

ABSTRACT
Lamellipodia and ruffles are veil-shaped cell protrusions composed of a highly branched actin filament meshwork assembled by the Arp2/3 complex. These structures not only hallmark the leading edge of cells adopting the adhesion-based mesenchymal mode of migration but are also thought to drive cell movement.
Although regarded as textbook knowledge, the mechanism of formation of lamellipodia and ruffles has been revisited in the last years leveraging new technologies. Furthermore, recent observations have also challenged our current view of the function of lamellipodia and ruffles in mesenchymal cell migration.
Here, I review this literature and compare it with older studies to highlight the controversies and the outstanding open issues in the field. Moreover, I outline simple and plausible explanations to reconcile conflicting results and conclusions. Finally, I integrate the mechanisms regulating actin-based protrusion in a unifying model that accounts for random and ballistic mesenchymal cell migration.
KEYWORDS: actin, Arp2/3 complex, cancer, cell migration, formins, lamellipodia, ruffles
Introduction
Actin cytoskeleton allows cells to change shape and to form specialized protrusions in response to specific demands of the extracellular environment. These adaptive responses rely on the assembly of monomeric globular (G)-actin into filamentous (F)-actin to apply a deformation force on the plasma membrane [1]. Cells exert a tight spatiotemporal control over the reaction of actin polymerization to produce plasma-membrane protrusions with unique morphologies and functions. Lamellipodia, ruffles and filopodia are actin-based protrusions particularly relevant in the context of mesenchymal (fibroblast-like) cell motility [2].
Lamellipodia and ruffles are dynamic wave-like extensions of the plasma membrane (Figure 1A). The lamellipodium is a thin (0.1–0.3 μm) and usually long (1–5 μm) projection that adheres to the underlying substrate. This protrusion hallmarks the cell's leading edge, contains densely packed branched actin filaments and is devoid of organelles [3]. Ruffles are thin sheet-like structures extending above the advancing lamellipodium or on the dorsal plasma membrane (Figure 1A). Instead, circular dorsal ruffles protrude exclusively from the dorsal surface of cells to give rise to ring-shaped structures (Figure 1A). Alike lamellipodia, ruffles do not contain organelles and are made of a highly branched actin network with the fast-growing filament barbed ends facing the plasma membrane [3]. While the lamellipodium is regarded as the major force driving mesenchymal cell migration both in 2D and 3D environments [4], ruffles may play context-dependent roles.
Figure 1.
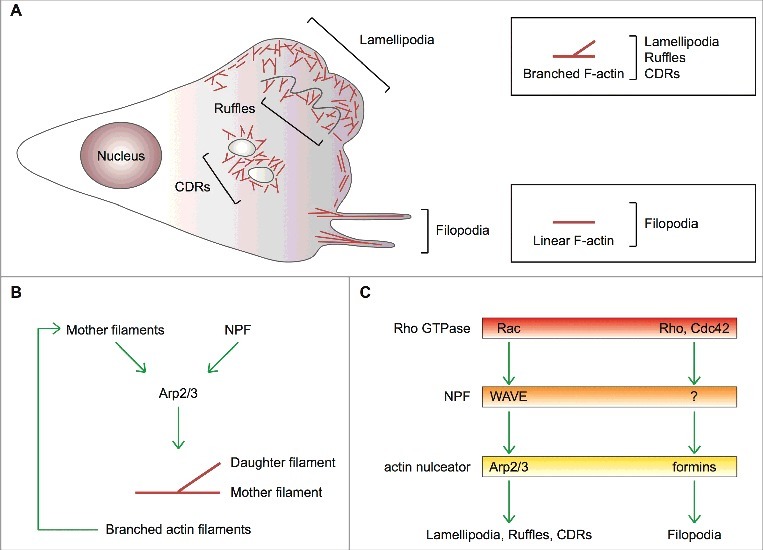
Actin-based protrusions in mesenchymal cells. (A) Schematic of the actin-based protrusions in a mesenchymal cell showing in red filamentous (F) actin. Lamellipodia, filopodia, (peripheral and dorsal) ruffles and circular dorsal ruffles (CDRs) are depicted. Boxes highlight that the geometry of F-actin distinguishes filopodia from all other actin-based cells protrusions formed on a 2D substrate. (B) Process diagram showing that 1) activation of the Arp2/3 complex requires two co-factors, namely a nucleation promoting factor (NPF) and pre-existing actin filaments (mother filaments), and 2) the Arp2/3 complex is an auto-catalytic actin nucleator because the product of the reaction (branched actin filaments) can be used as a substrate. (C) Dedicated pathways control actin nucleation by the Arp2/3 complex and formins thereby dictating the formation of different cell protrusions. Upstream regulatory Rho GTPases, NPFs and actin nulceators are shown. Question mark represents unknown formin-specific NPFs.
Filopodia project from the cell surface as tiny (about 200-nm wide) micrometer-long finger-like structures that can rotate and bend [5] (Figure 1A). They are composed of a bundle of 10–30 parallel linear actin filaments whose barbed ends push towards the tip [6]. Filopodia contain several types of receptors, including growth-factor receptors and integrins, and, for this reason, are regarded as cellular antennas that sense chemical and/or mechanical cues [7]. It is generally believed that filopodia may control directionality and persistence of movement by promoting cell-matrix adhesiveness at the leading edge to stabilize the advancing lamellipodium [5, 7, 8].
As actin polymerization guides lamellipodium, ruffle and filopodium formation, the geometry and supramolecular assembly of actin filaments determine the distinctive shape of each actin-based protrusion. Indeed, cells possess dedicated sets of proteins to control both the geometrical properties of actin filaments and the spatial relations among them [9].
Three decades of research on the molecular mechanisms controlling actin-based protrusion have identified Actin-related (Arp)2/3 complex as a key player in the making of lamellipodia and ruffles, and mesenchymal cell migration [9, 10]. Two actin-related proteins, Arp2 and Arp3, and other five subunits compose the Arp2/3 complex, which is intrinsically inactive [10]. Only when engaged by nucleation-promoting factor (NPF) proteins, does the Arp2/3 complex catalyze the polymerization of a new (daughter) actin filament that buds off a pre-existing (mother) filament with an angle of about 70 degrees [11] (Figure 1B). By promoting branched actin polymerization, the Arp2/3 complex contributes to cellular processes relying on movement, such as actin-based protrusion and cell migration, trafficking of intracellular membrane-delimited organelles, endo-exocytic vesicles and pathogenic viruses and bacteria [10, 12-16]. A set of dedicated NPFs acting at defined subcellular locations determines the remarkable functional versatility of the Arp2/3 complex [17].
Compelling evidence indicates that small GTPase Rac and WASP-family verprolin homologous NPF proteins (WAVE1, WAVE2 and WAVE3) direct Arp2/3-complex-mediated actin polymerization within lamellipodia and ruffles, and promote cell migration [18–21] (Figure 1C). It has been initially suggested that convergent elongation of branched actin filaments assembled by the Arp2/3 complex generates filopodia [6]. However, molecular genetic approaches have shown that the Arp2/3 complex is dispensable for filopodium formation, which is a formin-dependent process [7,22–26] (Figure 1C).
Surprisingly, recent observations have challenged our current view of the mechanism of formation and the function of lamellipodia and ruffles in crawling mesenchymal cells. Here, I review this new literature and compare it with older studies to highlight the controversies and the outstanding open issues in the field. Moreover, I outline simple and plausible explanations to reconcile conflicting results and conclusions. Finally, I integrate the mechanisms regulating actin-based protrusion in a unifying model that accounts for random and ballistic migration states of mesenchymal cells.
Lamellipodia, ruffles and circular dorsal ruffles: Common vs. different origin?
Lamellipodia and ruffles are a hallmark of the mesenchymal mode of cell migration [2]. Being composed of a dense network of branched actin filaments generated by the Arp2/3 complex (Figure 1C), a fundamental and debated question in the field is whether or not lamellipodia and ruffles are different manifestations of the same protrusion.
Lamellipodia contact the underlying matrix close to the leading edge by means of relatively weak adhesion sites. If these sites are sufficiently stable, the activity of non-muscle myosin II at (the more internal) focal adhesions can stimulate the retrograde flow of the lamellipodial actin network, but not its surrounding membrane [27]. If the contact between the lamellipodium and the substrate is instead broken, myosin II activity results in the rearward movement of the protrusion. Under these circumstances, myosin II-mediated pulling gives rise to a peripheral ruffle [27]. However, it has been shown that knockout of myosin IIA [28] and myosin inhibitor blebbistatin [29] increase ruffle formation. The opposite (negative vs. positive) role of myosin IIA and myosin IIB in the making of lamellipodia and ruffles [28,30], as well as their different sensitivity to blebbistatin [31], could partly explain these contrasting observations. In light of that, the autonomous movement of keratocyte fragments after acquisition of an acto-myosin-based asymmetric state [32] suggests that myosin II activity is finely balanced both in space and time within lamellipodia and ruffles.
In any case, peripheral ruffles lack adhesion sites and are unable to transmit traction forces when cells move on a 2D substrate and are sometimes used as a readout of lamellipodial adhesiveness [33].
Thus, most peripheral and dorsal ruffles can be certainly regarded as non-adhesive lamellipodia that retract into the plasma membrane, but some others fold back and fuse with the underlying membrane to form large vesicles (0.2–5 μm) called macropinosomes [34]. Therefore, the existence of a different type of Arp2/3-complex-dependent dorsal protrusions resembling ruffles cannot be discounted. In this regard, it is noteworthy that dorsal ruffles often protrude from the dorsal side of cells independently of lamellipodia [35].
Instead, there is no doubt that circular dorsal ruffles (CDRs) are distinct Arp2/3-complex-dependent cell protrusions. First, CDRs are ring-shaped structures arising exclusively on the dorsal surface of cells, whereas ruffles are curly and form also at the cell's periphery. Thus, CDRs differ morphologically from peripheral and dorsal ruffles, lamellipodia and filopodia. Second and at variance with the incessant protrusion and retraction of ruffles and lamellipodia, CDRs appear only once in cells stimulated with mitogenic and/or motogenic factors and have a life cycle typically encompassing the first 30 minutes of stimulation [36]. Third, mouse embryonic fibroblasts (MEFs) form CDRs and lamellipodia/ruffles through independent mechanisms that require WAVE1 and WAVE2, respectively [20]. Yet, WAVE1 and WAVE2 have been subsequently suggested to be dispensable for the making of CDRs, which may instead rely on the NPF Neural Wiskott-Aldrich syndrome protein (N-WASP) [37]. Nevertheless, CDRs were only reduced in N-WASP knockout fibroblast-like cells [37] and N-WASP does not regulate either lamellipodia or ruffles in mesenchymal and epithelial cells [13,38]. Hence, additional work is needed to decipher the exact mechanism of formation and physiological relevance of CDRs.
Lamellipodia and ruffles in mesenchymal cell migration
Mesenchymal cell migration is an actin- and adhesion-based mode of movement characterized by the presence of a lamellipodium at the cell's leading edge [39].
The lamellipodium is a highly dynamic actin-based structure undergoing cycles of edge protrusion and retraction [27]. During the protrusion phase, new integrin-mediated adhesions anchor the lamellipodial actin network to the underlying substratum. Interestingly, actin polymerization within the advancing lamellipodium drives clustering and positioning of activated, but unligated, integrins at the leading edge of crawling fibroblasts [40]. The ensuing site-restricted formation of cell-matrix adhesions makes the lamellipodium long-lived [39] and directs cell migration towards regions of high adhesiveness [41]. This adhesion-based mechanism to control cell directionality could be particularly important during haptotaxis and durotaxis. Furthermore, these frontal anchor points allow cells to move forward by building internal pulling forces that induce contraction and breakage of cell-matrix adhesions at the rear. Hence, the presence of adhesion sites within the lamellipodium provides also a way to integrate protrusion and contractility in the cell [41]. This makes it clear that the lamellipodium plays a two-fold function in crawling mesenchymal cells: it generates force for the advancement of the leading edge and couples it with the establishment of a regulated adhesion assembly-disassembly cycle required for productive cell movement [41,42].
Peripheral and dorsal ruffles lack adhesion sites when cells slide on a 2D substrate and, consequently, cannot promote displacement. Nevertheless, their constant protrusion and retraction accompanies mesenchymal cell migration. This suggests that peripheral and dorsal ruffles may have indirect pro-migratory functions, an idea supported by numerous clues. First, the rearward movement of peripheral and dorsal ruffles creates a constant flow of branched actin filaments that the cell can remodel to build stress fibers [43] mediating cell-body contraction and rear retraction. Second, ruffling is required for spatially restricted Rab5-dependent trafficking of activated GTP-bound Rac to sites of actin polymerization. This pathway stabilizes actin-based protrusion at the leading edge, in turn increasing directional movement of mesenchymal cells [44]. Third, membrane ruffling is associated with macropinocytosis, an non-selective endocytic route for extracellular components and signaling molecules, membrane receptors and portions of the plasma membrane [34]. In particular, surface integrins rapidly redistribute from ventral focal adhesions to CDRs in response to motogenic stimuli by moving along the plasma membrane [45]. At CDRs, they undergo macropinocytosis and recycling to new focal adhesions forming close to the leading edge when mesenchymal cells start to migrate [45]. These results show that CDRs are important to establish the front-rear axis in MEFs [45] and mark the transition from static to motile cellular states [36]. Yet, integrins are also enriched in ruffles [46] and macropinocytosis occurs at CDRs, peripheral and dorsal ruffles [34]. Moreover, CDRs disappear within 30 minutes of stimulation and are present only in some cell types [36,47]. Therefore, it is likely that all ruffle types participate in establishing the front-rear axis, whereas only peripheral and dorsal ruffles stabilize it during directed cell migration. The observation that reduced ruffling in WASP-interacting protein (WIP) knockout fibroblasts decreases directional persistence and chemotaxis without affecting cell speed strengthens this idea [48].
In summary, lamellipodia and ruffles have different functions when mesenchymal cell migration occurs on a bi-dimensional surface, but this distinction could fade away in 3D environments.
Role of the Arp2/3 complex in the dendritic nucleation/array treadmilling model of lamellipodium and ruffle protrusion
All types of actin-based protrusions possess a number of common features: i) their filaments' barbed ends face the distal part of the encasing plasma membrane, they show ii) polymerization-driven retrograde filament flow and iii) pointed-end filament depolymerization in the proximal part that, together, iv) produce treadmilling F-actin arrays. This explains why actin-based protrusion can only occur when the actin-filament assembly rate outpaces the retrograde flow. When these two rates equate, the protrusion does not further expand and pauses, whereas it retracts if the retrograde flow rate predominates.
The dendritic nucleation/array treadmilling model of lamellipodium and ruffle protrusion lays its foundations on the findings that the Arp2/3 complex assembles branched actin filaments in vitro [11], enriches in lamellipodia and ruffles [3,49] and is activated at the tip thereof where the WAVE complex accumulates [49–51]. This model postulates that the distal part of the lamellipodium contains short and highly branched actin filaments forming a narrow angle with the membrane, which are stiff enough and optimally oriented to push against the plasma membrane [3,49]. Given that the Arp2/3 complex creates Y-shaped branched filaments hallmarked by a 70° angle, most filaments contact the plasma membrane over a broad range of angles, the most common being ±35° normal of the leading edge. Years later, it became apparent that actin filaments in protruding lamellipodia subtend angles from 15° to 90° to the front, and that the proportion of filaments oriented more parallel to the cell edge is higher in paused than protruding lamellipodia [52]. This confirmed a previous perturbation study in which cell translocation negatively correlated with the incidence angle of actin filaments and the activity of anti-capping proteins [53].
Pushing in protruding lamellipodia and ruffles is effected by the elongating barbed ends of branched actin filaments at the interface with the plasma membrane [49,50]. Although the Arp2/3 complex is necessary also for the protrusion of CDRs, unique signaling, structural and regulatory components determine their circular geometry [47]. Given that CDRs are neither universally present nor persistent on the surface of crawling mesenchymal cells and have only an ancillary role in cell motility, CDR formation will not be discussed further. All interested readers are referred to a review presenting an inventory of the regulatory pathways and proteins involved in the making of CDRs [47]. The fairly well understood signaling events, post-translational modifications and conformational rearrangements controlling the activity of the WAVE complex have been already summarized elsewhere [18,54,55].
Conversely, the mechanism(s) regulating the activity of the Arp2/3 complex at the leading edge are much less understood. Landmark biochemical studies have established that NPF-primed Arp2/3 complex accelerates actin nucleation from the side of a pre-existing actin filament, creating a filament that elongates from its free barbed end. By crosslinking the new (daughter) and the pre-existing (mother) filaments end-to-side, the Arp2/3 complex forms branched actin filaments [10]. Moreover, actin polymerization by the Arp2/3 complex is an explosive auto-catalytic process in which the rate of the reaction increases as more filaments are assembled because daughter filaments can serve as mother filaments for further rounds of branching [10] (Figure 1B). Although newly polymerized actin filaments function as efficient mother filaments in vitro, the Arp2/3 complex hardly uses either aged or tropomyosin-bound actin filaments [11,56]. Consequently, forceful targeting of WAVE2 to the plasma membrane is insufficient to induce lamellipodium/ruffle formation without additional stimuli that trigger actin polymerization [57], partly because a number of actin-binding proteins prevent binding of the Arp2/3 complex to the cortical actin cytoskeleton [58]. Hence, biochemistry and cell biology concur on newly assembled actin filaments having a key role in the Arp2/3 complex-activation mechanism. Given that the Arp2/3 complex controls an auto-catalytic reaction, actin polymerization must then be involved in the formation of the first mother filaments that ignite the Arp2/3 complex.
Defining the mechanism of initiation of lamellipodia and peripheral ruffles
The origin of the mother filaments mediating initial activation of the Arp2/3 complex in lamellipodia and ruffles has long remained enigmatic. Theoretically, there are three ways whereby these filaments could be produced: 1) de novo polymerization from actin monomers, 2) severing and 3) uncapping of pre-existing actin filaments to generate free, elongation-competent barbed ends. In immobile cells, however, most barbed ends and actin filaments composing the actin cortex do not face the plasma membrane [52] or elongate spontaneously [49]. This has led to proposing both mother filament-based and mother filament-independent mechanisms of lamellipodium and ruffle formation.
Severing of existing actin filaments by cofilin [59,60] and mDia2-dependent actin polymerization have been suggested to provide the Arp2/3 complex with the first mother filaments in nascent lamellipodia and ruffles [61].
The notion that cofilin initiates branched actin polymerization in lamellipodia and ruffles stems from the observation that photo-activation of caged cofilin in advancing lamellipodia leads to the further protrusion of the leading edge [60]. Unfortunately, these experiments are open to multiple interpretations because, as previously argued [15], the cofilin activation area spun the entire lamellipodium breadth and photo-activation was performed in mature, but not nascent, lamellipodia. Thus, cofilin could have simply favored pointed-end depolymerization and debranching of actin filaments in the lamellipodial back to replenish the G-actin pool that sustains actin polymerization at the front, as described in the dendritic-nucleation/array treadmilling model for the generation of lamellipodia [15,49]. In keeping with this, cofilin severs primarily aged actin filaments as those located in the back of lamellipodia and ruffles [59]. Furthermore, both knockdown and instantaneous inactivation of cofilin result in enlarged lamellipodia and ruffles, as well as increased F-actin levels [62–65].
Formin mDia2 nucleates and elongates linear actin filaments [66] whose role in the initiation of lamellipodia and ruffles was inferred using RNA interference (RNAi) [61]. Notwithstanding, endogenous mDia2 is rarely present in these protrusions [61,67] and other RNAi studies concluded that the formation of lamellipodia and ruffles does not require mDia2 [23,51,67,68]. Consistent with these latter observations, Arp2/3 complex-dependent migration of melanoblasts is not compromised in mDia2 knockout mice as shown by the color of the belly fur [69,70]. Together, these results show that mDia2 is not essential for Arp2/3 complex-dependent membrane protrusion either in tissue culture or in vivo.
The finding that DIP, the yeast homologue of SPIN90 (also known as NCKIPSD), can activate the Arp2/3 complex without the need for pre-existing actin filaments has suggested that SPIN90 could provide an alternative filament-independent mechanism to initiate branched actin polymerization in nascent lamellipodia and ruffles [71]. However, this hypothesis clashes with the observations that SPIN90 knockout MEFs form lamellipodia and ruffles normally [72] and full-length SPIN90 fails to activate the Arp2/3 complex [68,73]. In summary, neither mechanism of initiation of lamellipodia and ruffles appears satisfactory.
A bunch of studies have recently solved this outstanding open issue by showing that Rho-family GTPase Rho recruits and activates formin mDia1, which polymerizes linear actin filaments near the plasma membrane in nascent lamellipodia and ruffles (Figure 2A-D). These new actin filaments serve as mother filaments enabling WAVE-primed Arp2/3 complex to make the first branch (Figure 2E-F) [51]. These findings demonstrate that the lamellipodium/ruffle-initiating machinery consists of two actin nucleators that act sequentially and integrate different upstream signals controlling membrane protrusion and mesenchymal cell migration [51]. This model is supported by correlative analyses in unperturbed Ptk1 showing that Rho activation precedes that of Rac and remains high during leading edge protrusion [74], as well as that mDia1 is recruited to the plasma membrane prior to the Arp2/3 complex [75]. Genetic evidence showing that murine dendritic and T cells isolated from mDia1-knockout animals fail to form lamellipodia and ruffles and migrate poorly [76,77] further strengthens the above observations. The finding that mDia1 +/− and −/− fibroblastoid cells overexpressing mDia2 show increased lamellipodium and filopodium formation [78] might superficially look at odds with the above model. At a closer look, this seems rather due to compensatory events as cell lines with high levels of WAVE and the Arp2/3 complex require mDia1 to form lamellipodia and ruffles only when the activity of the Arp2/3 complex is suboptimal [51]. Interestingly, this dose-dependent effect could involve mDia1's ability to sequester Arpin [79], a competitive inhibitor of Arp2/3 complex. Moreover, junction-mediating and regulatory protein (JMY), which polymerizes linear actin filaments through its WH2 domain and localizes within lamellipodia [80], could act together with mDia1 in some cell types or states. In line with this view, knockdown of JMY is detrimental for the migration of only certain cell lines [81]. Interestingly, new data suggest that mDia1 may polymerize actin filaments that are subsequently branched by the Arp2/3 complex also within the nucleus [82]. Hence, the mDia1-dependent mother filament-based mechanism of activation of the Arp2/3 complex might be important at multiple cellular locations.
Figure 2.
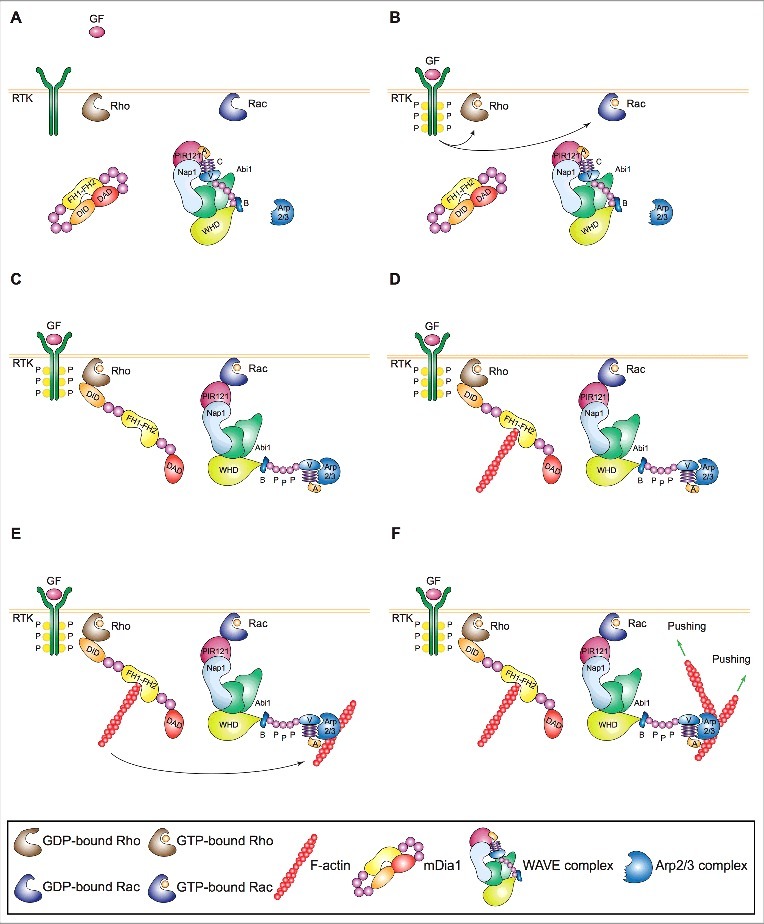
Mechanism of initiation of lamellipodia and ruffles. (A,B) Growth factors (GFs) activating receptor tyrosine kinases (RTKs) and other motogenic signals lead to the activation of the small GTPases Rac and Rho. (C) Active, GTP-bound Rho and Rac recruit mDia1 and the WAVE complex to the plasma membrane. Direct binding to Rho changes mDia1's conformation thereby converting it into the actin nucleation-proficient state. Similarly, a physical interaction between Rac and the WAVE complex results in the exposure of the VCA region of WAVE, which can then bind and prime the Arp2/3 complex. (D,E) mDia1 polymerizes linear actin filaments that can be used as a template by WAVE-primed Arp2/3 complex. (F) The Arp2/3 complex assembles the first actin branch thereby initiating lamellipodia and ruffles. Key proteins and complexes are not drawn in scale and are decoded in the box. Note that all WAVE-complex subunits but HSPC300, as well as the domains of WAVE, are depicted in the cartoon.
New insights into the expansion of lamellipodia and peripheral ruffles
Actin filaments elongate only transiently within lamellipodia and ruffles because growth is rapidly terminated by binding of capping proteins to the filament's barbed end [49]. Consequently, the expansion of the dendritic branched actin network relies on the auto-catalytic activity of the Arp2/3 complex. As activated WAVE complex and newly assembled actin filaments are enriched at the plasma membrane, actin polymerization by the Arp2/3 complex remains confined at the tip of lamellipodia and ruffles [11,19,49-51]. New evidence indicates that the role of WAVE goes beyond its NPF function and involves also tethering the branched actin network to the plasma membrane and accelerating branched filament elongation [83] (Figure 3A).
Figure 3.
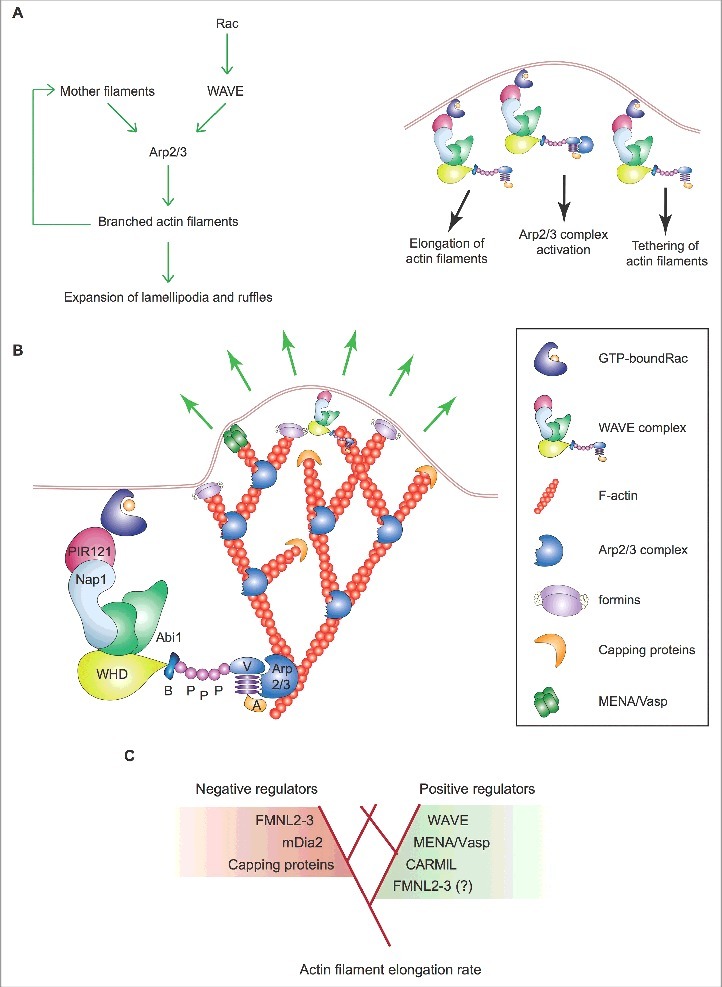
Mechanism of expansion of lamellipodia and ruffles. (A) Left: Process diagram showing how the Arp2/3 complex and WAVE and upstream regulators thereof sustain the expansion of lamellipodia and ruffles. Right: Cartoon depicting the enrichment of activated Rac and WAVE complex at the tip of lamellipodia and ruffles. The roles of WAVE as a 1) distributive polymerase promoting branched filament elongation, 2) tethering factor linking the actin network to the encasing plasma membrane and 3) nucleation promoting factor for the Arp2/3 complex are indicated. (B) The presence of active GTP-bound Rac within lamellipodia and ruffles is important for the enrichment and activity of the WAVE complex at the tip of these protrusions. The site-restricted cortical localization of the WAVE complex and the new branched actin filaments ensures persistent actin polymerization by the Arp2/3 complex in a narrow region close to the plasma membrane. This process and the proteins that regulate the elongation of the filaments' barbed ends pushing towards the plasma membrane control the protrusion of lamellipodia and ruffles, the latter indicated by green arrows. Key proteins and complexes are not depicted in scale and are decoded in the box. Note that all WAVE-complex subunits but HSPC300, as well as the domains of WAVE, are depicted in the cartoon. (C) Regulation of branched actin filament elongation within lamellipodia and ruffles. Inventory of the proteins that regulate either negatively (red box) or positively (green box) the filament elongation rate of the branched F-actin network. Question mark highlights that FMNL2 and FMNL3 might promote filament elongation only in some specific cell types.
Although this mechanism is sufficient to explain how lamellipodia and ruffles grow after the initiation phase, recent studies have proposed that actin nucleation and filament elongation by mDia and FMNL subfamilies of formins boost lamellipodium/ruffle formation [18] (Figure 3B). This idea is not far-fetched because anti-capping proteins like ENA/VASP and CARMIL favor the elongation of actin filaments thereby increasing the lamellipodial protrusion rate [18,53] (Figure 3B).
In particular, mDia2 has been suggested to facilitate lamellipodium/ruffle protrusion by polymerizing and/or protecting from capping new actin filaments [61]. At odds, overexpression of wild-type mDia2 inhibits lamellipodium/ruffle formation in different cell types [23,84] and that of constitutively active mDia2 reduces lamellipodial protrusion rate [61]. In line with these neglected observations, mDia2 is rarely present in lamellipodia and ruffles [61,67] and cannot substitute mDia1 during the initiation phase [51]. Moreover, mDia2 appears to be dispensable for the formation of lamellipodia and ruffles in both epithelial and mesenchymal cells [23,67]. Altogether, this makes it clear that mDia2 does not act within lamellipodia and ruffles to boost protrusion.
Alike mDia2, activation of formin FMNL2 by Cdc42 can accelerate actin filament elongation of branched actin filaments [22] and this might promote lamellipodium/ruffle protrusion and mesenchymal cell migration [67]. Unfortunately, there are a number of compelling observations that are incompatible with this claim. First, the knockout of Cdc42 does not prevent either lamellipodium/ruffle formation or the recruitment of FMNL2 (or its myristoylation-deficient mutant) to the leading edge [67]. This is indeed not surprising because FMNL2 binds to and is activated by Rho-family GTPase RhoC [85] and Rho, but not Cdc42, has high activity at the tip of protruding lamellipodia and ruffles [74]. Second, Cdc42 does not affect at all mesenchymal cell migration [86], thus explaining the marginal reduction in migration speed found in the FMNL2 knockdown cells [67]. Moreover, the emerging new roles of filopodia in the mesenchymal mode of migration [24,25] (see below) make it possible that reduced movement of FMNL2 knockdown cells is a consequence of impaired filopodium formation. Third, phosphorylated FMNL2 binds integrin β1 and regulates integrin trafficking [87], which affects force generation, the protrusion of lamellipodia and ruffles and cell migration. Fourth, there is evidence that FMNL2 and FMNL3 repress ruffle and lamellipodium formation in other cell types [88]. Fifth, mDia2, FMN1 [22,89,90] and FMNL1 [91] have actin-independent functions that regulate the actin cytoskeleton and cell migration [91,92], a possibility that holds also for FMNL2. In this regard, it is noteworthy that formins control G1 progression and entry into S phase of the cell cycle [90,93], as well as myocardin-related transcription factors [94] and p53 [90,92], well-known modulators of mesenchymal cell migration [80,92,94,95]. Sixth, downregulation and inhibition of formins impact the transcription of actin-regulatory genes [22,89,90,92,94]. Last but not least, functional inactivation of Mena/VASP proteins, which prevent filament capping and promote filament elongation alike FMNL2, increases lamellipodium persistence and cell migration [53]. Depletion of FMNL2 and FMNL3 diminishes also the number of microspikes, lamellipodial width and F-actin intensity [96]. Surprisingly, neither paralogue affects the rate of lamellipodial actin network treadmilling [96]. To explain these contradictory observations, it has been argued that FMNL2 and FMNL3 polymerize actin filaments that push orthogonally on the plasma membrane to optimize lamellipodial protrusion [96]. Yet, the distribution of filament angles within lamellipodia is largely independent of FMNL2 and FMNL3 [96] and does not support the above hypothesis.
In summary, the notion that formins facilitate the protrusion of lamellipodia and ruffles by nucleating and elongating actin filaments remains highly controversial and is certainly not universal. Strikingly, certain formins could even slow down lamellipodial protrusion rates and cell migration (Figure 3C). These latter observations might appear counterintuitive as enhanced filament elongation should speed up lamellipodial advancement and cell migration under conditions of sufficient adhesion to the substrate. However, formins could reduce the pool of Profilin-actin that enables WAVE, when clustered at high density on surfaces, to act as an actin polymerase that potently accelerates branched filament elongation [83]. Alternatively, the unexpected behavior of mDia and FMNL formins could be rooted in their ability to regulate cell-matrix adhesion [87,97]. Whatever the case, formins' contribution to the expansion of lamellipodia and peripheral ruffles warrants a less biased reassessment.
Retraction of lamellipodia and peripheral ruffles: one or multiple mechanisms?
Actin-based cell protrusion depends on the balance between the rate of actin polymerization and that of the actin retrograde flow [27,52] [41,42], the latter involving myosin II activity. Thus, altering either one or both parameters could trigger the retraction of lamellipodia and ruffles and, actually, there is solid experimental evidence for both regulatory modes to be at work in the cell.
Type-I Coronins, glia maturation factor (GMF), and Arp2/3 complex inhibitor (Arpin) are negative regulators of the Arp2/3 complex that control lamellipodia and ruffles.
Coronins are WD40 repeat-containing proteins that bind F-actin and inhibit the Arp2/3 complex [98]. The localization of type-I Coronins to subcellular regions of active actin polymerization seems to be coupled to the ATPase activity of actin on incorporation into filaments [49]. Ubiquitously expressed type-I Coronin human Coro1B has a 50-fold higher affinity of binding for newly polymerized (ATP/ADP + inorganic phosphate (Pi)-bound) than old (ADP-bound) F-actin. This contributes to targeting Coro1B to the lamellipodium [99] because the barbed-end proximal segment of actin filaments retains the Pi [49]. In addition, type-I Coronins bind the Arp2/3 complex keeping it in the open, inactive conformation in vitro [98,100]. Coro1B not only inhibits the activation of the Arp2/3 complex by antagonizing Cortactin, but it can also replace the Arp2/3 complex at the branch points thereby promoting filament debranching [101]. It should be noted that Coro1B has not been yet visualized in the branches at the back of lamellipodia and ruffles [101]. While type-I Coronins inhibit branched actin polymerization by the Arp2/3 complex at the leading edge and facilitate debranching at the lamellipodium/ruffle back, they stimulate the activity of cofilin either directly or by targeting Slingshot to the leading edge [102]. In turn, severing of actin filaments by activated cofilin promotes the disassembly of the branched actin network [102]. Therefore, type-I Coronins could provide a simple and efficient way to coordinate Arp2/3 complex-based actin assembly and cofilin-based actin disassembly within lamellipodia and ruffles. In this regard, it is tempting to speculate that the three-component ordered disassembly mechanism for linear actin filaments involving Coro1B, cofilin and Actin-interacting protein 1 (Aip1) [103] could potentially control also the disassembly of branched actin filaments. At any rate, Coro1B localizes with the Arp2/3 complex to the leading edge of migrating fibroblasts and regulates mesenchymal cell migration [102]. In fact, silencing of Coro1B results in a more densely branched lamellipodial actin network, reduced actin retrograde flow, enhanced lamellipodial protrusion rates and diminished lamellipodial persistence. Congruently with how Coro1B regulates lamellipodial dynamics, the knockdown of Coro1B has been shown to decrease cell speed [102].
GMF proteins are conserved from yeast to humans and structurally resemble ADF/cofilin proteins. However, GMF does not interact with actin and instead binds to the Arp2/3 complex [104]. Biochemically, GMF inhibits actin nucleation by the Arp2/3 complex and induces filament debranching by severing the junction between the Arp2/3 complex and the daughter filament [104,105]. This feature suggests a possible functional parallel between GMF and Coro1B. Surprisingly, silencing of GMF decreases the lamellipodial protrusion and retraction rates in mammalian cells, whereas it increases lamellipodial persistence [106]. Moreover, haptotaxis, but not chemotaxis, is dramatically impaired in GMF knockdown cells [106]. The fact that GMF mainly functions as a debranching factor in vivo could be related to these puzzling observations [106]. Most importantly, it is presently unknown whether GMF cooperates with ADF/cofilin to disassemble the lamellipodial actin network or simply remodels it into an unbranched network.
Arpin interacts physically with Rac, which allows it to be recruited to the tip of lamellipodia and ruffles in cells that use the mesenchymal mode of migration [107]. Arpin possesses also an acidic motif that competes with the VCA domain of WAVE for binding to the Arp2/3 complex. This enables Arpin to inhibit actin polymerization by the Arp2/3 complex in vitro [107]. In agreement with the competition between the Rac-Arpin-Arp2/3 inhibitory circuit and the Rac-WAVE-Arp2/3 activatory circuit, Arpin decreases the lamellipodium/ruffle lifetime thereby reducing speed and directional persistence of cell movement [107].
Taken together, these observations show that the activity of the Arp2/3 complex is subjected to both positive and negative regulation within lamellipodia and ruffles. However, it is not yet known whether Type-I Coronins, GMF and Arpin are gears of a complex retraction mechanism involving also ADF/cofilin or rather work independently of each other to reduce the rate of actin polymerization.
Anyway, it should not be overlooked that myosin II activity induces actin network disassembly through mechanical breakage of actin filaments [108]. Therefore, myosin II could regulate actin network turnover both by generating contraction forces that aliment the actin retrograde flow and by disassembling the actin network directly [108]. The finding that myosin II concentrates behind the leading edge in fibroblasts and epithelial cells [41] suggests that progressive incorporation of myosin II into the treadmilling actin network occurs at the back of the lamellipodium and results in the appearance of myosin II-positive anti-parallel filaments. The gradual conversion of the branched actin network into a more parallel organization further enhances myosin II activity, actin filament breakage and network depolymerization. Notably, this mechanism would provide a simple way to concomitantly increase the activity of myosin II and the actin retrograde flow as observed in paused and/or retracting lamellipodia and ruffles. In good agreement with this model, actin filaments shift from a more orthogonal to a more parallel angle to the front in paused versus advancing lamellipodia [52]. This was proposed to cause the transition from protrusion to pause while leaving unchanged the F-actin mass that support structurally the lamellipodia [52]. Yet, the increase in the number of filaments oriented more parallel to the cell edge in paused and/or retracting lamellipodia and ruffles is rather the consequence of the reduced Arp2/3 complex activity, the faster depolymerization of short (more perpendicular) than long (more parallel) filaments, and the stronger pulling action of myosin II during retraction [42].
Although the sum of these data suggests that multiple and interconnected pathways trigger and modulate the pause/retraction phase, a satisfactory molecular mechanism describing this aspect of the lifecycle of lamellipodia and ruffles is not yet available.
Revisiting the role of the Arp2/3 complex in mesenchymal cell migration
Efficient mesenchymal cell migration on two-dimensional surfaces is thought to require the small GTPase Rac, the WAVE and the Arp2/3 complexes, which compose the so-called core lamellipodium/ruffle-making machinery [19-21,23-25,51,109–111]. Cells lacking any component of this machinery have impaired migratory abilities [21,24,25]. This is consistent with the main difference in edge motion of fast- and slow-moving cells being the ratio between the amplitude of edge protrusion and amplitude of edge retraction [42]. Hence, it is also unsurprising that chemotaxis shows a similar impairment in Rac-deficient cells, as well as those lacking either the WAVE or the Arp2/3 complexes [21,24,111]. At the mechanistic level, genetic ablation (or inhibition) of Rac prevents the accumulation of the WAVE complex at the cell membrane, and likely also the activation of the Arp2/3 complex [112].
However, the idea that active Arp2/3 complex at the cell periphery and ensuing lamellipodium formation play an essential role in mesenchymal cell migration has been questioned by new observations.
Fibroblasts knocked out for the Arp2/3-complex subunit Arpc3 did not show any phenotype in random cell migration assays, even though they failed to form lamellipodia and ruffles [25]. These cells exhibited a severe defect in chemotaxis towards EGF [25] and PDGF [113]. As edge protrusion and retraction were unexpectedly similar in Arpc3 +/+ and Arpc3 −/− cells, this was attributed to a poor coordination in the protrusion and the retraction of neighboring lamellipodial regions [25]. Yet, an independent study carried out on Arpc2 and Arp2 double knockdown (2x KD) fibroblasts reached very different conclusions [24]. These cells were defective in random migration and haptotaxis consistent with the essential role of the Arp2/3 complex in the formation of lamellipodia and ruffles, but they showed normal chemotaxis towards PDGF [24]. Thus, these latter data would suggest that lamellipodia are not required for fibroblast chemotaxis. A follow-up study demonstrating that 2x KD cells secrete more hepatocyte growth factor, which increases EGF-mediated, but not PDGF-mediated, signaling and chemotaxis in a non-cell-autonomous manner has only partially solved these controversies [114]. In fact, downregulation of the Arp2/3 complex by 90% is insufficient to perturb key cellular functions [115] and thus the residual Arp2/3 complex present in the 2x KD fibroblasts might ensure efficient chemotaxis.
The role of the Arp2/3 complex in spreading fibroblasts remains also unsettled, albeit optimal spreading of epithelial cells requires the Arp2/3 complex [23]. In fact, while the Arpc3 −/− fibroblasts did not show any spreading phenotype [25], the 2x KD cells did [24]. Taking into account that Arpc3 limitedly contributes to the activity and the stability of the Arp2/3 complex in vitro [116], it is reasonable to assume that the Arp2/3 complex has spurious dominant-negative effects in the Arpc3 −/− cells. New data showing that knockout of Arpc4 results in Arp2/3-complex downregulation and different epidermal phenotypes compared to the knockout of Arpc3 corroborate the above idea [26,117].
On the contrary, it is unanimously accepted that epithelial and mesenchymal cells form more filopodia when the Arp2/3 complex is absent or cannot be activated [19–21,23–26,51,109,117,118]. Under these conditions, mesenchymal cells lack lamellipodia and ruffles and adopt a mode of migration whereby new flat cell surface area is created by filling the gaps between adjacent matrix-anchored filopodia [24,25]. However, there are contrasting results as to the efficiency of this filopodium-initiated “filling the gap” motility: Arpc3 −/− fibroblasts move as or even faster than their wild-type counterparts [25], whereas both 2x KD and Arpc2 −/− fibroblasts are much slower [24,118]. Of note, Wave2 KO fibroblasts also have dramatically reduced migratory abilities [20] and myosin II inhibition in the 2x KD cells increases both filopodium number and speed of movement [24]. These observations favor the view that the filopodium-initiated “filling the gap” motility is rather inefficient and cannot reach the same speed of the classical lamellipodium-based migration.
In any case, it is evident that neither the Arp2/3 complex nor lamellipodia are absolutely required for mesenchymal cells to move (Figure 4A). But, what is then the role of the Arp2/3 complex in crawling mesenchymal cells?
Figure 4.
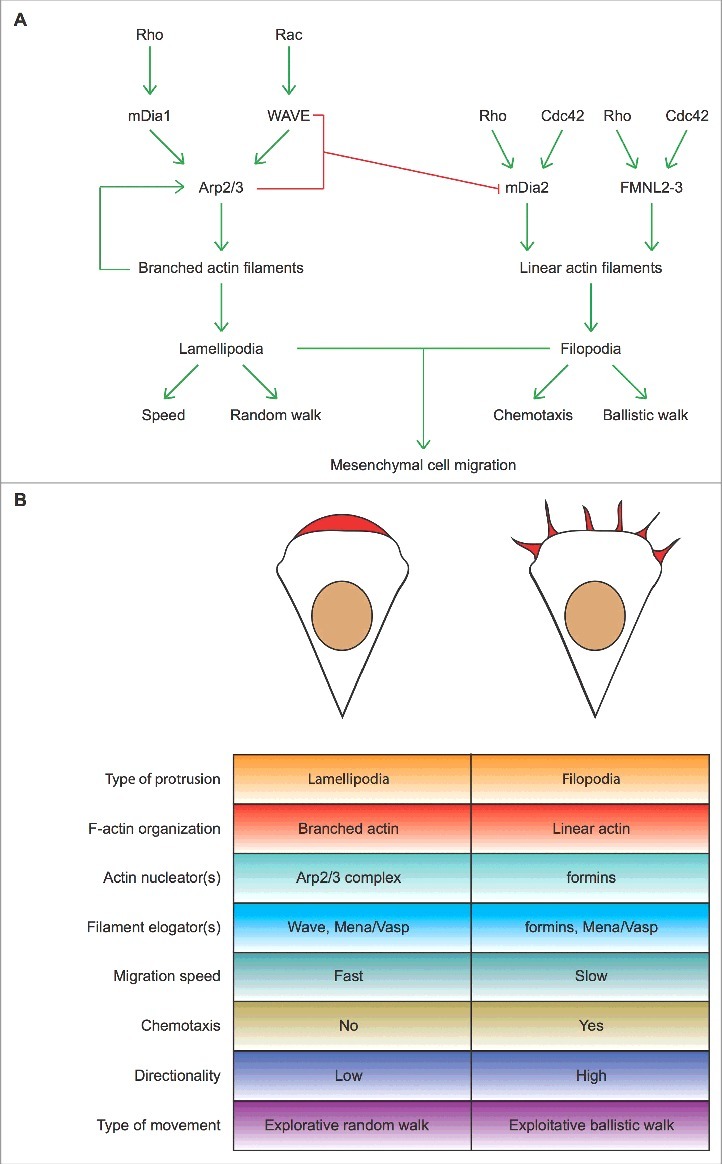
Lamellipodia and filopodia fulfill different exploratory functions but are not essential for mesenchymal cell migration. (A) Process diagram depicts the key players regulating actin polymerization for the formation of lamellipodia (left) and filopodia (right) and the main functions of either actin-based protrusion in mesenchymal cells migrating on a 2D uniform surface. Green and red lines denote activation and inhibition, respectively. The involvement of Rho proteins in filopodium formation has been recently reviewed [122]. (B) Top: Stereotypical representation of actin-based lamellipodial and filopodial protrusions at the leading edge of mesenchymal cells (F-actin arrays and nuclei are highlighted in red and brown, respectively). Bottom: The main molecular features and the functions of lamellipodia and filopodia in mesenchymal cell migration are compared side to side. Ruffles are not indicated because they affect cell migration on 2D surfaces only indirectly.
The above data relate it to the ability of cells to sense and respond to directional cues. Interestingly, this reveals an unexpected parallelism between mesenchymal and amoeboid modes of migration. In fact, inhibition of the Arp2/3 complex in leukocytes and other amoeboid cell types forming lamellipodia and ruffles reduces cell locomotion and the turning events, the latter dramatically increasing directional persistence [119]. In line with these observations, deletion of the WAVE complex in dendritic cells (DCs) eliminates lamellipodia and gives rise to unipolar pointed cells or cells with multiple filopodia in immature and mature DCs, respectively [120]. Unipolar cells migrate with increased speed and high directional persistence, although chemotaxis is defective. Cells with multiple filopodia instead retain chemotactic activity but move slowly [120]. These results confirm a previous study showing that immature DCs depend on mDia1-dependent actin polymerization at rear to facilitate forward locomotion, whereas the Arp2/3 complex and branched actin at the front limit migration while promoting antigen capture [121].
Thus, it appears that lamellipodia and filopodia fulfill explorative functions but are per se dispensable for cell locomotion (Figure 4A). Although the intrinsic chemotactic abilities of lamellipodia and ruffles await reassessment in cells that cannot form filopodia, they seem to enable a merely explorative random walk that does not (heavily) rely on extracellular cues. Conversely, filopodia promote a ballistic, more exploitative mode of migration guided by external inputs (Figure 4B). Direct inhibition of filopodium formation by the lamellipodium-making machinery [23] further suggests that lamellipodium-driven and filopodium-driven exploration are not functionally equivalent. This scenario allows to speculating that the filopodium-making machinery could also repress the formation of lamellipodia and ruffles. If so, this tag of war could be at the heart of the mechanism that determines the interconversion between common motile cell states, such as random and ballistic walk.
Conclusions and future directions
Recent advances have considerably increased our understanding of the mechanisms that govern the different phases of the lifecycle of lamellipodia and ruffles.
It is now clear that actin filaments polymerized by formin mDia1 along with the NPF activity of WAVE set in motion the Arp2/3 complex at plasma-membrane sites where lamellipodia and ruffles form.
Furthermore, new evidence shows that WAVE acts as a distributive polymerase that accelerates actin filament elongation in expanding lamellipodia and ruffles. This in conjunction with the NFP and tethering functions of WAVE and the well-known autocatalytic activity of the Arp2/3 complex make up a self-propagating mechanism that faithfully describes the growth of lamellipodia and ruffles. In view of that, the accumulating clues showing that formins oppose lamellipodium protrusion suggest a straightforward Rho-GTPase-mediated mechanism to harness the growth of lamellipodia and ruffles. Hence, p Pinpointing the molecular details of formins' action will certainly become a main research direction in years to come.
Although some of the key players that trigger and modulate pause/retraction of lamellipodia and ruffles have been identified, we only possess scattered and incomplete information. Our knowledge of this aspect of the lifecycle of lamellipodia and ruffles lags behind and we currently ignore whether one or more regulatory mechanisms control the pause/retraction phase. Given the high complexity of this process, future systematic and unbiased approaches would be needed to answer this outstanding question.
Finally, the realization that the Arp2/3 complex and lamellipodia are not essential for mesenchymal cells to move and the discovery of an unforeseen filopodium-based mode of cell motility raise new questions about the relationship between the lamellipodium/ruffle- and the filopodium-making machineries that challenge the textbook model of mesenchymal cell migration. We anticipate an exponential rise in our mechanistic understanding of this fundamental cellular behavior in the next few years. Leveraging precision medicine and new gene editing technologies, this knowledge will open the door for curative intervention on pathological migratory processes, first and foremost cancer metastasis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Dmitrieff S, Nedelec F. Amplification of actin polymerization forces. J Cell Biol. 2016;212:763–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. [DOI] [PubMed] [Google Scholar]
- [5].Bornschlogl T. How filopodia pull: what we know about the mechanics and dynamics of filopodia. Cytoskeleton (Hoboken). 2013;70:590–603. [DOI] [PubMed] [Google Scholar]
- [6].Yang C, Svitkina T. Filopodia initiation: focus on the Arp2/3 complex and formins. Cell Adh Migr. 2011;5:402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9:446–54. [DOI] [PubMed] [Google Scholar]
- [8].Heckman CA, Plummer HK. 3rd Filopodia as sensors. Cell Signal. 2013;25:2298–311. [DOI] [PubMed] [Google Scholar]
- [9].Bugyi B, Carlier MF. Control of actin filament treadmilling in cell motility. Annu Rev biophys. 2010;39:449–70. [DOI] [PubMed] [Google Scholar]
- [10].Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol. 2006;7:713–26. [DOI] [PubMed] [Google Scholar]
- [11].Blanchoin L, Amann KJ, Higgs HN, et al.. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 2000;404:1007–11. [DOI] [PubMed] [Google Scholar]
- [12].Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol. 2013;14:7–12. [DOI] [PubMed] [Google Scholar]
- [13].Innocenti M, Gerboth S, Rottner K, et al.. Abi1 regulates the activity of N-WASP and WAVE in distinct actin-based processes. Nat Cell Biol. 2005;7:969–76. [DOI] [PubMed] [Google Scholar]
- [14].Galovic M, Xu D, Areces LB, et al.. Interplay between N-WASP and CK2 optimizes clathrin-mediated endocytosis of EGFR. J Cell Sci. 2011;124:2001–12. [DOI] [PubMed] [Google Scholar]
- [15].Kedziora KM, Isogai T, Jalink K, et al.. Invadosomes – shaping actin networks to follow mechanical cues. Front Biosci (Landmark Ed). 2016;21:1092–117. [DOI] [PubMed] [Google Scholar]
- [16].Leyton-Puig D, Isogai T, Argenzio E, et al.. Flat clathrin lattices are dynamic actin-controlled hubs for clathrin-mediated endocytosis and signalling of specific receptors. Nat Commun. 2017;8:16068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rottner K, Hanisch J, Campellone KG. WASH, WHAMM and JMY: regulation of Arp2/3 complex and beyond. Trends Cell Biol. 2010;20:650–61. [DOI] [PubMed] [Google Scholar]
- [18].Krause M, Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol. 2014;15:577–90. [DOI] [PubMed] [Google Scholar]
- [19].Innocenti M, Zucconi A, Disanza A, et al.. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–27. [DOI] [PubMed] [Google Scholar]
- [20].Suetsugu S, Yamazaki D, Kurisu S, et al.. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell. 2003;5:595–609. [DOI] [PubMed] [Google Scholar]
- [21].Vidali L, Chen F, Cicchetti G, et al.. Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol Biol Cell. 2006;17:2377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Isogai T, Innocenti M. New nuclear and perinuclear functions of formins. Biochem Soc Trans. 2016;44:1701–8. [DOI] [PubMed] [Google Scholar]
- [23].Beli P, Mascheroni D, Xu D, et al.. WAVE and Arp2/3 jointly inhibit filopodium formation by entering into a complex with mDia2. Nat Cell Biol. 2008;10:849–57. [DOI] [PubMed] [Google Scholar]
- [24].Wu C, Asokan SB, Berginski ME, et al.. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Suraneni P, Rubinstein B, Unruh JR, et al.. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol. 2012;197:239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].van der Kammen R, Song JY, de Rink I, et al.. Knockout of the Arp2/3 complex in epidermis causes a psoriasis-like disease hallmarked by hyperactivation of transcription factor Nrf2. Development. 2017;144:4588–4603. [DOI] [PubMed] [Google Scholar]
- [27].Giannone G, Dubin-Thaler BJ, Rossier O, et al.. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128:561–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Even-Ram S, Doyle AD, Conti MA, et al.. Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol. 2007;9:299–309. [DOI] [PubMed] [Google Scholar]
- [29].Shutova M, Yang C, Vasiliev JM, et al.. Functions of nonmuscle myosin II in assembly of the cellular contractile system. PLoS One. 2012;7:e40814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Betapudi V. Myosin II motor proteins with different functions determine the fate of lamellipodia extension during cell spreading. PLoS One. 2010;5:e8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Limouze J, Straight AF, Mitchison T, et al.. Specificity of blebbistatin, an inhibitor of myosin II. J Muscle Res Cell Motil. 2004;25:337–41. [DOI] [PubMed] [Google Scholar]
- [32].Verkhovsky AB, Svitkina TM, Borisy GG. Self-polarization and directional motility of cytoplasm. Curr Biol. 1999;9:11–20. [DOI] [PubMed] [Google Scholar]
- [33].Borm B, Requardt RP, Herzog V, et al.. Membrane ruffles in cell migration: indicators of inefficient lamellipodia adhesion and compartments of actin filament reorganization. Exp Cell Res. 2005;302:83–95. [DOI] [PubMed] [Google Scholar]
- [34].Bloomfield G, Kay RR. Uses and abuses of macropinocytosis. J Cell Sci. 2016;129:2697–705. [DOI] [PubMed] [Google Scholar]
- [35].Yamazaki D, Suetsugu S, Miki H, et al.. WAVE2 is required for directed cell migration and cardiovascular development. Nature. 2003;424:452–6. [DOI] [PubMed] [Google Scholar]
- [36].Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–57. [DOI] [PubMed] [Google Scholar]
- [37].Legg JA, Bompard G, Dawson J, et al.. N-WASP involvement in dorsal ruffle formation in mouse embryonic fibroblasts. Mol Biol Cell. 2007;18:678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Snapper SB, Takeshima F, Anton I, et al.. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat Cell Biol. 2001;3:897–904. [DOI] [PubMed] [Google Scholar]
- [39].Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009;10:538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Galbraith CG, Yamada KM, Galbraith JA. Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science. 2007;315:992–5. [DOI] [PubMed] [Google Scholar]
- [41].Gardel ML, Schneider IC, Aratyn-Schaus Y, et al.. Mechanical integration of actin and adhesion dynamics in cell migration. Annu Rev Cell Dev Biol. 2010;26:315–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Burnette DT, Manley S, Sengupta P, et al.. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 2011;13:371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol. 2006;173:383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Palamidessi A, Frittoli E, Garre M, et al.. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell. 2008;134:135–47. [DOI] [PubMed] [Google Scholar]
- [45].Gu Z, Noss EH, Hsu VW, et al.. Integrins traffic rapidly via circular dorsal ruffles and macropinocytosis during stimulated cell migration. J Cell Biol. 2011;193:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bretscher MS. On the shape of migrating cells–a 'front-to-back' model. J Cell Sci. 2008;121:2625–8. [DOI] [PubMed] [Google Scholar]
- [47].Hoon JL, Wong WK, Koh CG. Functions and regulation of circular dorsal ruffles. Mol Cell Biol. 2012;32:4246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Banon-Rodriguez I, Saez de Guinoa J, Bernardini A, et al.. WIP regulates persistence of cell migration and ruffle formation in both mesenchymal and amoeboid modes of motility. PLoS One. 2013;8:e70364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–65. [DOI] [PubMed] [Google Scholar]
- [50].Leyton-Puig D, Kedziora KM, Isogai T, et al.. PFA fixation enables artifact-free super-resolution imaging of the actin cytoskeleton and associated proteins. Biology open. 2016;5:1001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Isogai T, van der Kammen R, Leyton-Puig D, et al.. Initiation of lamellipodia and ruffles involves cooperation between mDia1 and the Arp2/3 complex. J Cell Sci. 2015;128:3796–810. [DOI] [PubMed] [Google Scholar]
- [52].Koestler SA, Auinger S, Vinzenz M, et al.. Differentially oriented populations of actin filaments generated in lamellipodia collaborate in pushing and pausing at the cell front. Nat Cell Biol. 2008;10:306–13. [DOI] [PubMed] [Google Scholar]
- [53].Bear JE, Svitkina TM, Krause M, et al.. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–21. [DOI] [PubMed] [Google Scholar]
- [54].Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. [DOI] [PubMed] [Google Scholar]
- [55].Stradal TE, Rottner K, Disanza A, et al.. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol. 2004;14:303–11. [DOI] [PubMed] [Google Scholar]
- [56].Blanchoin L, Pollard TD, Hitchcock-DeGregori SE. Inhibition of the Arp2/3 complex-nucleated actin polymerization and branch formation by tropomyosin. Curr Biol. 2001;11:1300–4. [DOI] [PubMed] [Google Scholar]
- [57].Abou-Kheir W, Isaac B, Yamaguchi H, et al.. Membrane targeting of WAVE2 is not sufficient for WAVE2-dependent actin polymerization: a role for IRSp53 in mediating the interaction between Rac and WAVE2. J Cell Sci. 2008;121:379–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Biro M, Romeo Y, Kroschwald S, et al.. Cell cortex composition and homeostasis resolved by integrating proteomics and quantitative imaging. Cytoskeleton (Hoboken). 2013;70:741–54. [DOI] [PubMed] [Google Scholar]
- [59].Ichetovkin I, Grant W, Condeelis J. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr Biol. 2002;12:79–84. [DOI] [PubMed] [Google Scholar]
- [60].Ghosh M, Song X, Mouneimne G, et al.. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–6. [DOI] [PubMed] [Google Scholar]
- [61].Yang C, Czech L, Gerboth S, et al.. Novel roles of formin mDia2 in lamellipodia and filopodia formation in motile cells. PLoS Biol. 2007;5:e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hotulainen P, Paunola E, Vartiainen MK, et al.. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol Biol Cell. 2005;16:649–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rogers SL, Wiedemann U, Stuurman N, et al.. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J Cell Biol. 2003;162:1079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sidani M, Wessels D, Mouneimne G, et al.. Cofilin determines the migration behavior and turning frequency of metastatic cancer cells. J Cell Biol. 2007;179:777–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vitriol EA, Wise AL, Berginski ME, et al.. Instantaneous inactivation of cofilin reveals its function of F-actin disassembly in lamellipodia. Mol Biol Cell. 2013;24:2238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11:62–74. [DOI] [PubMed] [Google Scholar]
- [67].Block J, Breitsprecher D, Kuhn S, et al.. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr Biol. 2012;22:1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Eisenmann KM, Harris ES, Kitchen SM, et al.. Dia-interacting protein modulates formin-mediated actin assembly at the cell cortex. Curr Biol. 2007;17:579–91. [DOI] [PubMed] [Google Scholar]
- [69].Damiani D, Goffinet AM, Alberts A, et al.. Lack of Diaph3 relaxes the spindle checkpoint causing the loss of neural progenitors. Nat Commun. 2016;7:13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Li A, Ma Y, Yu X, et al.. Rac1 drives melanoblast organization during mouse development by orchestrating pseudopod- driven motility and cell-cycle progression. Dev Cell. 2011;21:722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wagner AR, Luan Q, Liu SL, et al.. Dip1 defines a class of Arp2/3 complex activators that function without preformed actin filaments. Curr Biol. 2013;23:1990–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fukumi-Tominaga T, Mori Y, Matsuura A, et al.. DIP/WISH-deficient mice reveal Dia- and N-WASP-interacting protein as a regulator of cytoskeletal dynamics in embryonic fibroblasts. Genes Cells. 2009;14:1197–207. [DOI] [PubMed] [Google Scholar]
- [73].Fukuoka M, Suetsugu S, Miki H, et al.. A novel neural Wiskott-Aldrich syndrome protein (N-WASP) binding protein, WISH, induces Arp2/3 complex activation independent of Cdc42. J Cell Biol. 2001;152:471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Machacek M, Hodgson L, Welch C, et al.. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lee K, Elliott HL, Oak Y, et al.. Functional hierarchy of redundant actin assembly factors revealed by fine-grained registration of intrinsic image fluctuations. Cell Syst. 2015;1:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sakata D, Taniguchi H, Yasuda S, et al.. Impaired T lymphocyte trafficking in mice deficient in an actin-nucleating protein, mDia1. J Exp Med. 2007;204:2031–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Tanizaki H, Egawa G, Inaba K, et al.. Rho-mDia1 pathway is required for adhesion, migration, and T-cell stimulation in dendritic cells. Blood. 2010;116:5875–84. [DOI] [PubMed] [Google Scholar]
- [78].Peng J, Wallar BJ, Flanders A, et al.. Disruption of the Diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr Biol. 2003;13:534–45. [DOI] [PubMed] [Google Scholar]
- [79].Sundaram GM, Ismail HM, Bashir M, et al.. EGF hijacks miR-198/FSTL1 wound-healing switch and steers a two-pronged pathway toward metastasis. J Exp Med. 2017;214:2889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zuchero JB, Coutts AS, Quinlan ME, et al.. p53-cofactor JMY is a multifunctional actin nucleation factor. Nat Cell Biol. 2009;11:451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Firat-Karalar EN, Hsiue PP, Welch MD. The actin nucleation factor JMY is a negative regulator of neuritogenesis. Mol Biol Cell. 2011;22:4563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Sen B, Uzer G, Samsonraj RM, et al.. Intranuclear Actin Structure Modulates Mesenchymal Stem Cell Differentiation. Stem Cells. 2017;35:1624–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bieling P, Hansen SD, Akin O, et al.. WH2 and proline-rich domains of WASP-family proteins collaborate to accelerate actin filament elongation. EMBO J. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Barzik M, McClain LM, Gupton SL, et al.. Ena/VASP regulates mDia2-initiated filopodial length, dynamics, and function. Mol Biol Cell. 2014;25:2604–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kitzing TM, Wang Y, Pertz O, et al.. Formin-like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene. 2010;29:2441–8. [DOI] [PubMed] [Google Scholar]
- [86].Czuchra A, Wu X, Meyer H, et al.. Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol Biol Cell. 2005;16:4473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang Y, Arjonen A, Pouwels J, et al.. Formin-like 2 Promotes beta1-Integrin Trafficking and Invasive Motility Downstream of PKCalpha. Dev Cell. 2015;34:475–83. [DOI] [PubMed] [Google Scholar]
- [88].Vega FM, Fruhwirth G, Ng T, et al.. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J Cell Biol. 2011;193:655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Isogai T, van der Kammen R, Bleijerveld OB, et al.. Quantitative Proteomics Illuminates a Functional Interaction between mDia2 and the Proteasome. J Proteome Res. 2016;15:4624–37. [DOI] [PubMed] [Google Scholar]
- [90].Isogai T, van der Kammen R, Goerdayal SS, et al.. Proteomic analyses uncover a new function and mode of action for mouse homolog of Diaphanous 2 (mDia2). Mol Cell Proteomics. 2015;14:1064–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Miller MR, Miller EW, Blystone SD. Non-canonical activity of the podosomal formin FMNL1gamma supports immune cell migration. J Cell Sci. 2017;130:1730–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Isogai T, van der Kammen R, Innocenti M. SMIFH2 has effects on Formins and p53 that perturb the cell cytoskeleton. Sci Rep. 2015;5:9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yamada K, Ono M, Perkins ND, et al.. Identification and functional characterization of FMN2, a regulator of the cyclin-dependent kinase inhibitor p21. Mol Cell. 2013;49:922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11:353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Gadea G, de Toledo M, Anguille C, et al.. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol. 2007;178:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kage F, Winterhoff M, Dimchev V, et al.. FMNL formins boost lamellipodial force generation. Nat Commun. 2017;8:14832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Gupton SL, Eisenmann K, Alberts AS, et al.. mDia2 regulates actin and focal adhesion dynamics and organization in the lamella for efficient epithelial cell migration. J Cell Sci. 2007;120:3475–87. [DOI] [PubMed] [Google Scholar]
- [98].Chan KT, Creed SJ, Bear JE. Unraveling the enigma: progress towards understanding the coronin family of actin regulators. Trends Cell Biol. 2011;21:481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Cai L, Makhov AM, Bear JE. F-actin binding is essential for coronin 1B function in vivo. J Cell Sci. 2007;120:1779–90. [DOI] [PubMed] [Google Scholar]
- [100].Volkmann N, Amann KJ, Stoilova-McPhie S, et al.. Structure of Arp2/3 complex in its activated state and in actin filament branch junctions. Science. 2001;293:2456–9. [DOI] [PubMed] [Google Scholar]
- [101].Cai L, Makhov AM, Schafer DA, et al.. Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell. 2008;134:828–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cai L, Marshall TW, Uetrecht AC, et al.. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell. 2007;128:915–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jansen S, Collins A, Chin SM, et al.. Single-molecule imaging of a three-component ordered actin disassembly mechanism. Nat Commun. 2015;6:7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Gandhi M, Smith BA, Bovellan M, et al.. GMF is a cofilin homolog that binds Arp2/3 complex to stimulate filament debranching and inhibit actin nucleation. Curr Biol. 2010;20:861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Luan Q, Nolen BJ. Structural basis for regulation of Arp2/3 complex by GMF. Nat Struct Mol Biol. 2013;20:1062–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Haynes EM, Asokan SB, King SJ, et al.. GMFbeta controls branched actin content and lamellipodial retraction in fibroblasts. J Cell Biol. 2015;209:803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Dang I, Gorelik R, Sousa-Blin C, et al.. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 2013;503:281–4. [DOI] [PubMed] [Google Scholar]
- [108].Wilson CA, Tsuchida MA, Allen GM, et al.. Myosin II contributes to cell-scale actin network treadmilling through network disassembly. Nature. 2010;465:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ridley AJ, Paterson HF, Johnston CL, et al.. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–10. [DOI] [PubMed] [Google Scholar]
- [110].Guo F, Debidda M, Yang L, et al.. Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. J Biol Chem. 2006;281:18652–9. [DOI] [PubMed] [Google Scholar]
- [111].Liu S, Kapoor M, Leask A. Rac1 expression by fibroblasts is required for tissue repair in vivo. Am J Pathol. 2009;174:1847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Millius A, Watanabe N, Weiner OD. Diffusion, capture and recycling of SCAR/WAVE and Arp2/3 complexes observed in cells by single-molecule imaging. J Cell Sci. 2012;125:1165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Suraneni P, Fogelson B, Rubinstein B, et al.. A mechanism of leading-edge protrusion in the absence of Arp2/3 complex. Mol Biol Cell. 2015;26:901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Wu C, Haynes EM, Asokan SB, et al.. Loss of Arp2/3 induces an NF-kappaB-dependent, nonautonomous effect on chemotactic signaling. J Cell Biol. 2013;203:907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Di Nardo A, Cicchetti G, Falet H, et al.. Arp2/3 complex-deficient mouse fibroblasts are viable and have normal leading-edge actin structure and function. Proc Natl Acad Sci U S A. 2005;102:16263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Gournier H, Goley ED, Niederstrasser H, et al.. Reconstitution of human Arp2/3 complex reveals critical roles of individual subunits in complex structure and activity. Mol Cell. 2001;8:1041–52. [DOI] [PubMed] [Google Scholar]
- [117].Zhou K, Muroyama A, Underwood J, et al.. Actin-related protein2/3 complex regulates tight junctions and terminal differentiation to promote epidermal barrier formation. Proc Natl Acad Sci U S A. 2013;110:E3820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Rotty JD, Wu C, Haynes EM, et al.. Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev Cell. 2015;32:54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Fritz-Laylin LK, Riel-Mehan M, Chen BC, et al.. Actin-based protrusions of migrating neutrophils are intrinsically lamellar and facilitate direction changes. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Leithner A, Eichner A, Muller J, et al.. Diversified actin protrusions promote environmental exploration but are dispensable for locomotion of leukocytes. Nat Cell Biol. 2016;18:1253–9. [DOI] [PubMed] [Google Scholar]
- [121].Vargas P, Maiuri P, Bretou M, et al.. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat Cell Biol. 2016;18:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Jacquemet G, Hamidi H, Ivaska J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr Opin Cell Biol. 2015;36:23–31. [DOI] [PubMed] [Google Scholar]