

ABSTRACT
Type I collagen and DDR1 axis has been described to decrease cell proliferation and to initiate apoptosis in non-invasive breast carcinoma in three-dimensional cell culture matrices. Moreover, MT1-MMP down-regulates these effects. Here, we address the effect of type I collagen aging and MT1-MMP expression on cell proliferation suppression and induced-apoptosis in non-invasive MCF-7 and ZR-75-1 breast carcinoma. We provide evidence for a decrease in cell growth and an increase in apoptosis in the presence of adult collagen when compared to old collagen. This effect involves a differential activation of DDR1, as evidenced by a higher DDR1 phosphorylation level in adult collagen. In adult collagen, inhibition of DDR1 expression and kinase function induced an increase in cell growth to a level similar to that observed in old collagen. The impact of aging on the sensitivity of collagen to MT1-MMP has been reported recently. We used the MT1-MMP expression strategy to verify whether, by degrading adult type I collagen, it could lead to the same phenotype observed in old collagen 3D matrix. MT1-MMP overexpression abrogated the proliferation suppression and induced-apoptosis effects only in the presence of adult collagen. This suggests that differential collagen degradation by MT1-MMP induced a structural disorganization of adult collagen and inhibits DDR1 activation. This could in turn impair DDR1-induced cell growth suppression and apoptosis. Taken together, our data suggest that modifications of collagen structural organization, due to aging, contribute to the loss of the growth suppression and induced apoptosis effect of collagen in luminal breast carcinoma. MT1-MMP-dependent degradation and aging of collagen have no additive effects on these processes.
KEYWORDS: Aging, Apoptosis, Breast carcinoma, DDR1, Type I collagen
Introduction
Several studies have shown the role of the cross-talk between cancer cells and their microenvironment in regulating tumor progression [1]. Type I collagen, a major component of interstitial tissues, represents up to 90% of the extracellular matrix (ECM) proteins. The type I collagen receptors which have been most studied are the integrins [2]. The GFOGER consensus sequence found in fibrillar collagens is essentially recognized by the integrins α1β1, α2β1 and α11β1 [3]. In the past two decades, DDR (Discoidin Domain Receptor) 1 and DDR2 receptors, which belong to the family of Discoidin-Domain Receptors have been largely explored [4]. DDRs differ from integrins in the fact that they belong to the tyrosine kinase receptor (RTKs) family and recognize the GVMGFO consensus sequence [5]. Unlike conventional RTK that have a rapid and transient activation, DDR1 and DDR2 exhibit a relatively late and prolonged activation [6].
Breast carcinoma is a major cause of death among women [7,8]. DDR1 has recently been demonstrated to induce apoptosis in luminal and poorly invasive breast carcinoma cells upon activation by type I collagen in three dimensional (3D) matrices. Membrane-type 1 matrix metalloproteinase (MT1-MMP, MMP-14), a major collagenolytic proteinase, inhibits this process suggesting that the possible degradation of type I collagen by MT1-MMP could be the cause of its inability to activate DDR1 and to induce apoptosis [9]. Before discovering the involvement of collagen-DDR1 pathway in cell proliferation suppression and induced apoptosis, previous works have reported the importance of the structural properties of collagen in this process. In fact, the degradation of collagen by MT1-MMP has been shown to allow the cells to escape this apoptosis and to proliferate at a level close to that observed without collagen [10,11]. Similar observations have been reported when heat-denatured collagen was used [9,12,13]. In agreement with these data, Vogel and co-workers reported that proteolysis and thermal denaturation of collagen inhibits DDR1 and DDR2 activation [6].
Because of its long half-life, type I collagen undergoes post-translational changes during biological aging. These modifications can, for example, generate advanced glycation endproducts (AGE) [14], and an increase in crosslinks level [15]. Our group has recently reported that these biochemical alterations impact the structural organization of type I collagen [16–18]. Previous works have shown that changes in structural properties of collagen during aging affects its degradation by matrix metalloproteinases [19, 20]. Since DDR1 and DDR2 are very sensitive to the structural organization of type I collagen, these changes could also influence the level of DDR1 and DDR2 activation and consequently their "cell growth suppressive” and “pro-apoptotic” functions. In agreement with this hypothesis, we have recently shown that collagen-aging decreased the activation of DDR2, resulting in the loss of its growth suppressive function in human fibrosarcoma cells [21].
Breast carcinoma incidence increases with age. In fact, more than 50% of patients with breast cancer are 65 years old or older, and 30% are more than 70 years old [7,8]. Moreover, structural organization of type I collagen, called “Tumor Associated Collagen Signature” (TACS) is now considered as an important marker in the prognosis of this pathology [22,23]. In the present study, we evaluated the effect of adult and aged (3D) collagen matrices on the proliferation and apoptosis of MCF-7 and ZR-75-1 cells, two poorly invasive luminal breast cancer cell lines. Our data demonstrate that adult collagen inhibited cell proliferation and induced apoptosis when compared to the old one. DDR1 phosphorylation was higher in the presence of adult collagen and inhibition of DDR1 promoted cellular proliferation and decreased adult type I collagen-induced apoptosis. Altogether, these results suggest that physiological collagen aging fosters tumor cell growth in poorly invasive breast carcinoma by reducing the activation of DDR1, one of the main matrix sensors, in addition to integrin receptors family.
Results
Collagen aging promotes cell proliferation
First, we compared the growth of MCF-7 and ZR-75-1 cells cultivated for 6 days in three different experimental settings: (i) plated on plastic (used as a control), (ii) plated on a thin coat of dried collagen or (iii) suspended within a 3D collagen gel. Native type I collagens extracted from either adult (2-month-old) or old (2-year-old) rats were compared with 2D plastic condition. Both cell lines exhibited a significantly lower cell growth in adult collagen 3D matrices when compared to 2D plastic condition (Fig. 1A). We then compared cell growth between adult and old collagen. As shown in Fig. 1A, the inhibitory effect on cell growth observed in 3D adult collagen matrices is abrogated in 3D old collagen matrices. This age-dependent cell proliferation was only observed in 3D, since no significant difference was observed in the 2D coating experiments (Fig. 1B). These data demonstrate that collagen aging promotes cell growth, and that this process occurs only in a 3D environment.
Figure 1.
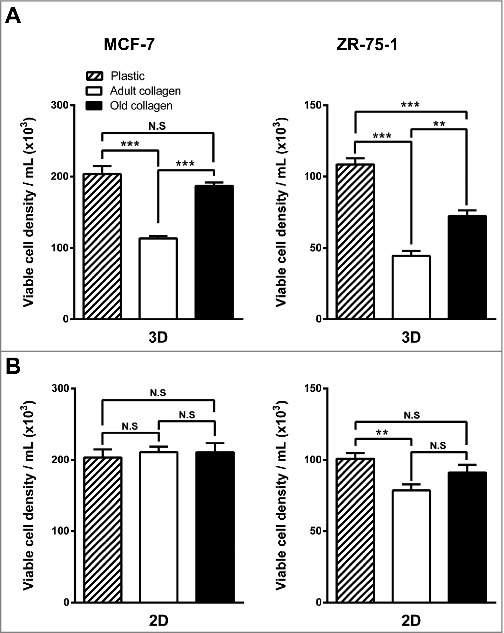
Effect of collagen aging on MCF-7 and ZR-75-1 luminal breast cancer cells growing on type I collagen 2D coating vs. 3D matrices. A. MCF-7 (left panel) and ZR-75-1 (right panel) cells were seeded on plastic (control) or in adult and old type I collagen 3D matrices at a density of 1.5 × 104 cells/ml. B. MCF-7 (left panel) and ZR-75-1 (right panel) cells were seeded on plastic or on adult and old type I collagen 2D coating (5 µg/cm²) at a density of 1.5 × 104 cells/ml. After 6 days of culture, viable cell density was evaluated by phase contrast microscopy. Values represent the mean ± S.E.M. of three independent experiments (**p < 0,01, ***p < 0.001, N.S = not significant).
DDR1 depletion promotes cell growth in 3D matrices
DDR1 and DDR2 are tyrosine kinase receptors with the unique ability to be activated only by type I collagen in its fibrillary state. Recently, DDR1 has been shown to initiate apoptosis in MCF-7 and ZR-75-1 cells embedded within 3D native type I collagen [9]. Here, we analyzed the expression of both DDR1 and DDR2 in MCF-7 and ZR-75-1 cells. To this end, HT-1080 cells were used as a positive control for DDR2 [21]. We showed that compared to HT-1080 cells, MCF-7 and ZR-75-1 strongly express DDR1 whereas they exhibit very small amount of DDR2 (Fig. 2A). Then, we studied the effect of DDR1 depletion on the growth of MCF-7 and ZR-75-1 cells in adult and old type I collagen matrices. To this end, DDR1 siRNAs were used to knockdown DDR1 in both cell lines. DDR1 siRNA reduced the expression of DDR1 by 78% and 65% in MCF-7 and ZR-75-1 cells 24 hours after transfection (Fig. 2B). 6 days after the transfection, DDR1 expression is still reduced by 72% in MCF-7 (Fig. 2C). DDR1-silenced cells were then seeded in adult and old 3D collagen matrices. As shown in Fig. 2D, DDR1 depletion promotes cell growth in adult but not in the old collagen, suggesting that DDR1 could be involved in the differential regulation of cell growth observed in the presence of the two collagens. DDR1 silencing has not been compensated by expressing DDR2 (Fig. 2E).
Figure 2.
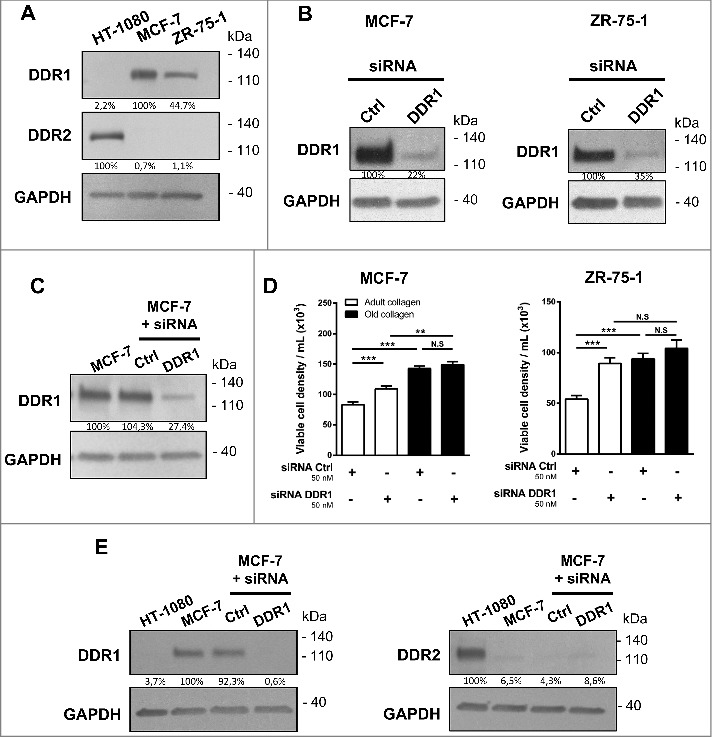
Effect of DDR1 silencing on the growth response of MCF-7 and ZR-75-1 cells in adult and old type I collagen 3D matrices. A. DDR1 and DDR2 expression was analyzed by immunoblotting. HT-1080 cells were used as a positive control for DDR2. GAPDH was used as a loading control. B. MCF-7 (left panel) and ZR-75-1 (right panel) cells were transfected with control or DDR1 siRNA. DDR1 expression was analyzed B. 24 hours and C. 6 days after transfection by immunoblotting. GAPDH was used as a loading control. D. DDR1-silenced cells were embedded in adult or old 3D collagen matrices at a density of 1.5 × 104 cells/ml. After 6 days of culture, viable cell density was assessed by phase contrast microscopy. E. MCF-7 cells were transfected with control or DDR1 siRNA. After 36 hours, DDR1 and DDR2 expression was analyzed by immunoblotting. HT-1080 cells were used as a positive control for DDR2. Data are representative of three independent experiments. Values represent the mean ± S.E.M. of three independent experiments (**p < 0.01, ***p < 0.001, N.S = not significant).
Collagen aging reduces DDR1 activation
We evaluated the effect of collagen aging on DDR1 expression and activation in MCF-7 (Fig. 3A) and ZR-75-1 (Fig. 3B) cells. While total DDR1 expression levels were similar in both collagens, DDR1 phosphorylation was reduced in old collagen when compared to the adult one, suggesting that aging impairs the capacity of type I collagen to induce DDR1 activation.
Figure 3.
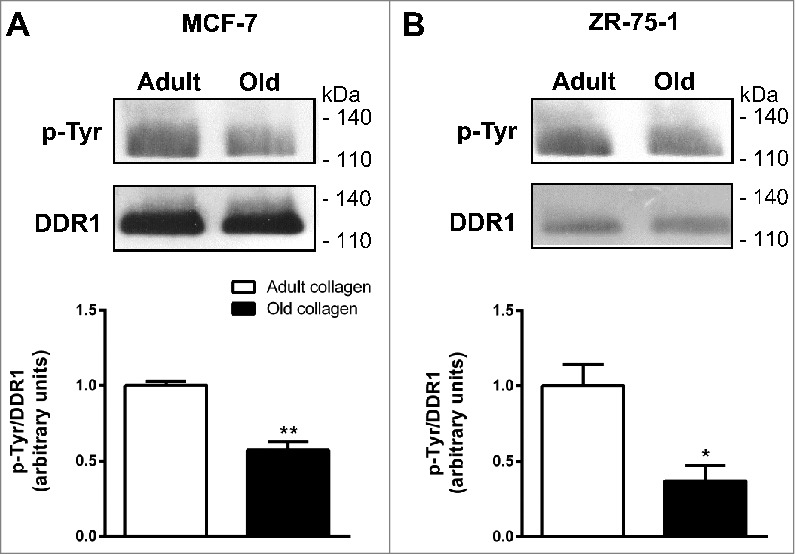
Effect of collagen aging on DDR1 expression and activation. A. MCF-7 cells, B. ZR-75-1 cells. Cells were starved during 12 hours, and then cultured 6 hours in adult and old 3D collagen matrices. Immunoprecipitation was performed using anti-DDR1 antibody, then a western blot was realized with a phosphotyrosine antibody to detect DDR1 activation. The membrane was then stripped, and total DDR1 was visualized using a specific DDR1 antibody (upper panel). Blots are representative of three independent experiments (for MCF-7) and two independent experiments (for ZR-75-1). The histogram shows the ratio of phosphotyrosine relative to total DDR1 (lower panel). Values represent the mean ± S.D. (*p < 0.05, **p < 0.01).
DDR1 tyrosine kinase activity inhibits cell growth
To evaluate the involvement of DDR1 kinase activity in the adult 3D collagen-mediated inhibition of cell growth, nilotinib and DDR1-IN-1, two tyrosine kinase inhibitors with high potency against DDR1 were used. Nilotinib, when used at non-toxic concentration (100 nM), restored cell proliferation in adult collagen to a level similar to that observed in the old one (Fig. 4A). As nilotinib also inhibits other kinases in addition to DDR1 [24], we investigated whether the DDR1 specific inhibitor DDR1-IN-1 was also able to restore cell growth. DDR1-IN-1, when used at non-toxic concentration (10 µM), similarly restored cell growth in adult collagen to a level identical to that observed in the old one (Fig. 4B). Taken together, these data demonstrate the involvement of DDR1 tyrosine kinase activity in the differential regulation of cell proliferation induced by collagen aging.
Figure 4.
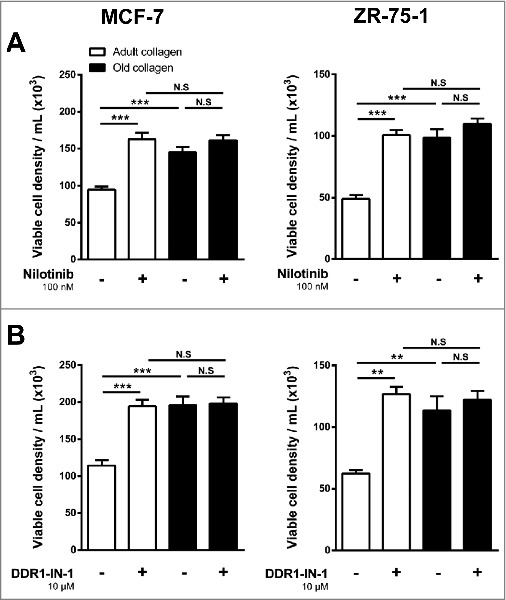
Effect of the inhibition of DDR1 kinase activity on the growth response of MCF-7 and ZR-75-1 cells in adult and old type I collagen 3D matrices. MCF-7 (left panel) and ZR-75-1 (right panel) cells were embedded in adult or old 3D collagen matrices at a density of 1.5 × 104 cells/ml. Cells were incubated in absence (-) or presence (+) of (A) nilotinib (100 nM) or (B) DDR1-IN-1 (10 µM). After 6 days of culture, viable cell density was assessed by phase contrast microscopy. Values represent the mean ± S.E.M. of three independent experiments (**p < 0,01, ***p < 0.001, N.S = not significant).
Collagen aging impairs DDR1-induced apoptosis
The negative regulation of MCF-7 and ZR-75-1 cell growth induced by adult 3D type I collagen has been previously ascribed to a strong DDR1-dependent induction of apoptotic cell death [9,11]. To evaluate whether collagen aging influences collagen/DDR1-induced apoptosis, two apoptosis markers were analyzed. First, the expression of BCL-2 interacting killer (BIK), a pro-apoptotic BH3-only member of the BCL-2 family known to be up-regulated by 3D type I collagen [11], was quantified by RT-qPCR. Adult collagen was significantly more potent than the old one to induce BIK expression in both MCF-7 and ZR-75-1 cells (Fig. 5A). Secondly, the levels of cytoplasmic histone-associated DNA fragments, used as a surrogate of apoptotic cell death, were strongly increased in MCF-7 and ZR-75-1 cells cultured in 3D collagen gels when compared with cells plated on plastic. However, this increase was significantly greater in adult than in old collagen (Fig. 5B). Taken together, these data suggest that type I collagen aging impairs DDR1 induced apoptosis.
Figure 5.
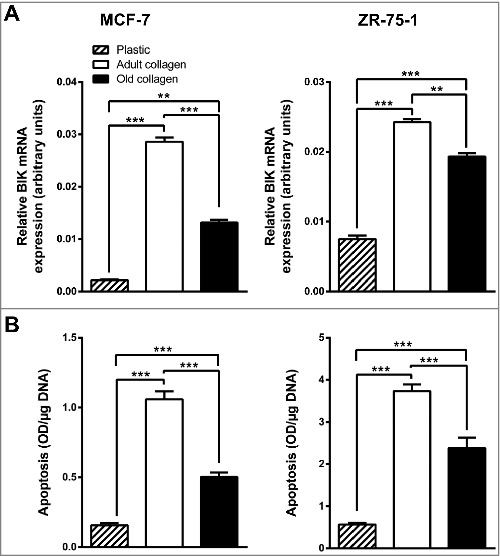
Effect of collagen aging on BIK expression and apoptosis. MCF-7 (left panel) and ZR-75-1 (right panel) cells were seeded on plastic or in adult or old 3D type I collagen matrices for 6 days before harvesting the cells. BIK expression was assessed by RT-qPCR (A) and apoptosis was quantified by ELISA (B). Values represent the mean ± S.E.M. of three independent experiments (**p < 0.01, ***p < 0.001, N.S = not significant).
DDR1 depletion decrease adult collagen-induced apoptosis
To analyze whether DDR1 is implicated in adult collagen-increased apoptosis, MCF-7 cells were treated with siRNA against DDR1 and cultured in adult and old collagen 3D matrices. DDR1 siRNA reduced the expression of DDR1 by 88% in MCF-7 (Fig. 6A). After 36h in collagens 3D matrices, the cells were harvested and percentage of annexin V positive cells was quantified. As shown in Fig. 6B, DDR1 depletion decrease the percentage of apoptotic cells in adult collagen compared to the cells treated with control siRNA, to a level similar to that observed in old collagen. These data suggest that DDR1 play a crucial role in adult collagen induced apoptosis, but not in the old one.
Figure 6.
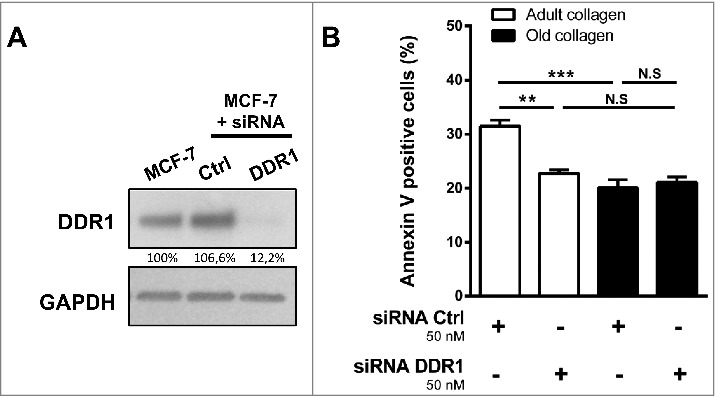
Effect of DDR1 silencing on apoptosis in adult and old collagen 3D matrices. MCF-7 cells were transfected with control or DDR1 siRNA. A. DDR1 expression was analyzed by immunoblotting. GAPDH was used as a loading control. C. DDR1-silenced cells were embedded in adult or old 3D collagen matrices at a density of 1 × 105 cells/ml. After 36 hours of culture, the percentage of apoptotic cells was assessed using the Muse® annexin V and dead cell assay kit. Values represent the mean ± S.E.M. of three independent experiments (**p < 0.01, ***p < 0.001, N.S = not significant).
DDR1 kinase activity mediates the collagen-induced BIK up-regulation
In order to investigate whether DDR1 kinase function was involved in adult and old collagen-induced apoptosis, MCF-7 and ZR-75-1 cells were treated with nilotinib in 3D collagen matrices. As shown in Fig. 7, the pharmacological inhibition of DDR1 prevented the increase of BIK expression induced by both adult and old collagens. These data confirmed a causal role for collagen-activated DDR1 and the induction of apoptosis.
Figure 7.
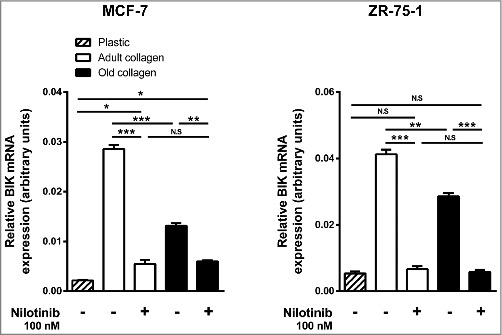
Effect of the inhibition of DDR1 kinase activity on BIK expression in adult and old collagen. MCF-7 (left panel) and ZR-75-1 (right panel) cells were seeded on plastic or in 3D type I collagen matrices for 6 days, with or without 100 nM of nilotinib. The cells were harvested and BIK expression was quantified by RT-qPCR. Values represent the mean ± S.E.M. of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
MT1-MMP prevents collagen-mediated cell growth suppression and apoptosis
Since MT1-MMP is the primary enzyme used by stromal and cancer cells to cleave and migrate through fibrillar collagen [25–27], we investigated whether MT1-MMP is involved in the differential regulation of cell growth and apoptosis observed between adult and old collagens. For that purpose, MCF-7 cells, which lack endogenous MT1-MMP, were stably transfected with a full-length MT1-MMP expression vector (MCF-7 MT1-MMP) or an empty vector (MCF-7 VEC). In MCF-7 MT1-MMP cells, MT1-MMP was expressed as a 60-kDa mature form and a 43-kDa autoproteolytic degradation product [28], demonstrating that the enzyme was active (Fig. 8A). Consistent with the data obtained with wild-type MCF-7 cells (Figs. 1A, 5A and 5B), MCF-7 VEC cells exhibited a lower cell growth, a higher BIK mRNA level and an increased DNA fragmentation in adult collagen when compared to the old one (Fig. 8B-8D). Expression of MT1-MMP increased cell growth in adult collagen to a level similar to that observed in the old one. In contrast, MT1-MMP did not further increase cell growth in the presence of old collagen (Fig. 8B). MT1-MMP significantly reduced BIK mRNA in cells cultivated in adult and old collagen by 6.4-fold and 3.6-fold, respectively (Fig. 8C). It also decreased the adult collagen-induced apoptosis to a level similar to that observed in the old one (Fig. 8C). However, despite its inhibitory effect on BIK mRNA level, MT1-MMP induced only a slight decrease of apoptosis in old collagen (Fig. 8D).
Figure 8.
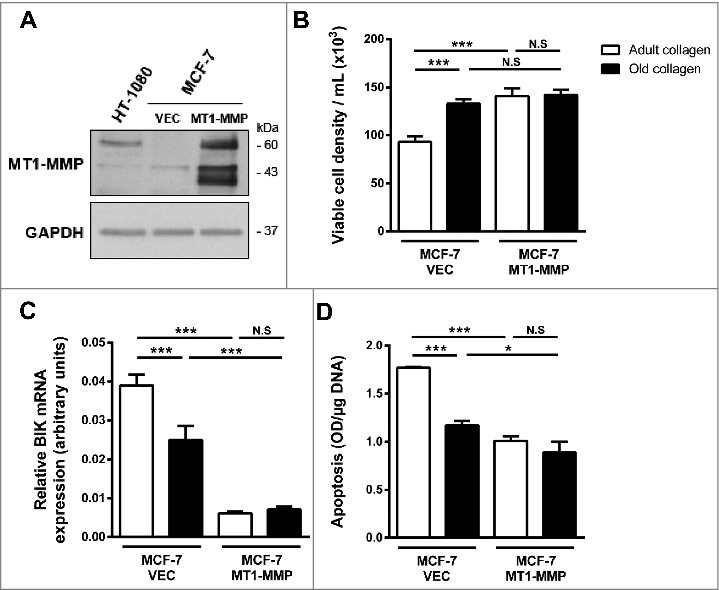
Effect of MT1-MMP expression on adult and old collagen I-induced apoptosis. A. Western blot analysis was performed using anti-MT1-MMP specific antibody. HT1080 cell lysate was used as a positive control. GAPDH was used as a loading control. B. MCF-7 VEC (empty vector) and MCF-7 MT1-MMP (full length MT1-MMP expression vector) cells were seeded in adult and old type I collagen 3D matrices at a density of 1.5 × 104 cells/ml. After 6 days of culture viable cell density was evaluated by phase contrast microscopy. C. MCF-7 VEC and MCF-7 MT1-MMP cells were seeded in adult or old 3D type I collagen matrices for 6 days before harvesting the cells. Apoptosis was assessed by ELISA. D. MCF-7 VEC and MCF-7 MT1-MMP cells were seeded on plastic or in adult or old 3D type I collagen matrices for 6 days. The cells were harvested and BIK expression was assessed by RT-qPCR. Values represent the mean ± S.E.M. of three independent experiments (*p < 0.05, ***p < 0.001, N.S = not significant).
Discussion
Collagens represent the most abundant proteins in the ECM and have very low turnover rates [29]. This turnover rate decreases with age as a consequence of the natural aging process, which correlates with alterations in the composition, the structural and mechanical integrity of the connective tissue. Aged collagen fibers show an increase in non-enzymatic AGE-dependent cross-links [14,15,30] and mineral deposits [31], leading to a loss in the fibrillar organization of the protein [16–18], a stiffening of the tissues and a reduced functionality.
One of the features of many carcinomas, particularly breast, is the presence of a dense collagenous stroma located in close contact with cancer cells. The interactions between these cells and the surrounding ECMs play key roles during cancer progression [22,23,32–35]. More than 50% of patients with breast cancer are 65 years old or older, suggesting that cancer cells of these patients are confronted to aged collagen fibers. Therefore, it is fundamental to better understand how aging processes affect the interactions between cancer cells and collagen molecules.
Several studies have shown the crucial role of cross-talks between cancer cells and components of the ECM in the regulation of tumor progression [1]. To understand these processes, especially for type I collagen, most studies were performed in 2D culture model, where cancer cells were plated either on a thin coat of collagen or on plastic with soluble pepsinized collagen added to the culture medium [36]. However, these experimental settings do not faithfully reproduce the cellular microenvironment observed in-vivo.
By using 2D type I collagen coats, different groups have shown that this protein downregulates the proliferation of M24met melanoma and HT-1080 fibrosarcoma cells [37,38]. In contrast, our recent data show that the proliferation of HT-1080 and A204 fibrosarcoma cell lines was downregulated by native type I collagen when present in 3D but not in 2D conditions [21]. Different studies, including our own, have reported that DDR2 was involved in this process [12,13], while integrins, which have been previously described to regulate cell proliferation and survival [39], were not involved in cell growth regulation by 3D matrix [9,21]. Maquoi group has shown recently that the growth of MCF-7 and ZR-75-1 breast carcinoma cells was also reduced when these cells were maintained in 3D type I collagen gels but not when plated on a 2D matrix [9]. Moreover, type I collagen was able to induce apoptosis in these cells. In fact, type I collagen is able to activate DDR1 and to induce the expression of BIK, a pro-apoptotic member of the BCL-2 protein family, thereby triggering apoptotic cell death in these breast cancer cell lines [11]. However, in a recent work, Badaoui et al. have reported that type I collagen was able to contribute also to survival in MCF-7 cells and this process also involved type I collagen/DDR1 axis [40]. In this case, it is important to note that as for other receptors, such as c-Met [41], the function of DDR1 is deeply dependent on cell culture model (2D coating or 3D matrices) and on the presence or not of the growth factors. In fact, in this study, 2D collagen coating has been used and the cells have been serum-deprived. Thus, the observed cell survival suggests that DDR1 can induce survival as well as apoptosis, depending on experimental settings.
Because of its long half-life, type I collagen is able to undergo post-translational modification such as an increase in AGEs [14], which in turn, can induce an increase in cross-link level [15] and a loss in the fibrillar organization of the protein [16–18]. We have recently demonstrated that biologically aged type I collagen is able to upregulate proliferation of fibrosarcoma cells. Moreover, DDR2 has been identified as a key matrix aging sensor responsible for the regulation of this process [21]. In the present study, we evaluated the effect of biological aging of type I collagen on the growth and apoptosis of MCF-7 and ZR-75-1 cell lines in a 3D model. Adult collagen was able to impede cell growth and to induce apoptosis when compared to the old one. We show that DDR1 phosphorylation in the presence of adult collagen was higher than that observed in the presence of the old one. These data are consistent with those obtained on DDR2 activation in fibrosarcoma cells [21]. The increase in cell growth and the reduction of apoptosis rate in the presence of DDR1 kinase function inhibitors demonstrate that this function is essential in this process. Concerning the specificity of the inhibitors, whereas the kinase inhibitor DDR1-IN-1 is specific for DDR1, with a reported EC50 value of 86.76 nM and a selectivity (S(1) at 1 µM) of 0.01 using the KinomeScan [42], this has not been demonstrated for nilotinib. However, it is important to note that we have demonstrated earlier, using cells expressing DDR2 harboring a gatekeeper mutation, that nilotinib did not induce any increase in the proliferation of fibrosarcoma cells, suggesting that this inhibitor should be at least specific to DDR1 and DDR2 and does not inhibit other kinase receptors [21]. As shown in our data, MCF-7 and ZR-75-1 cells express DDR1 but not DDR2. Thus, the differential effect of aging on cell proliferation rate and type I collagen induced-apoptosis suggests that the impact of matrix aging has to be taken into account when designing in-vitro experiments to understand cell/microenvironment interactions, and to evaluate their consequences on tumor cell responses.
Interestingly, Maquoi group has reported that the expression of MT1-MMP luminal breast carcinoma cells, a potent collagenolytic matrix metalloproteinase frequently overexpressed in basal invasive ones, decreased DDR1 activation and prevented the onset of apoptosis [9, 11]. This suggested that alterations of the structural integrity of collagen fibers might impair the collagen-evoked DDR1 signaling. The inhibitory effect of MT1-MMP on collagen I/DDR1-induced BIK expression might result from the proteolytic cleavage of collagen fibers but also from the capacity of active MT1-MMP to shed the extracellular domain of DDR1 [9, 43], thereby mitigating the collagen-induced DDR1 signaling. In a similar way, the post-translational changes (e.g. glycations, crosslinkings) which affect type I collagen during aging might reduce the capacity of DDR1 to recognize and interact with collagen, thereby alleviating its capacity to induce BIK expression and the resulting apoptosis.
Further investigation should consist in the study of the effect of the two collagens on invasive breast tumor cells which express MT1-MMP. Basal-like breast cancers are among the most aggressive and deadly breast cancer subtypes, displaying a high metastatic ability associated with mesenchymal features. These mesenchymal features are acquired as a consequence of an epithelial to mesenchymal transition (EMT). EMT is classically characterized by the dedifferentiation from an epithelial to mesenchymal phenotype, marked by the decreased expression of E-cadherin and increased expression of vimentin as well as expression of cellular proteases, allowing carcinoma cells to dissociate from the primary tumor thereby facilitating tumor cell invasion and metastasis. Therefore, the EMT appears as a critical step in the progression of cancers from both the pre-invasive to invasive state and from organ confined to metastatic disease. Decreased DDR1 expression has been associated with the EMT process in breast cancer and its overexpression in aggressive mesenchymal-like triple-negative breast cancer cells reduced their invasiveness in 3D cultures and in-vivo, supporting an anti-migratory function DDR1 in this cancer [44]. To further examine this correlation in multiple breast cancer cell lines, we interrogated the Broad-Novartis Cancer Cell Line Encyclopedia (CCLE) database for expression levels of E-cadherin (CDH1), vimentin (VIM), DDR1, DDR2, MT1-MMP (MMP14) and BIK mRNAs in 58 breast cancer cell lines. High DDR1 expression was clearly observed in the E-cadherin-high and vimentin-low epithelial cell lines. In contrast, the more mesenchymal breast cancer cell lines (E-cadherin-low and vimentin-high) were characterized by a weak DDR1 level, a high MT1-MMP and a low BIK expression. Our data support the hypothesis that the acquisition of mesenchymal features including the downregulation of DDR1 and the overexpression of collagenolytic proteinases such as MT1-MMP provides breast carcinoma cells with an increased capacity to resist to apoptosis induced by adult collagen (Fig. 9) [11].
Figure 9.
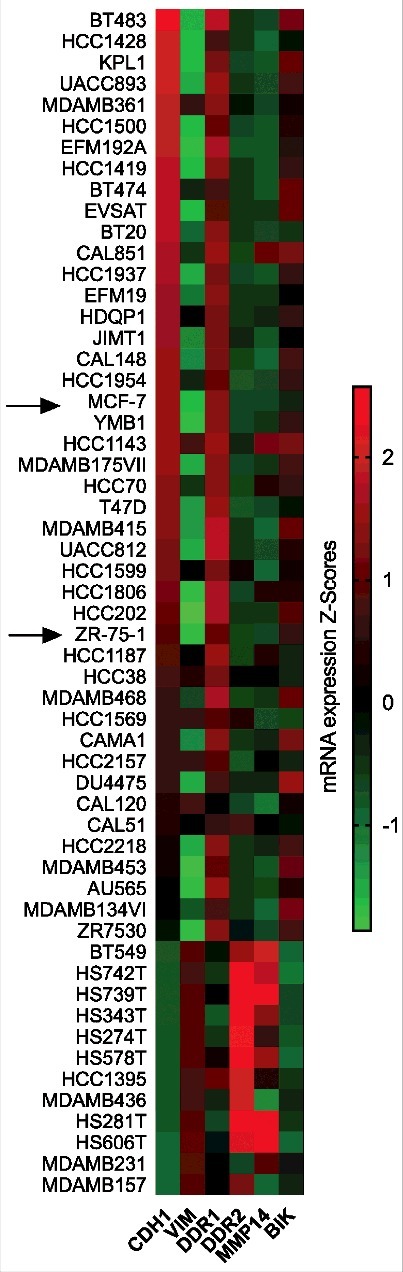
Heatmap of the expression levels of E-cadherin (CDH1), vimentin(VIM), DDR1, DDR2, MT1-MMP (MMP14) and BIK mRNAs in 58 breast cancer cell lines.
Taken together, our data show that changes in the fibrillar structure of type I collagen, whether during aging or due to proteolytic degradation, lead to a loss of its suppressive effect of proliferation and induction of apoptosis, and thus participate to tumor growth. As for DDR2 in the case of fibrocarcoma [21], we identify for the first time DDR1 as a key matrix aging sensor responsible for the regulation of cell proliferation and apoptosis processes, but in the case of breast carcinoma. Most of the patients with breast cancer are 65 years old or older and structural organization of collagen type I in breast tissue is now believed to be an important marker in the prognosis of this pathology [22,23]. Thus, age-dependent structural modifications of type I collagen may also contribute to better address the tumor behavior in elderly patients [45,46]. Finally, this suggests also that aging of type I collagen and its degradation by proteolysis could be potential therapeutic targets.
Materials and methods
Cell culture
The human breast adenocarcinoma cell lines MCF-7 (HTB-22) and ZR-75-1 (CRL-1500) were purchased from the American Type Culture Collection (ATCC). MCF-7 cells stably transfected with the full-length MT1-MMP vector (MCF-7 MT1-MMP) and MCF-7 cells transfected with the empty vector (MCF-7 VEC), were obtained as previously described [11]. MCF-7 cell line was cultured in MEM with Earle salts and Glutamax I (Invitrogen, 41090) supplemented with 10% fetal bovine serum, (Invitrogen, 10270) and 1% penicillin-streptomycin (Invitrogen, 15140). ZR-75-1 cell line was cultured in DMEM (4,5 g/l glucose) with Glutamax I (Invitrogen, 61965) supplemented with 10% fetal bovine serum, and 1% penicillin-streptomycin. Cultures were maintained at 37˚C in a humidified atmosphere containing 5% CO2 (v/v). Cells were routinely passaged at preconfluency using 0.05% trypsin, 0.53 mM EDTA (Invitrogen, 25300) and screened for the absence of mycoplasma using PCR methods.
Preparation and characterization of type I collagen
Fibrillar native type I collagen was extracted from tail tendons of 2 months (adult) and 2 years old (old) rats and prepared as already described [47]. Briefly, type I collagen was extracted from tail tendons of Wistar rats (Janvier) aged of 10 weeks for the “adult” group and 25 months for the “old” group using 0.5 M acetic acid at 4°C, in the presence of protease inhibitors. Then type I collagen was specifically precipitated with NaCl 0.7 M. After centrifugation, the precipitate was re-suspended in 18 mM acetic acid, and dialyzed against distilled water for 1 week at 4°C, to eliminate the salts used during the precipitation step. Before being used all collagens are characterized as described in our previous work [21]. The presence of the characteristic α1 and α2 chains is observed by SDS-PAGE, and spectrofluorimetric analysis are performed on adult and old collagen to detect AGEs-specific fluorescence using a spectrofluorimeter (Shimadzu model RF-500). Finaly, pentosidine and CML concentrations are measured using LC/MS/MS, and cross-links quantification is performed by ion exchange chromatography (Hitachi L-8800).
2D and 3D cell culture
The effects of the aged type I collagen on breast adenocarcinoma cells proliferation were studied using 24-well plates. For 2D cell cultures, each well was coated with 5 µg/cm² of adult or old collagens solubilized in 0.018 M acetic acid. Coated substrates were dried overnight at room temperature under sterile conditions and rinsed twice in cold PBS (Invitrogen, 14190) before cell plating. Cells were seeded on the coated surfaces at a density of 1.5 × 104 cells/well (1 ml/well). For 3D cell culture, 1.5 × 104 cells were resuspended in 100 μl fetal bovine serum and mixed with a solution containing 100 μl of 10X culture medium (MEM (Sigma-Aldrich, M3024) for MCF-7 cells and DMEM (Gibco, 52100) for ZR-75-1 cells), 100 μl NaHCO3 (0.26 M for MCF-7 cells, and 0.44 M for ZR-75-1 cells), 100 μl H2O, 90 μl NaOH 0.1 M, 10 μl glutamine 200 mM and 500 μl collagen 3 mg/ml. This solution was deposited in 24-well plates (1 ml/well). After polymerization at 37°C during 10 min, gels were covered with 1 ml of complete culture medium and incubated during 6 days. After removal of the culture medium, cell populated gels were digested with collagenase P (2 mg/ml – Roche, 11213873001). Viability and cellular density of this suspension were determined by phase contrast microscopy using Kova® Glasstic® Slides (Kova International Inc, 87144). In some 3D culture experiments, cells were treated with DDR1 pharmacological inhibitors at the indicated concentrations: nilotinib (100 nM, Selleckchem, No.S1033), and DDR1-IN-1 (10 µM), kindly provided by Dr. Gray N. (Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, USA).
siRNA transfections
siRNA oligonucleotides were transfected in MCF-7 and ZR-75-1 cells with Lipofectamine RNAiMax (Invitrogen, 11668) according to the manufacturer's instructions. A Pool of 3 target-specific 19–25 nucleotides DDR1 siRNA (sc-35187; Santa Cruz Biotechnology) were used at 50 nM. Negative control siRNA was purchased from Santa Cruz Biotechnology (sc-37007). Cells were allowed to grow 24 hours after transfection before use. DDR1 knock-down was controlled by western blotting, then cells were seeded in type I collagen 3D matrices as described above.
Quantification of apoptosis by ELISA
Cells were cultured for 6 days on plastic or in adult and old type I collagen 3D matrices as described in the “2D and 3D cell culture”. Then, for each condition, 4 × 105 cells were harvested using collagenase P at 2 mg/ml, washed twice with PBS, and separated in two tubes. The first tube was used to quantify the cytoplasmic histone-associated DNA fragments by enzyme-linked immunosorbent assay (Cell Death Detection ELISAPLUS, Roche, 11774425001), according to the manufacturer's instructions. The second tube was used to normalize the enzyme-linked immunosorbent assay absorbance with the total DNA content. DNA was quantitated by spectrofluorimetry, with the Hoechst 33342 dye.
Quantification of annexin V positive cells
Cells were cultured for 36 hours in adult and old type I collagen 3D matrices as described in the “2D and 3D cell culture”. Then, for each condition, 1 × 105 cells were harvested using collagenase P at 2 mg/ml, washed twice with PBS and analyzed using the (Muse® Annexin V and Dead Cell Assay Kit, Millipore, MCH100105), according to the manufacturer's instructions.
Western blotting
Cells were cultured for 6 days in adult and old type I collagen 3D matrices as described in the “2D and 3D cell culture”. Then, cells were harvested using collagenase P at 2 mg/ml, washed twice with PBS, then lysed with RadioImmunoPrecipitation Assay (RIPA) buffer (Thermo Fisher Scientific, 89900), supplemented with Halt™ Protease and Phosphatase Inhibitor Cocktail 1X (Thermo Scientific, 78442). Cell lysates were sonicated and then clarified by centrifugation at 14 000 × g at 4˚C for 15 min. Total protein content was estimated by bicinchoninic acid (BCA) assay method (Thermo Scientific, 23227). Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blocked with Tris Buffered Saline (TBS) (0.02 M Tris-HCl, 0.137 M NaCl, pH 7.6) containing 0.1% Tween (TBS-T) and 5% Bovine Serum Albumin (BSA) at room temperature during 1 hour and incubated overnight at 4˚C with anti-DDR1 (Cell signaling Technology, #5583), anti-DDR2 (R&D Systems, AD2538), anti-GAPDH antibodies (Cell signaling Technology, #5174), and anti-MT1-MMP, obtained from Dr. Tomasetto C.L (IGBMC, Illkirch, France) [48]. Membranes were washed with TBS-T and incubated with peroxidase conjugated anti-rabbit secondary antibody (Cell signaling Technology, #7074) or anti-goat secondary antibody (R&D Systems, HAF017) at room temperature for 1 hour. Chemiluminescent detection was performed by using an ECL Prime Kit (GE Healthcare, RPN2236). Electrophoretic images were analyzed with ImageJ software.
Immunoprecipitation
MCF-7 and ZR-75-1 cells were washed with DPBS and incubated overnight in serum-free media prior 6 hours of stimulation in adult and old type I collagen 3D matrices. Cells were then lysed with RIPA buffer supplemented with Halt™ Protease and Phosphatase Inhibitor Cocktail 1X, and cell lysates were clarified by centrifugation at 14 000 × g at 4˚C for 15 min. Then 300 µg of whole-cell extracts was immunoprecipitated with anti-DDR1 (1:100, Cell Signaling Technology) at 4°C for 12 hours and then bound to protein G agarose beads (GE Healthcare, 17–0618) and finally washed three times with TBS. The proteins were separated by SDS-PAGE, and the immunoprecipitates were blotted with anti-phosphotyrosine antibody, clone 4G10 (Millipore, 05–321). The blots were then stripped using a stripping buffer (200 mM glycine, 1% SDS, 0.02% sodium azide, pH 2.5) and re-probed with anti-DDR1 antibody (Cell Signaling Technology).
Quantitative RT-PCR
Total RNA purification was performed using TRI Reagent® (MRC, TR118) according to the manufacturer's instructions and 400 ng of RNA was converted to cDNA by reverse transcription using the Maxima First Strand cDNA Synthesis kit (Thermo Fisher Scientific, #K1671), and a PCR MasterCycler® (Eppendorf). Real-time PCR was performed using a Maxima SYBR GREEN/ROX qPCR Master Mix (Thermo Fisher Scientific, #KO222) on the Stratagene Mx3005P qPCR detection system (Agilent Technologies). Polymerase chain reaction conditions were 15 min at +95 °C, followed by 35 cycles each consisting of 15 s at +95 °C (denaturation) and 30 s at +60 °C (annealing/extension). Results were standardized to the eEF1a1 gene expression by calculating ΔCt using the formula ΔCt = (Ctgene of interest – CteEF1A). Gene expression was represented as 2−ΔCt. The forward primer for DDR1 transcript was 5′-TGCTCTCCAATCCAGCCTAC-3′ and the reverse primer was 5′-ATTATGCCGAGGCTGACATT-3′ with a 203 bp product. The forward primer for BIK transcript was 5′-AGGACCTGGACCCTATGGAG-3′ and the reverse primer was 5′- CCCTGATGTCCTCAGTTGGG -3′ with a 195 bp product. The forward primer for eEF1A transcript was 5′-CTGGAGCCAAGTGCTAACATGCC-3′ and the reverse primer was 5′-CCGGGTTTGAGAACACCAGTC-3′ with a 221 bp product.
Expression level of EMT marker in breast cancer cell lines
The expression levels of E-cadherin (CDH1), vimentin (VIM), DDR1, DDR2, MT1-MMP (MMP14) and BIK mRNAs in 58 breast cancer cell lines was obtained by interrogating the Broad-Novartis Cancer Cell Line Encyclopedia (CCLE) database.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) from three independent experiments. Statistical significance was assessed with Student's t-test, or with one-way ANOVA, followed by Tukey's multiple comparison test. p values lower than 0.05 were considered as significant (*p < 0.05; **p < 0.01; ***p < 0.001).
Funding Statement
This work was supported by grants from Ligue Contre le Cancer 2012 and 2016 (CCIR Grand-Est), 16ARC035 and FEDER/Emergence CELLnanoFLUO Program 2012 (Région Champagne-Ardenne), 12PRC120. Charles Saby is recipient of a doctoral fellowship from the French Ministry of Higher Education and Research. Erik Maquoi is a Research Associate from the Fund for Scientific Research – FNRS (Belgium).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to acknowledge the help Céline Charpentier for preparation and characterization of the different type I collagens. We also thank Dr. Nathanael Gray (Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, USA) for providing us DDR1-IN-1, and Dr. Catherine-Laure Tomasetto (IGBMC, Department of Functional Genomics and Cancer, Université de Strasbourg, Illkirch, France) for providing us the MT1-MMP antibody.
References
- [1].Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006;119(19):3901–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang WM, Kapyla J, Puranen JS, et al.. alpha 11beta 1 integrin recognizes the GFOGER sequence in interstitial collagens. J Biol Chem. 2003;278(9):7270–7. [DOI] [PubMed] [Google Scholar]
- [4].Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. 2014;310:39–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Carafoli F, Hohenester E. Collagen recognition and transmembrane signalling by discoidin domain receptors. Biochim Biophys Acta. 2013;1834(10):2187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vogel W, Gish GD, Alves F, et al.. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1(1):13–23. [DOI] [PubMed] [Google Scholar]
- [7].Yancik R, Ries LA. Cancer in older persons: an international issue in an aging world. Semin Oncol. 2004;31(2):128–36. [DOI] [PubMed] [Google Scholar]
- [8].Kohler BA, Sherman RL, Howlader N, et al.. Annual Report to the Nation on the Status of Cancer, 1975–2011. Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J Natl Cancer Inst. 2015;107(6):djv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Assent D, Bourgot I, Hennuy B, et al.. A Membrane-Type-1 Matrix Metalloproteinase (MT1-MMP) – Discoidin Domain Receptor 1 Axis Regulates Collagen-Induced Apoptosis in Breast Cancer Cells. PLoS One. 2015;10(3):e0116006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hotary KB, Allen ED, Brooks PC, et al.. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114(1):33–45. [DOI] [PubMed] [Google Scholar]
- [11].Maquoi E, Assent D, Detilleux J, et al.. MT1-MMP protects breast carcinoma cells against type I collagen-induced apoptosis. Oncogene. 2012;31(4):480–93. [DOI] [PubMed] [Google Scholar]
- [12].Wall SJ, Werner E, Werb Z, et al.. Discoidin domain receptor 2 mediates tumor cell cycle arrest induced by fibrillar collagen. J Biol Chem. 2005;280(48):40187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Iwai LK, Payne LS, Luczynski MT, et al.. Phosphoproteomics of collagen receptor networks reveals SHP-2 phosphorylation downstream of wild-type DDR2 and its lung cancer mutants. Biochem J. 2013;454(3):501–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Simm A, Muller B, Nass N, et al.. Protein glycation – Between tissue aging and protection. Exp Gerontol. 2015;68:71–5. [DOI] [PubMed] [Google Scholar]
- [15].Monnier VM, Mustata GT, Biemel KL, et al.. Cross-linking of the extracellular matrix by the maillard reaction in aging and diabetes: an update on “a puzzle nearing resolution”. Ann N Y Acad Sci. 2005;1043:533–44. [DOI] [PubMed] [Google Scholar]
- [16].Ait-Belkacem D, Guilbert M, Roche M, et al.. Microscopic structural study of collagen aging in isolated fibrils using polarized second harmonic generation. J Biomed Opt. 2012;17(8):080506–1. [DOI] [PubMed] [Google Scholar]
- [17].Wilson SL, Guilbert M, Sule-Suso J, et al.. A microscopic and macroscopic study of aging collagen on its molecular structure, mechanical properties, and cellular response. FASEB J. 2014;28(1):14–25. [DOI] [PubMed] [Google Scholar]
- [18].Guilbert M, Roig B, Terryn C, et al.. Highlighting the impact of aging on type I collagen: label-free investigation using confocal reflectance microscopy and diffuse reflectance spectroscopy in 3D matrix model. Oncotarget. 2016;7(8):8546–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Panwar P, Butler GS, Jamroz A, et al.. Aging-associated modifications of collagen affect its degradation by matrix metalloproteinases. Matrix Biol. 2017. [DOI] [PubMed] [Google Scholar]
- [20].Panwar P, Lamour G, Mackenzie NC, et al.. Changes in Structural-Mechanical Properties and Degradability of Collagen during Aging-associated Modifications. J Biol Chem. 2015;290(38):23291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Saby C, Buache E, Brassart-Pasco S, et al.. Type I collagen aging impairs discoidin domain receptor 2-mediated tumor cell growth suppression. Oncotarget. 2016;7:24908–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bredfeldt JS, Liu Y, Conklin MW, et al.. Automated quantification of aligned collagen for human breast carcinoma prognosis. J Pathol Inform. 2014;5(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].McConnell JC, O'Connell OV, Brennan K, et al.. Increased peri-ductal collagen micro-organization may contribute to raised mammographic density. Breast Cancer Res. 2016;18(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Manley PW, Drueckes P, Fendrich G, et al.. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochimica et biophysica acta. 2010;1804(3):445–53. [DOI] [PubMed] [Google Scholar]
- [25].Sabeh F, Ota I, Holmbeck K, et al.. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167(4):769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Holmbeck K, Bianco P, Yamada S, et al.. MT1-MMP: a tethered collagenase. J Cell Physiol. 2004;200(1):11–9. [DOI] [PubMed] [Google Scholar]
- [27].Wolf K, Wu YI, Liu Y, et al.. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9(8):893–904. [DOI] [PubMed] [Google Scholar]
- [28].Maquoi E, Peyrollier K, Noel A, et al.. Regulation of membrane-type 1 matrix metalloproteinase activity by vacuolar H+-ATPases. Biochem J. 2003;373(Pt 1):19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Verzijl N, DeGroot J, Thorpe SR, et al.. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275(50):39027–31. [DOI] [PubMed] [Google Scholar]
- [30].Snedeker JG, Gautieri A. The role of collagen crosslinks in ageing and diabetes – the good, the bad, and the ugly. Muscles Ligaments Tendons J. 2014;4(3):303–8. [PMC free article] [PubMed] [Google Scholar]
- [31].Li Q, Jiang Q, Uitto J. Ectopic mineralization disorders of the extracellular matrix of connective tissue: molecular genetics and pathomechanisms of aberrant calcification. Matrix Biol. 2014;33:23–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Levental KR, Yu H, Kass L, et al.. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Castello-Cros R, Cukierman E. Stromagenesis during tumorigenesis: characterization of tumor-associated fibroblasts and stroma-derived 3D matrices. Methods Mol Biol. 2009;522:275–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cukierman E, Bassi DE. Physico-mechanical aspects of extracellular matrix influences on tumorigenic behaviors. Semin Cancer Biol. 2010;20(3):139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Oskarsson T. Extracellular matrix components in breast cancer progression and metastasis. Breast. 2013;22(Suppl 2):S66–72. [DOI] [PubMed] [Google Scholar]
- [36].El Azreq MA, Naci D, Aoudjit F. Collagen/beta1 integrin signaling up-regulates the ABCC1/MRP-1 transporter in an ERK/MAPK-dependent manner. Mol Biol Cell. 2012;23(17):3473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Henriet P, Zhong ZD, Brooks PC, et al.. Contact with fibrillar collagen inhibits melanoma cell proliferation by up-regulating p27KIP1. Proc Natl Acad Sci U S A. 2000;97(18):10026–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wall SJ, Zhong ZD, DeClerck YA. The cyclin-dependent kinase inhibitors p15INK4B and p21CIP1 are critical regulators of fibrillar collagen-induced tumor cell cycle arrest. J Biol Chem. 2007;282(33):24471–6. [DOI] [PubMed] [Google Scholar]
- [39].Wozniak MA, Modzelewska K, Kwong L, et al.. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692(2–3):103–19. [DOI] [PubMed] [Google Scholar]
- [40].Badaoui M, Mimsy-Julienne C, Saby C, et al.. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10.1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget. 2018;9:24653–24671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tulasne D, Foveau B. The shadow of death on the MET tyrosine kinase receptor. Cell Death Differ. 2008;15(3):427–34. [DOI] [PubMed] [Google Scholar]
- [42].Kim HG, Tan L, Weisberg EL, et al.. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem Biol. 2013;18(10):2145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fu HL, Sohail A, Valiathan RR, et al.. Shedding of discoidin domain receptor 1 by membrane-type matrix metalloproteinases. J Biol Chem. 2013;288(17):12114–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Koh M, Woo Y, Valiathan RR, et al.. Discoidin domain receptor 1 is a novel transcriptional target of ZEB1 in breast epithelial cells undergoing H-Ras-induced epithelial to mesenchymal transition. Int J Cancer. 2015;136(6):E508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411(6835):375–379. [DOI] [PubMed] [Google Scholar]
- [46].de Magalhães JP. How ageing processes influence cancer. Nat Rev Cancer. 2013;13(5):357–365. [DOI] [PubMed] [Google Scholar]
- [47].Garnotel R, Rittie L, Poitevin S, et al.. Human blood monocytes interact with type I collagen through alpha x beta 2 integrin (CD11c-CD18, gp150-95). J Immunol. 2000;164(11):5928–34. [DOI] [PubMed] [Google Scholar]
- [48].Buache E, Thai R, Wendling C, et al.. Functional relationship between matrix metalloproteinase-11 and matrix metalloproteinase-14. Cancer Med. 2014;3:1197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]