

With years of experience in modeling and simulation (M&S), and model‐informed drug development (MIDD) as one of the goals in Prescription Drug User Fee Act (PDUFA) VI authorization, it is now the right time to apply MIDD for high‐impact decisions. Over the past two decades, current applications of MIDD in drug discovery, dose selection, benefit–risk assessment, and labeling have been useful. The value proposition of M&S is acknowledged in the PDUFA VI authorization, which provides an excellent opportunity for industry and regulators to collaborate in further advancing the applications of MIDD, potentially changing drug development paradigms.
The Preamble
Approximately 20 years ago, the US Food and Drug Administration (FDA) guidance on Providing Clinical Evidence of Effectiveness for Human Drug and Biologic Products 1 laid out fundamental principles with opportunities for the role of M&S in satisfying the regulatory requirements for the evidence of effectiveness. Subsequent guidances, such as the guidance on exposure–response analysis, detailed applications of model‐based approaches in regulatory decision making. Therefore, a natural extension would be its application in various regulatory submissions, including marketing approval based on a single efficacy trial with M&S providing additional primary “confirmatory” evidence. Throughout these years, there has been significant growth in applications of M&S by industry from discovery to clinical development and by regulatory agencies to support approval and labeling decisions. However, these are generally supportive and often post hoc rather than the primary basis of approval. Inclusion of MIDD in PDUFA VI authorization is an opportunity to reflect on prior successes and determine regulatory scenarios in which MIDD can play a more pivotal role in substantiating the evidence required for approval and labeling decisions uniformly across therapeutic areas. The current senior leadership at the FDA, including the FDA commissioner Dr. Scott Gottlieb and Center for Drug Evaluation and Research Director Dr. Janet Woodcock, stands in full support of leveraging M&S to increase the efficiency of drug development.2 The decision makers in industry should also leverage opportunities presented by PDUFA VI for greater integration of M&S in development programs to improve efficiency and enable faster access to new treatments. PDUFA VI presents an excellent opportunity for drug developers and regulators to come together to achieve the full potential of MIDD.
Contrasting the Current vs. MIDD‐Based Decision Making
Although there are many promising examples of application of MIDD approaches, as summarized in a white paper from the European Federation of Pharmaceutical Industries and Associations (EFPIA) Model Informed Drug, Discovery and Development (MID3) Workgroup3 and by the American Society for Clinical Pharmacology and Therapeutics Quantitative Pharmacology Impact and Influence Initiative,4 few examples in the current state go beyond supportive information to something that would be truly transformative for drug development. The published examples cover a broad spectrum of approaches and development stages, demonstrating how MIDD can increase confidence in decisions (notably around the appropriate dose and dose adjustments), provide support for assessment of benefit–risk, and support labeling. Despite the evident success of MIDD approaches in some categories, it is striking that few of the examples described in the literature fall into the category of providing confirmatory evidence of efficacy/safety in lieu of clinical data for regulatory submissions, and none describes true replacement of a clinical trial, the “gold standard,” for initial registration purposes. Although, in itself, this is not the only application of MIDD, an overarching goal of MIDD, as outlined in PDUFA VI, is to use the value of predictive attributes of M&S to reduce the need to generate clinical trial data where possible.
With years of experience in M&S approaches, the time is now right to move to a paradigm in which M&S is routinely leveraged to integrate early‐phase data and exposure–response relationships to predict the dosing regimens for clinical use, with a requirement for only a single registration trial to confirm the model predictions5 (Figure 1). M&S should be considered equally in the mix of options in thinking about how to generate evidence for efficacy, safety, and benefit–risk for approval decisions. Indeed, the application of M&S to fill the knowledge gaps should be driven by consideration of scientific merit and limitations, and experts with proper know‐how of M&S methods should be made integral members of the teams making development decisions to properly articulate the merits and limitations of these methods. For example, in some scenarios, in which prior information is robust enough, approvals based only on M&S (without any additional registration trial) should be considered, such as approval in pediatrics after approval in adults.
Figure 1.
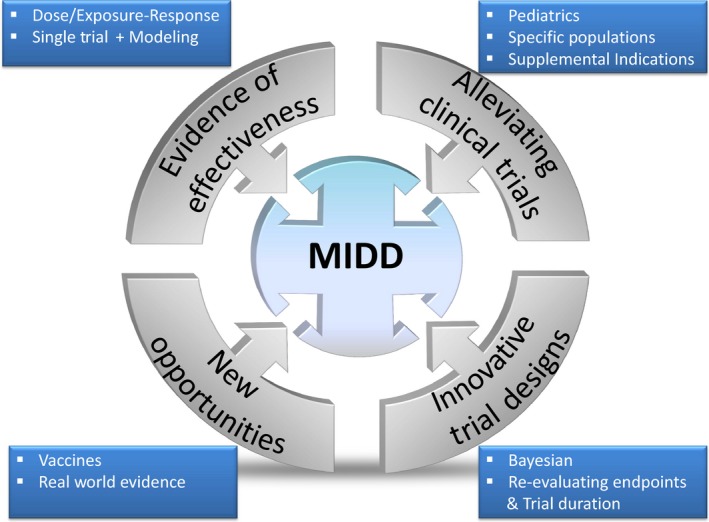
Opportunities for application of model‐informed drug development.
Barriers
There are many applications of MIDD, ranging from new and efficient designs of clinical trials, to incorporation of new end points, to how the data are analyzed to provide evidence of effectiveness (Table 1). These applications have been discussed for years by academia, industry, and regulatory agencies, but there remain barriers to realization of their true potential.
Table 1.
Opportunities for application of MIDD‐based approaches in clinical drug development
| Typical current paradigm | MIDD‐based paradigm | Advantages of MIDD paradigm |
|---|---|---|
| Sequential phase II– III trial approach | Innovative designs assisted with Clinical Trial Simulations (e.g., seamless phase II–III designs, adaptive designs, and biomarker‐based designs) | Better (integrated) use of prior information to make the subsequent steps more efficient |
| Clinical trials in general population with post hoc analysis in subgroups | More targeted clinical trials to fill gaps in evidence for clinical use, based on predictions of benefit–risk in population subgroups | Use of resources to address the relevant questions in a more efficient and timely manner |
| Primary hypothesis in dose‐ranging trials based on pairwise comparison of two doses or a dose and placebo | Primary hypothesis based on demonstration of positive slope in dose–response or exposure–response analysis |
|
| Traditionally designed pediatric studies (e.g., fully powered to demonstrate efficacy and safety end points) | Either replace the need for pediatric studies with evidence from M&S analysis or develop efficient designs to minimize burden on pediatric patients | Fast access to treatments in pediatric population |
| Specific population labeling based on studies evaluating drug–drug interaction, renal impairment, and hepatic impairment | Leverage integrated understanding of systems and drug PK characteristics to make predictions and evaluate only the extreme scenarios |
|
| Evidence for approval from replicate randomized trials | Evidence of approval from predictions using M&S, which are confirmed with single‐efficacy and safety study if necessary |
|
MIDD, model‐informed drug development; M&S, modeling and simulation; PK, pharmacokinetic.
First, medical and biostatistical experts have traditionally played a central role in making key decisions on what is considered acceptable evidence of effectiveness at both regulatory agencies and industry, and these decisions often hinge on prior established precedent. Because of this, although the information collected during drug development has increased exponentially, the fundamental view on what is routinely considered as evidence by regulators (replicate randomized controlled trials) has not changed since the 1970s. Lack of (or limited) familiarity with M&S approaches by medical and biostatistical experts often leads to situations in which the contributions of M&S are not judged on the basis of the value offered but considered as being secondary or less compelling. Although modeling is based on observed clinical data, it is often perceived with skepticism. A change in this paradigm will only occur if key decision makers in the regulatory agencies and industry, including medical experts, show more openness in discussing the utility of M&S, rely on appropriate experts for technical know‐how, are willing to deviate from precedent based on the scientific merits of a new approach, and are transparent about why M&S is not accepted for certain decisions. The call to action by the FDA leadership has to be met with actions at the drug review division levels. Similarly, industry should advance proposals to use M&S to support key regulatory decisions in addition to using M&S to support internal decisions. New regulatory mechanisms, such as the FDA MIDD Pilot Meeting, offer opportunities to obtain specific feedback; and sponsors should leverage these opportunities to work closely with regulators in thinking about innovative approaches and not restrict themselves to following precedent.
Second, there is a lack of consensus in standards among key regulatory agencies from different geographic regions. Because companies design clinical programs for global submissions, the full potential of M&S cannot be realized if all key regulatory agencies do not accept the approach. To this end, existing mechanisms of communication would need to be maximized to support worldwide acceptance. This may not lead to full harmonization, but recognition of the reasons for differences and transparent communication can inspire sponsors to propose innovative solutions. In addition, methods and models for conducting M&S and communicating results should also be standardized. This standardization would enable greater acceptance of M&S among decision makers, particularly for those with less experience in M&S approaches.
Third, another main challenge is acceptance of M&S‐based results by physicians and payers. There is a general perception that it is difficult to get drugs reimbursed by payers if observed clinical trial data do not exist and that physicians would not prescribe the drugs in absence of observed clinical data, which limits the utility of M&S approaches. The community will need to work together to resolve this challenge and enable acceptance through systematic education and communication of applications.
New Opportunities for Innovation
The applications of MIDD have progressed in some therapeutic areas more than others and are often limited to certain types of decisions. Applications have been more in areas with better understanding of pharmacodynamic end points and biomarkers and how they relate to clinical outcomes. Continued efforts are needed to harness untapped and new opportunities (Figure 1).
For example, MIDD has not been routinely used in vaccines development. M&S can be used to develop an understanding of the relationship between biomarkers (e.g., antibody titers) and efficacy to establish a correlate of protection.6 Such analysis can enable early drug development decisions, such as selection of vaccine platforms and end points, and inform dosing regimens and clinical strategy to use biomarkers, thus presenting an opportunity to accelerate the traditionally protracted timeline for vaccine development.
Real‐world data/real‐world evidence, a commitment under PDUFA VI, is an emerging area for extending the application of M&S. This field is rapidly evolving with ongoing refinements in terms of standardization of data collection and use. Such data from postmarketing registries are already using M&S approaches to answer questions that cannot be answered during routine drug development.
Other applications of M&S include evaluation and development of novel end points, trial design considerations for duration, or need for separate trial in population subgroups by conducting integrated analysis of data from across trials and drug classes. In Alzheimer’s disease area, M&S analysis using item response theory has been shown to provide more power to detect treatment effect than traditional method of analysis using the composite score.7 In oncology, M&S approaches have been applied to evaluate the adequacy of Response Evaluation Criteria in Solid Tumors (RECIST) in determining disease progression in patients receiving immunotherapy treatments.8 Recently, the FDA used M&S analysis to demonstrate that in children with partial‐onset seizures, aged ≥4 years, efficacy can be fully extrapolated from adults, which obviated the need for efficacy trials in that pediatric subgroup.9 Similarly, M&S analysis of integrated data from multiple trials for acute schizophrenia indication led to recommendation that the duration of registration trials for atypical antipsychotics for the treatment of schizophrenia can be shortened to 4 weeks instead of the typical duration of 6 weeks.10
The Call for Action
With technological advances and years of experience with M&S approaches, now is a better time than ever to make disruptive changes in how M&S is used to support regulatory decision making. PDUFA VI and 21st Century Cures Act are providing the legislative push with the FDA leadership strongly supporting application of MIDD. Through the MIDD Pilot Meeting program, PDUFA VI has also opened new avenues for the FDA–industry interaction to obtain specific feedback on M&S‐informed decisions. Although MIDD cannot be an approach in every situation, experts have to continue communicating the value and limitations with key stakeholders, including the medical community, to ensure long‐term success. In this enabling environment, it is time to unleash the full potential of MIDD and maximize the use of M&S in approval and labeling decisions.
Funding
Funding for this work was provided by Merck & Co., Inc. (Kenilworth, NJ).
Conflict of Interest
L.J., N.M., L.W., and V.S. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. (Kenilworth, NJ), and may own stock/stock options in the company. V.S. also reports stock/stock options and patents from Eli Lilly and Co. As an Associate Editor for CPT: Pharmacometrics & Systems Pharmacology, V.S. was not involved in the review or decision process for this article.
References
- 1. FDA guidance for industry: providing clinical evidence of effectiveness for human drug and biologic products. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072008.pdf> (May 1998)
- 2. US FDA voice by Commissioner Dr. Scott Gottlieb article on “How FDA Plans to Help Consumer Capitalize on Advances in Science.” <https://blogs.fda.gov/fdavoice/index.php/2017/07/how-fda-plans-to-help-consumers-capitalize-on-advances-in-science/>
- 3. Marshall, S.F. et al Good practices in model‐informed drug discovery and development: practice, application, and documentation. CPT Pharmacometrics Syst. Pharmacol. 5, 93–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nayak, S. et al Getting innovative therapies faster to patients at the right dose: impact of quantitative pharmacology towards first registration and expanding therapeutic use. Clin. Pharmacol. Ther. 103, 378–383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prevision Policy article . A new model for drug development: FDA sees chance to use modeling to shortcut early development: workshop is “first‐step” on “long journey.” <https://www.previsionpolicy.com/a-new-model-for-drug-development-fda-sees-chance-to-use-modeling-to-shortcut-early-development-workshop-is-first-step-on-long-journey> (February 8, 2018)
- 6. A report published by initiative for vaccine research of the Department of Immunization, Vaccines, Biologicals, World Health Organization: correlates of vaccine‐induced protection: methods and implications. <http://apps.who.int/iris/bitstream/handle/10665/84288/WHO_IVB_13.01_eng.pdf;jsessionid=8BA83129956843D52AABCB9E6688425D?sequence=1> (May 2013)
- 7. Ueckert, S. et al Improved utilization of ADAS‐cog assessment data through item response theory based pharmacometric modeling. Pharm. Res. 31, 2152–2165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwartz, L.H. et al Correlation of survival outcomes with progression heterogeneity in patients (pts) treated with pembrolizumab (pembro): ASCO 2018 poster. J. Clin. Oncol. 36, 3017 (2018). [Google Scholar]
- 9. Draft FDA guidance: drugs for treatment of partial onset seizures: full extrapolation of efficacy from adults to pediatric patients 4 years of age and older guidance for industry. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM596731.pdf>
- 10. Gopalakrishnan, M. et al Shortening the duration of acute schizophrenia registration trials is a possibility. Poster presentation at the American Conference on Pharmacometris 8 (ACoP 8), Fort Lauderdale, Florida October 15‐18, 2017.