J Virol 80 (2006) 11055–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stray SJ; Johnson JM; Kopek BG; Zlotnick A, An in vitro Fluorescence Screen to Identify Antivirals That Disrupt Hepatitis B Virus Capsid Assembly. Nat. Biotechnol 24 (2006) 358–362. [DOI] [PubMed] [Google Scholar]
- 51.Zlotnick A; Lee A; Bourne CR; Johnson JM; Domanico PL; Stray SJ, In vitro Screening for Molecules That Affect Virus Capsid Assembly (and Other Protein Association Reactions). Nat. Protoc 2 (2007) 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zlotnick A; Cheng N; Conway JF; Booy FP; Steven AC; Stahl SJ; Wingfield PT, Dimorphism of Hepatitis B Virus Capsids Is Strongly Influenced by the C-Terminus of the Capsid Protein. Biochemistry-US 35 (1996) 7412–7421. [DOI] [PubMed] [Google Scholar]
- 53.Pantoliano MW; Petrella EC; Kwasnoski JD; Lobanov VS; Myslik J; Graf E; Carver T; Asel E; Springer BA; Lane P; Salemme FR, High-Density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. J. Biomol. Screening 6 (2001) 429–440. [DOI] [PubMed] [Google Scholar]
- 54.Lo MC; Aulabaugh A; Jin G; Cowling R; Bard J; Malamas M; Ellestad G, Evaluation of Fluorescence-Based Thermal Shift Assays for Hit Identification in Drug Discovery. Anal. Biochem 332 (2004) 153–159. [DOI] [PubMed] [Google Scholar]
- 55.Huber AD; Michailidis E; Tang J; Puray-Chavez MN; Boftsi M; Wolf JJ; Boschert KN; Sheridan A; Leslie MD; Kirby KA; Singh K; Mitsuya H; Parniak MA; Wang ZQ; Sarafianos SG, 3-Hydroxypyrimidine-2,4-Diones as Novel Hepatitis B Virus Antivirals Targeting the Viral Ribonuclease H. Antimicrob. Agents Chemother. 61 (2017) e00245–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huber AD; Wolf JJ; Liu DD; Gres AT; Tang J; Boschert KN; Puray-Chavez MN; Pineda DL; Laughlin TG; Coonrod EM; Yang QY; Ji J; Kirby KA; Wang ZQ; Sarafianos SG, The Heteroaryldihydropyrimidine Bay 38–7690 Induces Hepatitis B Virus Core Protein Aggregates Associated with Promyelocytic Leukemia Nuclear Bodies in Infected Cells. Msphere 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baka E; Comer JEA; Takacs-Novak K, Study of Equilibrium Solubility Measurement by Saturation Shake-Flask Method Using Hydrochlorothiazide as Model Compound. J. Pharm. Biomed. Anal 46 (2008) 335–341. [DOI] [PubMed] [Google Scholar]
- 58.Waters NJ; Jones R; Williams G; Sohal B, Validation of a Rapid Equilibrium Dialysis Approach for the Measurement of Plasma Protein Binding. J. Pharm. Sci 97 (2008) 4586–4595. [DOI] [PubMed] [Google Scholar]
- 59.Houston JB, Utility of in-Vitro Drug-Metabolism Data in Predicting in-Vivo Metabolic-Clearance. Biochem. Pharmacol 47 (1994) 1469–1479. [DOI] [PubMed] [Google Scholar]
- 60.Obach RS, Prediction of Human Clearance of Twenty-Nine Drugs from Hepatic Microsomal Intrinsic Clearance Data: an Examination of in vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos 27 (1999) 1350–1359. [PubMed] [Google Scholar]
- 61.Hubatsch I; Ragnarsson EGE; Artursson P, Determination of Drug Permeability and Prediction of Drug Absorption in Caco-2 Monolayers. Nat. Protoc 2 (2007) 2111–2119. [DOI] [PubMed] [Google Scholar]
- 62.Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W, Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des 27 (2013) 221–234. [DOI] [PubMed] [Google Scholar]
- 63.Jorgensen WL; Maxwell DS; TiradoRives J, Development and Testing of the OPLS Allatom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc 118 (1996) 11225–11236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.