

ABSTRACT
In the last decades increasing importance has been attributed to the Insulin/Insulin-like Growth Factor signaling (IIGFs) in cancer development, progression and resistance to therapy. In fact, IIGFs is often deregulated in cancer. In particular, the mitogenic insulin receptor isoform A (IR-A) and the insulin-like growth factor receptor (IGF-1R) are frequently overexpressed in cancer together with their cognate ligands IGF-1 and IGF-2. Recently, we identified discoidin domain receptor 1 (DDR1) as a new IR-A interacting protein. DDR1, a non-integrin collagen tyrosine kinase receptor, is overexpressed in several malignancies and plays a role in cancer progression and metastasis.
Herein, we review recent findings indicating that DDR1 is as a novel modulator of IR and IGF-1R expression and function. DDR1 functionally interacts with IR and IGF-1R and enhances the biological actions of insulin, IGF-1 and IGF-2. Conversely, DDR1 is upregulated by IGF-1, IGF-2 and insulin through the PI3K/AKT/miR-199a-5p circuit. Furthermore, we discuss the role of the non-canonical estrogen receptor GPER1 in the DDR1-IIGFs crosstalk. These data suggest a wider role of DDR1 as a regulator of cell response to hormones, growth factors, and signals coming from the extracellular matrix.
KEYWORDS: breast cancer, discoidin domain receptor 1, insulin-like growth factor axis, insulin receptor isoform A, insulin-like growth factor-2
Introduction
The insulin-like growth factor (IGF) axis has an important role in cellular proliferation and differentiation as well as in protection from apoptosis [1,2]. The ligands of the IGF system are insulin and the two related factors IGF-1 and IGF-2 (IGFs), which interact with specific cell-surface receptors [2]. The insulin receptor (IR) and the IGF-1 receptor (IGF-1R) belong to the receptor tyrosine-kinase (RTK) class 2 (insulin receptor family). They are heterotetrameric receptors with two extracellular subunits (alpha subunits), which include ligand binding domains and two transmembrane beta subunits, which contain the kinase domain. Differential splicing of INSR gene generates two isoforms, named IR-A and IR-B, which differ for the exclusion (IR-A) or the inclusion (IR-B) of exon 11. The differential expression of these two IR isoforms has complex implications in physiology and disease [2]. The two IR isoform have similar high affinity for insulin, but differ for their affinity for IGF-2 and proinsulin, which is high for IR-A and very low for IR-B [3–6]. Notably, the IR-A has been named the ‘fetal IR isoform’ being predominantly expressed during pre-natal life. The IR-A has a prevalent proliferative/survival function and it is overexpressed in numerous tumors where it is activated by autocrine/paracrine IGF-2 [7–10]. On the other end, the IR-B, which is mostly expressed in liver and adipose tissue, binds only insulin with high affinity and prevalently mediates metabolic role [2]. Both IGF-1 and IGF-2 bind to the IGF-1R with higher affinity than insulin. IGF-2, but not IGF-1, binds also the IGF-2 receptor (IGF-2R), which has no tyrosine-kinase activity and works as a scavenger receptor titrating IGF-2 levels in the plasma [11].
In addition to homodimeric IR and IGF-IR, the insulin receptor family includes hybrid IR/IGF-1R receptors (HRs) formed by one IR and one IGF-1R alpha-beta dimer. HRs containing IR-A are predominantly expressed in fetal and neoplastic cells and likely have a prevalent mitogenic potential [12].
Cell proliferation, adhesion, and migration are fundamental biological processes for cancer cell growth and metastasis. The extracellular matrix (ECM) controls cancer cell behavior by regulating several signaling pathways through cell membrane receptors. In addition to confer structural properties to the tissues surrounding the tumor, the ECM can influence cell cancer proliferation, survival, migration, and invasion.
The proteoglycan decorin binds ligands and receptors of the IGF system and modulate IGF-1R and IR-A downstream action [13–15]. Significantly, Decorin preferentially inhibits IGF-2-mediated IR-A biological responses but does not affect insulin- or proinsulin-dependent signaling. Thus, decorin loss may contribute to tumor initiation and progression in neoplasms, which depend on an IGF-2/IR-A autocrine loop [13].
The integrins family constitutes a major class of receptors mediating cell interactions with extracellular matrix components. Several studies have now deepened our knowledge of the molecular basis of the interaction between integrin receptors and the IGF axis. Signals transmitted by integrins and receptors of the IGF axis originate in the presence of their cognate ligands [16]. Although studies have shown that IGF receptors and integrins can co-immunoprecipitate in different cell types, it is unclear whether this interaction is direct or requires the engagement of additional proteins [17].
Discoidin domain receptors (DDRs) are collagen-binding tyrosine-kinase receptors involved in cell adhesion [18]. Recent data have demonstrated that significant changes in the expression of DDR1 occur in many neoplasms, including lung, esophagus, breast, ovary, and pediatric brain neoplasia, suggesting a putative role of DDR1 in positively regulating tumor progression [19]. Findings showing that DDR1 is particularly expressed in highly-proliferative invasive tumors have suggested that DDR1 might be implicated in the proliferation and invasion of tumor cells [19].
The first evidence of a possible biological connection between DDR1 and the IR/IGF-1R signaling (IIGFs) was provided by a study with SILAC proteomics aimed at the identification of novel IR-A molecular partners, which would preferentially interact after stimulation with IGF-2 compared to insulin [20]. As mentioned above, the IR-A is frequently overexpressed in cancer [7,8,10,21] where is often constitutively activated by autocrine IGF-2 production [2]. However, given the very small structural difference between IR-A and the ‘metabolic’ isoform IR-B, selective inhibition of IR-A is a difficult task. Thus, the identification of molecular partners uniquely or preferentially recruited to phosphotyrosine protein complexes after IGF-2 stimulation could open the possibility of targeting the IR-A pathway without affecting glucose metabolism regulated by IR-B and insulin [22]. Indeed, several molecules, including DDR1 and DDR2, were preferentially recruited with IR containing phosphotyrosine protein complexes after IGF-2 stimulation [23] (Figure 1).
Figure 1.
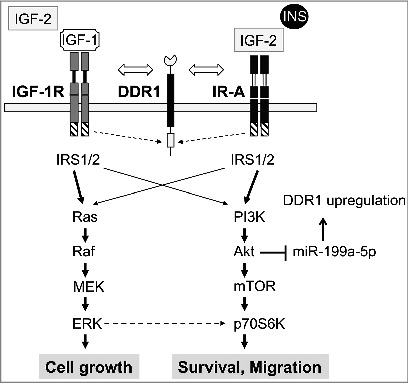
Schematic representation of the crosstalk involving the IIGFs and DDR1. In cells showing IIGFs overactivation, the stimulation of the IR and IGF-1R by their natural ligands induces a functional interaction with other molecular partners including DDR1. This crosstalk with DDR1 regulates IGF-1R and IR trafficking, expression, function and affects the activation of IIGFs downstream pathways such as ERK1/2 and PI3K/AKT. AKT activation, in turn, triggers miR-199a-5p inhibition, which stimulates DDR1 expression and consequent IGF-1R and IR upregulation. Overall, this positive feedback plays a key role in modulating IIGFs biological actions.
The involvement of DDR2 in the IR pathway was confirmed by an independent study [24] but not investigated in detail, while subsequent studies in human breast cancer cells have indicated that DDR1 is indeed a signaling partner of both the IR and the IGF-1R [23,25]. Moreover, DDR1 enhances the expression of both the IR and IGF-1R and potentiates the biological effects of insulin, IGF-1 and IGF-2. In turn, insulin and IGFs stimulate the expression of DDR1, thus establishing a positive feedback mechanism between DDR1 and the IR/IGF-1R axis that might contribute to the upregulation of both molecules in the same cancer cell population (Figure 1). Remarkably, these actions of DDR1 are totally independent from its collagen binding activity.
DDR1 functionally interacts with IR and IGF-1R and enhances the biological actions of insulin, IGF-1 and IGF-2
Studies have been performed in a panel of human breast cancer cells including both estrogen receptor (ER) positive and ER negative cells, which have been well characterized for IR and IGF-1R expression and biological response to insulin, IGF-1 and IGF-2.
The IR-DDR1 crosstalk was investigated in two ER+ breast cancer cell lines MCF-7 and BT-474 and in the triple negative cell line MDA-MB157. IR was expressed in all three cell lines, and IR-A relative abundance was approximately 60% in MCF-7 and BT-474 and 90% in MDA-MB157. In MCF-7 cells DDR1 rapidly co-internalized with the IR after stimulation of either insulin or IGF-2 and co-localized in the cytoplasm and perinuclear region in the absence of exogenous collagen. DDR1 also co-immunoprecipitated with the IR after ligand stimulation. IGF-2 and insulin were equipotent, although the binding affinity of IR for IGF-2 is approximately 7-fold lower than for insulin. These data are in line with previous studies showing that, upon IR-A binding, IGF-2 may be as effective as insulin or even more in regulating specific downstream signaling pathways and gene expression [4,5,20]. DDR1 modulated the main signaling cascades downstream of the activated IR, as in fact DDR1 silencing significantly reduced both AKT and ERK1/2 activation in response to insulin and IGF-2, while DDR1 overexpression enhanced it. Accordingly, breast cancer cell proliferation, invasion and colony formation in response to insulin and IGF-2 was enhanced by DDR1 overexpression and inhibited by DDR1 depletion. Similar results on cell proliferation and invasion were also obtained by DDR1 modulation in non-transformed murine fibroblasts lacking the IGF-1R and solely expressing the human IR-A. The effects mediated by DDR1 modulation were lost after IR knock-down, confirming the specific role of IR (our unpublished data). Further studies are needed to evaluate possible differences between the two IR isoforms with regard to their crosstalk with DDR1.
The IR homologous IGF-1R behaved similarly to IR. In breast cancer cell lines, as well as in murine fibroblasts transfected with the human IGF-1R, DDR1 associated with the IGF-1R at the cell membrane and co-internalized with IGF-1R within minutes after cell stimulation with IGF-1. IGF-IR co-localized with DDR1 in early endosomes and perinuclear regions but was not observed at the nuclear level [25]. Similarly, DDR1 has been shown to rapidly internalized into early endosomes after collagen stimulation suggesting that DDR1 internalization is not temporally linked to collagen-induced phosphorylation, which is a slow event and occurs after hours of collagen binding [26]. However, rapid DDR1 co-internalization after cell stimulation with either insulin or IGFs had not previously described. Noteworthy, IGF-IR localization in early endosomes has recently emerged as an important mechanism in the regulation of receptor signal transduction [27], suggesting that DDR1 co-internalization might critically modulate this step. After ligand induced internalization, a small fraction of both IR and IGF-1R enter the nucleus where they stimulate the transcription of specific genes. Nuclear localization of DDR1 following cell stimulation with either insulin or IGFs was not observed [25]. However, additional studies should be undertaken to evaluate whether minute amounts of DDR1 can enter the nucleus and elicit nuclear functional activity after co-internalization with either IR or IGF-1R.
DDR1 is tyrosine-phosphorylated after collagen binding with a slow kinetics [26]. However, recent data have suggested that the IGF-1R might have a peculiar role in DDR1 phosphorylation. Indeed, IGF-1R depletion severely impaired collagen-induced DDR1 phosphorylation [25]. Moreover, cell stimulation with IGF-1 induced rapid DDR1 phosphorylation in the absence of collagen [25]. Similarly to what observed with insulin and IGF-2, DDR1 also modulates IGF-1-dependent downstream signaling affecting both the Akt and the ERK1/2 pathways and IGF-1-dependent biological actions, namely cell proliferation, migration and colony formation [25]. This modulation was only partially dependent on DDR1 kinase activity, as it also occurred, albeit at a lower extent, in cells transfected with a kinase-inactive DDR1/K618A mutant.
Taken together, these data support a possible scaffolding role for DDR1 in regulating IR and IGF-1R signals (Figure 2), independently of collagen-induced DDR1 kinase activity. However, at least after IGF-1 stimulation, rapid DDR1 phosphorylation positively contributed to DDR1-IGF-IR crosstalk. Interestingly, DDR1 overexpression enhanced cell proliferation, migration and colony formation also in the absence of insulin or IGFs. However, either a functional IR (our unpublished data) or IGF-1R was a necessary requirement for DDR1 action [25]. These observations might be explained by a basal low level of tyrosine kinase activity of IR and IGF-1R, which might occur in cancer cells and in cells expressing high receptor numbers. More studies are required to establish whether collagen binding might affect the functional DDR1 interaction with either IR or IGF-1R.
Figure 2.
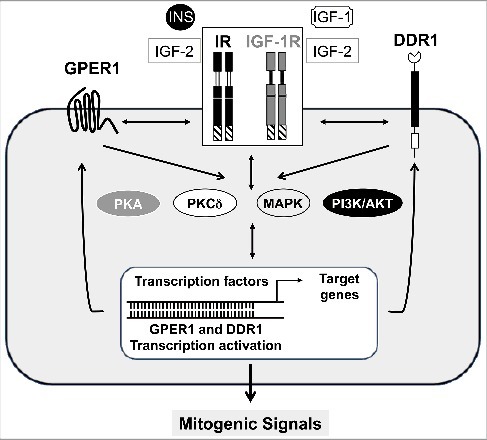
Cross-talk among GPER1, DDR1 and receptors of the IGF axis. The activation of the IGF-1R and IR by their natural ligands, triggers the crosstalk with DDR1 and GPER1 and stimulates a rapid signaling converging on PKA, PKCdelta, MAPK and PI3K/AKT networks. These pathways, in turn, induce the activation of GPER1 and DDR1 transcription factors and target genes which stimulate a loop responsible of GPER1, DDR1, IR and IGF-1R regulation at both mRNA and protein levels. The resulting effects of these functional signaling crosstalks and transcriptional events lead to mitogenic signals and potentiation of IIGFs biological responses.
DDR1 is a novel modulator of IR and IGF-1R expression
A major mechanism by which DDR1 affected intracellular signaling and biological responses to insulin and IGFs is linked to the modulation of both IR and IGF-1R expression levels but the mechanisms of regulation of the two receptors are partially different.
DDR1 silencing was associated with a decrease in IR protein levels whereas DDR1 overexpression caused IR upregulation in both ER+ and ER- human breast cancer cells [23]. IR protein changes were sensitive to inhibitors of the proteasome but not to lysosomal inhibition, suggesting that DDR1 may preferentially regulate the intracellular pool of IR protein degraded via the proteasome, without affecting IR turnover dependent on lysosomal degradation.
Likewise, DDR1 silencing and overexpression was accompanied by either a reduction or an increase, respectively, of IGF-1R protein levels in all three breast cancer cell lines evaluated. Similar results were observed in non-transformed mouse fibroblasts stably overexpressing the human IGF-1R (R+ cells) and transfected with DDR1 [25]. The kinase-inactive DDR1/K618A mutant was slightly less effective, indicating that DDR1 kinase activity is not required for the modulation of IGF-1R protein expression. Ubiquitination of tyrosine-kinase receptors, including the IR and IGF-1R [28,29], plays an important role in regulating ligand-dependent receptor endocytosis and sorting for degradation. However, whether DDR1 might affect IR and/or IGF-1R ubiquitination remains to be elucidated.
These data indicate that DDR1 modulates both IR and IGF-1R protein expression by regulating receptor internalization and intracellular sorting.
However, DDR1 differently affected steady-state levels of IR and IGF-1R mRNA. In fact, DDR1 silencing and overexpression similarly affected IR mRNA and protein levels but did not significantly regulate IGF-1R mRNA [23,25]. Experiments with the protein synthesis inhibitor cycloheximide or the transcriptional inhibitor actinomycin D suggested that IR up-regulation by DDR1 partially involves de novo synthesis and transcriptional activity. In accordance to these findings, DDR1 silencing in MCF-7 cells was associated with a reduced IR promoter activity accompanied by a reduction in Sp1 and HMGA1, two positive regulators of IR transcription, and with an increase of p53, which is instead a negative regulator of IR transcription. On the contrary, DDR1 overexpression was associated with opposite changes in IR promoter activity and in the expression of Sp1, HMGA1 and p53. Both mRNA and the protein expression profiles of these factors were modulated by DDR1 silencing or overexpression.
Taken together, these data indicate that DDR1 regulates IR expression at multiple levels by modulating protein degradation and stability, gene transcription and post-transcriptional mRNA regulation, whereas it regulates IGF-1R protein expression levels prevalently through post-translational mechanisms [23,25].
DDR1 is upregulated by IGFs and insulin through the PI3K/AKT/miR-199a-5p signaling cascade
Having shown that DDR1 is an important regulator of IR and IGF-1R expression, it was then asked whether insulin and IGFs might affect DDR1 expression. Indeed, in human breast cancer cells including both ER+ and ER- cells, exposure to IGF-1 (but also to IGF-2 and insulin) resulted in significant, time- and dose-dependent upregulation of DDR1 protein that was accompanied by less marked changes in steady state levels of DDR1 mRNA. Similar results were obtained in mouse fibroblasts lacking the IGF-1R but overexpression IR-A after stimulation with IGF-2 and insulin, indicating that IR-A stimulation upregulates DDR1 (our unpublished data). DDR1 protein upregulation by IGF-1 required IGF-1R tyrosine-kinase activity and PI3K/AKT signaling, as confirmed by using pharmacological inhibitors of PI3K and AKT. Additionally, the transfection with a constitutively active form of AKT (myr-AKT) caused marked DDR1 upregulation. In contrast, DDR1 expression was not affected by inhibitors of either the ERK1/2, the mTOR/p70S6K or the PKC pathways [30].
A major mechanism modulating DDR1 upregulation by insulin and IGFs was demonstrated as Akt-dependent downregulation of miR-199-5p [30]. Accordingly, transfection of breast cancer cells with miR-199-5p markedly downregulated DDR1 protein and blunted IGF-1 signaling and biological responses including cell proliferation and migration, while miR-199a-5p antagomir elicited opposite effects.
DDR1 is a known target gene of miR-199a-5p, which shows only partial complementarity to the 3’-untranslated region (3’UTR) of DDR1 mRNA [31]. For instance, in hepatoma cells, miR-199a-5p down-regulated DDR1 protein without inducing changes in DDR1 mRNA expression [31]. In accordance with these findings, in human breast cancer cells, miR-199a-5p reduced the activity of a luciferase construct containing the DDR1 3’-UTR, an effect significantly blunted by IGF-1 stimulation. It is worth mentioning that the resulting effects of specific miRNAs depend on their degree of complementarity with the 3’-UTR of target mRNAs, with perfect complementarity resulting in mRNA degradation and gene silencing, while partial complementarity resulting in modest degradation of target mRNA. The final effect on mRNA degradation is also affected by the set of proteins bound to the 3’-UTR of the target mRNA. Although the specific mechanism by which Akt causes miR-199a-5p down-regulation is poorly understood, studies in cardiac myocytes suggest that hypoxia-dependent AKT activation inhibits miR-199a-5p without requiring transcriptional activity [32]. Therefore, is it likely that IGF-1 downregulates miR-199a-5p by involving miRNAs processing and/or stability. Treatment of MCF-7 cells with cycloheximide, an inhibitor of translational elongation, completely blocked IGF-1-dependent DDR1 upregulation, suggesting that this process is dependent on protein synthesis and confirming that translation mechanisms have a role in the upregulation of DDR1 protein [30].
Constitutive AKT activation is a common occurrence in cancer and may derive from activating AKT mutations, inactivating mutation/deletions of the PTEN oncosuppressor, but also from autocrine and/or paracrine production of IGF-1 and/or IGF-2 [33]. Indeed, AKT activation resulting by either stable transfection of breast cancer cells with the IGF-2 gene as well as cell treatment with IGF-2 containing-conditioned medium derived from cancer-associated fibroblasts (CAFs) markedly upregulated DDR1 expression through inhibition of miR-199-5p expression [30].
These results indicate that activation of the AKT/miR-199a-5 pathway by insulin or IGFs, is an important feed-forward mechanism leading to upregulation of DDR1 and enhanced IR and IGF-1R expression (Figure 2). It could be hypothesized, therefore, that in embryonic/fetal or cancer cells producing autocrine IGFs, this feed-forward mechanism might be aimed at maximizing insulin and IGFs actions by counteracting ligand-dependent receptor down-regulation [8,34]. In addition, this possible regulatory mechanism might also explain the frequent concomitant overexpression of DDR1 with the IR or IGF-1R (see paragraph: “DDR1 is a novel modulator of IR and IGF-1R expression”).
Analysis of DDR1 correlation with IR and IGF-1R in large databases
The correlation between DDR1, IR or IGF-1R expression was analyzed in public domain databases. The first analysis was performed by using the Affymetrix GeneChip Human Genome U133 plus 2.0 arrays, comprising 1794 datasets, and the beta MEM method. Statistically relevant results were then validated in The Cancer Genome Atlas (TCGA) breast cancer dataset. DDR1 positively correlated with both IR and IGF-1R, but the correlation with IGF-1R was stronger [25] (Table 1). The positive correlation of DDR1 with the IGF-1R, but not with the IR, was confirmed in the TCGA breast cancer dataset. However, to further analyze the correlation between DDR1 and IR expression in human breast cancer specimens, Pearson correlation between probes for DDR1 and IR was calculated by analyzing various GEO datasets [23]. These computational data revealed a general positive correlation between DDR1 and IR transcripts. This correlation was stronger in human breast carcinomas than in normal breast tissues. Data stratification according to the bio-pathological features of tumors showed that the DDR1-IR correlation was stronger in breast cancers with aggressive characteristics, including the lack of estrogen and progesterone receptors, basal-like phenotype, metastatic lymph-nodes, elevated proliferative index and tumor grading. Moreover, using StarBase Pan-Cancer analysis, DDR1 expression inversely correlated with miR-199a-5p in several cancer histotypes, including cancers from breast, lung, ovary, thyroid, endometrium and acute myeloid leukemia (Table 2). IGF-1R also inversely correlated with miR-199a-5p. Taken together, these data support the concept that the regulatory network involving DDR1, IR/IGF-1R and the AKT/miR-199a-5p pathway plays an in vivo role in several cancer histotypes, and particularly in breast cancer.
Table 1.
Correlation between DDR1 and IGF-1 (IGF-1R) or insulin (IR) receptor expression from TCGA breast cancer data set. Pearson correlations and P-values between three different probes for DDR1 (i.e., 208779_X_AT, 207169_X_AT, 210749_X_AT) and probes for IGF-IR and IR are reported in the last column.
| Gene Probes | Pearson coefficient (P-value) |
|---|---|
| IGF-1R | 0.29 (<0.00001) |
| IR | 0.09 (0.035) |
Table 2.
Analysis of anticorrelation between miR-199a-5p and DDR1 or IGF-1R expression across 14 cancer types from TCGA data set. Data analysis was performed according to starBase Pan-Cancer Platform. The table shows only the statistically significant data. P-values are shown in brackets.
| Pearson coefficient |
||
|---|---|---|
| Cancer type | DDR1 (P- value) | IGF-1R (P- value) |
| Urothelial bladder cancer | ||
| Breast cancer | −0.17 (0.000001) | |
| Colon and rectal adenocarcinoma | ||
| Glioblastoma multiforme | −0.37 (0.000001) | |
| Head and neck squalous cell carcinoma | −0.11 (0.01) | |
| Chromophobe renal cell carcinoma | −0.66 (0.000000000001) | −0.43 (0.00001) |
| Clear cell kidney carcinoma | ||
| Acute myeloid leukemia | ||
| Lung adenocarcinoma | ||
| Lung squamous cell carcinoma | ||
| Ovarian serous cystadenocarcinoma | −0.19 (0.001) | |
| Cutaneous melanoma | −0.22 (0.00003) | −0.18 (0.0004) |
| Papillary thyroid carcinoma | −0.11 (0.008) | |
| Uterine corpus endometrial carcinoma | ||
A possible role for GPER1 in the DDR1-IIGFs crosstalk
Recently, the non-classical estrogen receptor GPER1 has been shown to play a role in the IIGFs – DDR1 signaling network (Figure 2). First, by using human breast and endometrial cancer cells, it has been shown that that both insulin and IGF-1 transactivate the GPER1 promoter and upregulate GPER1 by activating the PKCδ/ERK/c-fos/AP1 transduction pathway. In response to insulin, both IR-A and IR-B shared the same effects. Remarkably, cell migration in response to insulin and IGF-1 required GPER1 and its main target gene CTGF. Moreover, insulin, through GPER1, boosted glucose uptake stimulated by estrogens. In agreement with these data, insulin resistant patients with breast cancer, showed high expression levels of GPER1 in CAFs from tumor specimens [35–37].
Most interestingly, in both mesothelioma and lung cancer cells, DDR1 was part of a signaling network complex comprising IR/IGF-1R and GPER1. In fact, activation of the IIGFs led to up-regulation of GPER1 and its main target genes CTGF and EGR1 as well as the induction of DDR1 target genes like MATN-2, FBN-1, NOTCH 1 and HES-1. These events mediated cell chemotaxis and migration in response to IGF-1 [38]. VEGF is another gene stimulated by the IIGFs-GPER1 crosstalk, especially in hypoxic conditions [39], and although not specifically addressed, a role of DDR1 in this context could also be hypothesized.
Together, these data indicate that, in cancer cell models, IR, IGF-1R and DDR1 are components of a multiprotein signaling platform, which may also include GPER1, which integrates signals from growth factors/hormones and extracellular matrix, thus playing a crucial role in cancer cell biology.
DDR1 involvement in growth factors other than IGFs
As already mentioned, DDR1 activity has been implicated in multiple physiological processes such as cell migration [40], differentiation [41] and ECM remodeling [42], whereas dysregulated DDR1 has been linked to the progression of fibrosis, arthritis and cancer [43]. DDR1 actions may be also context dependent [43]. It can be supposed, therefore, that DDR1 may partially act by interacting with and regulating the signaling pathways of various growth factors, some of them could have tissue specificity. However, data demonstrating a DDR1 crosstalk with growth factors other than the insulin and IGFs are scanty. As a key feature of DDR1 activation by phosphorylation is a delayed and prolonged response upon collagen binding, it has been assumed that activated DDR1, in close proximity of classical receptor tyrosine kinases (RTKs), may act as a scaffold molecule for adaptors/signaling proteins, such as SHC and PI3K, thus facilitating the propagation of signaling networks by ligand-stimulated RTKs that compete for the recruitment of these adaptors/signaling proteins [44].
A functional interplay between DDR1 and the signaling pathways of Trasforming-Growth Factor-β1 (TGF-β1), Hepatocyte Growth Factor (HGF), and Wnt molecules have been described.
Transforming-Growth Factor-β1 (TGF-β1): A link between DDR1 and the inflammation factor TGF-β1 has been observed during invadosome formation in hepatocellular carcinoma (HCC) cells. Invadosomes are actin-based structures involved in matrix degradation and cell invasion through metalloproteinases (MMP) activity [45]. TGF-β1 is known to enhance the invadosome formation of HCC cells by increasing type I collagen production. Moreover, Ezzoukhry et al. demonstrated that this activity involves DDR1, as DDR1 depletion reduces invadosome formation. These findings highlight a critical role of DDR1 in the invasive capability of liver cancer cells that is modulated by TGF-β1 [46]. Accordingly, DDR1 is frequently overexpressed in HCC cells [47] and is associated with advanced tumor stages [31]. A functional crosstalk between DDR-1 and the TGF-β pathway has been also demonstrated in the progression of atherosclerotic plaque formation. Indeed, DDR1 regulates collagen deposition and release of calcifying extracellular vesicles by vascular smooth muscle cells (vSMCs) influencing the TGF-β signaling pathway [42,48].
Hepatocyte Growth Factor (HGF): DDR1 is an important element in the regulation of branching tubulogenesis induced by HGF, the cMet ligand. Branching tubulogenesis is an important feature of developmental process in many organs, including kidney, lung, mammary gland, pancreas, and salivary gland. DDR1 levels are critical for HGF-induced branching tubulogenesis as both over-expression and functional disruption of DDR1 resulted in severe disturbance of branching tubulogenesis. These activities of DDR1 are closely connected with the regulation of cell proliferation, cell survival and cell migration [49]. However, the molecular mechanisms involved in the crosstalk between DDR1 and the HGF/cMet pathway deserve further studies.
Wnt proteins: Wnt proteins coordinate a variety of cell biological and developmental processes by binding to the Frizzled family of receptors and to the coreceptors of the LDL-receptor related proteins family [50]. Previous studies have identified a role for Wnt-5a, a non-transforming Wnt protein, in mediating the activation of DDR1 phosphorylation, possibly through the Wnt/Ca2 pathway, which in turn leads to PKC activation [51]. Src tyrosine kinase was also found to play a role in allowing the effects of Wnt-5a on collagen-induced activation of DDR1 [51]. However, Wnt-5a – mediated DDR1 activation and cell adhesion to collagen was context dependent. In fact, it reduced motility and metastatic potential in breast cancer cells [51,52] that are firmly adherent, whereas it was prometastatic in the more loosely adherent melanoma cells [53].
Summary and future perspectives
Taken together, these studies strongly suggest that crosstalk between DDR1 and the IIGFs has several biological and potential practical implications. First, in addition to its role as a collagen receptor, DDR1 has non-canonical actions, which do not require its collagen binding function. Interestingly, IIGFs and DDR1 appear linked by feed-forward mechanisms. In fact, DDR1 has emerged as a potent regulator of the expression of both IR and IGF-IR, by acting through multiple and diverse mechanisms. Overall, DDR1 strongly potentiates the effects of the three ligands of the IGF axis, insulin, IGF-1 and IGF-2. Strikingly, in cells knockdown for DDR1, some biological responses to these ligands are exceedingly weak, underlying the important role of DDR1 in this context. In turn, IIGFs is an important positive modulator of DDR1 expression via the AKT/miR-199a-5p/DDR1 pathway, and a newly recognized factor modulating DDR1 phosphorylation and internalization. Moreover, DDR1 phosphorylation in response to collagen is impaired by IGF-1R knockdown.
Second, these studies support the concept that, both IR and IGF-1R, by functionally interacting with other membrane receptors may undergo an unexpected degree of diversification in intracellular signaling and biological actions, which is modulated by cell and tissue specific receptor expression and by the abundance of cognate ligands in the microenvironment. This signaling diversification may help understanding the pleiotropic functions of the IIGFs [2].
The IIGFs–DDR1 crosstalk has been shown in human breast cancer cells but also in non-transformed mouse fibroblasts. Indeed, cancer cell growth, colony formation in semi-solid agar, and especially cells invasion in response to insulin and IGFs were strongly enhanced by DDR1 expression while inhibited by DDR1 knockdown. As IIGFs is often dysregulated in cancer, the activation of the IGF-IR/AKT/miR-199a-5p/DDR1 pathway may play a relevant role in cancer EMT, cell stemness, invasion and metastasis as well as in resistance to oncologic therapies [30,54].
As the IGF-2/IR-A loop, which is activated in several malignancies, has proven to be a difficult target, DDR1 silencing/inhibition could represent a useful adjunct to anti-IR/IGF-1R therapies [23]. In particular, findings showing that DDR1 and IR expression levels are strongly correlated in breast cancers with aggressive characteristics do support the quest for further studies aiming at validating DDR1 and IGF-IR/IR as cotargets for therapy.
The relevance of IIGFs–DDR1 crosstalk in physiology is currently unknown. However, studies performed in non-transformed fibroblasts suggest that DDR1 might play a role as a modulator of IIGFs diversification in normal physiology, including regulation of homeostatic actions such as cell metabolism and survival, development and somatic growth [25]. As the IGF-IR plays a significant role in DDR1 phosphorylation by collagen, IGF-1R might contribute in regulating cell–matrix interactions. IIGFs plays an essential role in the biology of stem cells [34] and a cooperative role of DDR1 in this setting would be possible. It is worth mentioning that collagen-stimulated DDR1 has been implicated in stem cell self-renewal [55]. Moreover, both IGF-IR and DDR1 have been implicated in the induction and maintenance of epithelial-mesenchymal transition (EMT), one hallmark of stem cells and a critical process allowing tissue remodeling during embryogenesis, tumor invasion and metastasis [54,56]. MiR-199a-5p downregulation itself may also play a role in EMT by promoting E-cadherin loss [56].
However, many questions still remain to be addressed in future studies. The modalities of interaction between DDR1 and IR or IGF-1R need to be further elucidated. DDR1 has been shown to internalize with IR and IGF-1R following stimulation with insulin or IGFs. Interestingly, a small percentage of activated IR and IGF-1R reach the nucleus and activate transcription [2]. It could be hypothesized that DDR1 may also enter the nucleus. This hypothesis is not supported by available studies, but could be more specifically addressed in future studies.
In adult tissues the major role of insulin is to regulate metabolism. Therefore, future studies will address whether DDR1 may play any role in affecting the metabolic role of insulin. Insulin resistance and hyperinsulinemia are characteristic of several disorders, such as obesity, type 2 diabetes and metabolic syndrome, which are associated with several severe chronic complications, such as microangiopathy, atherosclerosis and cancer [57]. Whether insulin resistance may affect DDR1 expression either globally or in specific organs and tissues, and whether altered DDR1 expression is associated with such chronic complications is also worth exploring.
Finally, the non-canonical estrogen receptor GPER1 has also been shown to interact with DDR1 [38]. GPER1 is upregulated by insulin and IGFs [38] and favors the biological effects of these factors, including insulin-dependent glucose uptake [38]. These studies add complexity to the IIGFs-DDR1 crosstalk [23,25] and suggest a wider role of DDR1 as a regulator of the integrated cell response to cues from hormones and growth factors as well as the extracellular matrix itself in both physiology and disease.
Funding Statement
Italian Association for Cancer Research (AIRC)(IG 19242).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by a grant from the Italian Association for Cancer Research (AIRC IG 19242 to AB).
References
- [1].Belfiore A, Frasca F, Pandini G, et al.. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009. October;30(6):586–623. doi: 10.1210/er.2008-0047. PMID:19752219. [DOI] [PubMed] [Google Scholar]
- [2].Belfiore A, Malaguarnera R, Vella V, et al.. Insulin receptor isoforms in physiology and disease: An updated view. Endocr Rev. 2017. October 01;38(5):379–431. doi: 10.1210/er.2017-00073. PMID:28973479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Frasca F, Pandini G, Scalia P, et al.. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999. May;19(5):3278–88. PMID:10207053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pandini G, Medico E, Conte E, et al.. Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J Biol Chem. 2003. October 24;278(43):42178–89. doi: 10.1074/jbc.M304980200. PMID:12881524. [DOI] [PubMed] [Google Scholar]
- [5].Pandini G, Conte E, Medico E, et al.. IGF-II binding to insulin receptor isoform A induces a partially different gene expression profile from insulin binding. Ann N Y Acad Sci. 2004. December;1028:450–6. doi: 10.1196/annals.1322.053. PMID:15650270. [DOI] [PubMed] [Google Scholar]
- [6].Malaguarnera R, Sacco A, Voci C, et al.. Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology. 2012. May;153(5):2152–63. doi: 10.1210/en.2011-1843. PMID:22355074. [DOI] [PubMed] [Google Scholar]
- [7].Sciacca L, Costantino A, Pandini G, et al.. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999. April 15;18(15):2471–9. doi: 10.1038/sj.onc.1202600. PMID:10229198. [DOI] [PubMed] [Google Scholar]
- [8].Vella V, Pandini G, Sciacca L, et al.. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metab. 2002. January;87(1):245–54. doi: 10.1210/jcem.8718142. PMID:11788654. [DOI] [PubMed] [Google Scholar]
- [9].Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012. February 16;12(3):159–69. doi: 10.1038/nrc3215. PMID:22337149. [DOI] [PubMed] [Google Scholar]
- [10].Sciacca L, Mineo R, Pandini G, et al.. In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene. 2002. November 28;21(54):8240–50. doi: 10.1038/sj.onc.1206058. PMID:12447687. [DOI] [PubMed] [Google Scholar]
- [11].Scott CD, Kiess W. Soluble M6P/IGFIIR in the circulation. Best Pract Res Clin Endocrinol Metab. 2015;29(5):723–33. doi: 10.1016/j.beem.2015.08001. PMID:26522457. [DOI] [PubMed] [Google Scholar]
- [12].Pandini G, Frasca F, Mineo R, et al.. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002. October 18;277(42):39684–95. doi: 10.1074/jbc.M202766200. PMID:12138094. [DOI] [PubMed] [Google Scholar]
- [13].Morcavallo A, Buraschi S, Xu SQ, et al.. Decorin differentially modulates the activity of insulin receptor isoform A ligands. Matrix Biol. 2014;35:82–90. doi: 10.1016/j.matbio.2013.12010. PMID:24389353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tanimoto R, Palladino C, Xu SQ, et al.. The perlecan-interacting growth factor progranulin regulates ubiquitination, sorting, and lysosomal degradation of sortilin. Matrix Biol. 2017;64:27–39. doi: 10.1016/j.matbio.2017.04001. PMID:28433812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Iozzo RV, Buraschi S, Genua M, et al.. Decorin antagonizes IGF receptor I (IGF-IR) function by interfering with IGF-IR activity and attenuating downstream signaling. J Biol Chem. 2011. October 07;286(40):34712–21. doi: 10.1074/jbc.M111.262766. PMID:21840990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Takada Y, Takada YK, Fujita M. Crosstalk between insulin-like growth factor (IGF) receptor and integrins through direct integrin binding to IGF1. Cytokine Growth Factor Rev. 2017. April;34:67–72. doi: 10.1016/j.cytogfr.2017.01003. PMID:28190785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Canonici A, Steelant W, Rigot V, et al.. Insulin-like growth factor-I receptor, E-cadherin and alpha v integrin form a dynamic complex under the control of alpha-catenin. Int J Cancer. 2008. February 01;122(3):572–82. doi: 10.1002/ijc.23164. PMID:17955485. [DOI] [PubMed] [Google Scholar]
- [18].Vogel WF, Abdulhussein R, Ford CE. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal. 2006;18(8):1108–16. doi: 10.1016/j.cellsig.2006.02012. PMID:16626936. [DOI] [PubMed] [Google Scholar]
- [19].Valiathan RR, Marco M, Leitinger B, et al.. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012;31(1–2):295–321. doi: 10.1007/s10555-012-9346-z. PMID:22366781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Morcavallo A, Genua M, Palummo A, et al.. Insulin and insulin-like growth factor II differentially regulate endocytic sorting and stability of insulin receptor isoform A. J Biol Chem. 2012. March 30;287(14):11422–36. doi: 10.1074/jbc.M111.252478. PMID:22318726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Belfiore A, Malaguarnera R. Insulin receptor and cancer. Endocr Relat Cancer. 2011. August;18(4):R125–47. doi: 10.1530/ERC-11-0074. PMID:21606157. [DOI] [PubMed] [Google Scholar]
- [22].Malaguarnera R, Belfiore A. The insulin receptor: a new target for cancer therapy. Front Endocrinol (Lausanne). 2011;2:93. doi: 10.3389/fendo.2011.00093. PMID:22654833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vella V, Malaguarnera R, Nicolosi ML, et al.. Discoidin domain receptor 1 modulates insulin receptor signaling and biological responses in breast cancer cells. Oncotarget. 2017. June 27;8(26):43248–43270. doi: 10.18632/oncotarget.18020. PMID:28591735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Iwai LK, Chang F, Huang PH. Phosphoproteomic analysis identifies insulin enhancement of discoidin domain receptor 2 phosphorylation. Cell Adhesion & Migration. 2013. Mar-Apr;7(2):161–4. doi: 10.4161/cam.22572. PMID:23154445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Malaguarnera R, Nicolosi ML, Sacco A, et al.. Novel cross talk between IGF-IR and DDR1 regulates IGF-IR trafficking, signaling and biological responses. Oncotarget. 2015. June 30;6(18):16084–105. doi: 10.18632/oncotarget.3177. PMID:25840417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vogel W, Gish GD, Alves F, et al.. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997. December;1(1):13–23. PMID:9659899. [DOI] [PubMed] [Google Scholar]
- [27].Romanelli RJ, LeBeau AP, Fulmer CG, et al.. Insulin-like growth factor type-I receptor internalization and recycling mediate the sustained phosphorylation of Akt. J Biol Chem. 2007. August 03;282(31):22513–24. doi: 10.1074/jbc.M704309200. PMID:17545147. [DOI] [PubMed] [Google Scholar]
- [28].Monami G, Emiliozzi V, Morrione A. Grb10/Nedd4-mediated multiubiquitination of the insulin-like growth factor receptor regulates receptor internalization. J Cell Physiol. 2008. August;216(2):426–37. doi: 10.1002/jcp.21405. PMID:18286479. [DOI] [PubMed] [Google Scholar]
- [29].Vecchione A, Marchese A, Henry P, et al.. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol Cell Biol. 2003. May;23(9):3363–72. PMID:12697834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mata R, Palladino C, Nicolosi ML, et al.. IGF-I induces upregulation of DDR1 collagen receptor in breast cancer cells by suppressing MIR-199a-5p through the PI3K/AKT pathway. Oncotarget. 2016. February 16;7(7):7683–700. doi: 10.18632/oncotarget.6524. PMID:26655502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shen Q, Cicinnati VR, Zhang X, et al.. Role of microRNA-199a-5p and discoidin domain receptor 1 in human hepatocellular carcinoma invasion. Mol Cancer. 2010. August 27;9:227. doi: 10.1186/1476-4598-9-227. PMID:20799954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rane S, He M, Sayed D, et al.. An antagonism between the AKT and beta-adrenergic signaling pathways mediated through their reciprocal effects on miR-199a-5p. Cell Signal. 2010. July;22(7):1054–62. doi: 10.1016/j.cellsig.2010.02008. PMID:20193759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Vella V, Puppin C, Damante G, et al.. DeltaNp73alpha inhibits PTEN expression in thyroid cancer cells. Int J Cancer. 2009. June 01;124(11):2539–48. doi: 10.1002/ijc.24221. PMID:19173293. [DOI] [PubMed] [Google Scholar]
- [34].Malaguarnera R, Frasca F, Garozzo A, et al.. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab. 2011. March;96(3):766–74. doi: 10.1210/jc.2010-1255. PMID:21123448. [DOI] [PubMed] [Google Scholar]
- [35].De Marco P, Bartella V, Vivacqua A, et al.. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene. 2013. February 07;32(6):678–88. doi: 10.1038/onc.2012.97. PMID:22430216. [DOI] [PubMed] [Google Scholar]
- [36].De Marco P, Romeo E, Vivacqua A, et al.. GPER1 is regulated by insulin in cancer cells and cancer-associated fibroblasts. Endocr Relat Cancer. 2014. October;21(5):739–53. doi: 10.1530/ERC-14-0245. PMID:25012984. [DOI] [PubMed] [Google Scholar]
- [37].De Marco P, Cirillo F, Vivacqua A, et al.. Novel aspects concerning the functional cross-talk between the Insulin/IGF-I system and estrogen signaling in cancer cells. Front Endocrinol (Lausanne). 2015;6:30. doi: 10.3389/fendo.2015.00030. PMID:25798130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Avino S, De Marco P, Cirillo F, et al.. Stimulatory actions of IGF-I are mediated by IGF-IR cross-talk with GPER and DDR1 in mesothelioma and lung cancer cells. Oncotarget. 2016. August 16;7(33):52710–52728. doi: 10.18632/oncotarget.10348. PMID:27384677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rigiracciolo DC, Scarpelli A, Lappano R, et al.. Copper activates HIF-1alpha/GPER/VEGF signalling in cancer cells. Oncotarget. 2015. October 27;6(33):34158–77. doi: 10.18632/oncotarget.5779. PMID:26415222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hou G, Vogel WF, Bendeck MP. Tyrosine kinase activity of discoidin domain receptor 1 is necessary for smooth muscle cell migration and matrix metalloproteinase expression. Circ Res. 2002. June 14;90(11):1147–9. PMID:12065315. [DOI] [PubMed] [Google Scholar]
- [41].Yeh YC, Lin HH, Tang MJ. A tale of two collagen receptors, integrin beta1 and discoidin domain receptor 1, in epithelial cell differentiation. Am J Physiol Cell Physiol. 2012. December 15;303(12):C1207–17. doi: 10.1152/ajpcell.00253.2012. PMID:23015544. [DOI] [PubMed] [Google Scholar]
- [42].Ferri N, Carragher NO, Raines EW. Role of discoidin domain receptors 1 and 2 in human smooth muscle cell-mediated collagen remodeling: potential implications in atherosclerosis and lymphangioleiomyomatosis. Am J Pathol. 2004. May;164(5):1575–85. doi: 10.1016/S0002-9440(10)63716-9. PMID:15111304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. 2014;310:39–87. doi: 10.1016/B978-0-12-800180-6.00002-5. PMID:24725424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gordus A, Krall JA, Beyer EM, et al.. Linear combinations of docking affinities explain quantitative differences in RTK signaling. Mol Syst Biol. 2009;5:235. doi: 10.1038/msb.2008.72. PMID:19156127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Leitinger B. Transmembrane collagen receptors. Annu Rev Cell Dev Biol. 2011;27:265–90. doi: 10.1146/annurev-cellbio-092910-154013. PMID:21568710. [DOI] [PubMed] [Google Scholar]
- [46].Ezzoukhry Z, Henriet E, Piquet L, et al.. TGF-beta1 promotes linear invadosome formation in hepatocellular carcinoma cells, through DDR1 up-regulation and collagen I cross-linking. Eur J Cell Biol. 2016. November;95(11):503–512. doi: 10.1016/j.ejcb.2016.09003. PMID:27720259. [DOI] [PubMed] [Google Scholar]
- [47].Jian ZX, Sun J, Chen W, et al.. Involvement of discoidin domain 1 receptor in recurrence of hepatocellular carcinoma by genome-wide analysis. Med Oncol. 2012. December;29(5):3077–82. doi: 10.1007/s12032-012-0277-x. PMID:22752569. [DOI] [PubMed] [Google Scholar]
- [48].Krohn JB, Hutcheson JD, Martinez-Martinez E, et al.. Discoidin Domain Receptor-1 regulates calcific extracellular vesicle release in vascular smooth muscle cell Fibrocalcific response via transforming growth factor-beta signaling. Arterioscler Thromb Vasc Biol. 2016. March;36(3):525–33. doi: 10.1161/ATVBAHA.115.307009. PMID:26800565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang CZ, Hsu YM, Tang MJ. Function of discoidin domain receptor I in HGF-induced branching tubulogenesis of MDCK cells in collagen gel. J Cell Physiol. 2005. April;203(1):295–304. doi: 10.1002/jcp.20227. PMID:15468059. [DOI] [PubMed] [Google Scholar]
- [50].Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14159. PMID:9891778. [DOI] [PubMed] [Google Scholar]
- [51].Dejmek J, Dib K, Jonsson M, et al.. Wnt-5a and G-protein signaling are required for collagen-induced DDR1 receptor activation and normal mammary cell adhesion. Int J Cancer. 2003. January 20;103(3):344–51. doi: 10.1002/ijc.10752. PMID:12471617. [DOI] [PubMed] [Google Scholar]
- [52].Jonsson M, Andersson T. Repression of Wnt-5a impairs DDR1 phosphorylation and modifies adhesion and migration of mammary cells. J Cell Sci. 2001. June;114(Pt 11):2043–53. PMID:11493640. [DOI] [PubMed] [Google Scholar]
- [53].Weeraratna AT, Jiang Y, Hostetter G, et al.. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002. April;1(3):279–88. PMID:12086864. [DOI] [PubMed] [Google Scholar]
- [54].Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (Lausanne). 2014;5:10. doi: 10.3389/fendo.2014.00010. PMID:24550888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Suh HN, Han HJ. Collagen I regulates the self-renewal of mouse embryonic stem cells through alpha2beta1 integrin- and DDR1-dependent Bmi-1. J Cell Physiol. 2011. December;226(12):3422–32. doi: 10.1002/jcp.22697. PMID:21344393. [DOI] [PubMed] [Google Scholar]
- [56].Hu Y, Liu J, Jiang B, et al.. MiR-199a-5p loss up-regulated DDR1 aggravated colorectal cancer by activating epithelial-to-mesenchymal transition related signaling. Dig Dis Sci. 2014. September;59(9):2163–72. doi: 10.1007/s10620-014-3136-0. PMID:24711074. [DOI] [PubMed] [Google Scholar]
- [57].Hua F, Yu JJ, Hu ZW. Diabetes and cancer, common threads and missing links. Cancer Lett. 2016. April 28;374(1):54–61. doi: 10.1016/j.canlet.2016.02006. PMID:26879686. [DOI] [PubMed] [Google Scholar]