

Abstract
Background
Methyltransferase like 3 (METTL3) is an RNA methyltransferase implicated in mRNA biogenesis, decay, and translation control through N6-methyladenosine (m6A) modification.
Methods
To find new treatment strategies for lung cancer and to elucidate the mechanism underlying the phenomenon, we treated the human lung cancer cell lines A549 and H1299 to investigate the effect of METTL3 on lung cancer.
Results
We observed that knockdown of METTL3 inhibited the survival and proliferation of A549 and H1299 cells. The migration and proliferation of both cell lines were significantly decreased, and the apoptosis was induced in comparison with control cells. These results were further confirmed by the transfection of miRNA of METTL3 increased the Bax/Bcl-2 ratio in A549 and H1299 cells, which is a sign that mitochondrial apoptotic pathway was triggered. The PI3K/Akt pathway is implicated in cell growth and survival and we also observed that knockdown of METTL3 changed the expression and phosphorylation of proteins of PI3K signaling pathway members. Further, our results demonstrated that miR-600 inhibited the expression of METTL3 and reversed the positive effect of METTL3 on NSCLC progression, indicating an miR-600/METTL3 pathway in NSCLC.
Conclusion
These data suggested that miR-600 inhibited lung cancer via down-regulating METTL3 expression, and knockdown of METTL3 might be used as a novel strategy for lung cancer therapy.
Keywords: METTL3, miRNA, lung cancer, A549, H1299, apoptosis, PI3K pathway
Introduction
Lung cancer is the most common primary malignant tumor of the lung that causes cancer-related deaths for both women and men.1–3 Despite the optimal surgical management, the probability of 5-year survival remains low and is closely related to the stage of disease, ranging from about 90%, for stage IA, to 60%, for stage II.4,5
Methyltransferase like 3 (METTL3) is an RNA methyltransferase that modifies the biological occurrence, decay, and translation control of mRNA by n6-methylglycine (m6A).6 By immunofluorescence analysis, METTL3 (and other complex components) was mainly observed in nuclear spots, although m6A methyltransferase activity was detected in both nuclear extract and cytoplasmic extract.6–8 METTL3 promotes translation of certain mRNAs including epidermal growth factor receptor (EGFR) and the Hippo pathway effector TAZ in human cancer cells.9 The combination of METTL3 and ribosome promotes the translation of cytoplasm. Depletion of METTL3 inhibits translation, while both wild-type and catalytic activity of METTL3 promotes translation when combines with reporter gene mRNA. Mechanically, METTL3 enhances mRNA translation by interacting with the translation initiation mechanism.9 The Cancer Genome Atlas datasets http://cancergenome.nih.gov show that the expression of METTL3 mRNA is significantly elevated in lung adenocarcinoma (LUAD) and colon adenocarcinoma compared with the normal tissues. Also, Lin et al showed that METTL3 expression is elevated in LUAD via promoting growth, survival, and invasion of human lung cancer cells.9 These reports make METTL3 to be a potential target to suppress oncoprotein expression and work as a possible key for cancer therapy.9,11,12 In our study, we reported that knockdown of METTL3 can inhibit the growth of lung cancer by inducing apoptosis via PI3K/AKT pathway. These data suggest that silencing METTL3 may be a novel strategy for lung cancer therapy. miRNAs are a class of endogenous small noncoding RNAs of 18–25 nucleotides in length that negatively regulate gene expression at the post-transcriptional level. The miRNA binds complementarily to the 3′-UTR region of the target gene mRNA, resulting in mRNA reduction or inhibition of translation.13 So far, nearly 2,000 miRNAs have been found in the human genome, and 60% of gene expression is regulated by miRNAs.14,15 In recent years, the role of miR-600 has gradually been discovered in a variety of tumors, including acute myeloid leukemia, colorectal, cervical, and breast cancer.16–19 In this research, we proved that miR-600 inhibited the expression of METTL3 and reversed the positive effect of METTL3 on NSCLC progression.
Materials and methods
Agents
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis kit was purchased from 4A Biotech Company (Cat# FXP018-100, Beijing, People’s Republic of China). Ultrapure RNA extraction kit, HiFiScript cDNA Synthesis Kit, fluorescence quantitative PCR kit UltraSYBR Mixture, RIPA Lysis Buffer, BCA Protein Assay Kit, Protease Inhibitor Cocktail were all purchased from Beijing Kangwei Century company (CwBio, Beijing, People’s Republic of China). Primers were synthesized from Genewiz company (Beijing, People’s Republic of China). Matrigel was ordered from Becton, Dickinson and Company (Cat# 356234, Franklin Lakes, NJ, USA).
Primary antibodies, including anti-METTL3 (Cat# ab195352, 1:1,000); ß-catenin (Cat# ab32572, 1:1,000); Bcl (Cat# ab32124, 1:1,000); Caspase3 (Cat# ab2302, 1:1,000); Bax (Cat# ab32503, 1:1,000); AKT (Cat# ab8805, 1:1,000); p-AKT (Cat# ab38449, 1:1,000); Cyclin D1 (Cat# ab134175, 1:1,000); P70 (Cat# ab109393, 1:1,000); and GAPDH (Cat# ab9485, 1:5,000), were purchased from Abcam (Cambridge, United Kingdom). Horseradish peroxidase sheep anti-rabbit/mouse secondary antibodies (1:5,000) were ordered from PTG Company (Bellevue, WA, USA).
Cell culture and transfection
Human lung cancer cell lines A549 and H1299 were ordered from the cell bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). Cells were cultured in DMEM medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 0.1 mg/mL streptomycin at 37°C with 5% CO2. When the cells in the six-well plate reach logarithmic stage, the transfection was carried out according to the instructions of Lipofectamine2000 transfection kit. The METTL3 interfere sequence is 5′- GCTGCACTTCAGACGAATT-3′. After 24 hours, the expression of transferred plasmid can be observed or subsequent experiments can be carried out.
Fluorescence quantitative PCR
Total RNA was extracted with Ultrapure RNA extraction kit and cDNA was synthesized with HiFiScript cDNA Synthesis Kit. The expression of METTL3 was detected by using fluorescence quantitative PCR. The primers are listed as follows:
METTL3:
Forward: 5′- CAAGCTGCACTTCAGACGAA-3′
Reverse: 5′- GCTTGGCGTGTGGTCTTT-3′.
The expression volume was calculated by 2−DCt method from three independent experiments.
Western blot
The transfected cells were incubated on a six-well plate for 90%–95% fusion. After 48 hours of transfection, cells were collected and lysed with RIPA buffer. Twenty micrograms of protein samples were loaded in each lane containing 10% SDS-PAGE gel. After transfer, the polyvinylidene fluoride (PVDF) membrane was blocked with 5% of skim milk for 1 hour at room temperature and then incubated with primary antibody overnight at 4°C. The PVDF membrane was incubated at room temperature with secondary antibody for 1 hour. After flushing, the film was developed with the ECL substrate. Images were taken using chemiluminescent darkroom development.
Cell proliferation and viability assays
Cultured cells were made into a suspension by trypsin. About 1,000 cells were seeded in each well of the 96-well plate. Cell viability was detected by adding 10 µL CCK8 reagent every 24 hours. An enzyme standard instrument was used to detect OD value of the excitation light at 450 nautical miles. Diffusion curve with OD value was drawn.
Cell invasion assays
For invasion assay, the chilled Matrigel was diluted with serum-free medium at 1:6 and 100 µL was applied to the transwell chamber, and incubated for 4 hours.
After transfection, cells were resuspended in serum-free medium. 1×104 cells in 100 µL serum-free medium were seeded into the upper chamber. Five hundred microliters completed medium was added to the bottom wells to stimulate migration or invasion. After incubation, the cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature and stained with 0.1% crystal violet for 5 minutes. The cells were observed, imaged, and counted under a microscope.
Cell apoptosis analysis with flow cytometer
After 24 hours of serum-free culture, the cells were resuspended in 1× binding buffer at a concentration of 3−5 × 105 cells/mL. Five microliters of Annexin V FITC was added to the 100 µL cell suspension, brewing in the darkness at 25°C, followed by adding 10 µL 20 µg/mL of double staining of PI for 2 minutes. The analysis of results was performed by BD FACSCanto II, BD Biosciences, San Jose, CA, USA. Apoptosis rate was calculated by BD FACSDiva software.
Statistical analysis
SPSS 18.0 software was used for statistical analysis. Our results were denoted as plus or minus standard deviation. The data represented three separate experiments. The student’s t-test was used to determine the meaning of all pairs of interest comparisons. P<0.05 was statistically significant.
Results
Knockdown of METTL3
We first detected the efficiency of METTL3 knockdown in both A549 and H1299 cells. The mRNA expressions of METTL3 with the transfection of METTL3-specific siRNA were decreased significantly compared with that of negative siRNA (the NC group) in both A549 (Figure 1A, left) and H1299 (Figure 1A, right) cells. Accordingly, the protein levels of METTL3 in the METTL3-KD groups were also decreased compared with the NC group in both A549 and H1299 cells (Figure 1B, left and right).
Figure 1.

miR-600 inhibits the expression of METTL3.
Notes: (A)The mRNA expression of METTL3 in A549 and H1299 cells with the transfection of METTL3-specific siRNA (the METTL3-KD group) decreasing significantly compared with that of negative siRNA (the NC group). (B) METTL3 protein level of the METTL3-KD group also decreases in comparison to the NC group in both A549 and H1299 cells. (C) The predicted binding sites of METTL3 and miR-600. (D) Luciferase reporter gene assay was performed to confirm the combination of METTL3 and miR-600. (E) The METTL3 mRNA level was detected by using qPCR. (F) The METTL3 protein level was detected by using Western blot. (G) The values of the band intensity represent the densitometry estimation of each band normalized to GAPDH. Experiments in this figure were all performed in triplicate (*P<0.05).
Bioinformatics analysis from online software target-scan revealed that miR-600 had a putative binding site for METTL3 3′-UTR. To verify it, METTL3 3′-UTR mutant (METTL3-mut) and wild-type (METTL3-WT) vectors were constructed to transfect into miR-600 upregulated cells or negative control cells. The verification results of bi-luciferase showed that METTL3 was a target gene of miR-600. The binding sites of METTL3 and miR-600 are shown in Figure 1C. To confirm this result in our experiment, we detected the effect of miR-600 on the METTL3 overexpressed cells. Luciferase reporter gene assay was performed to confirm the combination of METTL3 and miR-600. Mir-600 significantly decreased the expression of exogenous wild-type METTL3 but had no effect on mutated METTL3 (Figure 1D). The qPCR and Western blot results showed that miR-600 inhibited the expression of METTL3 in both A549 and H1299 cells (Figure 1E–G).
Silencing METTL3 inhibits mRNA m6A level in lung cancer cells
We next detected the m6A RNA methylation levels. Our results showed the m6A content in the total RNA of the METTL3-KD group was significantly reduced compared with that of the NC group in both A549 and H1299 cells (Figure 2A, B). These results indicated that we successfully knocked down the METTL3 in both cell lines.
Figure 2.
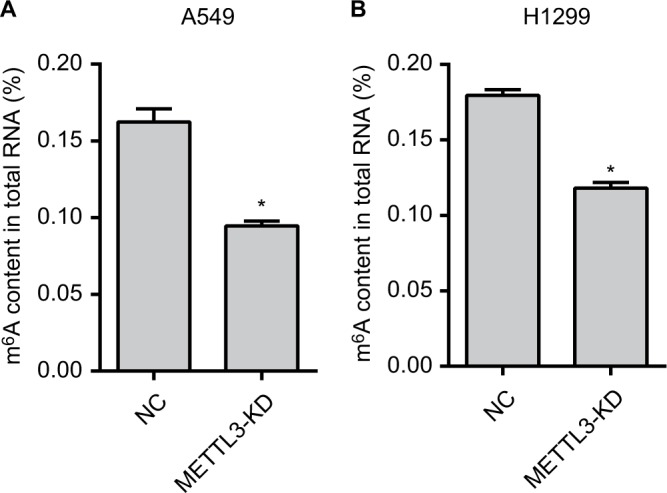
The m6A RNA methylation levels are reduced.
Notes: The m6A content in the total RNA of the METTL3-KD group was significantly reduced than that of the NC group in A549 (A) and H1299 (B) cells. Experiments in this figure were all performed in triplicate (*P<0.05).
Reduction of METTL3 inhibited the proliferation of lung cancer cells
We investigated the effect of METTL3 on miR-600 expression. Results of qPCR showed that knocking down METTL3 did not affect the level of miR-600 in both A549 and H1299 cells (Figure 3A).
Figure 3.

Knockdown of METTL3 reduces the viability of lung cancer cells.
Notes: (A) Results of qPCR showed that knocking down METTL3 did not affect miR600 expression in both A549 and H1299 cells. (B) The proliferation curves of A549 (left) and H1299 (right) cells transfected with METTL3-specific siRNA were detected by using CCK8. (C, D) Plate clonality assay. Experiments in this figure were all performed in triplicate (*P<0.05 vs NC; #P<0.05 vs METTL3-KD).
As siRNA of METTL3 efficiently decreased the expression of METTL3, we then detected the effect of siRNA of METTL3 on the proliferation of lung cancer cells. CCK8 and colony formation assays were performed to evaluate the effect of the reduction of METTL3 in cell proliferation. As shown by CCK8 assay, knockdown of METTL3 significantly decreased the viability of both A549 and H1299 cells (Figure 3B). Also, colony formation assay showed that, transfection of siRNA of METTL3 caused lower rate proliferation of both A549 and H1299 cells than si-NC transfection control (Figure 3C, D). Furthermore, we reexpressed METTL3 in METTL3-KD cells to clearly demonstrate that the observed effects were due to METTL3 silencing. Consistent with the expected results, METTL3 overexpression reversed the inhibition effect of METTL3 on NSCLC progression (Figure 3B–D).
Knockdown of METTL3 inhibited migration and invasion of lung cancer cells
Transwell assays were performed to investigate the effect of siRNA of METTL3 on cancer cell migration and invasion in vitro. The migration and invasion assay provides an in vitro system to study cell invasion of malignant and normal cells. Crystal violet stained cells on siRNA of METTL3 transfected wells were much less than siNC groups (Figure 4A left and B left). The numbers of migration cells and invasion cells were significantly decreased in siRNA of METTL3 transfected wells in both A549 and H1299 cells (Figure 4A right and B right).
Figure 4.

Transwell assay of lung cancer cells.
Notes: Knockdown of METTL3 inhibited the invasion of A549 (A) and H1299 (B) cells. Experiments in this figure were all performed in triplicate (*P<0.05).
Knockdown of METTL3 induced apoptosis of lung cancer cells by affecting expression of apoptosis-related proteins
The apoptosis of A549 and H1299 cells was analyzed by cell flow cytometry. Cells were transfected with siRNA of METTL3 or control siRNA for 48 hours and then double-stained by Annexin V and PI. Annexin V-positive and PI-negative staining identified apoptotic cells. The transfection of siRNA of METTL3 significantly increased the apoptosis rate of both A549 and H1299 cells compared with those transfected with control siRNA (Figure 5A, B).
Figure 5.

Knockdown of METTL3 induced apoptosis in lung cancer cells.
Notes: Knockdown of METTL3 promoted the apoptosis of A549 (A) and H1299 (B) cells. (C) Western blot was performed to analyze the expression of apoptosis-related protein, including BCL2, active caspase 3, Bax, pro- and cleaved-PARP. GAPDH served as a loading control. (D) The values of the band intensity represent the densitometry estimation of each band normalized to GAPDH (*P<0.05).
Abbreviation: PI, propidium iodide.
To determine the mechanism of apoptosis induced by siRNA of METTL3, we performed Western blot analysis to observe the effect of siRNA of METTL3 on apoptosis-related protein expression. Previous studies have shown that the expression of anti-apoptotic Bcl2 and pro-apoptotic Bax is the key factor to initiate apoptosis through mitochondrion.15,16 The expression of Bcl2 and Bax in the siRNA of METTL3 in A549 and H1299 cells was detected by Western blotting. Compared with the control group, nuclear METTL3 transfection reduced Bcl-2 expression and increased expression of Bax (Figure 5C, D). These results suggest that nuclear METTL3 increases Bax/Bcl2 ratio in lung cancer cells, and knockdown of METTL3 may induce mitochondrial apoptotic pathways in lung cancer cells. Next, we tested the expression of Caspase 3 and PARP. Transfected siRNA of METTL3 strongly increased the expression of cleaved Caspase3 and PARP in both A549 and H1299 cells (Figure 5C, D).
Knockdown of METTL3 suppressed the activation of the PI3K/Akt and ß-catenin STAT3 signaling pathways in lung cancer cells
To elucidate the molecular mechanism of METTL3 inhibition in lung cancer cells, we examined the effect of siRNA of METTL3 on the PI3K/AKT pathway of A549 and H1299 cells. The PI3K pathway is involved in many cellular processes, such as the promotion of cell growth, survival, and angiogenesis. Once activated on the cell membrane, PI3K phosphorylates PIP2, resulting in accumulation of PIP3.17 This lipid second messenger recruits AKT and PDK1 to the cell membrane, where AKT is phosphorylated by PDK1.18 Phosphorylation of AKT regulates cellular processes through the phosphorylation of a series of substrates, including the Bax/bcl-2-related death promoter.19 The PI3K/AKT pathway can be negatively regulated by the tumor suppressor gene PTEN.20
Our results showed that, in the siRNA of METTL3 transfected group, no change was observed in the expression of AKT while the phosphorylated form of p-AKT decreased (Figure 6A, B). These data suggested that knockdown of METTL3 inhibited PI3K and caused the decrease of phosphorylated form of AKT. The expression of P70 was also decreased by the transfection of siRNA of METTL3 (Figure 6A, B). To further confirm this conclusion, we detected the effect of siRNA of METTL3 on the expression levels of ß-catenin signaling pathway members including ß-catenin and Cyclin D1. Our results showed that the levels of ß-catenin and Cyclin D1 were decreased (Figure 6A, B). The decreased Cyclin D1 expression in METTL3 siRNA transfected group is consistent with the inhibition of AKT phosphorylation.
Figure 6.

Knockdown of METTL3 induced apoptosis through the AKT signaling pathway.
Notes: (A) The expression of AKT pathway related proteins was analyzed by Western blot. (B) The values of the band intensity represent the densitometry estimation of each band normalized to GAPDH. Experiments in this figure were all performed in triplicate (*P<0.05).
It has been reported that METTL3 promotes the translation of certain mRNAs including EGFR and the Hippo pathway effector TAZ in human cancer cells and METTL3 depletion results in decreased protein levels of EGFR, TAZ, and DNMT3A.9 To further confirm the inhibitory effect of siRNA of METTL3 in lung cancer cell lines, we detected the expressions of its target genes, EGFR, TAZ, and DNMT3A. Our results showed that knockdown of METTL3 significantly inhibited the expression levels of EGFR, TAZ, and DNMT3A in both A549 and H1299 cells (Figure 7A, B).
Figure 7.

Knockdown of METTL3 decreased the expressions of its target genes.
Notes: (A) The expressions of EGFR, TAZ, and DNMT3A proteins were analyzed by Western blot. (B) The values of the band intensity represent the densitometry estimation of each band normalized to GAPDH. Experiments in this figure were all performed in triplicate (*P<0.05).
Abbreviation: EGFR, epidermal growth factor receptor.
MiR-600 reversed the positive effect of METTL3 on NSCLC progression
We detected the effect of miR-600 on the proliferation and invasion in the METTL3 overexpressed A549 and H1299 cells. Overexpression of exogenous METTL3 robustly increased the proliferation of both A549 and H1299 cells (Figure 8A, B, red), while the miR-600 significantly reversed the increased effect of METTL3 (Figure 8A, B, blue). Overexpression of exogenous METTL3 also increased the invasion of both A549 and H1299 cells and the miR-600 decreased this positive effect of METTL3 (Figure 8C–F). Furthermore, we performed the functional assays using miR-600 mimic alone and miR-600 mimic combined with METTL3-KD cells to verify the miR-600/METTL3 pathway. As shown in Figure 9, miR-600 inhibited the proliferation and invasion as well as the expression levels of the related genes in the PI3K/AKT and ß-catenin stat3 signaling pathways. Moreover, miR-600 transfection in METTL3-KD cells has no such effect, suggesting that miR-600 suppressed cell growth by the inhibition of METTL3 expression. Based on the above results, we predict that miR-600 inhibits METTL3 expression and regulates cell proliferation, metastasis, and apoptosis by regulating AKT and β-catenin signaling pathways (Figure 9E).
Figure 8.

miR-600 reversed the positive effect of exogenous METTL3 on lung cancer cells progression.
Notes: The proliferation of A549 (A) and H1299 (B) cells was confirmed by CCK8 assay. (C–F) Transwell assay was performed to confirm the cell invasion in METTL3 and METTL3+ miR-600 cells. Experiments in this figure were all performed in triplicate (*P<0.05 vs NC cells; #P<0.05 vs METTL3 overexpression cells).
Figure 9.

miR-600 suppressed cell growth by the inhibition of METTL3 expression in human lung cancer.
Notes: (A) The proliferation of A549 cells was confirmed by CCK8 assay. (B, C) Transwell assay was performed to confirm the cell invasion in miR-600 and miR-600+METTL3-KD cells. (D) The expression levels of proteins were analyzed by Western blot. The values of the band intensity represent the densitometry estimation of each band normalized to GAPDH. Experiments in this figure were all performed in triplicate (*P<0.05). (E) A model depicting the mechanism of miR-600 suppressed cell growth by the inhibition of METTL3 expression in human lung cancer.
Abbreviation: EGFR, epidermal growth factor receptor.
Discussion
Lung cancer is the most common primary malignant tumor of the lung that causes cancer-related deaths for both women and men.1,2 To better understand the mechanisms underlying lung tumor growth and to find new drugs will result in new diagnostic tools and therapeutic strategies for this deadly disease.
For the first time, we reported the effect of METTL3 on human lung cancer cell lines A549 and H1299 and elucidate the underlying mechanisms. Our results showed that knockdown of METTL3 significantly inhibited the proliferation and migration of both A549 and H1299 cells, which were confirmed by the results from CCK8 and invasion assays. The knockdown of METTL3 induced apoptosis of both A549 and H1299 cells via PI3K/AKT pathway. In addition, another microRNA miR-600 suppressed the increased effects of overexpressed exogenous METTL3 on lung cancers, which double confirmed the conclusion that silencing of METTL3 inhibited lung cancer cells. Our results reflected a potential pathologic role of METTL3 in lung cancer, and it is reported for the first time that knockdown of METTL3 could inhibit the lung cancer cells A549 and H1299 proliferation and migration, and promote apoptosis.
Our Western blot results showed that in the METTL3 knockdown cells, the expression of BCL2 family proteins changed. BAX (BCL2 associated X) is a proapoptotic protein belonging to the BCL2 family. Overexpression of BAX triggers the release of mitochondrial proteins that cleave and thereby activate caspase-3 resulting in apoptosis.20 In our study, we found that the transfection of siRNA of METTL3 increased the Bax/Bcl-2 ratio in lung cancer cells, and the expressions of active Caspase-3 and PARP were increased. These data suggest that loss of METTL3 triggers the apoptosis in a mitochondrial pathway.
To further elucidate the role of METTL3 in lung cancer cells, we studied the PI3K pathway members. The PI3K/Akt/mTOR pathway is implicated in numerous cellular processes ranging from cell growth and survival to the promotion of angiogenesis.21,22 The serine/threonine kinase AKT inhibits apoptosis and mediates cell survival by activating phosphatidylinositol 3-kinase.23 Wnt proteins are a family of secreted proteins that play important roles in the development and maintenance of many tissues. Wnt proteins also control kinase-signaling pathways.24,25 Notably, canonical Wnt3a increases PI3K/Akt activity and increased free β-catenin levels.26 We found that knockdown of METTL3 decreased the amount of phosphorylated form p-AKT, which happened at upstream PI3K.
Our results first reported an important role of METTL3 in promoting translation of oncogenes in human lung cancer cell lines. Knockdown of METTL3 could inhibit the proliferation and migration of lung cancer cells by inducing apoptosis via inhibiting the activation of PI3K signaling pathway. Our findings indicated that miR-600 might be considered as a potent lung cancer inhibitor in therapy. The deep mechanism of METTL3 and conventional cancer therapies also requires deep further study in the future.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Santarpia M, Altavilla G, Pitini V, Rosell R. Personalized treatment of early-stage non-small-cell lung cancer: the challenging role of EGFR inhibitors. Future Oncol. 2015;11(8):1259–1274. doi: 10.2217/fon.14.320. [DOI] [PubMed] [Google Scholar]
- 4.Kris MG, Gaspar LE, Chaft JE, et al. Adjuvant systemic therapy and adjuvant radiation therapy for stage I to IIIA completely resected non-small-cell lung cancers: American Society of Clinical Oncology/Cancer Care Ontario Clinical Practice Guideline Update. J Clin Oncol. 2017;35(25):2960–2974. doi: 10.1200/JCO.2017.72.4401. [DOI] [PubMed] [Google Scholar]
- 5.Chansky K, Detterbeck FC, Nicholson AG, et al. IASLC Staging and Prognostic Factors Committee, Advisory Boards, and Participating Institutions The IASLC lung cancer staging project: external validation of the revision of the TNM stage groupings in the eighth edition of the TNM classification of lung cancer. J Thorac Oncol. 2017;12(7):1109–1121. doi: 10.1016/j.jtho.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3(11):1233–1247. [PMC free article] [PubMed] [Google Scholar]
- 7.Harper JE, Miceli SM, Roberts RJ, Manley JL. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990;18(19):5735–5741. doi: 10.1093/nar/18.19.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase METTL3 promotes liver cancer progression through YTHDF2 dependent post-transcriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 12.Cai X, Wang X, Cao C, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–19. doi: 10.1016/j.canlet.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 13.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edelstein LC, Bray PF. Small RNAs as potential platelet therapeutics. Handb Exp Pharmacol. 2012;210:435–445. doi: 10.1007/978-3-642-29423-5_17. [DOI] [PubMed] [Google Scholar]
- 15.Muhammad N, Bhattacharya S, Steele R, Ray RB. Anti-miR-203 suppresses ER-positive breast cancer growth and stemness by targeting SOCS3. Oncotarget. 2016;7(36):58595–58605. doi: 10.18632/oncotarget.11193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang P, Zuo Z, Wu A, et al. miR-600 inhibits cell proliferation, migration and invasion by targeting p53 in mutant p53-expressing human colorectal cancer cell lines. Oncol Lett. 2017;13(3):1789–1796. doi: 10.3892/ol.2017.5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng Y, Wang KX, Xu H, Hong Y. Integrative miRNA analysis identifies hsa-miR-3154, hsa-miR-7-3, and hsa-miR-600 as potential prognostic biomarker for cervical cancer. J Cell Biochem. 2018;119(2):1558–1566. doi: 10.1002/jcb.26315. [DOI] [PubMed] [Google Scholar]
- 18.El Helou R, Pinna G, Cabaud O, et al. miR-600 acts as a bimodal switch that regulates breast cancer stem cell fate through WNT signaling. Cell Rep. 2017;18(9):2256–2268. doi: 10.1016/j.celrep.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Xing C, Zhou B, et al. A regulatory circuitry between miR-193a/ miR-600 and WT1 enhances leukemogenesis in acute myeloid leukemia. Exp Hematol. 2018;61:59–68.e5. doi: 10.1016/j.exphem.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Eskes R, Antonsson B, Osen-Sand A, et al. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J Cell Biol. 1998;143(1):217–224. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rameh LE, Cantley LC. The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem. 1999;274(13):8347–8350. doi: 10.1074/jbc.274.13.8347. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Wang N, Han S, et al. Dietary compound isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 signaling pathway. PLoS One. 2013;8(7):e68566. doi: 10.1371/journal.pone.0068566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stål O, Pérez-Tenorio G, Åkerberg L, et al. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. 2003;5(2):R37–R44. doi: 10.1186/bcr569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14(15):1837–1851. [PubMed] [Google Scholar]
- 25.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 26.Huang T-S, Li L, Moalim-Nour L, et al. A regulatory network involving β-Catenin, e-Cadherin, PI3k/Akt, and slug balances self-renewal and differentiation of human pluripotent stem cells in response to Wnt signaling. Stem Cells. 2015;33(5):1419–1433. doi: 10.1002/stem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]