

ABSTRACT
Peptide vaccines derived from tumour-associated antigens have been used as an immunotherapeutic approach to induce specific cytotoxic immune response against tumour. We previously identified that MAGED4B and FJX1 proteins are overexpressed in HNSCC patients; and further demonstrated that two HLA-A2-restricted 9–11 amino acid peptides derived from these proteins were able to induce anti-tumour immune responses in vitro independently using PBMCs isolated from these patients. In this study, we evaluated the immunogenicity and efficacy of a dual-antigenic peptide vaccine (PV1), comprised of MAGED4B and FJX1 peptides in HNSCC patients. We first demonstrated that 94.8% of HNSCC patients expressed MAGED4B and/or FJX1 by immunohistochemistry, suggesting that PV1 could benefit the majority of HNSCC patients. The presence of pre-existing MAGED4B and FJX1-specific T-cells was detected using a HLA-A2 dimer assay and efficacy of PV1 to induce T-cell to secrete cytotoxic cytokine was evaluated using ELISPOT assay. Pre-existing PV1-specific T-cells were detected in all patients. Notably, we demonstrated that patients’ T-cells were able to secrete cytotoxic cytokines upon exposure to target cells expressing the respective antigen post PV1 stimulation. Furthermore, patients with high expression of MAGED4B and FJX1 in their tumours were more responsive to PV1 stimulation, demonstrating the specificity of the PV1 peptide vaccine. Additionally, we also demonstrated the expression of MAGED4B and FJX1 in breast, lung, colon, prostate and rectal cancer suggesting the potential use of PV1 in these cancers. In summary, PV1 could be a good vaccine candidate for the treatment of HNSCC patients and other cancers expressing these antigens.
Keywords: peptide vaccine, immunotherapy, MAGED4B, FJX1, head and neck cancer
Introduction
Globally, head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer with more than 1 million cases diagnosed annually.1 The majority of the cases occur n developing countries, where therapeutic options are limited to surgery, radiotherapy and chemotherapy2-4 with overall 5-year survival rate of 66%.5 Furthermore, locoregional recurrence is common in HNSCC patients, and about 20–30% of the patients will experience distant metastasis where treatment options become very limited.6
Recently, two monoclonal antibodies (pembrolizumab and nivolumab) targeting programmed cell death 1 (PD-1), an immune checkpoint protein, were approved for the treatment of chemo-refractory HNSCCs. Whilst these offer significant improvements to the treatment outcome for HNSCC patients, it benefit only ~18% of HNSCC patients7,8 under-scoring the urgent need for strategies to improve the overall response rate in HNSCC patients.
The immune system ensures a check and balance between immune activation and suppression.9 Blocking immune checkpoint alone might not be sufficient to fully re-activate the dampened immune system in cancer patients, as reflected by percentage of patients responding to immune checkpoint inhibitors.7,8 A rational way of improving could be the addition of immunotherapeutic approaches for example antigen-specific peptide vaccine to enhance the activity of existing T-cell repertoire. Peptide vaccines derived from immunogenic tumour antigens have the potential to stimulate cytotoxic immune responses against tumour cells bearing the same antigens. There are several advantages in using peptide vaccines over other immunotherapies including the ease of peptide synthesis, the ability to induce T-cell response and the safety of peptide vaccines that have been demonstrated in many clinical trials.10–12
Our laboratory has demonstrated melanoma antigen family D, 4B (MAGED4B) and four-jointed box 1 (FJX1) are two important antigens that play a functional role in carcinogenesis. MAGED4B promotes migration, proliferation and conferred resistant to apoptosis in vitro,13 whereas FJX1 enhances proliferation, invasion and increases metastatic ability in vitro.14 Recently, we showed that MAGED4B and FJX1 peptide induces anti-tumour immune responses in vitro using PBMCs collected from oral squamous cell carcinoma (OSCC) and nasopharyngeal carcinoma (NPC) patients respectively and elicits anti-tumour immune responses against MAGED4B or FJX1 overexpressing cell lines.15,16
As combination of immunogenic peptides from multiple tumour antigens could overcome the limitation of single antigen peptide vaccine, enhancing the immune response to eradicate cancer cells. In this study, we investigated the immunogenicity and efficacy of a dual-antigenic peptide vaccine that is comprised of both MAGED4B and FJX1 peptides (named as PV1), using PBMCs collected from OSCC and NPC patients, and compared their efficacy to respective single-peptide.
Results
Patient demographics
Peripheral blood from 41 HNSCC patients were collected. Of these, ~49% (20/41) were HLA-A2 positive and were used for subsequent analysis (Table 1A). A total of 80% (16/20) of these patients were from advanced disease stages (stage III and stage IV) reflecting the disease stage of patients who are typically diagnosed in Malaysia, and the demographics of these patients are shown in Table 1B. Samples from 15 patients were subjected to the dimer assay while 16 were subjected to the ex vivo and cytotoxic ELISPOT assays, other samples were excluded due to insufficient PBMCs or sub-optimal sample quality.
Table 1.
(A) Disease type and HLA-A2 status of patients recruited in this study. (B) Demographic data of HLA-A2 patients.
| Variables | Sample size, n (%) |
|---|---|
| A | |
| Total patients | 41 (100.0) |
| Disease | |
| NPC | 29 (70.7) |
| OSCC | 12 (29.3) |
| HLA type | |
| A2 | 20 (48.8) |
| Non A2 | 21 (51.2) |
| B | |
| Disease | |
| NPC | 16 (80.0) |
| OSCC | 4 (20.0) |
| Age (median, 48.5; range, 30–67) | |
| ≤ 40 | 3 (15.0) |
| > 40 | 17 (85.0) |
| Ethnicity | |
| Chinese | 16 (80.0) |
| Malay | 2 (10.0) |
| Indian | 2 (10.0) |
| Gender | |
| Male | 17 (85.0) |
| Female | 3 (15.0) |
| Overall staging | |
| Early stage (Stage I and II) | 4 (20.0) |
| Late stage (Stage III and IV) | 16 (80.0) |
MAGED4B and FJX1 are tumour antigens overexpressed in HNSCC
We have previously showed that MAGED4B and FJX1 are overexpressed in OSCC and NPC respectively13,17,18 and their expression is minimal or negligible in normal organs (Figure 1A). To demonstrate that these antigens are also relevant to HNSCC specimens other than those from our patients, RNAseq data of 43 HNSCC patients and their matched normal from the TCGA database19 were analysed. We found that the majority (31/43) of these patients had significantly elevated levels of FJX1 (Figure 1B, p < 0.001) and about 40% (17/43) of the cohort had elevated levels of MAGED4B (Figure 1C, p < 0.001) when compared to their matched normal.
Figure 1.
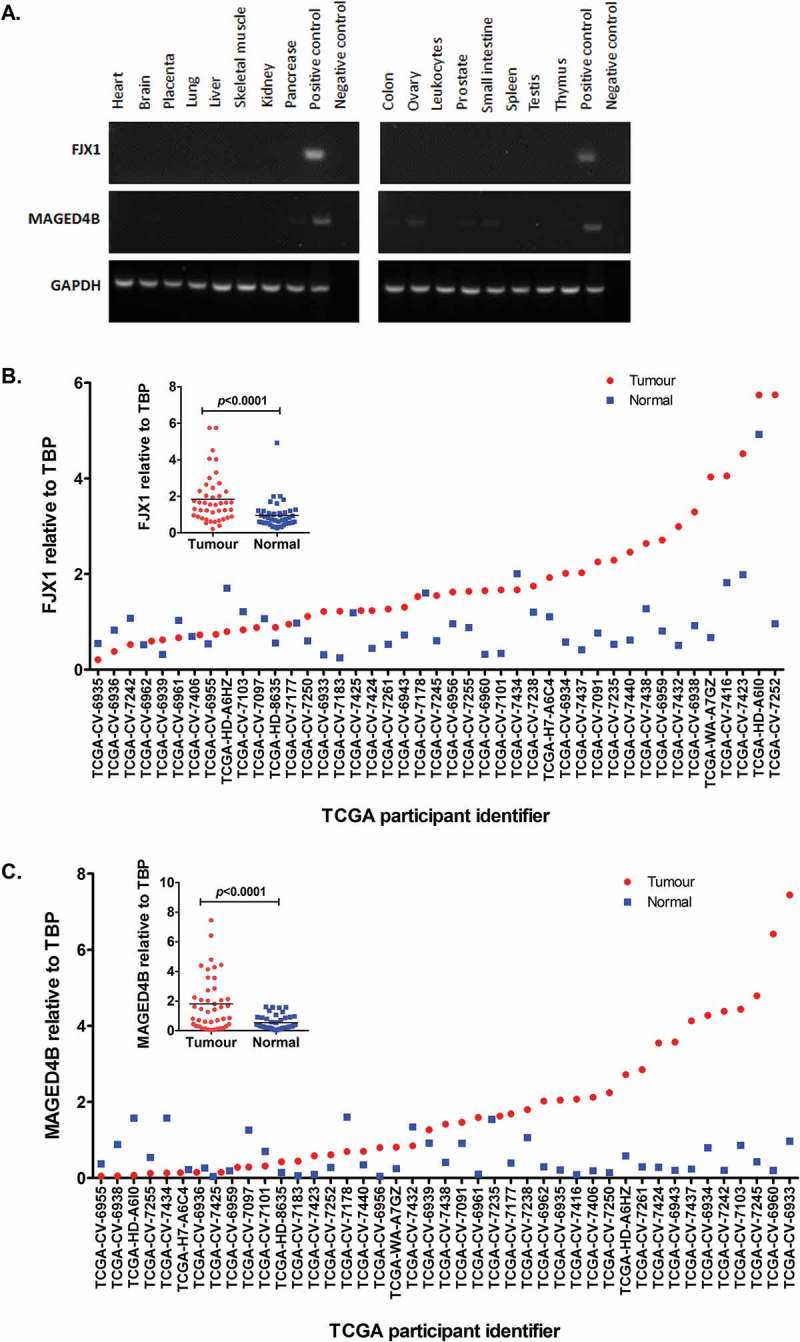
Levels of MAGED4B and FJX1 in normal human organs and HNSCC.
(A) Semi quantitative PCR using commercially available normal human cDNA template (Human MTCTM Panel I and II) showed that both MAGED4B and FJX1 were expressed at low/negligible level in various human organs. cDNA from HeLa/T expressing FJX1 was used as positive control for FJX1 and cDNA from ORL-195 expressing MAGED4B was used as positive control for MAGED4B. (B) FJX1 expressions are elevated at transcriptomic levels in more than 60% of HNSCC cancer samples (n = 43), while C, MAGED4B expressions are elevated in about 40% in the HNSCC samples when compared to the matched normals. The relative expression of MAGED4B and FJX1 in all 43 HNSCC samples were normalised against TBP (TATA-Box Binding Protein).
We also validated the expression of MAGED4B and FJX1 on commercially available TMAs consisting of HNSCC tissue specimens by IHC. The expression of MAGED4B and FJX1 were detected in 94.8% and 89.7% of HNSCC specimens respectively and the presence of MAGED4B and FJX1 expression is independent of disease stage (Table 2A). Representative images of intensity for IHC staining is as shown in Figure 2. Although FJX1 was reported to be secreted protein,20 the expression of FJX1 protein can also be detected in the cytoplasmic of cells using IHC, as reported previously.21
Table 2.
Expression of MAGED4B and FJX1 in: (A) HNSCC and (B) 5 most common cancers worldwide (breast, colon, lung, prostate and rectal cancers).
| HNSCC | MAGED4B | FJX1 | MAGED4B and/or FJX1 | MAGED4B and FJX1 | Both negative |
|---|---|---|---|---|---|
| A | |||||
| Stage I (n = 15) | 15 (100.0%) | 14 (93.3%) | 15 (100.0%) | 14 (93.3%) | 0 (0.0%) |
| Stage II (n = 22) | 22 (100.0%) | 20 (90.9%) | 22 (100.0%) | 20 (90.9%) | 0 (0.0%) |
| Stage III (n = 15) | 15 (100.0%) | 14 (93.3%) | 15 (100.0%) | 14 (93.3%) | 0 (0.0%) |
| Stage IV (n = 15) | 14 (93.3%) | 14 (93.3%) | 14 (93.3%) | 14 (93.3%) | 1 (6.7%) |
| Unknown stage (n = 30) | 26 (86.7%) | 25 (83.3%) | 26 (86.7%) | 25 (83.3%) | 4 (13.3%) |
| Total (n = 97) |
92 (94.8%) |
87 (89.7%) |
92 (94.8%) |
87 (89.7%) |
5 (5.2%) |
| Cancer type |
MAGED4B |
FJX1 |
MAGED4B and/or FJX1 |
MAGED4B and FJX1 |
Both negative |
| B | |||||
| Breast | 41/41(100.0%) | 36/41 (87.8%) | 41/41 (100.0%) | 36/41 (87.8%) | 0/41 (0.0%) |
| Colon | 25/30 (83.3%) | 15/30 (50.0%) | 27/29 (93.1%) | 11/29 (37.9%) | 2/29 (6.9%) |
| Lung | 42/42 (100.0%) | 37/48 (77.1%) | 42/42 (100.0%) | 32/42 (76.2%) | 0/42 (0.0%) |
| Prostate | 34/42 (81.0%) | 37/44 (84.1%) | 38/40 (95.0%) | 29/40 (72.5%) | 2/40 (5.0%) |
| Rectal | 13/16 (81.2%) | 12/14 (85.7%) | 13/14 (92.9%) | 11/14 (78.6%) | 1/14 (7.1%) |
| Total | 155/171 (90.6%) |
137/177 (77.4%) |
161/166 (97.0%) |
119/166 (71.7%) |
5/166 (3.0%) |
Figure 2.
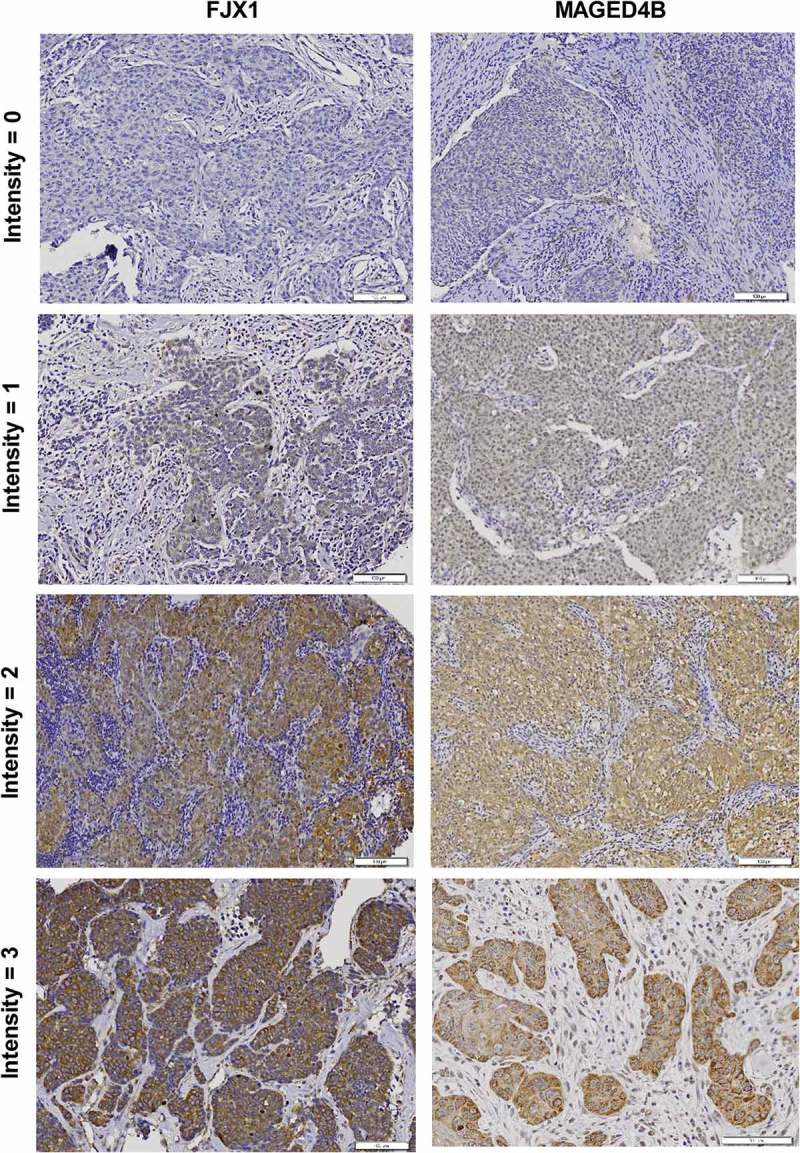
Expression of MAGED4B and FJX1 in HNSCC samples.
Representative images of intensity scores for IHC staining, ranging from negative (intensity = 0), to weak (intensity = 1) and strong (intensity = 2–3) staining.
MAGED4B and FJX1 proteins are overexpressed in several other cancer types
In order to investigate MAGED4B and FJX1 expression in other cancers and to identify potential patient cohorts that might benefit from PV1 therapy, we investigated the expression of MAGED4B and FJX1 using a set of TMA consist of 5 most common cancers worldwide (breast, lung, colon, prostate and rectal cancer). We show that more than 80% of patients in all five cancers expressed MAGED4B, and a similar observation was seen for FJX1 expression except for colon cancer patients where 50% were positive for FJX1 (Table 2B). When the expression of both MAGED4B and FJX1 were analysed collectively, more than 90% of breast, colon, lung, prostate and rectal tumour tissues had either MAGED4B or FJX1 expression (Table 2B). Consistently, western blot analysis on cell lines representing these 5 cancer types showed that MAGED4B expression is high in lung (A549) and prostate (PC3) cell lines, while the expression level is weaker in colorectal (HCT-116) and breast cancer cell lines (SKBR-3 and MCF-7). On the other hand, FJX1 was shown to be expressed in 5 of the 6 cell lines tested (Figure 3A).
Figure 3.
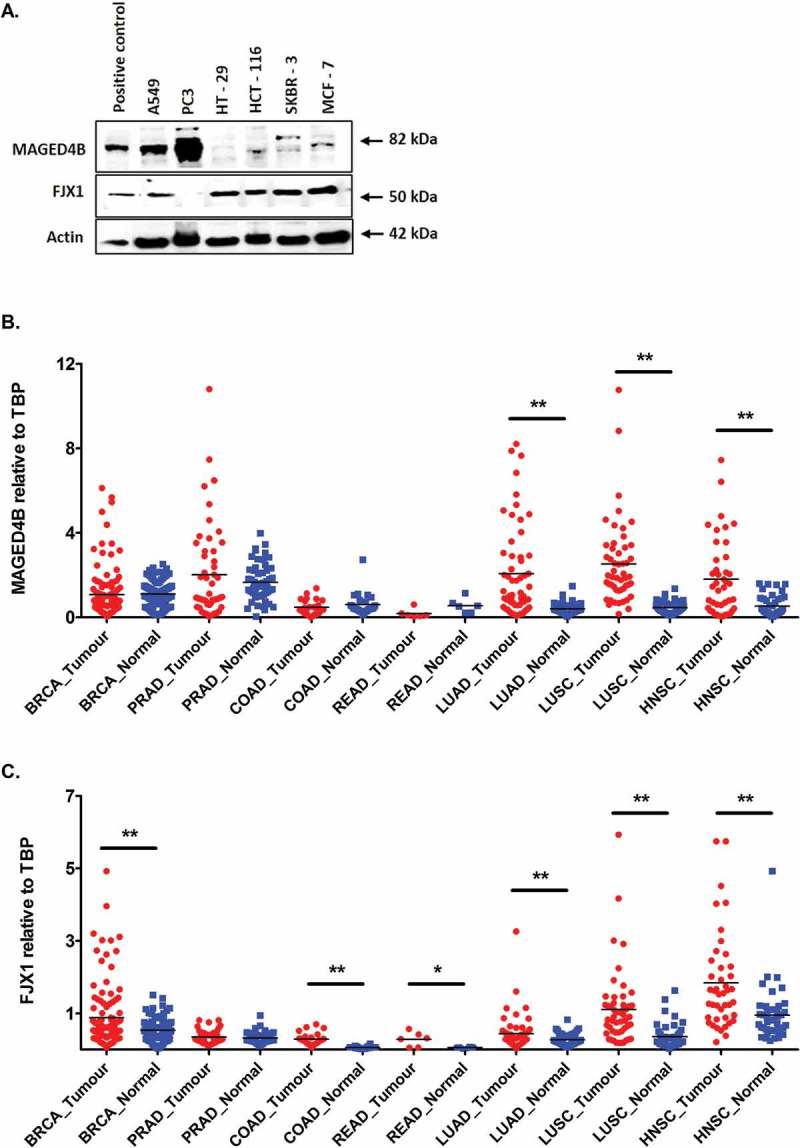
The expression of MAGED4B and FJX1 in 5 most common cancers.
(A) Western blot analysis showed the expression of 2 target antigens in cell lines including lung (A549), prostate (PC3), colorectal (HT-29, HCT-116) and breast (SKBR-3, MCF-7). Lysates from ORL-195 overexpressing MAGED4B and HeLa/T overexpressing FJX1 were used as positive controls for the western blot analyses. RNA-seq data for (B) MAGED4B and (C) FJX1 expression in 5 most common cancers comparing to head and neck cancer data sets derived from TCGA (BRCA: breast invasive carcinoma, PRAD: prostate adenocarcinoma, COAD: colon adenocarcinoma, READ: rectal adenocarcinoma, LUAD: lung adenocarcinoma, LUSC: lung squamous carcinoma, HNSC: head and neck squamous carcinoma). The symbol * denotes p value < 0.05, while ** denotes that p value < 0.001.
In addition, TCGA data set of matched tumour and normal controls from these 5 common cancers showed that mRNA levels of MAGED4B is higher in prostate adenocarcinoma (PRAD, p = 0.188), lung adenocarcinoma (LUAD, p < 0.001) and lung squamous carcinoma (LUSC, p < 0.001) but remained similar levels in breast, colorectal and renal cancer when compared to respective matched normal samples (Figure 3B). While mRNA levels for FJX1 was seen elevated in breast invasive carcinoma (BRCA, p < 0.001), colon adenocarcinoma (COAD, p < 0.001), rectal adenocarcinoma (READ, p = 0.036), lung adenocarcinoma (LUAD, p < 0.001) and lung squamous carcinoma (LUSC, p < 0.001) but remained similar levels in prostate adenocarcinoma when compared to matched normal samples (Figure 3C). Our data, together with RNAseq information from TCGA suggesting that these cancers expressing MAGED4B/FJX1 could potentially be targeted by PV1.
PV1-specific CD8 T-cells are present in HNSCC patients
Since both MAGED4B and FJX1 were overexpressed in HNSCC patients, we hypothesised that MAGED4B- and/or FJX1- specific CD8 + T-cells would also be present in the peripheral blood of these patients. Using the dimer assay, we determined the percentage of CD8 + T-cells that were able to recognise the PV1 peptides (FJX1 and/or MAGED4B peptides). We demonstrated the presence of FJX1- and/or MAGED4B-specific CD8 + T-cells in all analysed HNSCC samples (15/15), ranging from 0.4%-22.1% (mean = 7.1%) after subtracting negative control values obtained with mock-peptide loaded dimer (Figure 4). Representative plot of gating strategy for flow cytometry is shown in Supplementary Fig. S1. The average FJX1 and/or MAGED4B-specific T-cell population detected inherently in patients was higher (7.1%) compared to single FJX1 (2.6%; p < 0.001) or MAGED4B-specific T-cell (4.6%; p = 0.177) population, demonstrating stronger immunogenic responses when both peptides were used in combination. T-cell populations recognizing the positive control, FluM peptide-loaded dimer was significantly higher when compared to the mock-loaded dimer control (p < 0.001).
Figure 4.
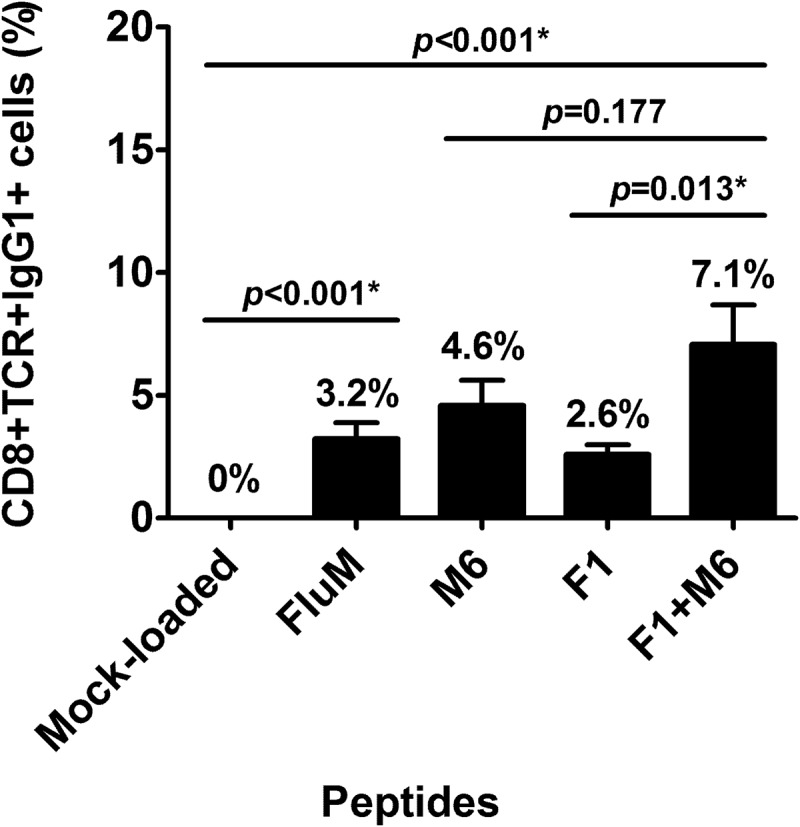
PV1 is immunogenic.
The graph shows the average population of antigen-specific T-cells that recognise the peptide-loaded dimers (CD8+ TCR+ IgG1+ %), after subtracting mock-peptide loaded control. Dimer loaded with F1, M6 and PV1 peptides can be recognised by the inherent T-cells from HNSCC patients where PV1 peptides can be recognised by higher population of T-cells compared to single peptide (F1 or M6) alone.
PV1 induces secretion of cytotoxic cytokines from HNSCC patients’ T-cells
Next, we sought to determine whether stimulation with the PV1 peptide vaccine would be able to increase the number of peptide-specific CD8 + T-cells in vitro. Direct exposure of fresh PBMC with the peptides in an ex vivo ELISPOT assay revealed that, the population of CD8 + T-cells that can secrete IFNγ and granzyme B were low (Figure 5A and 5B). PBMC samples (n = 16) were then stimulated with either M6, F1 or PV1, and the expanded T-cells were co-cultured with target cells expressing MAGED4B (195M) or FJX1 (C666.1F) in a cytotoxic ELISPOT assay. As seen in Figure 5A and 5B, the expanded peptide-specific T-cells showed increased cytokine secretion for both IFNγ and granzyme B compared to the cytokine secreting cells in the ex vivo ELISPOT assay. Overall, T-cells activated by either M6, F1, PV1 or FluM peptides demonstrated 2.4 to 39.7-fold increment (Figure 5C and 5D) of cytokine secretion in the cytotoxic ELISPOT assay when compared to T-cells that were not stimulated with peptides, suggesting of the capability of PV1 peptide vaccine in stimulating and expanding the precursor antigen specific CD8 T-cells present in these patients.
Figure 5.
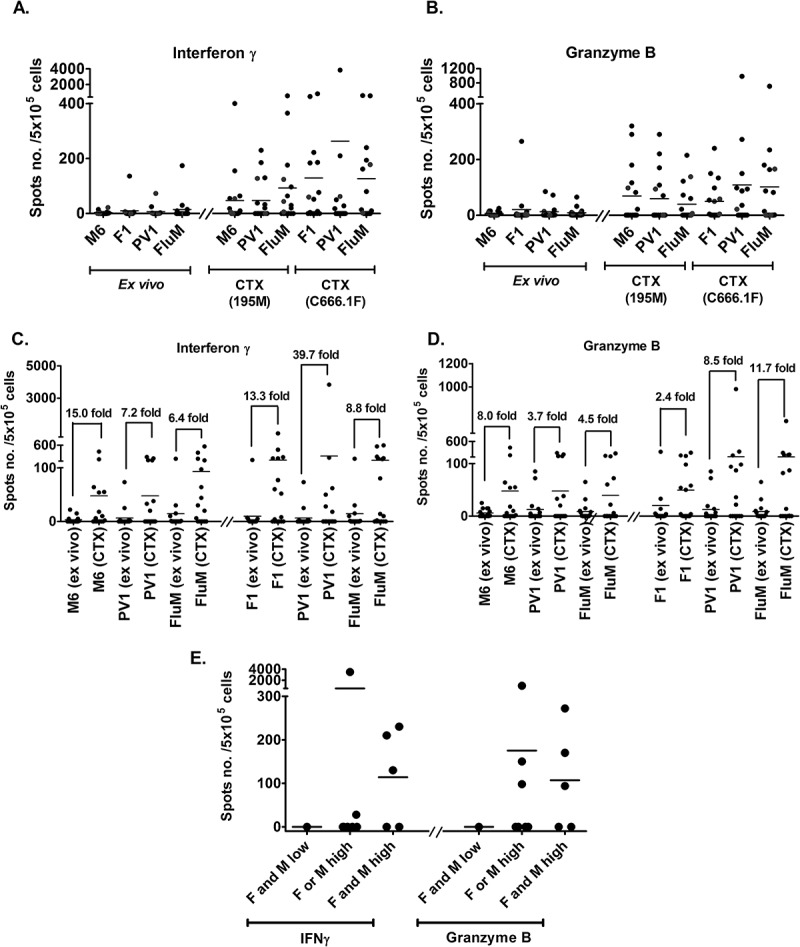
In vitro stimulation with PV1 peptides increases cytokine secretion in PMBC obtained from HNSCC patients (n = 16).
The graphs show the: (A) IFNγ and (B) granzyme B secretion from PBMC of patients in the ex vivo and cytotoxic (CTX) ELISPOT assays. Prior to peptide vaccine stimulation (ex vivo), inherent T-cell response against M6, F1 and PV1 is low. The level of cytokine secretion increased after the T-cells were stimulated by peptide-pulsed DCs (CTX). Ex vivo assays were conducted by exposing PBMCs to peptides in overnight cultures and measuring the response. In the CTX assay, T cells were co-cultured with autologous peptide-pulsed DC and expanded with IL-2 prior to incubation with the respective target cell lines in overnight cultures. Fold change increment of: (C) IFNγ and (D) granzyme B secreting T-cell post M6, F1, PV1 and FluM peptides stimulation is shown. (E) Patients were placed into 3 groups based on the intensity of MAGED4B and FJX1 staining of their tumour tissues. Patients with high expression of MAGED4B/FJX1 in their tumours have better responses when stimulated with PV1, and secrete higher levels of IFNγ and granzyme B in the cytotoxic ELISPOT assay, compared to patients with low expression of these proteins. 195M cells were used as target cells for T cells stimulated with MAGED4B peptide and C666.1F was used for T cells stimulated with FJX1 peptide.
Level of T-cell activation is higher in HNSCC patients whose tumours express high MAGED4B and FJX1
Among the 15 patient samples used in the dimer and ELISPOT assays, we were able to obtain 11 archived FFPE tumour blocks from these patients. The expression levels of MAGED4B and FJX1 in these tumours were determined by IHC and patients were divided into 3 groups (FJX1 and MAGED4B low, FJX1 or MAGED4B high, FJX1 and MAGED4B high) based on staining intensity (Figure 1; intensity 0 and 1: low; intensity of 2 and 3: high).
We demonstrated that patients with high expression of FJX1 or MAGED4B have higher number of cytokine secreting cells (either IFNγ or granzyme B) when compared to patients with low expression of FJX1 or MAGED4B (Figure 5E). Success rate of T-cell recognition and mounting of immune response is higher in patients whose tumour has expression of either MAGED4B or FJX1. Patients with no expression of both MAGED4B and FJX1 have very low antigen-specific T-cells that recognizes F1 and M6 peptides and are unable to mount an immune response after being stimulated with F1 and M6 peptides in the ELISPOT assays suggesting the specificity of PV1 peptide vaccine (Table 3).
Table 3.
Peptide-specific T-cells (Dimer Assay) population and T-cell cytokine responses (ELISPOT Assay) after being stimulated with peptides, in patients with no expression of MAGED4B/FJX1, patients with either MAGED4B or FJX1 expression and patients with both MAGED4B and FJX1 expression. The percentage of peptide-specific population shown in dimer experiment was after subtracting the mock-dimer control (range: 0.0% – 3.0%).
| Antigen expression (IHC) |
% Population of peptide specific T-cells (Dimer) |
|||||
|---|---|---|---|---|---|---|
| Patient no. | MAGED4B | FJX1 | FluM | MAGED4B | FJX1 | Responses upon peptide stimulation (ELISPOT) |
| 195635 | - | - | 2.8 | 0.2 | 0.4 | N |
| 045497 | - | + | 0.8 | 0.9 | 3.5 | Y |
| 145091 | - | + | 3.9 | 12.7 | 1.5 | N |
| 156479 | - | + | 1.2 | 0.0 | 1.4 | Y |
| 205945 | - | + | 6.9 | 4.1 | 1.0 | N |
| 065217 | - | + | 1.8 | 4.2 | 3.4 | Y |
| 105203 | - | + | 8.8 | 9.3 | 3.6 | Y |
| 011815 | + | - | 6.9 | 4.1 | 1.0 | N |
| 055593 | + | + | 0.0 | 2.7 | 4.0 | Y |
| 135443 | + | + | 4.2 | 5.5 | 5.4 | Y |
| 060616 | + | + | 2.5 | 2.0 | 5.1 | N |
Discussion
Many tumours tend to harbour deregulated expression of proteins that are normally only expressed in immune-privileged sites such as the testis22,23 or those that are only expressed during embryogenesis.24,25 The unique expression or overexpression of these antigens offer an opportunity for them to be used as targets for cancer therapy. Our previous studies identified 2 such tumour antigens, MAGED4B and FJX1, with negligible expression in normal head and neck tissues, but are overexpressed in OSCC and NPC respectively.13,15
In this study, we expanded our study cohort and demonstrated that 94.8% of the OSCC and NPC tumours have overexpression of either MAGED4B and/or FJX1. Further correlative studies revealed that MAGED4B and FJX1 expression did not correlate with patients’ disease staging. This suggest that these proteins might be important for cancer initiation and progression, as their expression was present in both early and advanced stages. As both MAGED4B and FJX1 are functionally important,13,14 we hypothesised that both MAGED4B and FJX1 could be important targets for HNSCC treatment. In addition, we also showed that MAGED4B and FJX1 are expressed in other cancer types, including breast, colon, lung, prostate and rectal cancers, indicating that targeting these 2 antigens could also benefit patients suffering from these cancers as well. However, targeting these antigens separately might not be an efficient therapeutic approach as several studies showed that antigenic heterogeneity and antigen loss are common phenomena that could limit the effectiveness of single antigen peptide vaccines.10,26 In order to overcome such limitations, we chose to target multiple antigens to induce broader immune responses for tumour eradication. Importantly, multiple antigenic peptide vaccines could benefit a larger patient cohort.27 Previous studies on oesophageal cancer patients showed that multiple antigen peptide vaccines can elicit a more robust immune response in patients and resulted in better overall survival compared to those having CTL responses against single-antigen peptides.28 A recent clinical trial demonstrated HNSCC patients treated with a multiple-antigen peptide vaccine have prolonged overall survival from 3.5 months to 4.9 months compared to patients who received optimal supportive care.11
In the current study, we combined peptides from both MAGED4B and FJX1 (PV1) and sought to test the immunogenicity of PV1 as a dual-antigenic peptide vaccine using PBMCs from HNSCC patients. Our results showed that the dual-antigenic peptide vaccine PV1 is more immunogenic compared to the single peptides. We detected larger numbers of inherent T-cells that are able to recognise PV1-loaded dimers compared to dimers loaded with either M6 or F1 peptide alone in the dimer assay. We then evaluated the ability of these T-cells to secrete cytokines using the ELISPOT assay. Although ELISPOT is an indirect method to measure cytolytic activity via secretion of cytokines, it has been demonstrated to be sensitive in detecting the functional T-cells at single cell level.29 Although the ability of T-cells to secrete IFNγ and granzyme B was relatively weak in the direct ex vivo ELISPOT assay, in vitro stimulation of these T cells with PV1 or individual peptides was able to increase the numbers of cytokine secreting T-cells as detected in the cytotoxic ELISPOT assay. IFNγ and granzyme B secretion after PV1 stimulation was at least similar, if not better, compared to stimulation by single peptide. In our study, we observed that the FluM peptide-stimulated T cells also seemed to generate a positive response against the 195M or C666.1F cells in the cytotoxicity experiments. We believe this to be an aberrant response that is most probably be due to strongly activated flu-specific T cells, which may not have been fully rested from their original stimulations with FluM peptide. Another possibility is that it is due to M6 and/or F1-specific T cells present in the FluM cultures that have been non-specifically activated as bystanders (since the majority of the positive responders are from the same patient samples that have also high responses in their M6, F1 or PV1 stimulated cultures).
Notably, our data showed that patients with higher expression levels of MAGEDB4 or FJX1 in their tumour tissue also had higher levels of peptide-specific T-cells that were more readily activated and yielded strong immune responses upon PV1 stimulation. This encouraging data suggests the possibility that pre-existing PV1-specific T cells might only be sub-optimally activated ex vivo, resulting in low secretion of cytokines, but can be re-activated after stimulation with PV1. Patient screening based on the expression levels and/or the presence of either MAGED4B or FJX1 in the tumour would be an important inclusion criterion for targeting patients for PV1 vaccine therapy.
However, as shown in Table 3, levels of antigen expression in tumour this does not serve as the sole-criteria to determine the success of augmentation of immune response. We also noticed that despite the expression of MAGED4B or FJX1 in the tumour tissues, a small portion of recruited patients do not response to the PV1 re-stimulation. For example, despite having high expression of both antigens and detectable levels of MAGED4B and FJX1 peptide specific T-cells, the ELISPOT results of patient no. 060616 did not show any augmentation of immune response upon stimulation. We understand that one of the limitations of using peptide vaccine targeting self-antigen is the difficulties of mounting immune response due to tolerance. To overcome such limitation, enhancing the efficacy of vaccine by fusing it with non-self proteins could enhance the recruitment of T-helper cells and antibody response for better tumour elimination.30 Another method to circumvent the tolerance including the use of co-stimulatory agonistic antibodies to convey activation signals to T-cell.31 Another possibility for lack of immune response after re-stimulation, is the presence of other inhibitory mechanisms that block the re-sensitising of T cell response, such as the presence of immune checkpoint molecules. Although improved efficacy has been demonstrated with immune checkpoint inhibitors such as nivolumab and pembrolizumab, the full promise of cancer immunotherapy remains elusive as the majority of patients still do not benefit from these new therapies. One of the reasons proposed for these treatment failures is the lack of a strong pre-existing anti-tumour immune response in some patients. Therefore, combination of different immunotherapy approaches could improve response rate. Combining immune checkpoint inhibitors with an immunogenic immune activator such as cancer vaccines or co-stimulatory signals has been reported to alter the effector T-cells to regulatory T-cells ratio by increasing antigen-specific T cells and activating the immune response in a more efficient manner.32–36 We propose that a more robust immune response can be induced when immune checkpoint inhibitors are used in combination with PV1, as we have demonstrated the ability of PV1 in augmenting antigen-specific T-cells in most patients.
Although our data demonstrate the potential for a PV1 peptide vaccine to successfully augment the immune response of HNSCC patients in vitro, the current study is constrained by a few limitations. We acknowledge the small cohort of patients studied (41 in total and 20/41 are HLA-A2 positive) who were from a wide range of ages (30–67 years old). This opens up the possibility that the older patients might experience immune-senescence37,38 which could render their PBMCs to be non-responsive to PV1 peptide stimulation. Further, the associations between the T cell immune responses and patient survival status has yet to be made because the samples studied are from newly diagnosed HNSCC patients.
Nonetheless, although we do not observe significant expression of these 2 antigens in our normal samples, we are aware that there are marginally expression of these antigens in normal brain sample, as reported by BioGPS and Protein Atlas.39,40 As the nature of brain’s unique immunologically features, such as presence of blood-brain-barrier which do not allow crossing of blood cells into the brain, inherent immunosuppressive environment, low expression of MHCs and lack of antigen-presenting cells,41 hence, central nervous system could be protected from unwanted side effects from peptide vaccine treatment. However, as the immune regulation mechanism in brain is not fully understood, caution steps remain crucial and further investigation the safety of PV1 peptide vaccine in animal model is warranted.
In conclusion, the use of a dual-antigenic peptide vaccine comprising of both MAGED4B and FJX1 peptides against HNSCC is feasible and warrant further investigation using in vivo models for the safety, immunogenicity and efficacy of PV1.
Materials and methods
Patient samples
Forty-one newly diagnosed OSCC and NPC patients (collectively called HNSCC patients from here on) were enrolled into this study. This project was approved by the Institutional Review Board, Faculty of Dentistry, University of Malaya [DFOS0910/0049(L)] and Medical Research and Ethics Committee, Ministry of Health, Malaysia (NMRR-09–944-4848). Written informed consent was obtained from participating patients. Demographic information of patients included in this study are shown in Table 1A and 1B.
Cell lines
Cancer cell lines included in this study (from the oral cavity, nasopharynx, breast, colon, prostate and lung) and their respective culture media is summarized in Supplementary Table S1. Cell lines overexpressing target antigens, ORL-195-MAGED4B (referred as 195M) and HeLa/T-FJX1 (referred as HeLa/T-F), were used as positive controls in western blot experiments. C666.1-A0201 cell line overexpressing FJX1 (referred as C666.1F), and 195M were used as target cell lines in cytotoxic ELISPOT assays as described below. All cell lines used in this study have been authenticated.
Peptides
The PV1 peptide vaccine consists of two HLA-A2-restricted peptides derived from FJX1 (F1; WLLALGSLLAL), and MAGED4B (M6; RLSLLLVIL), which were selected based on their superior immunogenic characteristics in our previous studies.15,16 HLA-A2-restricted influenza peptide, FluM (GILGFVFTL) was used as positive control. All peptides used for this study were produced commercially by JPT Peptide Technologies with purity > 80%.
Polymerase chain reaction
Presence of the MAGED4B and FJX1 at transcriptomic level in normal human organs were determined by PCR using cDNA from Human MTC™ Panel I (Clontech, Cat. #636,742) and II (Clontech, Cat. #636,743) as template. The primers used were: FJX1 (5ʹ-ACTACCTGACGGCCAACTTC-3ʹ and 5ʹ-CGCCTCATTGTCCAGAAAG-3ʹ); MAGED4B (5ʹ-CCAGAATCAGAACCGAGA-3ʹ and 5ʹ-CCAAAATCTCCGTCCTCA-3ʹ. GAPDH (5ʹ-GAAGGTGAAGGTCGGAGTC-3ʹ and 5ʹ-GAAGATGGTGATGGGATTTC-3ʹ) was also amplified and served as an internal control.
Analysis of MAGED4B and FJX1 genes in the cancer genome atlas (TCGA)
The RNA-Seq gene expression profile of MAGED4B and FJX1 of matched tumour-normal head and neck cancers (HNSC, n = 43), breast invasive carcinoma (BRCA, n = 112), colon adenocarcinoma (COAD, n = 26), rectal adenocarcinoma (READ, n = 6), lung adenocarcinoma (LUAD, n = 58) and lung squamous carcinoma (LUSC, n = 51) samples, in the form of Fragments Per Kilobase of transcript per Million mapped reads upper quartile (FPKM-UQ) were extracted from GDAC Firehose (http://firebrowse.org/). The gene expression of MAGED4B and FJX1 relative to TBP (TATA-box binding protein) expression for each tumour-normal pairs was analysed. Paired t-test was used to assess the differential gene expression between match tumour normal sample pair, and p < 0.05 is considered as statistically significant.
Western blots
Lysates from cell lines were harvested and processed as described previously.42 Fifty micrograms of total protein was subjected to 12% and 10% SDS-polyacrylamide gel electrophoresis (for the detection of MAGED4B and FJX1 protein respectively) and transferred onto Immobilon-P membrane (Millipore, Cat. #IPVH00010). Briefly, blots were incubated with primary antibodies: anti-MAGED4B (1:1000; Sigma Aldrich, Cat. #HPA003554), anti-FJX1 (1: 2000; Aviva Systems Biology, Cat. #ARP47013) for overnight, then probed with respective secondary antibodies conjugated with horseradish peroxidase (1:10,000; Southern Biotech, Cat. #1036–05) for 1 hour. The detection was performed via enhanced chemiluminescence method, using the FluorChem HD2 Imaging System (ProteinSimple, USA). The blots were probed with anti-GAPDH (1:1000; Trevigen, Cat. #2275-PC-020) or anti-actin (1:1000; Milipore, Cat. #MAB1501) antibodies as housekeeping control for each experiment.
Immunohistochemistry
Tissue microarray (TMA) slides [consisting of NPC (n = 74), OSCC (n = 49) and 179 multiple cancers (breast, n = 41; colon, n = 30; lung, n = 48; prostate, n = 44; rectal, n = 16)] included in the study were purchased from Biomax (USA). Expression levels of the target proteins were detected by immunohistochemistry (IHC) using anti-MAGED4B (1:100; Sigma Aldrich, Cat. #HPA003554) and anti-FJX1 (1:200; Sigma Aldrich, Cat. #HPA059220) antibodies, and processed using Dakocytomation Envision+ Dual Link System HRP (DAB+) kit (Dako, Cat. #K4065) as described previously.42 Immunoreactivity of the two antibodies was scored by a board certified pathologist based on a 4-point intensity scoring system: 0 = negative expression; 1 = weak positive; 2 = moderate positive; 3 = strong positive, as reported previously.43 Score 0 and 1 expression were grouped as low expression, 2 and 3 expression were grouped as high expression in subsequent analysis.
Preparation of peripheral blood mononuclear cells (PBMC)
Thirty-five millilitres of blood from HNSCC patients was collected in CPT Vacutainer tubes (Becton Dickinson, USA) and PBMCs were isolated as per manufacturer’s instruction. As both the MAGED4B and FJX1 peptides are HLA-A2 specific, HLA-A2 status was determined by staining of PBMC samples with phycoerythrin (PE) tagged mouse anti-human HLA-A2 antibody (Clone BB7.2; BD Pharmingen, Cat. #558,570) as described previously.15,16 Only patients with positive HLA-A2 were used for subsequent HLA-A2:Ig dimer assay, ex vivo and cytotoxic ELISPOT assay as described below.
Dimer assay
The presence of MAGED4B and FJX1 specific CD8 + T-cells in PBMCs of HLA-A2 positive HNSCC patients was assessed using the HLA-A2:Ig dimer assay. Briefly, freshly isolated PBMCs were added to the respective peptide loaded DimerX human HLA-A2:Ig fusion protein (BD Biosciences, Cat. #551,263) as described previously.16 The presence of peptide/dimer specific CD8 + T-cells was determined by cells that are positive for IgG1, CD8 and TCR after subtracting the value obtained from mock-peptide loaded dimer.
Ex vivo ELISPOT
Ex vivo ELISPOT was used to determine the presence of inherent T-cells from patient PBMCs that recognise the peptides.16 Briefly, isolated PBMCs were incubated with 50 ng/ml of IL-7 and 10 ng/ml of IL-12 in T-cell culture medium containing RPMI (Gibco, Thermo Fisher, Cat. no. 21,870–100) supplemented with 5% heat-inactivated human AB serum (Gemini Bio-Product, Cat. 100–512), 1X penicillin/streptomycin (Gibco, Thermo Fisher, Cat. no. 15,140,122) and 2mM L-glutamine for two hours at 37°C. The cells were then mixed with 50 μg/ml of F1, M6, PV1 and FluM peptides and incubated overnight in the anti-human IFNγ (Mabtech RES-3420-4APW-10) and granzyme B (RES-3485-4APW-10) coated ELISPOT plates. After an overnight incubation, the ELISPOT assay was performed according to the manufacturer’s instructions. The detected spots were then quantitated using CTL ELISPOT Analyzer (Cellular Technology Limited, USA) and analysed using the ImmunoSpot Professional Software (Cellular Technology Limited, USA). Peptide-recognizing T-cells were calculated by subtracting spots formed in the background control wells with no exposure to peptides. The FluM peptide was used as positive control.
Dendritic cells (DCs) and cytotoxic T-lymphocytes (CTLs) isolation and culture
T-lymphocytes were expanded from non-adherent cells isolated from patient PBMC.15 The collected cells were cultured in T-cell culture medium containing 10 ng/ml IL-7 (R&D Systems, Cat. #207-IL-005) and 50 µg/ml of peptides for 2–5 days at 37°C. The adherent cells were cultured at 37°C for 2–5 days in M-SFM in the presence of 100 ng/ml GM-CSF (R&D Systems, Cat. #215-GM-010) and 25 ng/ml IL-4 (R&D Systems, Cat. #204-IL-010) to induce differentiation of dendritic cells (DCs). Differentiated DCs were further incubated with 50 μg/ml peptides (F1, M6 and PV1) for 2 hours at 37°C before pulsing to the T-cells derived from non-adherent PBMCs. After co-culture for 18–24 hours, 10 ng/ml of IL-2 (R&D Systems, Cat. #202-IL-010) was added to the DCs/T-cell culture and incubated for an additional 24 hours. Fresh media was replenished the next day and DCs/T-cells were further incubated in 37°C for 3–5 days to allow the activation of CTLs.
Cytotoxic ELISPOT assay
Cytotoxic ELISPOT assay was used to study the response of peptide-pulsed CTLs derived from the patients against target cell lines overexpressing the target antigens. CTLs were co-cultured with 195M and C666.1F at an effector to target ratio of 20:1, with the presence of 20 ng/ml of IL-7 and IL-2 on the IFNγ/granzyme B ELISPOT plate for 16 hours at 37°C. Then, ELISPOT assay was performed as described above.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding Statement
This study was funded by ScienceFund from Ministry of Science, Technology and Innovation, Malaysia (02-04-03-SF0017) and donors of Cancer Research Malaysia. The funders are involved in the project monitoring and milestone settings. Cancer Research Malaysia is a non-profit research organisation. We are committed to an understanding of cancer prevention, diagnosis and treatment through a fundamental research program.
Acknowledgments
The authors would like to thank Dr. Ricardo Dolcetti for providing the NPC cell line C666.1-A0201; members of the University Malaya Oral Cancer Research & Coordinating Centre (OCRCC); Ms. Rahmah Abdul Hamid from Penang General Hospital; Mr. Bernard Lee and Mr. Sean Wen Wei Xiong from Cancer Research Malaysia for technical assistance. The authors also thank all consented patients who participated in this study.
Supplementary material
Supplemental data for this article can be access on the http://dx.doi.org/10.1080/21645515.2018.1520584 publisher’s website.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F.. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Joshi P, Dutta S, Chaturvedi P, Nair S. Head and neck cancers in developing countries. Rambam Maimonides Med J. 2014;5:e0009. doi: 10.5041/RMMJ.20769172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilyoma JM, Rambau PF, Masalu N, Kayange NM, Chalya PL. Head and neck cancers: a clinico-pathological profile and management challenges in a resource-limited setting. BMC Res Notes. 2015;8:772. doi: 10.1186/s13104-015-1773-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wildeman MA, Fles R, Herdini C, Indrasari RS, Vincent AD, Tjokronagoro M, Stoker S, Kurnianda J, Karakullukcu B, Taroeno-Hariadi KW, et al. Primary treatment results of Nasopharyngeal Carcinoma (NPC) in Yogyakarta, Indonesia. PloS one. 2013;8:e63706. doi: 10.1371/journal.pone.0063706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pulte D, Brenner H. Changes in survival in head and neck cancers in the late 20th and early 21st century: a period analysis. Oncologist. 2010;15:994–1001. doi: 10.1634/theoncologist.2009-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vermorken JB, Specenier P. Optimal treatment for recurrent/metastatic head and neck cancer. Ann Oncol. 2010;21(Suppl 7):vii252–61. doi: 10.1093/annonc/mdq453. [DOI] [PubMed] [Google Scholar]
- 7.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–965. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 8.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. The New England journal of medicine. 2016. doi: 10.1056/NEJMoa1602252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slingluff CL., Jr. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer J. 2011;17:343–350. doi: 10.1097/PPO.0b013e318233e5b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshitake Y, Fukuma D, Yuno A, Hirayama M, Nakayama H, Tanaka T, Nagata M, Takamune Y, Kawahara K, Nakagawa Y, et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clin Cancer Res. 2015;21:312–321. doi: 10.1158/1078-0432.CCR-14-0202. [DOI] [PubMed] [Google Scholar]
- 12.Ohno S, Okuyama R, Aruga A, Sugiyama H, Yamamoto M. Phase I trial of Wilms’ Tumor 1 (WT1) peptide vaccine with GM-CSF or CpG in patients with solid malignancy. Anticancer Res. 2012;32:2263–2269. [PubMed] [Google Scholar]
- 13.Chong CE, Lim KP, Gan CP, Marsh CA, Zain RB, Abraham MT, Prime SS, Teo S-H, Silvio Gutkind J, Patel V, et al. Over-expression of MAGED4B increases cell migration and growth in oral squamous cell carcinoma and is associated with poor disease outcome. Cancer Lett. 2012;321:18–26. doi: 10.1016/j.canlet.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 14.Ahmad Zabidi MM. Identification of four-jointed box 1 as a potential oncogene in nasopharyngeal carcinoma. M.Sc. Thesis. Institute of Biological Sciences, Faculty of Science; Kuala Lumpur: (http://pendeta.um.edu.my/client/default/search/detailnonmodal/ent:$002f$002fSD_ILS$002f871$002fSD_ILS:871872/one;jsessionid=55866C8DEF2376DCA6A645D4DCFFDBE1.enterprise-20200?qu=%22871872%22&te=ILS):University of Malaya,2011:100. [Google Scholar]
- 15.Chai SJ, Yap YY, Foo YC, Yap LF, Ponniah S, Teo SH, Cheong SC, Patel V, Lim KP, Zhang L. Identification of Four-Jointed Box 1 (FJX1)-specific peptides for immunotherapy of Nasopharyngeal Carcinoma. PloS one. 2015;10:e0130464. doi: 10.1371/journal.pone.0130464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim KP, Chun NA, Gan CP, Teo SH, Rahman ZA, Abraham MT, Zain RB, Ponniah S, Cheong SC. Identification of immunogenic MAGED4B peptides for vaccine development in oral cancer immunotherapy. Hum Vaccin Immunother. 2014;10:3214–3223. doi: 10.4161/hv.29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheong SC, Chandramouli GV, Saleh A, Zain RB, Lau SH, Sivakumaren S, Pathmanathan R, Prime SS, Teo SH, Patel V, et al. Gene expression in human oral squamous cell carcinoma is influenced by risk factor exposure. Oral Oncol. 2009;45:712–719. doi: 10.1016/j.oraloncology.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Bose S, Yap LF, Fung M, Starzcynski J, Saleh A, Morgan S, Dawson C, Chukwuma MB, Maina E, Buettner M, et al. The ATM tumour suppressor gene is down-regulated in EBV-associated nasopharyngeal carcinoma. J Pathol. 2009;217:345–352. doi: 10.1002/path.2487. [DOI] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas N Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rock R, Heinrich AC, Schumacher N, Gessler M. Fjx1: a notch-inducible secreted ligand with specific binding sites in developing mouse embryos and adult brain. Dev Dyn. 2005;234:602–612. doi: 10.1002/dvdy.20553. [DOI] [PubMed] [Google Scholar]
- 21.Al-Greene NT, Means AL, Lu P, Jiang A, Schmidt CR, Chakravarthy AB, Merchant NB, Washington MK, Zhang B, Shyr Y, et al. Four jointed box 1 promotes angiogenesis and is associated with poor patient survival in colorectal carcinoma. PloS one. 2013;8:e69660. doi: 10.1371/journal.pone.0069660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inaoka RJ, Jungbluth AA, Baiocchi OC, Assis MC, Hanson NC, Frosina D, Tassello J, Bortoluzzo AB, Alves AC, Colleoni GW. An overview of cancer/testis antigens expression in classical Hodgkin’s lymphoma (cHL) identifies MAGE-A family and MAGE-C1 as the most frequently expressed antigens in a set of Brazilian cHL patients. BMC Cancer. 2011;11:416. doi: 10.1186/1471-2407-11-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 2015;6:15772–15787. doi: 10.18632/oncotarget.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawrence MG, Margaryan NV, Loessner D, Collins A, Kerr KM, Turner M, Seftor EA, Stephens CR, Lai j, Postovit L-M, et al. Reactivation of embryonic nodal signaling is associated with tumor progression and promotes the growth of prostate cancer cells. Prostate. 2011;71:1198–1209. doi: 10.1002/pros.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Becker D, Sfakianakis I, Krupp M, Staib F, Gerhold-Ay A, Victor A, Binder H, Blettner M, Maass T, Thorgeirsson S, et al. Genetic signatures shared in embryonic liver development and liver cancer define prognostically relevant subgroups in HCC. Mol Cancer. 2012;11:55. doi: 10.1186/1476-4598-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slingluff CL Jr., Colella TA, Thompson L, Graham DD, Skipper JC, Caldwell J, Brinckerhoff L, Kittlesen DJ, Deacon DH, Oei C, et al. Melanomas with concordant loss of multiple melanocytic differentiation proteins: immune escape that may be overcome by targeting unique or undefined antigens. Cancer Immunol Immunother. 2000;48:661–672. doi: 10.1007/s002620050015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerdemann U, Katari U, Christin AS, Cruz CR, Tripic T, Rousseau A, Gottschalk SM, Savoldo B, Vera JF, Heslop HE, et al. Cytotoxic T lymphocytes simultaneously targeting multiple tumor-associated antigens to treat EBV negative lymphoma. Mol Therapy. 2011;19:2258–2268. doi: 10.1038/mt.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kono K, Iinuma H, Akutsu Y, Tanaka H, Hayashi N, Uchikado Y, Badet L, Hauet T. Multicenter, phase II clinical trial of cancer vaccination for advanced esophageal cancer with three peptides derived from novel cancer-testis antigens. J Transl Med. 2012;10:141. doi: 10.1186/1479-5876-10-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shafer-Weaver K, Sayers T, Strobl S, Derby E, Ulderich T, Baseler M, Malyguine A. The Granzyme B ELISPOT assay: an alternative to the 51Cr-release assay for monitoring cell-mediated cytotoxicity. J Transl Med. 2003;1:14. doi: 10.1186/1479-5876-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huijbers EJM, van Beijnum JR, Le CT, Langman S, Nowak-Sliwinska P, Mayo KH, Griffioen AW. An improved conjugate vaccine technology; induction of antibody responses to the tumor vasculature. Vaccine. 2018. doi: 10.1016/j.vaccine.2018.03.064. [DOI] [PubMed] [Google Scholar]
- 31.Capece D, Verzella D, Fischietti M, Zazzeroni F, Alesse E. Targeting costimulatory molecules to improve antitumor immunity. J Biomed Biotechnol. 2012;2012:926321. doi: 10.1155/2012/926321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahrends T, Babala N, Xiao Y, Yagita H, van Eenennaam H, Borst J. CD27 agonism plus PD-1 blockade recapitulates CD4+ T-cell help in therapeutic anticancer vaccination. Cancer Res. 2016;76:2921–2931. doi: 10.1158/0008-5472.CAN-15-3130. [DOI] [PubMed] [Google Scholar]
- 33.Morse MA, Lyerly HK. Checkpoint blockade in combination with cancer vaccines. Vaccine. 2015;33:7377–7385. doi: 10.1016/j.vaccine.2015.10.057. [DOI] [PubMed] [Google Scholar]
- 34.Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, Cook L, Khleif SN. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol. 2011;41:2977–2986. doi: 10.1002/eji.201141639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Avogadri F, Zappasodi R, Yang A, Budhu S, Malandro N, Hirschhorn-Cymerman D, Tiwari S, Maughan MF, Olmsted R, Wolchok JD, et al. Combination of alphavirus replicon particle-based vaccination with immunomodulatory antibodies: therapeutic activity in the B16 melanoma mouse model and immune correlates. Cancer Immunol Res. 2014;2:448–458. doi: 10.1158/2326-6066.CIR-13-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, Behrens MD, Knutson KL. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res. 2014;74:2974–2985. doi: 10.1158/0008-5472.CAN-13-2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma S, Dominguez AL, Lustgarten J. Aging affect the anti-tumor potential of dendritic cell vaccination, but it can be overcome by co-stimulation with anti-OX40 or anti-4-1BB. Exp Gerontol. 2006;41:78–84. doi: 10.1016/j.exger.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J Pathol. 2007;211:144–156. doi: 10.1002/path.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu C, Jin X, Tsueng G, Afrasiabi C, Su AI. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 2016;44:D313–6. doi: 10.1093/nar/gkv1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 41.Lyon JG, Mokarram N, Saxena T, Carroll SL, Bellamkonda RV. Engineering challenges for brain tumor immunotherapy. Adv Drug Deliv Rev. 2017;114:19–32. doi: 10.1016/j.addr.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gan CP, Patel V, Mikelis CM, Zain RB, Molinolo AA, Abraham MT, Teo S-H, Abdul Rahman ZA, Gutkind JS, Cheong SC. Heterotrimeric G-protein alpha-12 (Galpha12) subunit promotes oral cancer metastasis. Oncotarget. 2014;5:9626–9640. doi: 10.18632/oncotarget.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Charafe-Jauffret E, Tarpin C, Bardou VJ, Bertucci F, Ginestier C, Braud AC, Puig B, Geneix J, Hassoun J, Birnbaum D, et al. Immunophenotypic analysis of inflammatory breast cancers: identification of an ‘inflammatory signature’. J Pathol. 2004;202:265–273. doi: 10.1002/path.1515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.