

Abstract
Suicide is one of the leading causes of mortality worldwide; it causes the death of more than one million patients each year. Suicide is a complex, multifactorial phenotype with environmental and genetic factors contributing to the risk of the forthcoming suicide. These factors first generally lead to mental disorders, such as depression, schizophrenia and bipolar disorder, which then become the direct cause of suicide. Here we present a high quality dataset (including processed BAM and VCF files) gained from the high-throughput whole-exome Illumina sequencing of 23 suicide victims – all of whom had suffered from major depressive disorder - and 21 control patients to a depth of at least 40-fold coverage in both cohorts. We identified ~130,000 variants per sample and altogether 442,270 unique variants in the cohort of 44 samples. To our best knowledge, this is the first whole-exome sequencing dataset from suicide victims. We expect that this dataset provides useful information for genomic studies of suicide and depression, and also for the analysis of the Hungarian population.
Subject terms: Molecular neuroscience, Next-generation sequencing, Medical genomics
Background & Summary
Suicide is the 10th leading cause of mortality in the world1,2. It is a complex, multifactorial behavioural condition in which proximal and distant risk factors, such as mental disorders, genetic and epigenetic factors, social, cultural and economic environment, gender, age, personality, eating disorders, as well as drug or alcohol abuse3–7 play important roles. The presence of early child abuse has also been described as a contributing factor to suicidal behaviour8. Studies have repeatedly shown that suicide is most common among patients with mood disorders, such as major depressive disorder and bipolar disorder9.
Whole-exome sequencing (WES) is a cost-effective and powerful tool for the analysis of complex and rare genetic diseases10. WES technique allows a base-pair comparison of exomes and consequently the examination of rare genetic variants, which may play a role in suicide. Alterations in copy number variations (CNVs) and short-tandem repeats (STRs), as well as the viral infections can be also analysed by WES.
We performed high-depth, paired-end, short-read WES on 23 suicide victims suffered from major depressive disorder (MDD; this information is available in EGA sample metadata) and 21 control patients who died from other diseases (EGA sample metadata). Harvesting of brain tissues was approved by the local ethics committee11. Informed consents from the next kin have not been obtained from the victims because the local regulations did not require consent for autopsy. All data were handled anonymously. Both of the cohorts were of Hungarian ethnicity. The Hungarian population is known to have had a very high annual suicide rate in the previous century12,13. The DNA samples derived from post-mortem brains were sequenced using the Agilent SureSelectXT Human All Exon V5 + UTRs kit and Illumina HiSeq 2000 sequencing platform resulting in an average of 117 million paired-end reads.
We obtained very high coverage per base position in both cohorts; in the suicide cohort 95.4–97.5% (quartiles) of target regions had higher than 20-fold and 82.8–90.5% of target regions had higher than 40-fold coverage. In the Hungarian control cohort these values were 96.3–97.5% for 20-fold and 81.6–91.6% for 40-fold coverage, respectively. We called on average ~130,000 variants per sample. Altogether we observed 442,270 unique variants in the control and suicide cohorts. Furthermore, we determined a total of 320,976 single nucleotide variants (SNVs) and 47,313 insertions/deletions (indels).
The library preparation kit used in this study, as well as the obtained very high coverage of sequencing reads that we obtained enabled us to capture information not only about the exons but also about the 5′- and 3′-UTRs, the promoters to a certain length, and even about off-target sequences, such as introns, intergenic regions and infecting viruses. Our dataset had 286,754 high coverage regions (exceeding 20-fold coverage) in all samples that can be suitable for downstream analysis of CNVs.
Here we describe the sample collection methods, the library preparation and sequencing method, the currently available data records, and technical validations for our dataset. A schematic overview of this study, including the experimental procedure and the bioinformatics workflow, is also presented (Fig. 1).
Figure 1. Data flow diagram shows the detailed overview of the study design, wet lab experiments and bioinformatics pipelines.
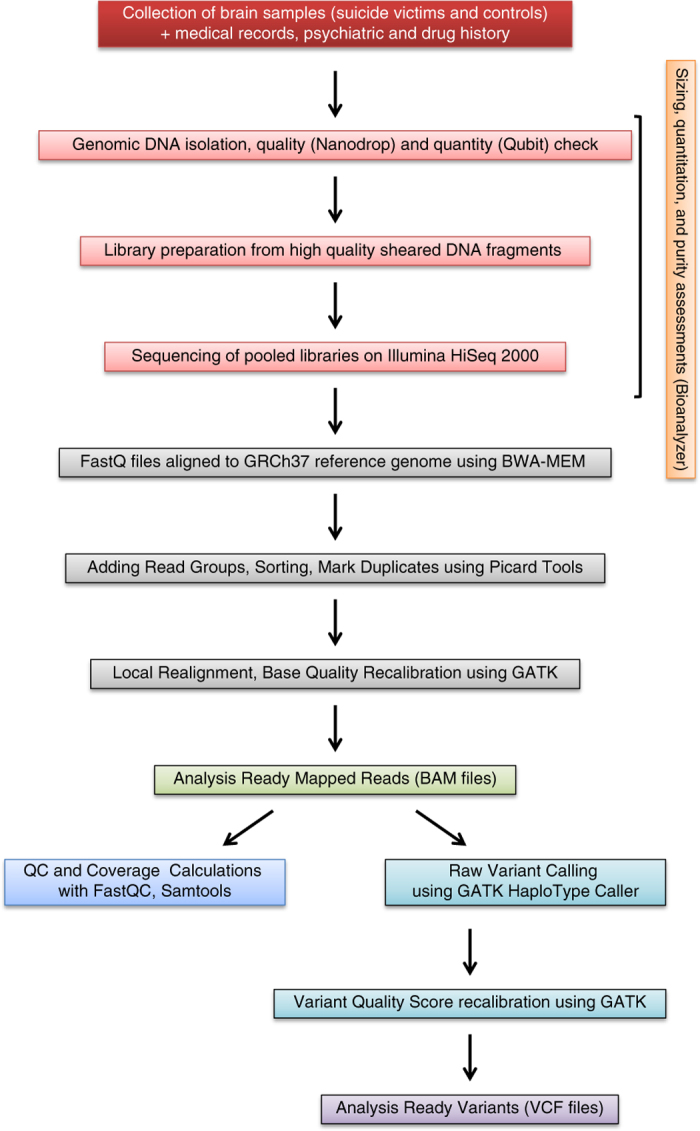
To our knowledge, this dataset is the first individual-level, open-access WES data release targeted to suicide. The dataset described here has been thoroughly analysed in our related manuscript6, where it has been shown that the TGF-β signalling pathway and three voltage-gated calcium ion channels may play an important role in the pathogenesis of suicidal behaviour. Our analysis proposed that suicide may be an umbrella-disease like disorder.
Here we provide the processed BAM files, as well as the processed Variant Call format (VCF) files for each of the samples containing the analysis ready variants of GATK HaplotypeCaller pipeline that allows for the testing of different variant filtration strategies that we had used in our original manuscript or for hypothesis testing of candidate genes (covered by SureSelect V5 All Exon plus UTR kit) without tedious bioinformatic processing.
Our dataset provided here in this manuscript can be re-used to investigate suicide and MDD. These data can be a valuable resource for investigating genetic variants, genes and signalling pathways to identify novel factors related to these disorders, and may provide novel information for the investigation of the Hungarian population or in general for studies of genetic polymorphisms of human population.
Methods
The methods described here are an expanded version of those described in our recently published, related publication6.
Medical history of the suicide victims
This study is based on whole-genome sequence data from occipital, cerebellar and somatomotor cortex regions of the brains of 23 suicide victims (15 males and 8 females) and 21 control patients (14 males and 7 females) who died suddenly from other causes (this information is available in EGA sample metadata). Samples were obtained by autopsy at the Department of Forensic Medicine of the Semmelweis University Medical School. For all of the suicide victims, a psychiatric diagnosis of major depression was on record. The diagnoses had been made by experienced psychiatrists on the basis of Diagnostic and Statistical Manual of Mental Disorders Fourth Edition (DSM-IV). Patients with a history of schizophrenia, epilepsy and other disorders were excluded from the study. The medical, psychiatric, and drug history of the suicide victims was obtained by psychological autopsy, which included interviews with the attending physician and with family members, as well as data obtained from medical records. The participants included in this study had no history of drug or alcohol abuse, and had not used antidepressant medication for at least two months prior to death. This information was confirmed by toxicology tests on blood samples. Suicide victims died by hanging (n = 16), drug overdose (n = 6), or jumping from height (n = 1). Causes of death in control subjects are presented in EGA sample metadata. Harvesting of tissues was approved by the local ethics committee.
Tissue collection, dissection, and storage
Brains were obtained 1 to 10 h after death (this information is available in EGA sample metadata). After removal from the skull, the brains were cut in six major pieces (four cortical lobes, basal ganglia – diencephalon, and lower brain stem – cerebellum), rapidly frozen on dry ice, and stored at −70 °C until dissection (2 days to 2 months). At the time of dissection, the brain samples were sliced into 1 mm-thick coronal sections at a temperature of 0 to 10 °C. The cortical areas were cut out of the sections by a fine microdissecting (Graefe’s) knife. In all cases, cortical samples were always taken from the right hemisphere. The samples were stored in airtight containers or plastic tubes at −70 °C until further use.
DNA purification
Genomic DNA (gDNA) samples were isolated from the occipital, cerebellar or somatomotor cortex regions (Table 1 (available online only)) using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s recommendations for the spin-column protocol, using 30 mg staring tissue material from each sample. In short, tissue samples were cut into small pieces, and then lysis buffer (provided by the kit) and proteinase K were added. Lysis reactions were carried out at 56°C until complete lysis was obtained. DNeasy Mini spin columns (kit’s component) were used for the isolation of gDNA from the lysate. Elution was carried out twice to a final volume of 100 μl per elution.
Table 1. This table summarizes the brain tissue regions that were used for preparation of genomic DNA, and the final concentrations (ng/ul) of the gDNA samples.
| SAMPLE | BRAIN REGION | Cc |
|---|---|---|
| Y919 | occipital cortex | 14.5 |
| Y166 | occipital cortex | 29.8 |
| Y645 | occipital cortex | 15.2 |
| BI269 | occipital cortex | 8.39 |
| Y558 | occipital cortex | 7.73 |
| Y591 | occipital cortex | 5.08 |
| Y520 | occipital cortex | 15.3 |
| Y440 | occipital cortex | 15.4 |
| Y426 | occipital cortex | 5.78 |
| Y787 | occipital cortex | 2.1 |
| Y988 | occipital cortex | 2.24 |
| Y482 | occipital cortex | 20.9 |
| BIB82 | occipital cortex | 1.8 |
| BI370 | occipital cortex | 6.08 |
| YA205 | occipital cortex | 15.3 |
| Y640 | occipital cortex | 10 |
| BI919 | occipital cortex | 21.6 |
| YA626 | occipital cortex | 11.9 |
| YA29 | occipital cortex | 5.13 |
| YB479 | occipital cortex | 2.23 |
| YB330 | occipital cortex | 7.18 |
| Y928 | occipital cortex | 6.68 |
| Y974 | occipital cortex | 4.38 |
| Y78 | occipital cortex | 14.5 |
| YA762 | occipital cortex | 2.55 |
| Y803 | cerebellar cortex | 18.9 |
| Y915 | cerebellar cortex | 18.6 |
| A438 | cerebellar cortex | 5.14 |
| Y880 | cerebellar cortex | 10.6 |
| YA243 | cerebellar cortex | 6.42 |
| Y967 | cerebellar cortex | 11.8 |
| Y726 | cerebellar cortex | 20.3 |
| B168 | cerebellar cortex | 6.46 |
| Y872 | cerebellar cortex | 35.5 |
| Y64 | cerebellar cortex | 6.1 |
| Y532 | cerebellar cortex | 4.91 |
| Y375 | cerebellar cortex | 2.44 |
| Y331 | cerebellar cortex | 2.25 |
| Y316 | cerebellar cortex | 2.51 |
| Y724 | cerebellar cortex | 16.1 |
| Y292 | cerebellar cortex | 11.8 |
| Y393 | cerebellar cortex | 7.23 |
| Y421 | cerebellar cortex | 7.42 |
| Y516 | cerebellar cortex | 8.77 |
| Br969 | somatomotor cortex | 33.2 |
| Br708 | somatomotor cortex | 6.11 |
| Br333 | somatomotor cortex | 31.3 |
| Br857 | somatomotor cortex | 21 |
| BrA206 | somatomotor cortex | 17.2 |
Quality and quantity check of DNA
The quality of the DNA was checked prior to downstream analysis by NanoDrop (Thermo Fischer Scientific) measurement. High-quality samples with an OD 260/280 ratio ranging from 1.8 to 2.0 were subjected to the next step. Qubit 2.0 fluorometer (Invitrogene) was used for genomic DNA quantification before library preparation (Table 1 (available online only)).
Whole-exome sequencing
Paired-end, indexed libraries for Illumina sequencing were prepared from the DNAs of the 23 suicide victims and the 21 controls using post-mortem brain cortex tissues as a source. Whole-exome sequencing was carried out as previously described14 with slight modifications. In short, 200 ng from each of the qualified gDNA samples in 100 μl volume were sheared into fragments of approximately 200–450 bps in Covaris microTUBEs (Covaris microTUBE AFA Fiber Pre-Slit Snap-Cap 6 × 16 mm) with Covaris s220 high-performance ultrasonicator. The quality of the DNA was checked prior to downstream analysis, using the Agilent Bioanalyser 2100 (Agilent Technologies) and the High Sensitivity DNA Analysis Kit (Agilent Technologies; Fig. 2). The end-repair, A-tailing (3′ adenylation) and ligation of the paired-end adaptors steps were executed with the Agilent SureSelect Human All Exon V5 + UTRs kit (Agilent Technologies) as was described in the following manual: SureSelectXT Target Enrichment System for Illumina Paired-End Sequencing Library Illumina HiSeq and MiSeq Multiplexed Sequencing Platforms Protocol version 1.6 (SureSelectXT). Amplification of the adaptor-ligated libraries was performed using the Herculase II Fusion DNA polymerase (Agilent Technologies) following instructions as set forth in the above mentioned manual. Purification after the enzymatic reactions was carried out by using Agencourt AMPure XP beads (Beckman Coulter). Quality and quantity of the libraries were determined by 2100 Bioanalyser instrument and DNA 1000 Assay (Agilent Technologies; Fig. 3). DNA library samples were hybridized with target-specific Capture Library using the SureSelectXT Reagent Kit, HSQ for the HiSeq platform (Agilent Technologies) based on the SureSelectXT protocol. Incubation of the hybridization mixture was carried out at 65 °C for 24 h. After hybridization, the targeted molecules were captured on streptavidin-coated magnetic beads. The SureSelect enriched libraries were amplified by PCR with 6-bp indexing primers (Table 2 (available online only)) following the SureSelectXT manual’s post-capture PCR recommendations using 12 cycles. The amplified libraries were purified using AMPure XP beads. The quality and quantity of the indexed libraries were checked by Agilent Bioanalyser and High Sensitivity DNA Assay (Fig. 4, Table 3). The quantities were also measured by Qubit 2.0 (Table 3). Two or three indexed libraries were pooled (Tables 2 (available online only) and 3) and the pooled samples were sequenced in one lane of a HiSeq 2000 flow cell, generating 101 bp paired-end reads.
Figure 2. Bioanalyzer (BA) electropherograms of a representative sample (# Y516) at the first step in the library preparation workflow.

The figure shows a representative example of the DNA fragment size distribution after sonication. The fragment size peak of this sample is 141 bps.
Figure 3. A representative sample peak is shown in this figure, which illustrates the quality of the amplified library.

This electropherogram shows a distribution with a DNA fragment size peak of 256 bps for sample Y516.
Table 2. This table shows the samples, which were mixed and sequenced together.
| RUN | SAMPLE | TYPE | BRAIN REGION | INDEX |
|---|---|---|---|---|
| Altogether 17 pooled libraries were run; three (or two, in two cases: “run 9” and “run 17”) barcoded libraries were sequenced in a HiSeq 2000 lane. The type of the samples (suicide or control), as well as their origin (brain tissue region) were the same in the pools, with the exception of “run 13”, in which a sample from cerebellar cortex was mixed with two samples from somatomotor cortex region. The following SureSelectXT 6-bp indexing primers were used: index 4 - TGACCA; index 6 – GCCAAT; index 12 – CTTGTA. | ||||
| 1 | Y919 | suicide | occipital cortex | 6 |
| Y166 | suicide | occipital cortex | 12 | |
| Y645 | suicide | occipital cortex | 4 | |
| 2 | Y375 | suicide | cerebellar cortex | 12 |
| Y316 | suicide | cerebellar cortex | 6 | |
| Y393 | suicide | cerebellar cortex | 4 | |
| 3 | Br333 | suicide | somatomotor cortex | 6 |
| Br857 | suicide | somatomotor cortex | 12 | |
| BrA206 | suicide | somatomotor cortex | 4 | |
| 4 | Y440 | suicide | occipital cortex | 12 |
| Y482 | suicide | occipital cortex | 6 | |
| Y558 | suicide | occipital cortex | 4 | |
| 5 | Y331 | suicide | cerebellar cortex | 12 |
| Y724 | suicide | cerebellar cortex | 6 | |
| Y292 | suicide | cerebellar cortex | 4 | |
| 6 | Y591 | suicide | occipital cortex | 12 |
| Y520 | suicide | occipital cortex | 6 | |
| Y787 | suicide | occipital cortex | 4 | |
| 7 | Y421 | suicide | cerebellar cortex | 12 |
| Y516 | suicide | cerebellar cortex | 6 | |
| Y532 | suicide | cerebellar cortex | 4 | |
| 8 | BIB82 | suicide | occipital cortex | 12 |
| Y988 | suicide | occipital cortex | 6 | |
| BI370 | suicide | occipital cortex | 4 | |
| 9 | BI269 | suicide | occipital cortex | 12 |
| Y426 | suicide | occipital cortex | 6 | |
| 10 | Y803 | control | cerebellar cortex | 12 |
| A438 | control | cerebellar cortex | 6 | |
| Y880 | control | cerebellar cortex | 4 | |
| 11 | YA243 | control | cerebellar cortex | 12 |
| Y915 | control | cerebellar cortex | 6 | |
| Y967 | control | cerebellar cortex | 4 | |
| 12 | Y64 | control | cerebellar cortex | 12 |
| Y726 | control | cerebellar cortex | 6 | |
| B168 | control | cerebellar cortex | 4 | |
| 13 | Y872 | control | cerebellar cortex | 12 |
| Br 969 | control | somatomotor cortex | 4 | |
| Br708 | control | somatomotor cortex | 6 | |
| 14 | YA 205 | control | occipital cortex | 6 |
| YB479 | control | occipital cortex | 12 | |
| YB330 | control | occipital cortex | 4 | |
| 15 | Y928 | control | occipital cortex | 12 |
| Y974 | control | occipital cortex | 6 | |
| Y78 | control | occipital cortex | 4 | |
| 16 | BI919 | control | occipital cortex | 12 |
| Y640 | control | occipital cortex | 6 | |
| YA762 | control | occipital cortex | 4 | |
| 17 | YA626 | control | occipital cortex | 12 |
| YA29 | control | occipital cortex | 6 | |
Figure 4. This figure represents the post-capture quality analysis of the PCR-amplified indexed library from the sample Y516.

The size peak of this sample was 289 bps, while the average size was 351 bps.
Table 3. Summary table of the quantitative data of sample pools for multiplexed sequencing.
| RUN | SAMPLE | C (nM)A | Volume (uL)B | C (pg/uL)C | C (ng/uL)D | C (pg/uL)E | C (ng/uL)F | Library sizeG | Average sizeH |
|---|---|---|---|---|---|---|---|---|---|
| The final concentration was 30 nM in each pool. A: initial concentration of the libraries; B: volume to use in the 30 nM final pool; C: final concentration of the libraries based on Bioanalyzer (BA) measurements; D: final concentration of the pooled libraries determined by BA; E: final concentration of the single libraries measured by Qubit; F: final concentration of the libraries based on Qubit measurement; G: The length of the single libraries were determined by BA; H: The average length of the mixed pools. | |||||||||
| 1 | Y166 | 199.2 | 1.51 | 41654.8 | 6.4 | 68000 | 8.4 | 343 | 356 |
| Y919 | 124.8 | 2.40 | 26722 | 26300 | 358 | ||||
| Y645 | 74.5 | 4.03 | 16024 | 21500 | 366 | ||||
| 2 | Y375 | 58.781 | 5.10 | 12521.73 | 6.4 | 8610 | 4.7 | 357 | 350 |
| Y316 | 57.027 | 5.26 | 11749.08 | 7200 | 338 | ||||
| Y393 | 86.632 | 3.46 | 18817.57 | 17100 | 356 | ||||
| 3 | Br857 | 111.258 | 2.70 | 24428.34 | 6.4 | 30700 | 6.7 | 375 | 369 |
| Br333 | 54.035 | 5.55 | 11475.42 | 13100 | 375 | ||||
| BrA206 | 131.504 | 2.28 | 27907.52 | 19500 | 357 | ||||
| 4 | Y440 | 39.245 | 7.64 | 8191.29 | 6.4 | 13300 | 7.8 | 352 | 363 |
| Y482 | 25.05 | 11.98 | 5482.86 | 5100 | 375 | ||||
| Y558 | 75.605 | 3.97 | 16207.71 | 18000 | 363 | ||||
| 5 | Y331 | 61.145 | 4.91 | 12795.49 | 6.4 | 13500 | 7.2 | 344 | 357 |
| Y724 | 60.31 | 4.97 | 12715.34 | 20500 | 362 | ||||
| Y292 | 147.258 | 2.04 | 31909.64 | 23500 | 365 | ||||
| 6 | Y591 | 103.25 | 2.91 | 22871.87 | 6.6 | 19500 | 7.3 | 376 | 374 |
| Y520 | 60.856 | 4.93 | 12793.51 | 17400 | 357 | ||||
| Y787 | 110.566 | 2.71 | 25104.72 | 28100 | 388 | ||||
| 7 | Y421 | 75.488 | 3.97 | 16289.39 | 6.5 | 14200 | 5.7 | 355 | 353 |
| Y516 | 61.276 | 4.90 | 13104.89 | 11000 | 351 | ||||
| Y532 | 69.348 | 4.33 | 14934.17 | 14300 | 352 | ||||
| 8 | BIB82 | 139.113 | 2.16 | 29501.49 | 6.0 | 27400 | 5.7 | 350 | 330 |
| Y988 | 68.642 | 4.37 | 13380.9 | 12600 | 315 | ||||
| BI370 | 43.857 | 6.84 | 8660.63 | 8200 | 325 | ||||
| 9 | BI269 | 81.736 | 5.51 | 15828.78 | 6.0 | 11800 | 4.4 | 313 | 329 |
| Y426 | 54.168 | 8.31 | 11230 | 8210 | 345 | ||||
| 10 | Y803 | 43.374 | 6.92 | 8970.21 | 6.4 | 6420 | 4.8 | 341 | 358 |
| A438 | 135.98 | 2.21 | 30004 | 22100 | 373 | ||||
| Y880 | 115.546 | 2.60 | 24433.7 | 19200 | 359 | ||||
| 11 | YA243 | 49.12 | 6.11 | 10428.1 | 6.4 | 9450 | 5.7 | 371 | 361 |
| Y915 | 48.499 | 6.19 | 10605 | 8720 | 363 | ||||
| Y967 | 123.886 | 2.42 | 26052 | 25000 | 349 | ||||
| 12 | Y64 | 139.449 | 2.15 | 29738.07 | 6.4 | 30500 | 7.1 | 357 | 357 |
| Y726 | 112.212 | 2.67 | 23744 | 28300 | 357 | ||||
| B168 | 135.267 | 2.22 | 29033.01 | 32100 | 357 | ||||
| 13 | Y872 | 149.778 | 2.00 | 31377.06 | 6.4 | 28100 | 6.5 | 350 | 361 |
| Br708 | 116.782 | 2.57 | 24462.06 | 33500 | 355 | ||||
| Br969 | 100.18 | 2.99 | 21967.36 | 17400 | 377 | ||||
| 14 | YB479 | 127.498 | 2.35 | 27282.84 | 6.3 | 25500 | 6.1 | 356 | 350 |
| YA205 | 61.118 | 4.91 | 12889.7 | 10030 | 354 | ||||
| YB330 | 95.388 | 3.15 | 19830.54 | 23900 | 340 | ||||
| 15 | Y928 | 67.261 | 4.46 | 14431.43 | 6.4 | 14700 | 6.9 | 358 | 356 |
| Y974 | 99.194 | 3.02 | 21249.16 | 24900 | 367 | ||||
| Y78 | 90.024 | 3.33 | 18648 | 19800 | 343 | ||||
| 16 | BI919 | 113.104 | 2.65 | 24609.88 | 6.4 | 24100 | 7.5 | 377 | 360 |
| Y640 | 161.115 | 1.86 | 34615.56 | 48300 | 369 | ||||
| YA762 | 119.56 | 2.51 | 24368.42 | 28200 | 335 | ||||
| 17 | YA626 | 94.57 | 4.76 | 20189.16 | 6.5 | 22400 | 6.6 | 352 | 360 |
| YA29 | 136.8 | 3.29 | 29716.84 | 28000 | 368 | ||||
Sequence QC
Illumina HiSeq paired-end BCL files were converted to FASTQ files by the standard Illumina protocol (bcl2fastq) in order to remove the adapters, the known Illumina artefacts.
Read Mapping, BAM file post processing
The trimmed, adapter removed FASTQ files were mapped to the GRCh37 reference genome by the Burrows-Wheeler Aligner (version 0.7.9a-r786) with the BWA MEM paired-end mapping algorithm using the following parameters: -M -w 150 -d 80 -c 2000. This mapper is generally recommended for high-quality queries as it is fast, and it allows gapped alignments which are essential for the accurate identification of SNV and insertion/deletions (indels; http://genestack-user-tutorials.readthedocs.io/tutorials/WES_data_analysis/index.html). Duplicates were marked by the Picard tools (version 1.113) MarkDuplicates algorithm. The raw BAM files (Data Citation 1) were realigned and base quality recalibrated by Genome Analysis Toolkit (GATK version: 3.3-0-g37228af).
Sequence QC
FastQC (version v0.11.2) was used to generate quality reports (Data Citation 2) using the aligned and base quality recalibrated BAM files.
Variant calling
The variants were called by the GATK HaplotypeCaller (version: 3.3-0-g37228af) algorithm using the GVCF mode cohort analysis according to the Best Practice Guidelines15–17. Raw Variants were recalibrated by GATK VariantRecalibrator according to the best practice recommendations using the HapMap, Omni, dbSNP, 1000 G, Mills datasets for training included in the GATK GRCh37 bundle resulting analysis ready variants (Data Citation 2).
Data records
The raw FASTQ files, the GRCh37 aligned BAM files, and the variants in VCF 4.1 format associated with the samples analysed in this study are available on request at EGA (Data Citation 1). We also deposited the QC metric files of Picard MarkDuplicate, and QC reports of FastQC of the analysis ready Bam files at Figshare (Data Citation 2) and in Supplementary Table 1.
Technical validation
Quantitation of the purified DNA samples
The isolated DNA samples were quantified by Qubit (Life Technologies) fluorometer using Qubit dsDNA HS (High Sensitivity) Assay Kit, which is highly selective for double-stranded DNA over RNA and is designed to be accurate for initial sample concentration ranging from 10 pg/μl to 100 ng/μl. DNA samples were diluted to 4 ng/μl with 1X Low TE Buffer. NanoDrop (Thermo Fischer Scientific) measurements were also performed to assess quantity and quality of DNA, 260:280 and 260:230 ratios greater to 1.8 were accepted.
Quality control of the sheared DNA samples
The quality of the sheared DNA samples (200 ng of each) were checked prior to downstream analysis, using the Agilent Bioanalyser 2100 (Agilent Technologies), and High Sensitivity DNA chip and reagent kit. The electropherogram showed a DNA fragment size peak (for each of the samples) at around 150 bps (Fig. 2).
Quality check of the amplified samples
Agilent Bioanalyser 2100 (Agilent Technologies) and DNA 1000 Assay were used for the quality and quantity control of the libraries after PCR. The sample fragments sizes were between 225 and 275 bps (Fig. 3).
Assessment of the quantity and quality of the indexed library DNAs
The quality of the amplified, indexed libraries were determined before multiplexing using the Agilent Bioanalyser 2100 (Agilent Technologies) and High Sensitivity DNA Assay (Table 3). The peak of the DNA fragment size was between 200–450 bps for each of the samples with an average of 355 bps (Fig. 4, Table 3).
Technical replicates
Two independent technical replicate libraries were prepared and sequenced from the same patients (three suicide victims and two controls; Table 4).
Table 4. Summary of the technical replicates.
| PATIENT | BRAIN REGION | ||
|---|---|---|---|
| Different brain tissue samples (originating from the same region, but from different tissue pieces or different regions) from five patients (3 suicide victims and 2 controls) were used as technical parallels for sequencing. | |||
| #194 | control | occipital cortex | |
| occipital cortex | |||
| #230 | control | somatomotor cortex | |
| occipital cortex | |||
| #148 | suicide | occipital cortex | |
| occipital cortex | |||
| #143 | suicide | occipital cortex | |
| occipital cortex | |||
| #158 | suicide | cerebellar cortex | |
| occipital cortex | |||
Quality control of raw reads
Illumina BCL files were converted to FASTQ files by the standard Illumina protocol to remove low-quality reads and adaptors.
Quality control of mapped reads
We used Picard tools MarkDuplicate algorithm to identify PCR or optical duplicates. According to MarkDuplicates statistics, we had very low percentages of duplicated reads (on average 0.624%) in our cohort. Genome Analysis Toolkit (GATK version 3.3) BaseRecalibrator base quality score recalibration tool was used to generate the final quality recalibrated BAM files for downstream analysis and variant calling according to best practices15–17. To check the quality of our mapped reads we used FastQC to generate quality reports of base quality score recalibrated BAM files. According to the FastQC reports we had no adapter contamination or overrepresented sequences indicating contamination in any of the analysed sequences. Furthermore, the majority of sequences were of high quality (average Phred quality score >34) and even the last two base pairs of the sequences had an average Phred quality score >30. On average 99.9% of reads were mapped, 99% were properly paired, and we had less than 0.1% singletons. The average read counts in the cohort were 117 013 694.9 while the average insert sizes were in the range of 157.7–190.0 base pairs (Table 5 (available online only)). According to the main quality indicators of our dataset all data files fall within acceptable parameters as shown in Table 5 (available online only).
Table 5. Quality control data and statistics from whole-exome sequencing.
| Sample ID | no. reads | mapped reads (%) | duplicates (%) | statistics of the analyzed region (Sure Select V5 All Exon Plus UTR, 15 bp added) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| >Q30 reads (%) | average insert size | insert size StdDev | average depth | >5x cov (%) | >10x cov (%) | >20x cov (%) | >40x cov (%) | ||||||||
| The sample ID of the technical replicates are marked with asterisk. | |||||||||||||||
| A438 | 138452577 | 99.92% | 5.26% | 92.01% | 190.8 | 67.7 | 119.9 | 99.60% | 99.20% | 97.60% | 90.60% | ||||
| B168 | 140611220 | 99.94% | 2.28% | 92.93% | 184.2 | 55.3 | 134.5 | 99.30% | 98.60% | 96.40% | 88.80% | ||||
| BI269* | 187542878 | 99.96% | 9.18% | 0.926137 | 157.93 | 45.71 | 168.597 | 99.10% | 98.40% | 96.40% | 90.50% | ||||
| BI370* | 138284674 | 99.96% | 8.02% | 0.925547 | 159.99 | 48.74 | 125.946 | 98.90% | 97.80% | 94.80% | 85.50% | ||||
| BI919* | 97057952 | 99.94% | 3.96% | 0.901908 | 183.16 | 63.17 | 88.512 | 99.10% | 97.70% | 93.30% | 78.00% | ||||
| BIB82 | 130560882 | 99.94% | 3.30% | 92.36% | 180.8 | 57.1 | 122.1 | 99.50% | 99.00% | 97.20% | 89.90% | ||||
| Br333 | 93171478 | 99.95% | 6.63% | 93.12% | 190.6 | 68.6 | 80.7 | 99.00% | 97.70% | 93.10% | 76.40% | ||||
| Br708 | 178033001 | 99.94% | 3.25% | 93.01% | 177.5 | 54.2 | 166.6 | 99.50% | 99.10% | 98.00% | 94.20% | ||||
| Br857 | 144136294 | 99.96% | 3.43% | 92.79% | 186.4 | 64.2 | 133.5 | 99.40% | 98.90% | 97.20% | 90.80% | ||||
| Br969 | 106686550 | 99.92% | 5.23% | 91.83% | 185.9 | 68.0 | 95.7 | 99.30% | 98.60% | 95.90% | 84.80% | ||||
| BrA206 | 80177041 | 99.96% | 3.44% | 93.32% | 185.3 | 62.6 | 72.9 | 98.80% | 97.20% | 91.40% | 71.70% | ||||
| Y166 | 76810758 | 99.96% | 4.51% | 93.30% | 174.9 | 52.4 | 69.8 | 99.10% | 97.60% | 91.80% | 71.30% | ||||
| Y292 | 95054488 | 99.81% | 2.32% | 92.12% | 180.9 | 57.4 | 88.5 | 99.20% | 98.10% | 94.30% | 80.00% | ||||
| Y316 | 87607856 | 99.95% | 9.49% | 92.09% | 174.1 | 56.7 | 74.9 | 99.20% | 98.20% | 93.90% | 76.60% | ||||
| Y331 | 96374496 | 99.82% | 7.61% | 90.97% | 179.4 | 57.5 | 85.0 | 99.30% | 98.40% | 94.80% | 80.30% | ||||
| Y375 | 99837105 | 99.93% | 7.35% | 91.82% | 181.6 | 63.8 | 86.9 | 99.40% | 98.60% | 95.60% | 82.20% | ||||
| Y393 | 126108409 | 99.94% | 6.30% | 93.40% | 189.4 | 59.8 | 110.9 | 99.60% | 99.00% | 97.00% | 88.10% | ||||
| Y421 | 100073380 | 99.93% | 5.74% | 92.01% | 186.1 | 58.5 | 89.2 | 99.20% | 98.30% | 94.80% | 81.10% | ||||
| Y426 | 165152957 | 99.94% | 24.86% | 90.08% | 175.2 | 60.6 | 113.5 | 99.60% | 99.20% | 97.90% | 91.50% | ||||
| Y440* | 120135303 | 99.78% | 13.84% | 0.900957 | 177.41 | 62.88 | 97.494 | 99.40% | 98.80% | 96.30% | 85.80% | ||||
| Y482 | 83982287 | 99.75% | 21.33% | 90.80% | 188.2 | 70.9 | 59.7 | 99.10% | 97.60% | 91.10% | 67.20% | ||||
| Y516 | 105766222 | 99.92% | 6.94% | 92.24% | 183.6 | 56.0 | 93.3 | 98.80% | 97.50% | 93.40% | 79.60% | ||||
| Y520 | 116261151 | 99.80% | 5.07% | 91.62% | 175.6 | 58.4 | 106.2 | 99.20% | 98.30% | 95.50% | 85.10% | ||||
| Y532 | 104607488 | 99.92% | 5.72% | 91.77% | 185.9 | 56.8 | 93.6 | 99.40% | 98.60% | 95.50% | 82.80% | ||||
| Y558 | 99200163 | 99.79% | 4.78% | 91.02% | 180.2 | 60.6 | 89.0 | 99.00% | 97.70% | 93.70% | 79.80% | ||||
| Y591 | 81128182 | 99.79% | 3.15% | 91.46% | 189.9 | 67.4 | 74.3 | 99.20% | 98.00% | 93.10% | 74.50% | ||||
| Y64 | 133401783 | 99.93% | 2.80% | 92.41% | 180.0 | 58.0 | 127.8 | 99.40% | 98.80% | 96.60% | 88.50% | ||||
| Y640 | 129387141 | 99.94% | 2.19% | 89.53% | 176.1 | 55.8 | 125.0 | 99.40% | 98.70% | 96.40% | 88.10% | ||||
| Y645 | 114470684 | 99.95% | 9.44% | 93.06% | 180.0 | 61.7 | 96.1 | 99.50% | 98.80% | 96.30% | 85.30% | ||||
| Y724 | 124302299 | 99.84% | 5.34% | 92.01% | 176.0 | 59.8 | 114.0 | 99.30% | 98.60% | 96.30% | 87.60% | ||||
| Y726 | 101306831 | 99.93% | 3.54% | 91.98% | 177.7 | 60.4 | 94.1 | 99.30% | 98.40% | 95.20% | 82.90% | ||||
| Y78 | 89614381 | 99.94% | 5.49% | 90.99% | 176.9 | 56.5 | 79.4 | 98.90% | 97.40% | 92.10% | 74.20% | ||||
| Y787 | 114795165 | 99.80% | 3.63% | 92.68% | 197.0 | 71.2 | 103.7 | 99.50% | 98.90% | 96.70% | 87.20% | ||||
| Y803 | 120331211 | 99.93% | 12.18% | 91.54% | 176.2 | 58.9 | 100.5 | 99.40% | 98.70% | 96.20% | 86.00% | ||||
| Y872 | 141008850 | 99.93% | 2.49% | 92.74% | 175.4 | 52.1 | 137.4 | 99.30% | 98.60% | 96.50% | 89.10% | ||||
| Y880 | 110514415 | 99.90% | 4.95% | 92.46% | 181.6 | 65.7 | 85.9 | 99.20% | 98.10% | 93.90% | 78.60% | ||||
| Y915 | 101630477 | 99.88% | 12.02% | 93.19% | 192.2 | 65.7 | 82.7 | 99.30% | 98.40% | 94.80% | 79.80% | ||||
| Y919 | 88647901 | 99.96% | 3.69% | 93.03% | 179.2 | 57.4 | 80.1 | 99.30% | 98.20% | 93.90% | 77.50% | ||||
| Y928 | 94852671 | 99.94% | 4.20% | 91.06% | 182.6 | 61.4 | 86.2 | 98.90% | 97.40% | 92.60% | 76.90% | ||||
| Y967 | 117471985 | 99.93% | 3.69% | 93.49% | 179.6 | 58.5 | 108.5 | 99.50% | 98.80% | 96.40% | 86.80% | ||||
| Y974 | 119874575 | 99.94% | 4.46% | 89.84% | 178.6 | 61.7 | 109.1 | 99.60% | 98.90% | 96.70% | 87.50% | ||||
| Y988 | 127546449 | 99.96% | 5.33% | 92.30% | 157.7 | 46.3 | 120.2 | 98.70% | 97.50% | 93.90% | 83.30% | ||||
| YA205 | 96160167 | 99.95% | 7.07% | 90.93% | 177.5 | 61.1 | 84.7 | 99.20% | 98.10% | 93.80% | 78.30% | ||||
| YA243 | 129654716 | 99.92% | 9.35% | 93.46% | 186.4 | 69.1 | 110.0 | 99.10% | 98.10% | 94.80% | 83.50% | ||||
| YA29 | 170816106 | 99.91% | 2.96% | 83.51% | 182.1 | 55.8 | 161.4 | 99.30% | 98.70% | 97.20% | 92.20% | ||||
| YA626 | 195722635 | 99.91% | 6.58% | 84.44% | 179.3 | 49.7 | 175.8 | 99.60% | 99.10% | 97.90% | 93.90% | ||||
| YA762* | 107955782 | 99.95% | 4.02% | 0.89896 | 168.69 | 49.99 | 101.205 | 99.20% | 98.30% | 95.30% | 84.20% | ||||
| YB330 | 106184066 | 99.94% | 4.54% | 91.56% | 178.1 | 51.5 | 98.6 | 99.00% | 98.00% | 94.70% | 82.90% | ||||
| YB479 | 105205966 | 99.94% | 2.95% | 90.97% | 181.0 | 56.4 | 98.6 | 99.40% | 98.50% | 95.30% | 83.10% | ||||
Variant calling
The identification of different genomic variants including SNVs, indels, multiple nucleotide variants (MNVs), Genome Analysis Toolkit (GATK version 3.3) HaplotypeCaller algorithm was used with the GVCF mode cohort analysis according best practice15–17. HaplotypeCaller was run with the “-stand_emit_conf 10 -stand_call_conf 30” parameters. Variants were recalibrated by GATK VariantRecalibrator according to the best practice recommendations using the HapMap, Omni, dbSNP, 1000 G, Mills datasets for training included in the GATK GRCh37 bundle. We used 100.0, 99.9, 99.0, 90.0 tranches for variant recalibration and the 99.0 truth sensitivity level was used at ApplyRecalibration algorithm to identify QC PASS-ed variants.
Usage Notes
Ethical approval for the study was obtained from the following institutional review boards:
The brain samples were collected by the Human Brain Tissue Bank (HBTB; Semmelweis University, Budapest). The activity of the HBTB has been authorized by the Committee of Science and Research Ethic of the Ministry of Health Hungary (ETT TUKEB: 189/KO/02.6008/2002/ETT) and the Semmelweis University Regional Committee of Science and Research Ethic (No. 32/1992/TUKEB) to remove human brain tissue samples, collect, store and use them for research.
The HBTB is a member of the BrainNet Europe II. consortium. The activity of the HBTB is in accordance with the ethic and safety rules and the scientific requirements of the consortium.
Human brain microdissection procedures were approved by the Regional and Institutional Committee of Science and Research Ethics of Scientific Council of Health (the number of the ethical license: 34/2002/TUKEB-13716/2013/EHR, Budapest, Hungary) and the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Conducting genetic testing on tissue samples and then sending them abroad has been authorized by the Semmelweis University Regional Committee of Science and Research Ethics (No. 34/2002/TUKEB).
The Institutional Review Board (IRB) at Stanford has made the following determination about the activity of the study, titled “Exome sequencing analysis of suicidal behavior using high-throughput DNA sequencing”, based on Office for Human Research Protections (OHRP) and Food and Drug Administration (FDA) regulations and guidance: this project does not require submission to the Stanford IRB, because the study does not involve human subjects, it is not about living individuals (NOT-H3 1/1; November 26, 2014).
The following explains how to access the dataset provided in this manuscript
The data is made available through the European Genome-phenome Archive (EGA). Search for the Data Access Committee (DAC) ID of our study (EGAD00001004184) at the homepage of EGA (https://ega-archive.org/). This will show detailed information on our dataset, data providers, DAC and any other related documentation. You can also find information about who to contact about access to this dataset. Access to data will be allowed to qualified researchers for appropriate health related studies.
A DAC group is responsible for the access of the datasets deposited in EGA.
Request for data access will be referred directly to our Data Access Committee: https://ega-archive.org/datasets/EGAD00001004184.
Data access decisions can be passed to the EGA in two ways: 1) By emailing helpdesk@ega-archive.org with the email address of each applicant and confirmation of the dataset/s to provide access. The EGA will then create an EGA account with the relevant access permissions. 2) By using the EGA DAC Admin tools (available to DAC’s dealing with more than 5 data access applications/month).
If you need to request access to this data set, please contact:
DAC for Hungarian human exome team (Department of Medical Biology, University of Szeged, Hungary)
Contact person: Dr. Zsolt Boldogkői
Email: boldogkoi.zsolt@med.u-szeged.hu
Applicants will be asked to complete the Data Access Agreement (DAA) (including a brief summary of the proposal, proposed usage of the dataset, the storage of data, so the DAC can determine if the planned usage falls within the consents) and to agree to the terms and conditions of the DAA. The DAA must be signed by the applicant and the relevant Head of Department, or equivalent. If applications include a named collaborator then the collaborator’s Institution must sign and submit a separate DAA. A template DAA can be found on the EGA website: https://www.ebi.ac.uk/ega/sites/ebi.ac.uk.ega/files/documents/Example%20DAA.doc.
Additional information
How to cite this article: Tombácz, D. et al. Whole-Exome Sequencing Data of Suicide Victims who had Suffered from Major Depressive Disorder. Sci. Data. 6:190010 https://doi.org/10.1038/sdata.2019.10 (2019).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was funded by the European Union and the Hungarian State, co‐financed by the European Social Fund under the framework of TÁMOP [4.2.2/B-10/1- 2010-0012] and the Swiss-Hungarian Cooperation Programme [SH/7/2/8] to ZBo. The work also received support from the Bolyai János Scholarship of the Hungarian Academy of Sciences to DT, and was supported by the NIH Centers of Excellence in Genomic Science (CEGS) Center for Personal Dynamic Regulomes [5P50HG00773502] to MS and by the High-throughput sequencer for multi-scale genomic studies [1S10OD02014101] to MS.
Footnotes
The authors declare no competing interests.
Data Citations
- 2018. European Genome-phenome Archive. EGAD00001004184
- Tombacz D., et al. . 2018. Figshare. [DOI]
References
- Hawton K. & van Heeringen K. Suicide. The Lancet 373, 1372–1381 (2009). [DOI] [PubMed] [Google Scholar]
- National Institute of Mental Health. Statistics/Suicide, https://www.nimh.nih.gov/health/statistics/suicide/leading-causes-of-death-ages-1-85-in-the-us.shtml (2018).
- Haw C. & Hawton K. Suicide is a complex behaviour in which mental disorder usually plays a central role. Aust. N. Z. J. Psychiatry 49, 13–15 (2015). [DOI] [PubMed] [Google Scholar]
- Platt S. Suicide in men: what is the problem? Trends - Urology & Men’s Health 8, 9–12 (2017). [Google Scholar]
- Haw C. et al. Suicide Clusters: A Review of Risk Factors and Mechanisms. Suicide Life Threat. Behav. 43, 97–108 (2013). [DOI] [PubMed] [Google Scholar]
- Tombácz D. et al. High-Coverage Whole-Exome Sequencing Identifies Candidate Genes for Suicide in Victims with Major Depressive Disorder. Sci. Rep. 7, 7106, 10.1038/s41598-017-06522-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradus J. L. et al. Acute stress reaction and completed suicide. Int. J. Epidemiol. 39, 1478–1484 (2010). [DOI] [PubMed] [Google Scholar]
- Brent D. What family studies teach us about suicidal behaviour: Implications for research, treatment, prevention. Eur. Psychiatry 25, 260–263 (2010). [DOI] [PubMed] [Google Scholar]
- Harris E. C. & Baraclough B. Suicide as an outcome for mental disorders. A meta-analysis. Br. J. Psychiatry 170, 205–228 (1997). [DOI] [PubMed] [Google Scholar]
- Tang D. et al. Reference genotype and exome data from an Australian Aboriginal population for health-based research. Sci. Data. 3, 160023, 10.1038/sdata.2016.23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merali Z. et al. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J. Neurosci. 24, 1478–1485 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo A. et al. The use of regression methods for the investigation of trends in suicide rates in Hungary between 1963 and 2011. Soc. Psychiatry Psychiatr. Epidemiol 50, 249–256 (2015). [DOI] [PubMed] [Google Scholar]
- Rihmer Z., Gonda X., Kapitany B. & Dome P. Suicide in Hungary-epidemiological and clinical perspectives. Annals of General Psychiatry 12, 21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7 A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J. Allergy Clin. Immunol. 132, 656–664.e17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G. A. et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinfrmatics. 43, 11.10.1–11.10.33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2018. European Genome-phenome Archive. EGAD00001004184
- Tombacz D., et al. . 2018. Figshare. [DOI]