

Model‐informed drug development (MIDD) has recently garnered much attention as a potential enabler of efficient drug development. In their recent article, Jain et al. 1 describe barriers and opportunities to achieve the full potential of MIDD. The authors point out well‐accepted and promising new applications of MIDD in drug development and regulatory evaluation. There is arguably a greater appreciation of the benefits and promise of MIDD now more than ever. As noted by the authors, this is reflected by the inclusion of MIDD‐related performance goals in the latest congressionally reauthorized Prescription Drug User Fee Act (PDUFA) VI. 2 Against this backdrop, the key question is how we capitalize on this unique opportunity to advance MIDD.
DEFINING IMPACT
Historically, the “impact” of MIDD has been demonstrated through: (i) highlighting a single, instructive drug development or regulatory review case, (ii) aggregating and disseminating the regulatory review experience with MIDD, and (iii) collating and publishing the return on investment/drug development impact of MIDD by the drug development community.3, 4, 5, 6 These approaches anecdotally, if not systematically, highlight the range of issues for which MIDD approaches have been successfully used. Jain et al.1 recommend continuing this approach by focusing on a paradigm in which determination of efficacy can be accomplished with “a single efficacy trial with modeling and simulation (M&S) providing additional primary “confirmatory evidence.” Although we appreciate the enthusiasm around MIDD for this purpose, there are several reasons why we find an emphasis on MIDD as a replacement to clinical trials potentially problematic.
We have noticed a recent trend in which replacement of a clinical trial by in silico approaches has been lauded as the ultimate goal of MIDD. From our standpoint, the goal of MIDD is to derisk drug development and public health decision making and robustly inform optimal pharmacotherapy. To that end, MIDD can be used for critical company decisions about if and how to develop an investigational new drug. MIDD can also be used in the regulatory context to inform clinical trial planning, drive approvability and labeling decisions, provide supportive (even pivotal) evidence of effectiveness and safety in difficult‐to‐study scenarios, and obviate the need for unnecessary clinical trials. Although we agree that, in some cases, MIDD can (and has) been used as a primary basis for efficacy determination, we also caution against placing disproportionate emphasis on replacing an efficacy trial with M&S as a paramount goal of MIDD. This valuation of MIDD's ultimate utility risks de‐emphasizing the breadth of opportunities in which MIDD can be leveraged. We also submit that overemphasizing replacement of clinical trials can have the unintended consequence of reinforcing what we feel is an artificial distinction between the importance of “pivotal” and “supportive” evidence of effectiveness. Perhaps most obviously, defining high impact as whether a regulatory requirement for a clinical trial is waived can set an unrealistically high standard against which the success of MIDD is measured. Given that multiple efficacy trials are often conducted in different clinical contexts (e.g., different stages of the same disease), one important impact of MIDD can be in the aiding of more informative clinical trial designs that not only elucidate risk/benefit balance but also inform therapeutic individualization. Notwithstanding, we appreciate the point made by Jain et al.1 and think a clear articulation of when M&S can stand in for an efficacy trial is a worthy topic for public discussion.
AN INTEGRATIVE APPROACH TO ADVANCING MIDD
We agree with the authors that this is an important time to “unleash the full potential of model‐informed drug development.” To that end, we feel a holistic, integrative approach is required, and the commitments laid out in PDUFA VI provide the community with several opportunities (Figure 1).
Figure 1.
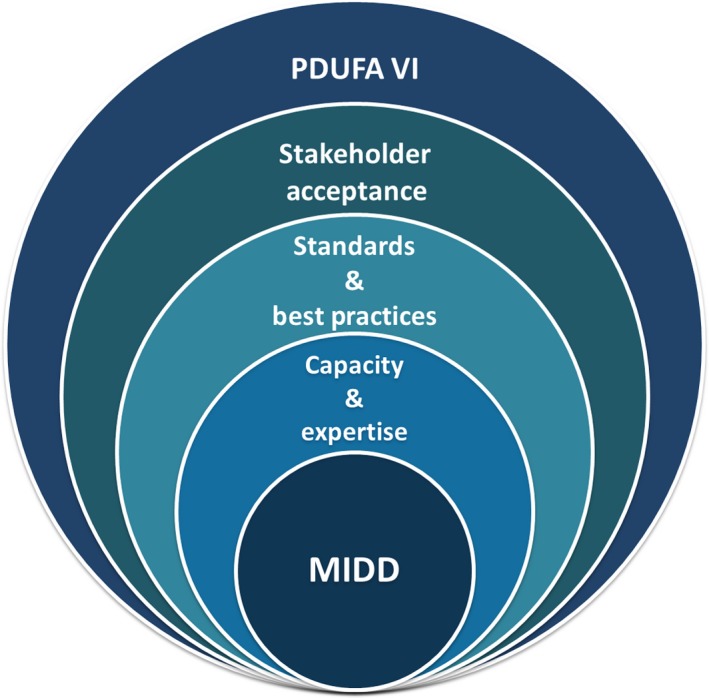
An integrative approach for advancing model‐informed drug development (MIDD) under Prescription Drug User Fee Act (PDUFA) VI. MIDD can be seen as foundational to efficient and effective drug development and regulatory evaluation of small molecule drugs and biological products. To advance more widespread and predictable application, MIDD requires an adequate staff capacity and expertise, community‐accepted standards and best practices, and multistakeholder acceptance eyond technical experts. The commitments laid out under PDUFA VI provide an opportunity to achieve these goals in a holistic and integrated manner.
Creating an environment that increases stakeholder acceptance of MIDD approaches
The importance of stakeholder familiarity with and acceptance of MIDD approaches cannot be overstated given the multidisciplinary nature of both drug development and regulatory evaluation. This was also highlighted in a recent survey of clinical pharmacology and pharmacometrics scientists across pharmaceutical industry and regulatory agencies.7 In our experience, successful application of MIDD occurs when there is clarity around the issue to be addressed with MIDD; there is sufficient opportunity to fully discuss the proposed approach, including its putative value and limitations; key decision makers have ample opportunity for dialogue within the setting of the multidisciplinary team. Essentially, rate‐limiting steps to uptake include insufficient opportunity for real‐time engagement between sponsors and regulators on the merits and constraints of a particular MIDD strategy in a specific drug development context. Some of the commitments under PDUFA VI provide powerful mechanisms to overcome this barrier. For example, the MIDD Meeting Pilot Program provides an avenue for direct engagement between drug developers and the US Food and Drug Administration (FDA) review teams on product‐specific issues in which MIDD approaches are being proposed.8 Learnings from the pilot program will undoubtedly allow us to better align future opportunities for direct interaction during all stages of drug development and increase consistency and transparency of the scenarios in which the FDA supports the use of MIDD.
Developing standards and best practices that lead to consistent application and evaluation
Community convergence on best practices for model development, qualification, and application is critical, as highlighted at a recent FDA advisory committee meeting.9 Development of standard methodologies and reporting practices can also increase efficiency during the regulatory interaction. The commitments to convene public workshops, publish guidances, and develop policy/procedures under PDUFA VI provide opportunities to realize this goal. Public workshops will allow us to explore past practices, current state, and aspirational goals for MIDD in various scenarios. Subject matter experts and nontechnical experts alike are afforded opportunities to come together and identify needs based on collective drug development, regulatory, scientific, and therapeutic area experience. In turn, these exchanges can inform standards and best practices for critical MIDD methodologies. In terms of policy development, the FDA is actively revising relevant MIDD guidances and expects MIDD‐related topics to be ripe for international harmonization in the coming years.
Increasing capacity and expertise to address growing demands and innovation
Historically, the regulatory review and application of MIDD approaches have been more frequent in the new drug application or biologic license application evaluation stage compared with during the drug development stage (i.e., investigational new drug) stage. Additionally, even during new drug application or biologic license application reviews, the FDA staff is under significant, often expedited, review timelines to address complex MIDD issues. Educational initiatives targeting nontechnical expert regulatory scientists have been sparse, limiting integration of MIDD principles into some therapeutic areas. Finally, development of standards and policies, as outlined above, has been opportunistic at best. These realities are, in part, byproducts of staff capacity limitations. Under PDUFA VI, the FDA will receive a nominal but important increase in new staff to work on MIDD reviews. The FDA has also committed to continuing education of both resident experts and nonexperts alike. It will be important to invest in comprehensive training programs, data standardization, and automation of analysis and reporting where possible, as a means for increasing staff capacity to handle the influx of increasingly complex MIDD applications throughout drug development. Freely available, widely accessible community resources for education and training would be greatly enabling, especially as the field contends with exciting and rapidly evolving methodologies and approaches (e.g., quantitative systems pharmacology, machine learning, and artificial intelligence). With an eye to the future, development of expertise on these fronts could be critical to understand how these approaches might be best incorporated to inform drug development.
ADDITIONAL CONSIDERATIONS
We believe the above comprehensive approach can help move toward an advanced vision for MIDD. We would also like to address several other important observations made by Jain et al.1 that may further the field. First, it is important to continuously calibrate expectations about the potential of MIDD as we gain more experience. For example, although we share the enthusiasm of Jain et al.1 about the ability of MIDD to result in a requirement of a single “registration” trial, we have learned through the lifecycle of scientific advances, such as pharmacogenomics, physiologic‐based pharmacokinetic modeling and simulation, predictive safety, and drug transporters, that the articulated promise of these sciences should not outsize the state of the science itself. An honest acknowledgment of the constraints of the science and how they can be overcome would add to the credibility of MIDD and serve as a driver to further implementation.
Jain et al.1 also highlight “…limited…familiarity with M&S approaches by medical and biostatistical experts often leads to situations where the contributions of M&S are not judged based on the value offered…” Although we support the need for socialization of MIDD concepts among multidisciplinary teams, we must also acknowledge our role (i.e., the role of the clinical pharmacology/pharmacometrics community), which may have contributed to heterogeneous uptake of MIDD approaches. We can ask ourselves if the existential drive to demonstrate “impact” has led to practices that limit our colleagues’ enthusiasm for MIDD approaches. For example, have we been reasonable and nuanced with our expectations for the role of M&S in decision making? Are we routinely transparent with our underlying assumptions? Have we communicated and implemented community‐wide standards for model development, validation, and application? Have we emphasized the importance of and committed resources to communication across disciplinary lines? Much can be done on our end to appropriately and responsibly advance the application of MIDD strategies in drug development and regulatory review.
We are approaching nearly 2 decades of experience in demonstrating the relevance and value of MIDD. With exciting innovations on the horizon and institutional support for MIDD across many sectors, we are at an important moment in which synergies can be brought to bear to achieve consistent and relevant application of MIDD for patient and societal benefit.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
Disclaimer
The opinions expressed in this article are those of the authors and should not be interpreted as the position of the US Food and Drug Administration.
References
- 1. Jain, L . et al PDUFA VI: it's time to unleash the full potential of model informed drug development. CPT Pharmacometrics Syst. Pharmacol. (2018), 1‐4; doi: 10.1002/psp4.12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2018 Through 2022. <https://www.fda.gov/downloads/forindustry/userfees/prescriptiondruguserfee/ucm511438.pdf>.
- 3. Huang, S. et al The utility of modeling and simulation in drug development and regulatory review. J. Pharm. Sci. 102, 2914–2923 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Lee, J.Y. et al Impact of pharmacometric analyses on new drug approval and labelling decisions: a review of 198 submissions between 2000 and 2008. Clin. Pharmacokinet. 50, 627–635 (2011). [DOI] [PubMed] [Google Scholar]
- 5. Marshall, S et al Good practices in model‐informed drug discovery and development (MID3): practice, application and documentation. CPT Pharmacometrics Syst. Pharmacol. 5, 93–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lalonde, R.L. et al Model‐based drug development. Clin. Pharmacol. Ther. 82, 21–32 (2007). [DOI] [PubMed] [Google Scholar]
- 7. Marshall, S . et al Model‐informed drug discovery and development (MID3): current industry good practice & regulatory expectations and future perspectives. CPT Pharmacometrics Syst. Pharmacol. (2018) doi: 10.1002/psp4.12372 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Model‐Informed Drug Development Pilot Program . <https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm600311.htm>.
- 9. Summary Minutes of the Advisory Committee for Pharmaceutical Science and Clinical Pharmacology Meeting 2017. <https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM559417.pdf>.