

ABSTRACT
Discoidin domain receptors 1 and 2 (DDR1 and DDR2) are members of the tyrosine kinase receptors activated after binding with collagen. DDRs are implicated in numerous physiological and pathological functions such as proliferation, adhesion and migration. Little is known about the expression of the two receptors in normal and cancer cells and most of studies focus only on one receptor. Western blot analysis of DDR1 and DDR2 expression in different tumor cell lines shows an absence of high co-expression of the two receptors suggesting a deleterious effect of their presence at high amount. To study the consequences of high DDR1 and DDR2 co-expression in cells, we over-express the two receptors in HEK 293T cells and compare biological effects to HEK cells over-expressing DDR1 or DDR2. To distinguish between the intracellular dependent and independent activities of the two receptors we over-express an intracellular truncated dominant-negative DDR1 or DDR2 protein (DDR1DN and DDR2DN). No major differences of Erk or Jak2 activation are found after collagen I stimulation, nevertheless Erk activation is higher in cells co-expressing DDR1 and DDR2. DDR1 increases cell proliferation but co-expression of DDR1 and DDR2 is inhibitory. DDR1 but not DDR2 is implicated in cell adhesion to a collagen I matrix. DDR1, and DDR1 and DDR2 co-expression inhibit cell migration. Moreover a DDR1/DDR2 physical interaction is found by co-immunoprecipitation assays. Taken together, our results show a deleterious effect of high co-expression of DDR1 and DDR2 and a physical interaction between the two receptors.
KEYWORDS: discoidin domain Receptors, DDR1, DDR2, Proliferation, Adhesion, Migration, Signalization, Interaction
Introduction
Tyrosine kinase receptors (RTK) are a large family of transmembrane receptors with diverse extracellular binding domains and a conserved intracellular tyrosine kinase domain. Based on the conserved kinase domain, two receptors with an extracellular discoidin domain were characterized and named DDR1 and DDR2 [1]. Discoidin domain is homologous to the lectin discoidin I, a protein secreted by the slime Dictyostelium discoideum [2]. Although DDRs share the tyrosine kinase activity with other RTK, they have also specificities. They are activated by collagens [3], the most abundant fibrous protein of the extracellular matrix [4], and not by soluble peptide growth factors. Receptor dimerisation is not induced by ligand binding but occurs during the transfer from the endoplasmic reticulum to the plasma membrane. Collagen binding induces modifications in the conformation of the receptor, releasing the autoinhibition mediated by the justamembrane domain and, probably, induces multimerisation of the already formed dimers [5,6]. After collagen binding, DDRs autophosphorylate intracellular docking sites with a slow and sustained kinetics [2]. Several intracellular pathways are activated such as ERK1/2, P38, JNK, PI-3 kinase/Akt, Notch-1 and NFκB [2]. DDRs can cooperate with the integrin pathway to enable migration or adhesion but in an opposite way depending on the cell type [7, 8]. In MDCK cells, a study demonstrated inhibition of α2β1 integrin activity by DDR1 [7], but in other cell lines DDR1 or DDR2 enhances integrin activation or increases integrin expression at the cell surface [8].
DDRs are implicated in many cell biological functions such as cell proliferation, adhesion and migration, extracellular matrix contraction and degradation [2, 9].
An increasing number of publications reports DDR1 collagen-independent or kinase independent functions. At cell-cell contacts DDR1 co-localizes with E-cadherin and it is sequestered away from collagen present at apical or basal membranes. When E-cadherin is down regulated, DDR1 re-localizes to the apical and basal membranes, binds collagen and induces cell spreading [10]. Through interaction with the Par3/Par6 cell polarity complex, DDR1 localizes RhoE at the cell-cell junctions and inhibits ROCK-driven actomyosin contractility allowing collective migration [11]. Neither DDR1 activity nor collagen binding is required to regulate collective migration. In breast cancer, collagen binding to DDR1 regulates the formation of linear invadosomes independently of the kinase activity [12]. In adipose stromal cells a non-collagen ligand of DDR1 activates JNK and consequently transcription of aromatase, an enzyme implicated in estrogen synthesis [13]. Little is known about DDR2 collagen-independent or kinase independent functions. Nevertheless, DDR2 mediates fibroblast spreading and migration independently of ligand binding and of its kinase activity [14].
DDR1 expression is mainly described in epithelial cells and DDR2 in mesenchymal cells [2], but little is known about the expression of the two receptors in normal and cancer cells. Indeed, most of studies focus only on one receptor. In this study, we investigated the expression of the two receptors in different tumor cell lines and found that most of the cell lines expressed predominantly only one or the other receptor. To study the consequences of DDR1 and DDR2 co-expression in cells, we over-expressed the two receptors in HEK 293T cells and found an inhibition of both cell proliferation and migration. For the first time we also evidenced an interaction between the two receptors which may explain the deleterious effect of the co-expression on cell proliferation and migration.
Results
DDR1 and DDR2 expression in different cell lines
First we studied DDR1 and DDR2 expressions in different tumor cell lines cultured on plastic dishes and analyzed by western blotting. These tumor cells are of different origins, including carcinoma from different tissues, glioma and pediatric tumor. These include 786-O, Renca and Caki-2 cells (renal carcinoma), C6 and U87 cells (glioma), the SU-DIPG-IV cell line (derived from a neuroblastoma), HepG2 and HuH7 cells (hepato-carcinoma), A375 cells (melanoma) and MDA-MB-231 cells (breast carcinoma). Most cell lines expressed DDR1 at variable levels with Caki-2, C6, U87, HepG2 and A375 cells having higher level (Figure 1). Only, SU-DIPG-IV and A375 cells expressed high amount of DDR2 (Figure 1). With exception of A375 cells, DDR1 and DDR2 expressions were inversely correlated. Three cell lines, including HEK 293T, Renca and HuH7, expressed both receptors at low amounts, Caki-2, C6, U87, HepG2, and SU-DIPG-IV expressed both receptors but only one at high level. Only A375 cells expressed high level of the two receptors.
Figure 1.
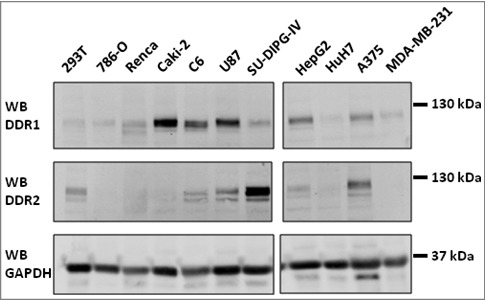
DDR1 and DDR2 expressions in different cell lines: cells cultured on plastic dishes were lysed, DDR1 and DDR2 expressions were analyzed by western blot using CST antibodies (DDR1, DDR2). GAPDH, loading control. N = 2.
In The Human Genome Atlas dataset, we found consistently that DDR1 and DDR2 mRNA expressions was mutually exclusive in the different cell lines (Supp Figure 1). In most of the cell lines DDR1 mRNA expression was high when DDR2 mRNA expression was low and vice versa. From all of cell lines found in the dataset, three were analyzed by western blotting. U87 cells expressed DDR1 and DDR2 mRNA and the two proteins were detected; HEK 293 cells expressed solely DDR1 mRNA but we found also the DDR2 protein and, inversely, HepG2 expressed only DDR2 mRNA but we found the DDR1 protein. This result suggests DDR regulation at the transcriptional and post-transcriptional level.
To study why in most cell lines only one DDR form is expressed at high level, we decided to modify HEK 293T cells and to over-express DDR1 or DDR2 in order to compare biological behaviors to that of cells over-expressing the two receptors. To distinguish between the intracellular dependent and independent activities of the two receptors we over-expressed WT or an intracellular truncated DDR1 or DDR2 protein, lacking the justamembrane and the kinase domains. These mutants are dominant-negative forms of DDR1 and DDR2 [15], and were, respectively, called DDR1DN and DDR2DN.
DDR1 and DDR2 over-expression in HEK 293T cells
First, we generated four HEK 293T cell populations that over-expressed DDR1, DDR1DN, DDR2 or DDR2DN, two populations (DDR1 DDR2-5 and -10, named DDR1&DDR2 in the text) that over-expressed both DDR1 and DDR2; and a control population (CTL) (Figure 2A and Table 1).
Figure 2.
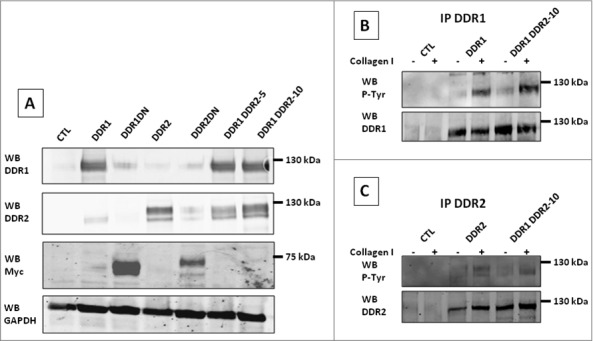
Characterization of the different HEK 293T cell populations: Using a combination of lentivirus transductions and plasmid transfections, HEK 293T cells were modified to over-express DDR1, DDR1DN, DDR2, DDR2DN or both DDR1 and DDR2. (A) Over-expressions were analyzed by western blot using anti DDR1 (SCB), DDR2 (SCB), Myc (DDRDN) or GAPDH (loading control) antibodies. N = 3. (B) and (C), DDR1 (B) or DDR2 (C) in cell lysates were immunoprecipitated with the SCB antibodies. Protein autophosphorylations were analyzed using anti P-Tyr antibody (4G10), immunoprecipitation control was analyzed using anti DDR1 (B) or anti DDR2 (C) antibodies from SCB. N = 2.
Table 1.
Cell lines: HEK 293T were modified by lentivirus transduction (pER220, MOI:10) and plasmid transfection (pCDNA4). Cell lines were selected by 5 µg/ml of puromycin and 200 µg/ml of Zeocin. DDR1 DDR2-5 and -10 derived from the DDR1 cell line, further transfected with, respectively 5 or 10 µg of pCDNA4 DDR2 in two independent experiments.
| Cell line | pER220 | pCDNA4 |
|---|---|---|
| DDR1 | DDR1 | Empty |
| DDR1DN | DDR1DN | Empty |
| DDR2 | DDR2 | Empty |
| DDR2DN | DDR2DN | Empty |
| DDR1 DDR2-5 | DDR1 | DDR2 |
| DDR1 DDR2-10 | DDR1 | DDR2 |
| CTL | Empty | Empty |
Using RT-qPCR experiments we found an increase in DDR1 mRNA 60 fold the amount of DDR1 mRNA in CTL cells; an increase in DDR1DN mRNA 80 fold the amount of DDR1 mRNA in CTL cells; an increase in DDR2 mRNA 6 fold the amount of DDR2 mRNA in CTL cells and an increase in DDR2DN mRNA 30 fold the amount of DDR2 mRNA in CTL cells (data not shown).
Upon stimulation with coated collagen I, DDR autophosphorylation was examined. Starved cells were cultured on collagen I-coated dishes for 16 h, after lysis, DDR1 or DDR2 were immunoprecipitated and tyrosine-autophosphorylation was analyzed by probing the blot with the anti phosphotyrosine antibody 4G10. Figure 2B shows DDR1 activation by collagen I in cells over-expressing DDR1 or DDR1&DDR2. Figure 2C depicts DDR2 activation by collagen I in cells over-expressing DDR2 or DDR1&DDR2. As expected, DDR1 or DDR2 autophosphorylation was not observed in cell lines expressing DDR1DN or DDR2DN (data not shown). These results showed that the presence of DDR1 and DDR2 in cells did not impede receptor autophosphorylation.
DDR1 and DDR2 intracellular signalization in the different HEK 293T cell populations
In order to study collagen I induced intracellular signalizations, cells were serum starved for 16 h and stimulated with collagen I for 2 h, 8 h and 24 h. Cells were then lysed and different intracellular pathways analyzed by western blot. DDR1, DDR2 and DDR1&DDR2 induced Jak2 phosphorylation on Tyr1007-1008 after 2 h and 8 h of collagen stimulation but with different electrophoretic mobility (Figure 3 and data not shown). Collagen I induced Jak2 activation, at 130 kDa molecular weight, in cells expressing DDR1 or DDR1&DDR2 (Figure 3). In cell expressing DDR2 or DDR1&DDR2, collagen I induced Jak2 tyrosine-phosphorylation but with a lower electrophoretic mobility (star in Figure 3) suggesting that, in addition to Tyr1007-1008 phosphorylation, Jak2 is modified on other residues. Alternatively, the antibody may cross-react with activated Jak1 or Tyk2. After 24 h of collagen I stimulation no Jak2 activation was observed (data not shown).
Figure 3.
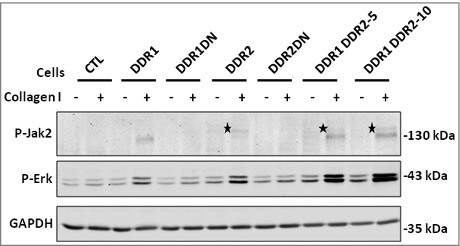
DDR1 and DDR2 intracellular signalization in the different HEK 293T cell populations. Cells were cultured or not on collagen I matrix for 8 h. Lysates were analyzed for the presence of activated Jak2 (P-Jak2) or Erk (P-Erk) by western blot. GAPDH, loading control. Star, low mobility P-Jak2 in DDR2 expressing cells. N = 3.
After 2 h of stimulation, collagen I induced Erk activation only in cells expressing DDR1&DDR2 (data not shown). After 8 h of collagen I stimulation, Erk was activated in both cell populations expressing DDR but the activation was higher in cells expressing DDR1&DDR2 (Figure 3). Quantification of the western blot signal indicated a 2.5 to 3.5 increase in cells over-expressing DDR1&DDR2 in comparison to DDR1 or DDR2 cells. After 24 h only a small increased in Erk phosphorylation persisted if cells expressed both DDR1&DDR2 (data not shown). These results indicate that Erk activation was increased if cells expressed DDR1&DDR2.
AKT, another signalization pathway downstream of collagen I induced DDR activation [9], was not activated in our conditions (data not shown).
Taken together, this indicates that Erk phosphorylation is highly increased in cells expressing both DDR1&DDR2, albeit Erk can also be activated by each of the DDRs.
DDR1 increases cell proliferation in HEK 293T cells in 3D conditions but decreases it when combined with DDR2
The role of DDR1 and DDR2 in cell proliferation was studied in 2D and 3D conditions.
In 2D conditions, cells were cultured on collagen I coated dishes for 72 h and counted. No difference in cell proliferation was observed if cells express DDR1 or DDR1DN, suggesting that DDR1 is not implicated in HEK 293T cell proliferation. Cell proliferation was significantly inhibited if cells expressed DDR2DN but no difference was observed in DDR2 cells. Expression of both DDR1 and DDR2 in cells significantly inhibited proliferation (Figure 4A).
Figure 4.
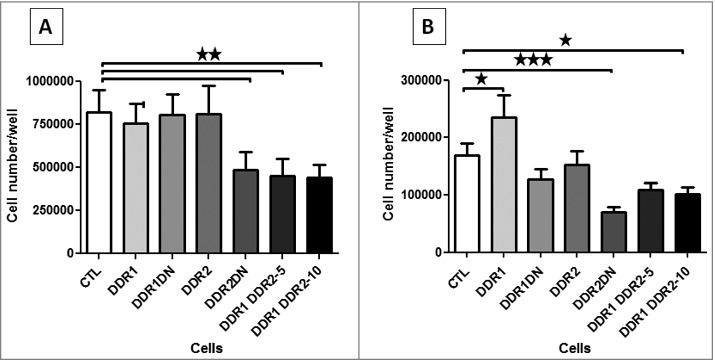
DDR1 and DDR2 roles in cell proliferation. (A) 2D cell proliferation, after 72 h of cell culture on a collagen I matrix HEK 293T cells were detached and counted. N = 5. (B) HEK 293T cells were embedded in collagen I matrix and counted after 7 days of culture. N = 6.
In 3D experiments, cells were embedded in 2 mg/ml of collagen I and cultured for 7 days. In these conditions DDR1 increased cell proliferation and DDR1DN had no effect, suggesting a role of the DDR1 intracellular kinase in 3D cell proliferation. As in 2D, DDR2DN but not DDR2 and the co-expression of DDR1&DDR2 inhibited cell proliferation (Figure 4B).
All these results showed that over-expression of both DDR1&DDR2 on HEK 293T cells inhibited cell proliferation. DDR1 alone increased cell proliferation only in 3D culture, and the effect was certainly mediated by the kinase activity of the receptor.
DDR1 and DDR1&DDR2 increase cell adhesion on collagen I matrix
To determine whether DDR1 or DDR2 were involved in cell adhesion, we quantified cells after 15 min adhesion on collagen I matrix as described [16]. Adhesion of DDR1, DDR1DN, DDR2DN and of DDR1&DDR2 expressing cells was increased compared to CTL (Figure 5A). No difference in cell adhesion was observed if cells over-expressed DDR2. Because this assay takes into account integrin and DDR adhesion to collagen I, we set-up a test in presence of 5 mM EDTA, a chelator of Ca++ and Mg++, two ions implicated in integrin activation and in their binding to collagen. DDR binding to collagen is independent of these ions, and their binding to collagen is not affected by the presence of EDTA. In this assay, CTL and DDR2 cells were unable to adhere on collagen I, suggesting that in absence of EDTA, CTL or DDR2 cell adhesion was mediated only by integrins (Figure 5B). DDR1, DDR1DN, DDR2DN and DDR1&DDR2 expressing cells adhered on collagen I matrix in presence of EDTA (Figure 5B).
Figure 5.
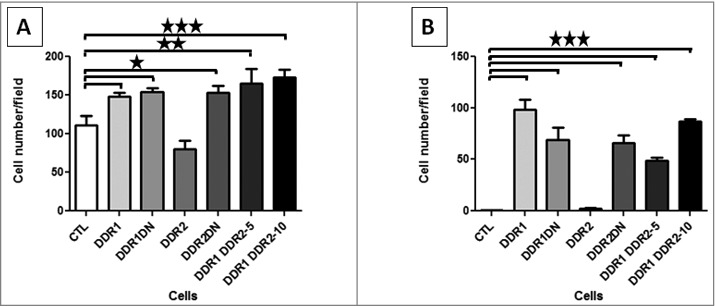
DDR1 and DDR2 roles in cell adhesion. HEK 293T cells were allowed to adhere on a collagen I matrix for 15 min, fixed and nuclei labeled. 5 photos per well were taken and nuclei counted using ImageJ software. (A) cell adhesion in presence of divalent ions. N = 6. (B) cell adhesion in presence of the divalent ion chelator EDTA. N = 4.
Altogether, these results showed that DDR1 participates in cell adhesion on collagen I matrix. DDR1 adhesion is independent of the intracellular domains since cell adhesion was maintained in DDR1DN cells. DDR2DN but not DDR2 cells adhere to the collagen I matrix, indicating that, in our conditions, DDR2 inhibited cell adhesion by a mechanism dependent of the intracellular domains. In DDR1&DDR2 cells, adhesion is mediated by DDR1 and is not inhibited by DDR2.
DDR1 and DDR1&DDR2 inhibit collagen I induced cell migration independently of the intracellular domains
The role of DDR1 and DDR2 in cell migration was evaluated using a wound healing assay. Cells were cultured on collagen I coated dishes until confluence and a scratch was performed. Closing of the wound area was recorded over a 24 h period (Figure 6A). The wound closure was delayed in cells expressing DDR1, DDR1DN or both DDR1&DDR2 (Figure 6A and B). DDR1 inhibition of cell migration was independent of the intracellular domains of the receptor. As unexpected DDR2 or DDR2DN were not implicated in collagen I induced cell migration (Figure 6A and B).
Figure 6.
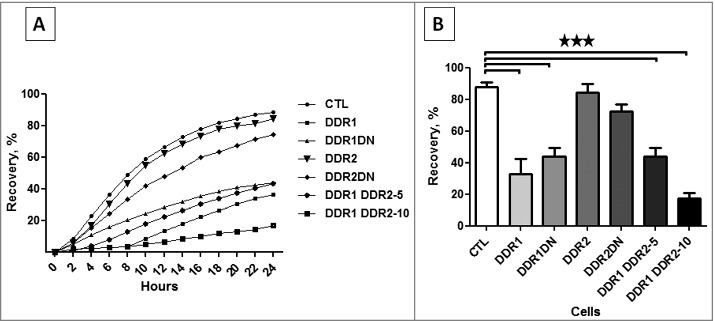
DDR1 and DDR2 roles in cell migration. HEK 293T cell migration was assessed in a wound healing assay. The kinetics of the cell migration into the wound was followed using an IncuCyte system (Essen BioScience). (A) kinetics of wound healing. (B) percentage of wound closure after 24 h of cell migration. N = 4.
These experiments showed that DDR1, independently of its intracellular activities inhibited HEK 293T cell migration in a wound healing assay.
Altogether, these results suggest an implication of DDR1 in HEK 293T cell adhesion and when cells adhere firmly to the matrix they stop migration. When the two receptors are over-expressed together, cells adhesion is predominant and cell migration is inhibited.
DDR1 and DDR2 immunocytolocalization and cell phenotypes
To study DDR1 and DDR2 localization and their roles in cell phenotypes we performed co-immunofluorescence experiments using anti DDR1 or DDR2 antibodies and phalloidin as filamentous actin marker on HEK 293T cells cultured or not on collagen I matrix for 24 h. In control cells or in cells over-expressing DDR1DN or DDR2DN, DDR1 or DDR2 were faintly detected (Figure 7A cells cultured on collagen I, Supp Figure 2A cells cultured on absence of collagen I and data not shown). In cells over-expressing DDR1, DDR2 or DDR1&DDR2, the proteins were found at cell-cell junctions when cells were cultured on plastic dishes (Supp Figure 2B-D) as it was previously described [10, 17, 11]. In presence of collagen I, the two proteins delocalized from cell-cell junctions, DDR1 bound to collagen fibers at basal membrane of the cell (Figure 7B and C); DDR2 aggregated and formed dots (Figure 7D and E).
Figure 7.
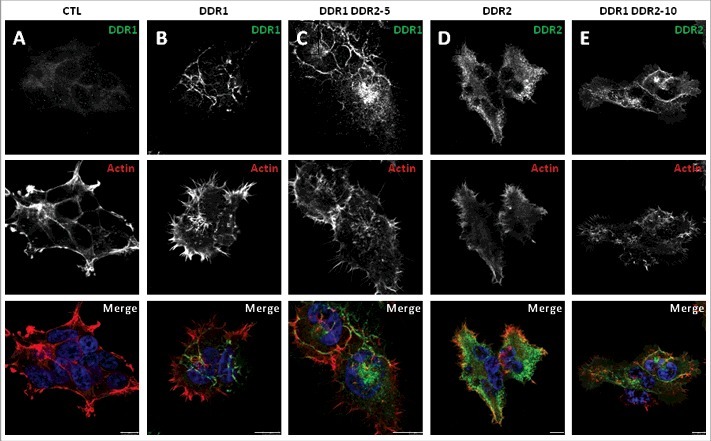
Immunocytofluorescence localization of DDR1, DDR2 and filamentous actin in the different HEK 293T cell populations cultured in presence of collagen I. Cells were cultured for 24 h on glass coverslips coated with collagen I, fixed and stained with DDR1 or DDR2 antibody (CST, green) as indicated and with phalloidin (filamentous actin, red). Images were captured using a confocal microscope (Leica SP5). Bar: 10 µm. N = 3.
Because our DDR1 and DDR2 antibodies are rabbit antibodies we were unable to show DDR1 and DDR2 co-localization. In order to study a co-localization between the two receptors, we used A375 cells stably expressing two fusion proteins (DDR1-GFP and DDR2-mCherry). In absence of collagen I stimulation, DDR1-GFP and DDR2-mCherry co-localized at cell periphery (Figure 8A). The co-localization was preserved when A375 cells were stimulated with collagen I, the two receptors were found together along collagen fibers (Figure 8B).
Figure 8.
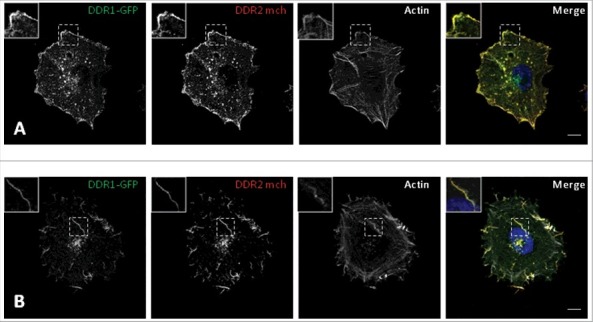
DDR1 and DDR2 localization in A375 cells. A375 cells expressed DDR1-GFP or DDR2-mCherry (DDR2 mch) and were cultured on gelatin (A) or Collagen I. (B) coated glass coverslips. The localization of the two fusion proteins was analyzed with a confocal microscope (Leica SP5). Bar: 5.6 µm.
All HEK 293T cell populations changed their phenotype when cells were stimulated with collagen I. On plastic, cells formed clusters with cell-cell junctions, cells at the periphery had some filopodia (Supp Figure 2). Cultured on collagen I, the phenotypes changed. Control cells were elongated with some membrane protrusions. DDR1 and DDR1DN cells were full of filopodia (Figure 7 and data not shown, actin labeling). DDR2, DDR2DN and DDR1&DDR2 cells were more elongated with full of filopodia (Figure 7 and data not shown, actin labeling).
DDR1 and DDR2 interaction
Because DDR1 and DDR2 were found at cell-cell junctions or along collagen fibers in HEK 293T cells or co-localized in absence or presence of collagen I in A375 cells we were interested to study the interaction between the two receptors. To this aim, we performed co-immunoprecipitation experiments for DDR1 and DDR2.
HEK 293T cells over-expressing DDR1 and DDR2 were stimulated or not for 16 h with collagen I, and cell lysates immunoprecipitated and immunoblotted. As shown in Figure 9A DDR1 was found associated with DDR2 in absence or in presence of collagen I stimulation, suggesting DDR1 DDR2 interaction at the cell membrane before collagen stimulation.
Figure 9.
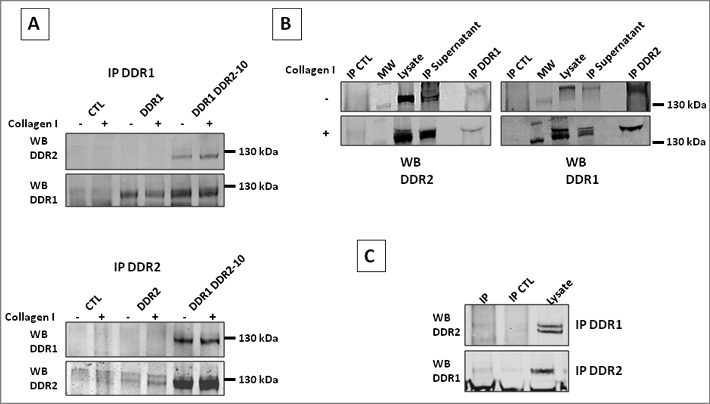
DDR1 and DDR2 interaction in different cell lines. DDR1 and DDR2 physical interaction was studied using co-immunoprecipitation (IP) assay. DDR1 or DDR2 were immunoprecipitated using anti DDR1 or DDR2 antibodies. Immunoprecipitation were analyzed in western blots using DDR2 or DDR1 antibodies. (A) IP of 293T cell lysates, SCB antibodies were used. (B) IP of DDR1-GFP and DDR2-mCherry transfected A375 cell lysates, CST antibodies used. IP CTL, IP with control rabbit IgG. (C) IP of C6 cell lysates, CST antibodies used. IP CTL, beads alone. N = 3 for each immunoprecipitation.
In a second experiment, we used A375 cells co-expressing the proteins DDR1-GFP and DDR2-mCherry. Cells were stimulated or not with collagen I, lysed and DDR1 or DDR2 was immunoprecipitated. DDR2-mCherry was found associated to DDR1 and DDR1-GFP was associated to DDR2. As in HEK 293T cells, the interaction between the two receptors was independent of collagen I stimulation (Figure 9B).
In a third experiment, we used unmodified C6 cells and carried out co-immunoprecipitation. C6 cells express moderate level of DDR1 and DDR2 (Figure 1). DDR2 was found associated to DDR1 when DDR1 was immunoprecipitated and vice versa, DDR1 was found associated to DDR2 when DDR2 was immunoprecipitated (Figure 9C). Again, the association between the two receptors was found in absence and in presence of collagen I.
Discussion
The two tyrosine kinase discoidin domain receptors, DDR1 and DDR2 have more than 52% of sequence identity [6]. They are both implicated in cell proliferation, adhesion and migration but most of the time these studies were performed with cell expressing only one receptor. In this study, we analyzed these biological activities in HEK 293T expressing high amount of both, DDR1 and DDR2. We first confirmed that the majority of tumor or non-tumor cell lines expressed mainly one receptor, DDR1 or DDR2 or low amount of both receptors (Figure 1 and Supp Figure 1). The reason for this preferential expression in both cell lines is not known. It may involve redundancy of the activities of the two receptors or a deleterious phenotype induced by the presence of the two receptors. To discriminate between these different hypotheses we over-expressed DDR1, DDR2 or both receptors in HEK 293T cells. To discriminate between the intracellular dependent or independent activities of DDRs we also over-expressed the intracellular truncated forms, DDR1DN or DDR2DN.
DDR1 and DDR2 can be phosphorylated on Tyr residues after activation by collagen I, as previously described for DDR1 over-expression in MDCK cells [15]. Nevertheless DDR2 autophosphorylation was lower than DDR1, maybe because of DDR2 lower expression than DDR1 as suggested by RT-qPCR analysis. Recently it was reported that ADAM 10 controls DDR1 activity by inducing ectodomain shedding of DDR1 [18]. As it was shown by Shitomi et al, a shedding and tyr phosphorylated form of DDR1 was immunoprecipitated in our experiments. Moreover collagen I stimulation increased the shedding of the tyr phosphorylated DDR1 (data not shown).
DDR1 and DDR2 were found associated to the cell-cell junctions, where they may participate at the stabilization of the E-cadherin junctions, as it was described for DDR1 [17]. The majority of the cells expressing DDR1, DDR2 and DDR1&DDR2 exhibited filopodia when cells were cultured on collagen I. This was independent of the intracellular domain and certainly mediated by the discoidin domains of the receptors because DDR1DN and DDR2DN cells harbored the same phenotype. Because filopodia formation is dependent of the Rho GTPase Cdc42 [19], Rho GTPase Cdc42 must be over activated in these cells. DDR1 activation of Cdc42 was shown to pass by the activation of the Cdc42 Gef Tuba [12]. Whether or not DDR2 activate Tuba or another Cdc42 Gef, is under investigation.
DDR1 and DDR2 were able to activate the Erk pathway when cells were cultured on collagen I. As previously described, the activation was very slow but with sustained kinetics [20, 21]. Moreover when the two receptors were expressed at the same time, Erk activation was increased. Collagen I binding to DDR1, such as to DDR2 [20], was found to activate Jak2. However, the electrophoretic mobility of Jak2 activated by DDR2 was lower than for DDR1, suggested an additional post-translational modification of Jak2 activated by DDR2 or an activation of Jak1 or Tyk2, two members of Jak family presenting some cross reactivity with the antibody used. Whether or not Jak2 activates Stat transcriptional factors, is under investigation.
As attempted, DDR1 induced HEK 293T cell proliferation at least in the 3D assay. DDR2 over-expression had no effect on cell proliferation but DDR2DN inhibited proliferation. The reason for this is not known, maybe endogenous DDR2 is enough to saturate downstream signaling involved in cell proliferation. More interestingly, co-expression of DDR1 and DDR2 inhibited proliferation. This may be explained by an over-activation of the Erk pathway that can inhibited cell proliferation, as it was shown with HGF inducing high Erk activation but inhibition of proliferation of HepG2 cells [22]. Nevertheless, DDR1 and DDR2 co-expression was deleterious for biological activity. A375 cells over-expressing DDR1 and DDR2 quickly declined in cultured, which is in agreement with results obtained with HEK 293T cells (Saltel, personal communication). The mechanism inducing inhibition of cell proliferation is under investigation, but DDR1 was implicated in cell apoptosis in cardiomyocytes after ischemia/reperfusion [23] and in breast cancer cells cultivated in 3D [24].
DDR1 was implicated in cell adhesion [2] and this is in agreement with our results. The role of DDR2 is more controversial. In smooth muscle cells DDR2 has no effect on cell adhesion [25], but in HEK 293 cells, DDR2 increases cell adhesion [7]. In our study, DDR2 was unable to induce HEK 293T cell adhesion in our experimental shorter kinetic conditions (15 min instead of 1 h) than the one used in Xu et al [7]. Alternatively very high amount of DDR2 may be necessary to induce cell adhesion to the collagen matrix.
DDR1 was shown to promote cell migration [2, 26] but in MDCK cells, DDR1 over-expression inhibited cell migration on a collagen matrix [27] which is in agreement with our results. Moreover, we found that this inhibition was not dependent of the intracellular domains and was certainly mediated by the strong adhesion of the discoidin domain with collagen I. Inhibition of cell migration was maintained when cells co-expressed DDR1 and DDR2.
Finally, our co-immunoprecipitation assay clearly demonstrated an association between DDR1 and DDR2. This association was found not only in HEK 293T and A375 cells over-expressing DDR1 and DDR2 but also in C6 cells expressing endogenous DDR1 and DDR2. Whether or not this interaction may cause the inhibitory effect of DDR1&DDR2 expressing cells is under investigation as well as the underlying mechanism.
In summary, we report that co-expression of high amount of DDR1 and DDR2 induces inhibition of cell proliferation and cell migration. Therefore, the co-expression of the two receptors may be deleterious in most of the cells, particularly to tumor cells and may explain why in most of the cell lines solely one receptor is predominantly expressed or why they expressed low amount of each receptor.
Materials and methods
Cell lines and cell culture
HEK 293T were a gift of Dr Richard Iggo (INSERM U1218, France) and cultured in complete medium (DMEM complemented with 10% Fetal Calf Serum (FBS); 100 U/ml penicillin and 100 µg/ml streptomycin) at 37°C and 5% CO2. Cells were modified to express DDR1, DDR2, DDR1 and DDR2, DDR1DN or DDR2DN by lentivirus transduction (pER220) or plasmid transfection (pCDNA4) as indicated in Table 1. All the cell lines were selected with 5 µg/ml of puromycin and 200 µg/ml of Zeocin.
The DDR1DN and the DDR2DN were truncated after the transmembrane domain and lack all the intracellular domains. To facilitate the detection, a Myc Tag was added in the intracellular part of the two dominant negative receptors.
Rat tail collagen I (Corning) was diluted at 250 µg/ml and used to coat plastic plates for at least 1 h at 37°C. After 3 washes with PBS, plates were immediately used.
The neuroblastoma SU-DIPG-IV cell line was a gift of Dr Michelle Monje Deisseroth (Stanford University, USA). All the other cell lines were cultured as recommended by American Type Culture Collection.
Cell lysis
Cells cultured in 6 cm culture dishes were washed in PBS (Phosphate Buffer Saline) and lysed in 200 µl of lysis buffer (50 mM Hepes pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 5 mM EDTA, 10% glycerol, 1% Triton X100, 0.5% NP40) containing proteases and phosphatases inhibitors (Roche) for 30 min at 4°C under agitation. Samples were centrifuged at 20800 g for 15 min and the protein concentration of the supernatant quantified with a Bradford assay using Bovine Serum Albumin (BSA) as a standard.
For cell signaling experiments, cells were starved overnight in cell medium containing 0.5% FBS. After trypsinization, 2.106 cells were cultivated on a 10 cm dish coated or not with 250 µg/ml of collagen I. After 2 h, 8 h and 24 h cells were lysed and proteins quantified.
Western blot
Proteins were separated on a 8% or 10% (signalization experiments) SDS-PAGE and transferred on a nitrocellulose membrane. Membranes were incubated in blocking buffer (5% BSA in TBS-Tween; 50 mM Tris HCl pH 7.4, 150 mM NaCl, 0.1% Tween 20) for 1 h at RT, then overnight at 4°C with primary antibody diluted in blocking buffer (Table 2). After extensive washes in TBS-Tween, membranes were incubated with appropriate secondary antibodies diluted in blocking buffer (dilution 1/5000, Li-Cor Biosciences). After extensive washes membranes were analyzed using an Odyssey infrared imaging system (Li-Cor Biosciences).
Table 2.
Primary antibodies used for western blot analysis.
| Antigen | From | Dilution |
|---|---|---|
| DDR1 | –Santa Cruz Biotechnology (SCB) (Rabbit) | 1/1000 |
| –Cell Signaling Technology (CST) (Rabbit) | 1/1000 | |
| DDR2 | –SCB (Goat) | 1/1000 |
| –CST (Rabbit) | 1/1000 | |
| c-Myc | –Clone 9E10 | 1/1000 |
| P-Tyr | –Clone 4G10 | 1/1000 |
| P-Jak2 (Tyr1007-1008) | –CST (Rabbit) | 1/1000 |
| P-Erk | –CST (Rabbit) | 1/1000 |
| GAPDH | –SCB (Rabbit) | 1/2000 |
Immunoprecipitation
1 mg/500µl of solubilized proteins were incubated with DDR1 or DDR2 antibody (1 µg, SCB; or 1/100 dilution, CST) for 3 h (SCB) or overnight (CST) at 4°C. 50 µl of sepharose conjugated protein G (1/2 in lysis buffer) were added for 1 h incubation at 4°C. After 3 to 5 washes in lysis buffer, the pellet was resuspended in Laemli buffer and submitted to gel electrophoresis.
Cell proliferation
2D proliferation: 5000 cells in 500 µl of complete medium were cultured in a 24 well plate coated with 250 µg/ml of collagen I for 72 h. Cells were trypsinized and counted using a Coulter Particle Counter.
3D proliferation: 1.105 cells were mixed with 500 µl of 2 mg/ml collagen I and deposed in a 24 well plate. After polymerization, 500 µl of complete medium were added. After 3 days in culture, the medium was changed and cells were cultured for another 4 days. The medium was removed and the collagen digested by 500 µl of collagenase (2 mg/ml). After centrifugation cells were separated by adding 250 µl of trypsin (Gibco) and counted using a Coulter Particle Counter.
Migration: Wound healing assay
125 000 cells in 100 µl of complete medium were cultivated in 96 well plate ImageLock coated with 250 µg/ml of collagen I. When cells were at confluence the wound was performed using a Wound Maker (Essen BioScience), the cells were washed with PBS and 100 µl of complete medium were added. Cell migration was followed for 24 h using an IncuCyte system (Essen BioScience).
Cell adhesion
Cell adhesion assay was derived from von Wichert et al [16]. Briefly, cells were quickly trypsinized and washed 5 times in adhesion buffer (DMEM containing 0.1% BSA). Cells were counted, the concentration adjusted to 50 000 cells in 500 µl of adhesion buffer containing or not 5 mM EDTA and left 1 h at 37°C. 500 µl of cells were deposed in a 24 well plate previously coated with collagen I and blocked 30 min with 500 µl of adhesion buffer. After 15 min of incubation, dishes were washed 3 times with DMEM containing or not 5 mM EDTA. Adherent cells were fixed with 4% paraformaldehyde for 10 min and nuclei were labeled 10 min with DAPI (2 µg/ml, FluoProbes). After washes, 5 pictures per dish were taken using a Zoe Fluorescent Cell Imager (Biorad). Nuclei were counted using ImageJ software.
Immunofluorescence
Cells were cultured for 24 h on a glass coverslip coated or not with 250 µg/ml of collagen I and fixed 10 min with 4% paraformaldehyde. Coverslips were blocked 1 h with 5% BSA 0.3% Triton X100 in PBS and incubated overnight at 4°C with primary antibody diluted 1/800 in 1% BSA 0.3% Triton X100 in PBS. After 5 washes in PBS, coverslips were incubated 1 h with appropriate FITC-conjugated secondary antibody, Alexa 556-conjugated Phalloidin (1/500 dilution, FluoProbes) and DAPI (1 µg/ml, FluoProbes) in 1% BSA 0.3% Triton X100 in PBS. After 3 washes in PBS coverslips were mounting for microscope observation using Fluoromount G. Cells were observed and pictures taken under a confocal microscope (Leica SP5).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 5 software. Multiple comparisons were performed with Repeated Measures ANOVA analysis of variance, followed by Dunnett's Multiple Comparison Test. ★, P <0.05; ★★, P <0.01; ★★★, P <0.001.
Supplementary Material
Funding Statement
University of Bordeaux, Ligue Contre Le Cancer, Comités Aquitaine et Charentes, National institute of Health and Medical Research (Inserm)
Disclosure of potential conflicts of interest
No conflicts of interest, financial or otherwise are declared by the authors.
Acknowledgments
We would like to thank Dr Michelle Monje Deisseroth and Dr Richard Iggo for providing us SU-DIPG-IV and HEK 293T cell lines. We thank Nathalie Senant for helps in confocal image acquisition. We are grateful to Dr Clotilde Billottet and to the MiRCaDe team (U1035) to helpful discussions. This work was supported by grants from Inserm and Bordeaux University and from La Ligue Contre Le Cancer, Comités Aquitaine et Charentes.
References
- [1].Johnson JD, Edman JC, Rutter WJ. A receptor tyrosine kinase found in breast carcinoma cells has an extracellular discoidin I-like domain. Proc Natl Acad Sci U S A. 1993;90:5677–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. 2014;310:39–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vogel W, Gish GD, Alves F, et al.. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23. [DOI] [PubMed] [Google Scholar]
- [4].Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mihai C, Chotani M, Elton TS, et al.. Mapping of DDR1 distribution and oligomerization on the cell surface by FRET microscopy. J Mol Biol. 2009;385:432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Carafoli F, Hohenester E. Collagen recognition and transmembrane signalling by discoidin domain receptors. Biochim Biophys Acta. 2013;1834:2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xu H, Bihan D, Chang F, et al.. Discoidin domain receptors promote α1β1- and α2β1-integrin mediated cell adhesion to collagen by enhancing integrin activation. PloS One. 2012;7:e52209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Staudinger LA, Spano SJ, Lee W, et al.. Interactions between the discoidin domain receptor 1 and β1 integrin regulate attachment to collagen. Biol Open. 2013;2:1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Valiathan RR, Marco M, Leitinger B, et al.. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012;31:295–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang C-Z, Yeh Y-C, Tang M-J. DDR1/E-cadherin complex regulates the activation of DDR1 and cell spreading. Am J Physiol Cell Physiol. 2009;297:C419–429. [DOI] [PubMed] [Google Scholar]
- [11].Hidalgo-Carcedo C, Hooper S, Chaudhry SI, et al.. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol. 2011;13:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Juin A, Di Martino J, Leitinger B, et al.. Discoidin domain receptor 1 controls linear invadosome formation via a Cdc42-Tuba pathway. J Cell Biol. 2014;207:517–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ghosh S, Ashcraft K, Jahid MJ, et al.. Regulation of adipose oestrogen output by mechanical stress. Nat Commun. 2013;4:1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Herrera-Herrera ML, Quezada-Calvillo R. DDR2 plays a role in fibroblast migration independent of adhesion ligand and collagen activated DDR2 tyrosine kinase. Biochem Biophys Res Commun. 2012;429:39–44. [DOI] [PubMed] [Google Scholar]
- [15].Wang C-Z, Hsu Y-M, Tang M-J. Function of discoidin domain receptor I in HGF-induced branching tubulogenesis of MDCK cells in collagen gel. J Cell Physiol. 2005;203:295–304. [DOI] [PubMed] [Google Scholar]
- [16].von Wichert G KD, Schmid H, et al.. Focal adhesion kinase mediates defects in the force-dependent reinforcement of initial integrin-cytoskeleton linkages in metastatic colon cancer cell lines. Eur J Cell Biol. 2008;87:1–16. [DOI] [PubMed] [Google Scholar]
- [17].Eswaramoorthy R, Wang CK, Chen WC, et al.. DDR1 regulates the stabilization of cell surface E-cadherin and E-cadherin-mediated cell aggregation. J Cell Physiol. 2010;224:387–397. [DOI] [PubMed] [Google Scholar]
- [18].Shitomi Y, Thøgersen IB, Ito N, et al.. ADAM10 controls collagen signaling and cell migration on collagen by shedding the ectodomain of discoidin domain receptor 1 (DDR1). Mol Biol Cell. 2015;26:659–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Amarachintha SP, Ryan KJ, Cayer M, et al.. Effect of Cdc42 domains on filopodia sensing, cell orientation, and haptotaxis. Cell Signal. 2015;27:683–693. [DOI] [PubMed] [Google Scholar]
- [20].Ruiz PA, Jarai G. Collagen I induces discoidin domain receptor (DDR) 1 expression through DDR2 and a JAK2-ERK1/2-mediated mechanism in primary human lung fibroblasts. J Biol Chem. 2011;286:12912–12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ongusaha PP, Kim JI, Fang L, et al.. p53 induction and activation of DDR1 kinase counteract p53-mediated apoptosis and influence p53 regulation through a positive feedback loop. EMBO J. 2003;22:1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tsukada Y, Miyazawa K, Kitamura N. High intensity ERK signal mediates hepatocyte growth factor-induced proliferation inhibition of the human hepatocellular carcinoma cell line HepG2. J Biol Chem. 2001;276:40968–40976. [DOI] [PubMed] [Google Scholar]
- [23].Ouyang F, Huang H, Zhang M, et al.. HMGB1 induces apoptosis and EMT in association with increased autophagy following H/R injury in cardiomyocytes. Int J Mol Med. 2016;37:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Assent D, Bourgot I, Hennuy B, et al.. A membrane-type-1 matrix metalloproteinase (MT1-MMP)-discoidin domain receptor 1 axis regulates collagen-induced apoptosis in breast cancer cells. PloS One. 2015;10:e0116006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hou G, Wang D, Bendeck MP. Deletion of discoidin domain receptor 2 does not affect smooth muscle cell adhesion, migration, or proliferation in response to type I collagen. Cardiovasc Pathol Off J Soc Cardiovasc Pathol. 2012;21:214–218. [DOI] [PubMed] [Google Scholar]
- [26].El Azreq M-A, Kadiri M, Boisvert M, et al.. Discoidin domain receptor 1 promotes Th17 cell migration by activating the RhoA/ROCK/MAPK/ERK signaling pathway. Oncotarget. 2016;7:44975–44990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang C-Z, Su H-W, Hsu Y-C, et al.. A discoidin domain receptor 1/SHP-2 signaling complex inhibits alpha2beta1-integrin-mediated signal transducers and activators of transcription 1/3 activation and cell migration. Mol Biol Cell. 2006;17:2839–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.