

Abstract
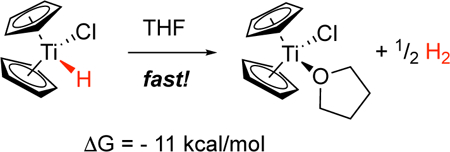
The role of Cp2Ti(H)Cl in the reactions of Cp2TiCl with trisubstituted epoxides has been investigated in a combined experimental and computational study. Although Cp2Ti(H)Cl has generally been regarded as a robust species, its decomposition to Cp2TiCl and molecular hydrogen was found to be exothermic (ΔG = −11 kcal/mol when the effects of THF solvation are considered). In laboratory studies, Cp2Ti(H)Cl was generated using the reaction of 1,2-epoxy-1-methylcyclohexane with Cp2TiCl as a model. Rapid evolution of hydrogen gas was demonstrated, indicating that Cp2Ti(H)Cl is indeed a thermally unstable molecule, which undergoes intermolecular reductive elimination of hydrogen under the reaction conditions. The stoichiometry of the reaction (Cp2TiCl:epoxide = 1:1) and the quantity of hydrogen produced (1 mole per 2 moles of epoxide) is consistent with this assertion. The diminished yield of allylic alcohol from these reactions under the conditions of protic versus aprotic catalysis can be understood in terms of the predominant titanium(III) present in solution. Under the conditions of protic catalysis, Cp2TiCl complexes with collidine hydrochloride and the titanium(III) center is less available for “cross-disproportionation” with carbon-centered radicals; this leads to by-products from radical capture by hydrogen atom transfer, resulting in a saturated alcohol.
Graphical Abstract
INTRODUCTION
Titanocene hydridochloride, 1, is an enigmatic molecule in organometallic chemistry: often invoked but never observed. On the one hand, 1 has been proposed as an intermediate in reactions as diverse as ethylene polymerization1 and the aminolysis of N-acyl carbamates.2 In particular, 1 is implicated as a product of the reaction between tertiary alkyl radicals and the titanium(III) reagent Cp2TiCl.3–5 Because of the expectation that 1 is a robust organometallic species, there has been concern that its formation might be an impediment in catalytic reactions using sub-stoichiometric amounts of Cp2TiCl. Specialized protocols have been developed to address this supposed issue.3,4
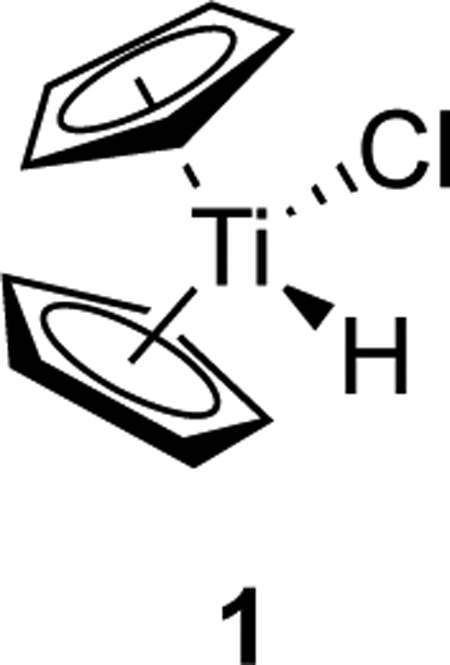
On the other hand, 1 has never been isolated or even observed in situ. Attempts to prepare 1 by treatment of Cp2TiCl2 with hydride reagents have instead consistently afforded the titanium(III) complex Cp2TiCl.6,7 In contrast, zirconocene hydridochloride, Cp2Zr(H)Cl (Schwartz’s Reagent) is a thermally stable organometallic compound with many useful applications in organic synthesis.8,9 Interestingly, the permethylated analogue of 1, bis-(pentamethylcyclopentadienyl)titanium hydridochloride has been synthesized and is stable in the solid state and in THF solution.10,11
Cp2TiCl is a broadly useful reagent in organic synthesis.12–14 Cp2TiCl is usually generated in situ by reduction of Cp2TiCl2 with a metal powder, typically zinc or manganese as shown in Eq. 1. We are particularly interested in the role of 1 in the reactions of Cp2TiCl with trisubstituted epoxides.
| (1) |
It is instructive to first consider the reaction of Cp2TiCl with a simple monosubstituted epoxide 2 in the presence of a hydrogen atom donor Q–H. (Examples of such donors include 1,4-cyclohexadiene and tert-butyl thiol.) When run under stoichiometric conditions,15 the reaction proceeds as shown in Scheme 1. Initial homolysis of one epoxide C–O bond results in formation of β-titanoxy radical 3. Carbon-centered radical 3 abstracts hydrogen from Q–H to afford alkoxide 4. Alkoxide 4 is then hydrolyzed in a separate step with dilute aqueous acid to afford the free alcohol.
Scheme 1.

Cp2TiCl-Mediated Opening of an Epoxide in the Presence of Hydrogen Atom Donor Q-H
In order to carry out such reactions in a catalytic manner, one must address the fact that titanium(IV) alkoxide 4 cannot be directly reduced to the +3 oxidation state. It is essential to first convert the alkoxide back to Cp2TiCl2 so that it may once again be reduced according to Eq. 1. In one approach, this is accomplished by the use of a buffered BrØnsted acid, most often 2,4,6-collidininium hydrochloride (coll•HCl).16,17 By employing stoichiometric amounts of coll•HCl and metal reductant with a sub-stoichiometric amount of Cp2TiCl2 as pre-catalyst, a catalytic cycle can be achieved as shown in Scheme 2.
Scheme 2.

Protic Catalysis of Ti(III)-Mediated Epoxide Ring Opening (TiIV = Cp2ClTi-)
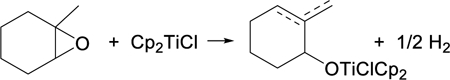
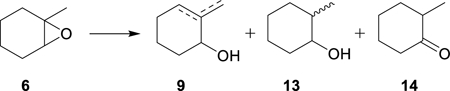
A different reaction path emerges when a trisubstituted epoxide such as 6 reacts with Cp2TiCl. In Scheme 3, the initial C–O bond homolysis gives rise to a tertiary alkyl radical 7. In the absence of a hydrogen atom donor, the observed product after hydrolysis is a mixture of allylic alcohols 9a and 9b. Examples of this type of reaction began to appear in the late 1990’s18–20 and the transformation was subsequently explored in detail by Bermejo and Sandoval.21
Scheme 3.

Formation of Allylic Alcohols via Cp2TiCl-Mediated Opening of Trisubstituted Epoxide
In Scheme 3, the reaction between the radical 7 and Cp2TiCl affords both 1 and either alkene 8a or 8b, depending on which β-hydrogen atom is transferred to Cp2TiCl. This type of process is proposed to proceed via a “mixed disproportionation” between the carbon-centered radical 7 and the “titanium-centered radical” Cp2TiCl in which a β-hydrogen atom is abstracted by the Ti(III) reagent.22 This is illustrated for the major pathway leading to 8a in Eq. 2. (Alternatively, formation of Cp2Ti(H)Cl has been suggested to involve the combination of 7 with Cp2TiCl to afford a 3° organotitanium complex, followed by β-hydride elimination.21,23a See Supporting Information for a more detailed discussion; however, this distinction does not impact the conclusions of the current paper.)
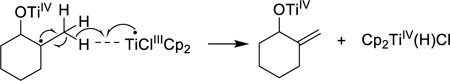 |
(2) |
Reactions of this type provide an efficient means for the isomerization of trisubstituted epoxides to allylic alcohols. Moreover, the loss of a β-hydrogen atom has also been observed as a radical termination step in other reactions of epoxides with Cp2TiCl that give rise to a 3° alkyl radical. Examples include cyclization of epoxyolefins,20,24 transannular cyclizations,25,26 and the cascade cyclization of epoxypolyenes.4,24
β-Scission of hydrogen and reduction by hydrogen abstraction are sometimes observed as competitive processes, as is the case in Scheme 4. When Bermejo and Sandoval carried out the stoichiometric reaction of epoxide 10 with Cp2TiCl (2.2 equiv), they discovered that different products were favored depending on the order of addition of the reactants (Scheme 4).21 When a 0.5 M solution of 10 in THF was added dropwise to 0.1 M solution of Cp2TiCl in THF (normal addition), the predominant product was 11 derived from β-scission of hydrogen. When the order of addition was reversed (inverse addition) so that Cp2TiCl was added to epoxide, the principal product was the saturated alcohol 12. Inverse addition results in a very low instantaneous concentration of Ti(III), which disfavors mixed disproportionation. This suggests that the kinetic order in titanium(III) for the product-forming step is higher for the formation of 11 than for 12 and provides evidence for the requirement for a second equivalent of Ti(III) in the β-scission of hydrogen.
Scheme 4.

Effect of Mode of Addition on Reduction vs β-Hydrogen Scission
With few exceptions,3 the Ti(III)-mediated rearrangement of trisubstituted epoxides to allylic alcohols has not been carried out under protic catalytic conditions. This is consistent with the prevailing view that Cp2Ti(H)Cl is a robust species that would not readily react with coll•HCl to regenerate Cp2TiCl2.4,5 To address this issue, Takahashi and coworkers proposed the use of triethylborane as an additive to promote the reduction of Cp2Ti(H)Cl.3 In another approach, Barrero and coworkers introduced an alternative catalytic protocol, which proceeds under aprotic conditions.26 The coll•HCl that is used in protic catalysis is replaced with a combination of chlorotrimethylsilane and 2,4,6-collidine. Under these conditions, the titanium alkoxide product will be converted to the corresponding trimethylsilyl ether, regenerating Cp2TiCl2 as illustrated for alkoxide 8a in Eq. 3.
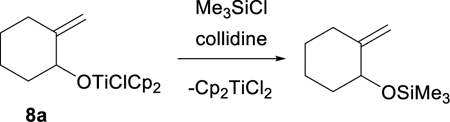 |
(3) |
In addition, it has been proposed that chlorotrimethylsilane is able to regenerate Cp2TiCl2 from Cp2Ti(H)Cl according to Eq. 4.4 This reaction is presumed to proceed by σ-bond metathesis and produces Me3SiH as a co-product (although no evidence for the formation of Me3SiH has been reported.)
| (4) |
Aprotic catalysis of titanium(III)-mediated epoxide opening has been successfully applied not only to simple reactions such as the rearrangement of trisubstituted epoxides but also to an array of cyclization and polycyclization reactions. A compelling example is the transannular cyclization of the simple germacrolide derivative in Eq. 5 which affords (after desilylative workup) a useful bicyclic product as a single stereoisomer in 68% isolated yield.27
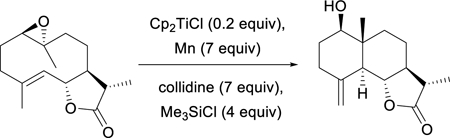 |
(5) |
RESULTS AND DISCUSSION
The fact that Cp2Ti(H)Cl has never been isolated caused us to wonder whether this complex is in actuality thermally stable. To address this question, we first examined the stability of 1 using DFT calculations. These studies revealed that the decomposition of Cp2Ti(H)Cl to Cp2TiCl and molecular hydrogen in solvent THF (Eq 6) is in fact exothermic (ΔG = −9.4 kcal/mol at the PW6B95-D3 level of theory). The effects of explicit THF solvation were not included in this preliminary study.
| (6) |
With this preliminary computational result in hand we proceeded to the laboratory for a closer examination of Scheme 3. A 0.33 M solution of Cp2TiCl was prepared by stirring a solution of Cp2TiCl2 (2.0 mmol) with excess zinc dust (6 mmol) in THF (6.0 mL). To this solution was added a solution of 1-methylcyclohexene oxide (2.0 mmol) in THF (2.0 mL) over the course of several minutes. During the addition, vigorous evolution of gas was observed. Rapid gas evolution ceased after 5 min. At this point the volume of gas produced could be determined by gas burette; after correction to standard temperature and pressure it corresponded to the release of 0.94 mmol of gas. The identity of the gas was confirmed to be hydrogen by gas chromatography.
GLC analysis after quenching with dilute acid indicated that the allylic alcohols 9a and 9b were formed in a 5:1 ratio (Scheme 3). Their identity was confirmed by GC-MS followed by isolation and spectroscopic analysis of the mixture. The combined yield of 9a and 9b was shown by GLC versus an internal standard to be 1.88 mmol (94%). On the basis of these data, the stoichiometry for the overall transformation is best described by Eq. 7.
 |
(7) |
Based on these experiments it appears that Cp2Ti(H)Cl is in fact an unstable compound which decomposes rapidly in THF solution at room temperature. A practical consequence of this fact is that it should not be necessary to employ 2 or 3 equiv of Cp2TiCl in the conversion of trisubstituted epoxides to allylic alcohols as is the general practice.21,22 The second equivalent of Cp2TiCl is unnecessary, since half of the reagent would be converted to Cp2Ti(H)Cl which then decomposes, regenerating Cp2TiCl. In fact we have confirmed this experimentally (see Table 3, entry 1 below).
Table 3.
| entry | Cp2TiCl2 (equiv) |
Coll•HCl (equiv) |
solvent | 9 (%) | 13 (%) | 14 (%) |
|---|---|---|---|---|---|---|
| lc | 1.00 | 0.00 | THF | 100 | 0 | 0 |
| 2 | 0.25 | 1.50 | THF | 44 | 53 | 3 |
| 3d | 0.25 | 1.50 | Et2O | 71 | 24 | 5 |
| 4 | 0.25 | 1.50 | toluene | 67 | 24 | 9 |
Reaction conditions 0.25 M, RT, 3.0 equiv Zn, 20 h unless otherwise indicated.
Product ratios determined by GC-MS and NMR.
Reaction complete in 15 min; gas evolution observed.
Reaction complete after 2.5 h.
The observed instability of 1 stands in contrast to the known stability of its sterically encumbered pentamethylcyclopentadienyl (Cp*) analogue. Treatment of (Cp*)2TiH with lead(II) chloride results in oxidation of titanium to the +4 oxidation state and affords (Cp*)2Ti(H)Cl.10 The analogue (PhC5Me4)2Ti(H)Cl was similarly prepared.11
It is reasonable to assume that decomposition of Cp2Ti(H)Cl follows a bimolecular pathway because of the requirement that H2 is generated in this process. A unimolecular pathway would require formation of atomic hydrogen, which is a high energy intermediate.28 Moreover, if formed, atomic hydrogen would be expected to rapidly abstract hydrogen from solvent THF, which would not be consistent with the observed stoichiometry. A bimolecular pathway also helps to explain the kinetic stability of sterically hindered (Cp*)2Ti(H)Cl.
The calculations in Table 1 compare the energetics of the decomposition process in THF for three different complexes at two different levels of theory but ignore the specific effects of THF solvation. The ΔGsolv term in the last column employs the COSMO-RS solvation model to incorporate the overall effect of the solvent (dielectric constant, etc.) but does not include explicit THF coordination. Table 1 suggests that, independent of substitution on the cyclopentadienyl rings, the decomposition of the hydridochloride complexes is exothermic.
Table 1.
Calculated Thermodynamic Parameters for Cp2Ti(H)Cl → Cp2TiCl + ½ H2
| Complex | Level | ΔE (kcal/mol) | ΔG (kcal/mol} | ΔGsolv (kcal/mol) |
|---|---|---|---|---|
| Cp2TiHCl | B3LYP-D3 | −3.25 | −8.95 | −8.62 |
| PW6B95-D3 | −4.00 | −9.70 | −9.37 | |
| (ClC5H4)2TiHCl | B3LYP-D3 | −4.25 | −10.06 | −11.06 |
| PW6B95-D3 | −5.12 | −10.93 | −11.93 | |
| Cp*2TiHCl | B3LYP-D3 | −2.68 | −8.77 | −7.96 |
| PW6B95-D3 | −3.36 | −9.45 | −8.63 |
B3LYP-D3-COSMO-RS/def2-QZVP//TPSS-D3/def2-TZVP in THF at 298.15 K or PW6B95-D3-COSMO-RS/def2-QZVP//TPSS-D3/def2-TZVP in THF at 298.15 K.
In comparing the various complexes, differences in ΔG arise primarily from differences in the change in electronic energy ΔE. The other contributions to ΔG (entropy and zero-point energy) are essentially constant for all complexes. The order of stability of the three complexes is consistent with their expected redox potentials. The electron-withdrawing chlorine atoms in (ClC5H4)2Ti(H)Cl significantly destabilize that complex relative to the parent Cp2Ti(H)Cl. The electron-donating methyl groups of (Cp*)2Ti(H)Cl stabilize it relative to Cp2Ti(H)Cl.
However, the results in Table 1 are problematic in that the decomposition of (Cp*)2Ti(H)Cl is predicted to be favorable. For this reason, we carried out additional calculations incorporating explicit THF solvation. It has been noted previously that explicit calculation of solvent molecules coordinating to a titanium(III) center can provide more reasonable energies.33 (Cp2TiCl exists in THF solution as a equilibrating mixture of monomeric Cp2TiCl(THF) and its dimer. For simplicity, we consider only the monomer and do not expect this simplification to significantly impact our conclusions.) These results are summarized in Table 2.
Table 2.
Calculated Thermodynamics for Cp2TiHCl + THF → Cp2Tl(THF)Cl + ½ H2
| Complex | Level | ΔE (kcal/mol) | ΔG (kcal/mol} | ΔGsolv (kcal/mol) |
|---|---|---|---|---|
| Cp2TiHCl | B3LYP-D3 | −15.25 | −6.60 | −10.50 |
| PW6B95-D3 | −16.30 | −7.65 | −11.55 | |
| (ClC5H4)2TiHCl | B3LYP-D3 | −16.25 | −7.52 | −11.67 |
| PW6B95-D3 | −17.03 | −8.30 | −12.45 | |
| Cp*2TiHCl | B3LYP-D3 | −7.54 | +2.19 | −0.04 |
| PW6B95-D3 | −8.64 | +1.09 | −1.15 |
B3LYP-D3-COSMO-RS/def2-QZVP//TPSS-D3/def2-TZVP in THF at 298.15 K or PW6B95-D3-COSMO-RS/def2-QZVP//TPSS-D3/def2-TZVP in THF at 298.15 K.
The data in Table 2 indicate that decomposition of Cp2Ti(H)Cl and (ClC5H4)2Ti(H)Cl are further promoted by the effect of THF solvation. Explicit solvation of the titanium atom is possible and has the expected effect (quenching of Lewis acidity).
Interestingly, the data in Table 2 indicate that decomposition of (Cp*)2Ti(H)Cl is approximately thermoneutral in THF solution. At first glance, it may seem strange that explicit solvation is actually unfavorable in the case of (Cp*)2Ti(H)Cl. However, in this case the THF is not strongly bonded in the (Cp*)2TiCl product and in a sense Ti is actually not solvated. We suggest that decomposition of (Cp*)2Ti(H)Cl is not observed in THF solution due to both kinetic and thermodynamic factors. The extreme steric bulk of the Cp* ligands precludes the necessary bimolecular pathway required for decomposition. In addition, there is little or no driving force for decomposition of (Cp*)2Ti(H)Cl to (Cp*)2TiCl and H2.
Thus the instability of Cp2Ti(H)Cl is confirmed both in the laboratory and computationally. It seems unlikely that the σ-bond metathesis in Eq. 4 plays a role in the mechanism of aprotic catalysis. In fact, our calculations suggest that Eq. 4 is approximately thermoneutral. The calculated ΔG is −0.18 kcal/mol at the PW6B95-D3 level. Given that no experimental evidence for the formation of Me3SiH has been reported,4 σ-bond metathesis (eq. 4) does not seem to be operating. Therefore, we limited our calculations to the thermodynamic features of both processes. Clearly, the H2-formation is largely favored thermodynamically. The higher driving force seems to lead to a faster H2-formation.
Aprotic catalysis has been successfully employed for the rearrangement of trisubstituted epoxides to allylic alcohols. In contrast, there is a single report where this transformation was attempted under the conditions of protic catalysis.3 It is not clear whether this situation simply reflects the assumption that the formation of Cp2Ti(H)Cl would interrupt the catalytic cycle under protic conditions. To clarify this situation, we returned to the laboratory to examine the reaction of epoxide 6 under the conditions of protic catalysis.
In these experiments, zinc powder was employed as stoichiometric reductant and 2,4,6-collidine hydrochloride as buffered BrØnsted acid. All of the reactions in Table 3 were carried out under similar conditions (THF, 20 h, RT, 0.25 M except as otherwise indicated.) When a sub-stoichiometric amount of Cp2TiCl2 was utilized, in addition to the allylic alcohols 9, the 2-methylcyclohexanols cis- and trans-13 and 2-methylcyclohexanone 14 were also produced as shown in Eq. 8.
 |
(8) |
When comparing entries 1 and 2 in Table 3, it is evident that the diminished yield of allylic alcohols 9 under protic catalytic conditions is not due to a failure of catalytic turnover since complete conversion of epoxide 6 is achieved in each case. Instead, a new pathway arises under protic catalytic conditions in which a significant proportion of the starting epoxide is converted to the reduction product, alcohols 13. This change in product distribution was also observed (although to a lesser extent) when the THF solvent was replaced with diethyl ether or toluene (Entries 3 and 4). We suggest that this result is related to the fact that, in the presence of excess coll•HCl (especially in THF), titanium(III) is largely present as complex 15. Recent studies have underscored the importance of complex 15 in stabilizing titanium(III) during protic catalytic reactions.29
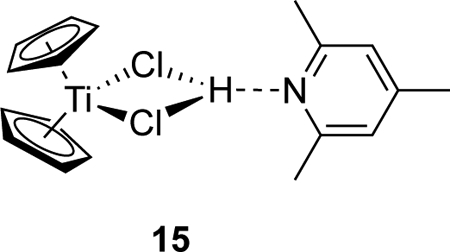
The role of 15 in other reactions of epoxides with Ti(III) has been delineated using cyclic voltammetry, kinetics, and computational studies.30 It was shown that reversible formation of 15 diminishes the concentration of active Cp2TiCl reagent, suppressing radical trapping by Ti(III) and therefore increasing radical lifetime. In reactions such as reduction to the corresponding alcohol or inter- or intramolecular addition, trapping of intermediate radicals by Cp2TiCl is an undesired intermolecular side-reaction and consequently the formation of 15 has a beneficial effect. However, mixed disproportionation (Eq. 2) requires interaction of the carbon-centered radical 7 with the active Cp2TiCl species. The above results suggest that, in the presence of excess coll•HCl, reaction of radical 7 with Ti(III) is inhibited. Consequently, the extent of mixed disproportionation is diminished and reduction of 7 by hydrogen atom abstraction increases.
This raises the question, what is the source of hydrogen in the formation of reduction product 13? That is to say that, what is serving as the hydrogen atom donor Q–H in Scheme 2? We considered five possible candidates: (1) the N–H hydrogen atom of 15, (2) the α-hydrogen of solvent THF, (3) the methyl hydrogen of collidine (4) the Ti–H hydrogen atom of Cp2Ti(H)Cl, or (5) disproportionation between two β-titanoxy radicals to give one molecule each of 9 and 13. These possibilities are illustrated in Scheme 5.31
Scheme 5.

Possible Sources of Hydrogen Atoms in Saturated Alcohols 13
The first possibility requires that the N–H bond of coll•HCl is activated (weakened) by coordination to titanium(III). This type of activation would be consistent with recent discoveries regarding the activation of heteroatom-hydrogen bonds by Ti(III). It has been shown that the O–H hydrogen of water,25,32,33 the O–H hydrogen of methanol,34 and the N–H hydrogen of amides35–37 can all be activated as hydrogen atom donors when complexed to Cp2TiCl.
The second possibility is that the α-hydrogen atoms in solvent THF are being abstracted by the intermediate β-titanoxy radicals. It can be noted that the BDE for the α-hydrogen of THF is 92 kcal/mol38 and these hydrogens might arguably be further activated when THF is coordinated to Ti(III).
The third candidate would be the “benzylic” methyl groups on 2,4,6-collidine. The concentration of collidine is low compared to that of THF, but HAT from collidine benefits from a statistical factor of 9 benzylic hydrogens per molecule. We are unaware of any reported BDE data for the C–H bonds of 2,4,6-collidine. However, the BDE for 2-picoline has been estimated to be 87.2 kcal/mol and that for 4-picoline as 86.5 kcal/mol.39 It might be argued that electron release from multiple methyl groups in collidine would further lower this number. In the early stages of a protic catalytic reaction, collidine will be present as its hydrochloride salt, which we expect will be less reactive as a hydrogen atom donor. However, as the reaction proceeds, more and more of the hydrochloride is converted to the free base. It has been noted that when collidine is added to the stoichiometric reactions of trisubstituted epoxides with Cp2TiCl, β-scission is greatly diminished and reduction via hydrogen abstraction is instead observed.23b
Fourth, it is conceivable that Cp2Ti(H)Cl is functioning as a hydrogen transfer agent. The instantaneous concentration of Cp2Ti(H)Cl will be low due to the fact that a sub-stoichiometric amount of titanium is utilized and especially due to the rapid decomposition of Cp2Ti(H)Cl under the reaction conditions. Balanced against this, the reaction of a carbon-centered radical with Cp2Ti(H)Cl results in the replacement of a weak Ti–H bond with a far more stable C–H bond and the process should be highly exothermic. For example, we calculated ΔG for the reaction of radical 7 (see Scheme 3) with Cp2Ti(H)Cl. At the PW6B95-D3 level of theory, after correction for the effect of bulk THF solvent, ΔG = −48 kcal/mol for this process. (See Supporting Information for details.)
Fifth, the final possibility would be disproportionation between two β-titanoxy groups. This process would necessarily produce equal amounts of unsaturated alcohols 9 and saturated alcohols 13.
In an effort to determine the identity of the hydrogen atom donor, isotopic labeling studies were carried out using isotopically labeled THF-d8 or collidine deuterochloride or both. These studies might militate in favor or against possibilities (1) and (2).
Table 4 summarizes GC-MS studies of the cis-13 obtained when Eq. 8 was carried out. The observed (M+1)/M ratios were adjusted for the 7.8% contribution to M+1 from natural abundance 13C. Up to 26% deuterium incorporation was observed when the reaction was carried out in THF-d8 using coll•DCl as the buffered acid.
Table 4.
GC-MS Results for Deuterium Labeling Studies
| Entry | Sample | (M+1)/Ma | D Incorporation (%)b |
|---|---|---|---|
| 1. | Authentic cis-10 | 0.072 | 0 |
| 2. | cis-10 prepared using coll•HCl in THF-d8 | 0.087 | 1 |
| 3. | cis-10 prepared using coll•DCl in protio THF | 0.25 | 14 |
| 4. | cis-10 prepared using coll•DCl in THF-d8 | 0.46 | 26 |
Ratio of m/z = 115 to m/z = 114 before correction for 7.8% contribution from 13C.
Calculated as % D = (ratio – 0.078)/(1.00 + ratio).
The location of the isotopic label in cis-13 produced in these experiments was confirmed to be the tertiary (methyl bearing) carbon using 600 MHz 1H NMR. The resonance for the tertiary hydrogen was located using a combination of COSY, HSQC, and 1D-decoupling and appears as a multiplet in the δ 1.61–1.64 region. The tertiary hydrogen resonance itself is poorly resolved due to overlapping resonances, which interferes with integration. However, the associated methyl resonance proved more useful. In protio-cis-13 the methyl group appears as a doublet (J = 6.8 Hz) at δ 0.94 ppm. For deuterium-labeled cis-10 this resonance collapses to a singlet, which exhibits the expected40 upfield isotopic shift to 0.93 ppm. Integration of these peaks was consistent with the results of GC-MS analysis.
Since the labeling studies in Table 4 account for only 26% of the saturated alcohol, the remaining hydrogen must originate from a source other than THF-d8 or coll•DCl. The remaining candidates are the methyl hydrogen atoms of collidine, the hydride of Cp2Ti(H)Cl, or direct disproportionation between β-titanoxy radicals. Distinguishing between these possibilities must await additional isotopic labeling studies, e.g. labeling of the epoxide (and indeed the final two possibilities would be difficult to distinguish based on labeling studies).
We believe that our results are best understood in terms of the different titanium(III) species present in solution during protic catalysis versus stoichiometric conditions (or aprotic catalysis). Under stoichiometric conditions, Ti(III) is mainly present as Cp2TiCl(THF).41,42 The weakly coordinated THF ligand is readily displaced so that titanium(III) is available for cross-disproportionation with a β-titanoxy radical such as 7. However, in protic catalysis, Ti(III) is complexed with coll•HCl in the form of 15.30,43 The titanium(III) is not as readily available to trap the intermediate β-titanoxy radical. Therefore, carbon-centered radical 7 survives and is ultimately quenched by hydrogen abstraction.
Earlier we noted that the order of addition of epoxide and Ti(III) impacted the products formed from epoxide 10. We observed a similar, though considerably smaller, effect in the stoichiometric reaction of epoxide 6 with Cp2TiCl. The ratio of the products of reduction (cis-13 + trans-13) to the products of H-atom loss (9a + 9b + 14) was different depending on the mode of addition. Normal addition under stoichiometric conditions gave less of the reduction product (ratio = 0.11) as compared with reverse addition of Cp2TiCl to the epoxide (ratio = 0.29). The corresponding ratio for the catalytic reactions was 0.39, although the reaction conditions are not strictly comparable, with excess coll•HCl also present in the catalytic reactions. It is not clear why the magnitude of this effect is lower for epoxide 6 versus epoxide 10 but presumably it reflects the greater steric encumbrance of 10.
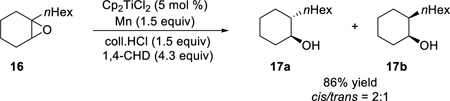
It is evident that aprotic catalysis will be superior to protic catalysis when the desired termination step from Ti(III)-catalyzed epoxide ring-opening is β-scission of hydrogen. However, it is worth reiterating that the diminished reactivity of 15 as a hydrogen atom acceptor can be beneficial for types of reactions where β-scission is an undesired side reaction.44 In particular, we note that the reduction of certain trisubstituted epoxides to the corresponding alcohol under protic catalysis is a clean and high-yielding transformation. For example, the reduction of epoxide 16 in Eq. 9 affords the secondary alcohol 17 in 86% yield as a 2:1 mixture of diastereomers using just 5% of Cp2TiCl2 as catalyst.45 In this reaction, 1,4-cyclohexadiene functions as the hydrogen atom donor.
 |
(9) |
Finally, several attempts were made to observe Cp2Ti(H)Cl, generated according to Scheme 3, using in situ infrared spectrometry. We hoped to observe the v(Ti–H) band in the 1600 cm−1 region by analogy to the strong band observed at 1605 cm–1 for Cp*2Ti(H)Cl.46 However, rapid gas evolution disrupted the operation of the IR detector of the spectrometer. It is noteworthy that gas evolution was rapid even when the reaction was carried out at –40 °C.
CONCLUSIONS
The combination of experimental work and computations used in this study has provided a series of surprises and has several practical consequences. The conventional wisdom that Cp2Ti(H)Cl is a robust species that resists reduction to the Ti(III) oxidation state is not correct. On the contrary, Cp2Ti(H)Cl spontaneously decomposes to regenerate Cp2TiCl and molecular hydrogen. The failure of protic catalytic conditions to cleanly promote the conversion of trisubstituted epoxides to allylic alcohols is the result of competing hydrogen atom transfer rather than an interruption in catalyst turnover. Aprotic catalysis does not require the putative σ-bond metathesis of Cp2Ti(H)Cl with Me3SiCl. The stoichiometric reactions of trisubstituted epoxides with Cp2TiCl have previously been carried out using 2 or 3 equivalents of the titanium(III) reagent. Our results demonstrate that a single equivalent of Cp2TiCl will suffice. (Nevertheless, it appears prudent to use a ca. 20% excess of Cp2TiCl to avoid potential side reactions when the titanium(III) concentrations becomes very low near the end of the reaction.) The previously undocumented, rapid evolution of hydrogen may represent a safety concern for emerging large scale applications of this chemistry.47
The fundamental difference between protic and aprotic catalysis of the reaction of Cp2TiCl with epoxides is the form of Ti(III) that is present. In aprotic catalysis, Ti(III) is present as Cp2TiCl(THF) where the titanium(III) center is available to participate in reactions with carbon-centered radicals. In protic catalysis, Cp2TiCl and coll•HCl form the stable complex 15 which less readily reacts with carbon-centered radicals. For these reasons, protic and aprotic catalysis of these reactions are in fact complementary protocols. Aprotic catalysis will generally prove superior when scission of β-hydrogen or deoxygenation is the desired termination step. Protic catalysis will be advantageous when reduction of the epoxide via hydrogen atom transfer is desired, especially with an added hydrogen atom donor such as 1,4-cyclohexadiene.
EXPERIMENTAL SECTION
Reaction of Cp2TiCl with 1,2-epoxy-1-methylcyclohexane (6).
Titanocene dichloride (0.498 g, 2.00 mmol) and zinc dust (0.393 g, 6.00 mmol) were placed in a Schlenk flask which was then flushed with argon. To the flask were added THF (6.0 mL) and tert-butyl methyl ether (238 mL, 0.177 g, 2.00 mmol) as an internal standard. The mixture was stirred for 30 minutes at which time the initial red color had discharged to lime green. The flask was closed to argon and a thin tube was fed from the flask into an inverted burette filled with water (see photograph in Supporting Information). A solution of epoxide (0.225 g, 2.00 mmol) in 1.5 mL of THF was added slowly and as gas evolved, it bubbled into the burette to displace 22.4 mL of water. The reaction was quenched with 2.5 mL of saturated aqueous sodium hydrogen carbonate. Gas chromatographic analysis indicated the formation of allylic alcohols 9a and 9b (1.88 mmol) in a 5:1 ratio. The solution was filtered over Celite, rinsing with THF, and was subjected to flash chromatography (4:1 pentane/diethyl ether). CDCl3 was added to the residue and removed at reduced pressure to obtain an approximately 3:2 mixture of 2-methylenecyclohexan-1-ol 9a (major) and 2-methylcyclohex-2-ene-1-ol 9b (minor) free of residual solvents. NMR analysis and comparison with literature data48,49 allowed the assignment of the spectra as follows. For the major product 9a: 1H NMR (400 MHz, CDCl3) δ 4.90 (s, 1H), 4.77 (s, 1H), 4.09–4.13 (m, 1H), 2.41–2.45 (m, 1H), 1.94–1.53 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 151.6, 105.0, 72.7, 36.7, 33.5, 27.7, 23.8 GC-MS m/z 112.10 ([M+]); calcd for C7H12O 112.09. For the minor product 9b: 1H NMR (400 MHz, CDCl3) δ 5.56 (br s, 1H), 4.00 (t, J = 4.4 Hz)(m, 1H), 1.94–1.53 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 135.3, 125.5, 68.5, 32.2, 25.4, 20.6, 18.1; GCMS m/z 112.10 ([M+]); calcd for C7H12O 112.09.
Procedure for Catalytic Reactions in Table 3.
In a Schlenk tube, 2,4,6-collidinium chloride (1.50 eq) is sublimed in vacuo under careful heating. After cooling to room temperature, the solids are flushed with argon and transferred back to the bottom of the flask. Zn powder (3.00 eq) and Cp2TiCl2 (0.05 to 1.00 eq) are added and the solids are evacuated and flushed with argon three times. Dry THF (substrate 0.25 M) is added via syringe and the reaction is stirred for 15 minutes at room temperature. The solution turns green within minutes. 1,2-Epoxy-1-methylcyclohexane (1.00 eq) is added via syringe and the reaction is stirred under argon at room temperature for the indicated time. Saturated aqueous NH4Cl solution is added (100% v/v THF) and the aqueous phase is extracted with ethyl acetate three times. The combined yellow organic phase is filtered through a short silica pad. The solvent is removed under reduced pressure (not below 150 mbar at 40 °C, due to volatility of the substrate and products) to yield the crude product that is subjected to GC-MS analysis.
A sample of the product obtained under the conditions of Table 3, entry 2 was carefully chromatographed on silica using 10% ether in pentane to collect various fractions for identifications. The order of elution was as follows: cis-13 Me at δ 0.939, J = 6.6 Hz, α-H at δ 3.77), 9a α-H at δ 4.089, 9b α-H at δ 3.987, trans-13 Me at δ 1.004, J =6.6 Hz, α-H at δ 3.10). The ketone 14 and the cis-13 and trans-13 alcohols were further confirmed by co-injection in the GC with authentic samples. Samples of the saturated alcohols were obtained by NaBH4 reduction of the commercially available 14 (ratio of cis-13 to trans-13 42:58). Pure samples of cis-13 to trans-13 were isolated by careful column chromatography on silica gel using 20:1 pentane:ether as eluent. Spectra for cis-13 and trans-13 match those in the literature.50 The C2-H in cis-13 was identified by 1H COSY and decoupling experiments. Irradiation of the CH3 (δ 0.939) doublet shows simplification of the multiplet centered around δ 1.61–1.64. Conversely, decoupling of the broad peak at δ 1.63 causes the CH3 signal to appear as a singlet.
Preparation of Collidine Deuterochloride for Isotopic Labeling Studies.
A Schlenk flask equipped with a stir bar was charged with 2,4,6-collidine (1 mL, 7.57 mmol, d = 0.917 g/mL, 1 equiv) in a N2 filled glove box before transferring to the Schlenk line. Diethyl ether (5 mL) was added followed by a commercial 1M deuterium chloride (96% D) in ether solution (9.0 mL, 9.0 mmol, 1.2 equiv). A white solid precipitate formed immediately and was stirred for several minutes. The remaining Et2O was decanted and removed with a syringe through the septum. The solid was washed with Et2O (3 × 10 mL) and decanted using a syringe. The flask was then returned to the glovebox and the remaining Et2O was removed under vacuum to yield coll•DCl (0.996 g, 83% yield). The deuterium content was established to be ca. 90% by integration of the residual NH resonance in the 1H NMR at δ 17.3 ppm. 1H NMR (600 MHz, CDCl3) δ 2.52 (s, 6H), 2.90 (s, 3H), 7.20 (s, 2H); 13C NMR (151 MHz, CDCl3) δ 19.3, 22.1, 125.2, 153, 157.9.
Procedure for Isotopic Labeling Studies in Table 4.
In a N2 filled glove box an oven-dried Schlenk flask (or a 20 mL screw-capped vial for small scale reactions) equipped with a stir bar was charged with collidine•DCl (238 mg, 1.5 equiv) or collidine•HCl (235 mg, 1.5 equiv), activated Zn (199 mg, 3 equiv), and Cp2TiCl2 (62 mg, 0.25 equiv) after which was added distilled THF or THF-d8 (4 mL, to make a 0.25 M solution in epoxide). The greenish turbid solution was stirred for 15 min before 1,2-epoxy-1-methylcyclohexane (124 μL, 1.00 mmol, 1 equiv, density = 0.905 g mL−1) was added via a micro-syringe. The solution showed a slight reddish color and gas evolution was observed. After stirring for 1 h, the reaction was quenched with saturated NH4Cl (4 mL) and extracted with Et2O. The yellow solution was filtered over a pad of silica, eluted with Et2O (100 mL), and the solvent evaporated under reduced pressure to obtain the crude product, which was analyzed by GC-MS (HP-5MS 5% Phenyl methyl silicone (HP-5) capillary column, 30 m length; 250 μm diameter; 0.25 μm film thickness; 35 °C 5 min, 5 °/min to 250 °C and hold 2 min). The relative intensity for the M and M+1 peaks was determined by integrating the total ion current for the cis-10 molecular ion (retention time 3.39 min) at m/Z = 114 and 115. For details of the determination of isotopic composition of products, see Supporting Information.
Computational Details.
DFT calculations were carried out with the TURBOMOLE 7.0 program package.51 Geometries were first optimized at the TPSS52-D353,54/def2-TZVP55 level of theory. Final reaction free energy values were obtained through single-point calculations on the PW6B9556-D3 or B3LYP57,58-D3 level in the gas phase. Solvation contributions to ΔG were included using the COSMO-RS continuum solvation model.59,60 For a full description of the computational methods and data, see Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the SFB 813 (“Chemistry at Spin Centers”) and Ga-619/12–1 to AG and the U. S. National Institutes of Health (R01 GM108762), and NSF (CHE-1362095) to TVR. S. H. and S. K. thank the Jürgen Manchot Stiftung for doctoral fellowships. J. G. and K. R. D acknowledge financial assistance by the Ohio State University.
Footnotes
Supporting Information
General experimental details, preparation of 6 and authentic cis- and trans-13, 1H and 13C NMR data, GC-MS chromatograms, mechanistic discussion of β-hydrogen scission, complete computational details and Cartesian coordinates for optimized structures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES AND NOTES
- (1).Sakai S Theoretical study on the reaction mechanisms of ethylene with Cp2Ti+R, Cp2Ti(Cl)R, and Cp2Ti(Cl:AlH2Cl)R {R = H and CH3). J. Mol. Struct.: Theochem, 2001, 540, 157–169. [Google Scholar]
- (2).Narasaka K; Iwasawa N; Inoue M; Yamada T; Nakashima M; Sugimori J Asymmetric Diels-Alder Reaction Catalyzed by a Chiral Titanium Reagent. J. Am. Chem. Soc 1989, 111, 5340–5345. [Google Scholar]
- (3).Fuse S; Hanochi M; Doi T; Takahashi T Ti(III) radical cyclization of 6,7-epoxygeranyl acetate. Tetrahedron Lett 2004, 45, 1961–1963. [Google Scholar]
- (4).Justicia J; Rosales A; Bunuel E; Oller-Lopez JL; Valdivia N; Haidour A; Oltra JE; Barrero AF; Cardenas DJ; Cuerva JM Titanocene-Catalyzed Cascade Cyclization of Epoxypolyprenes: Straightforward Synthesis of Terpenoids by Free-Radical Chemistry. Chem. Eur. J 2004, 10, 1778–1788. [DOI] [PubMed] [Google Scholar]
- (5).Justicia J; Oller-Lopez JL; Campana AG; Oltra JE; Cuerva JM; Bunuel E; Cardenas DJ 7-endo Radical Cyclization Catalyzed by Titanocene(III). Straightforward Synthesis of Terpenoids with Seven-Membered Carbocycles. J. Am. Chem. Soc 2005, 127, 14911–14921. [DOI] [PubMed] [Google Scholar]
- (6).Birmingham JM; Fischer AK; Wilkinson G The reduction of biscyclopentadienyl compounds. Naturwissenschaften 1955, 42, 96–96. [Google Scholar]
- (7).Green MLH; Lucas CR Some d1 Bis-π-cyclopentadienyl Titanium Complexes with Nitrogen or Phosphorus Ligands. J. Chem. Soc., Dalton Trans 1972, 1972, 1000–1003. [Google Scholar]
- (8).Schwartz J; Labinger JA Hydrozirconation: A New Transition Metal Reagent for Organic Synthesis. Angew. Chem. Int. Ed 1976, 15, 333–340. [Google Scholar]
- (9).Wipf P; Jahn H Synthetic applications of of organochlorozirconocene complexes. Tetrahedron 1996, 52, 12853–12910. [Google Scholar]
- (10).Luinstra GA; Teuben JH Lead Dichloride: a Mild Reagent for the Oxidation of Tervalent Titanium Compounds (η5-C5Me5]2TiR to Monochloride derivatives (η5-C5Me5)2R(Cl). J. Chem. Soc., Chem. Commun 1990, 1470–1471. [Google Scholar]
- (11).de Wolf JM; Meetsma A; Teuben JH Synthesis and Structure of Bis(phenyltetramethylcyclopentadienyl)titanium(III) Hydride: The First Monomeric Bis(cyclopentadienyl)titanium(III) Hydride. Organometallics 1995, 14, 5466–5468. [Google Scholar]
- (12).Rosales A; Rodríguez-García I; Muñoz-Bascón J; Roldan-Molina E; Padial NM; Morales LP; García-Ocaña M; Oltra JE The Nugent-RajanBabu Reagent: A Formidable Tool in Contemporary Radical and Organometallic Chemistry. Eur. J. Org. Chem 2015, 2015, 4567–4591. [Google Scholar]
- (13).Barrero AF; del Moral JFQ; Sanchez EM; Arteaga JF Titanocene-Mediated Radical Cyclization: An Emergent Method for the Synthesis of Natural Products. Eur. J. Org. Chem 2006, 2006, 1627–1641. [Google Scholar]
- (14).Cuerva JM; Justicia J; Oller-Lopez JL; Bazdi B; Oltra JE The Growing Impact of Titanocene(III)-Mediated Radical Epoxide Opening on the Synthesis of Natural Products. Mini-Rev. Org. Chem 2006, 3, 23–35. [Google Scholar]
- (15).RajanBabu TV; Nugent WA Selective Generation of Free Radicals from Epoxides Using a Transition-Metal Radical: A Powerful New Tool for Organic Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]
- (16).Gansäuer A Titanocenes as Electron Transfer Catalysts: Reagent Controlled Catalytic Radical Reactions. Synlett 1998, 801–809. [Google Scholar]
- (17).Gansäuer A; Bluhm H; Pierobon M Emergence of a Novel Catalytic Radical Reaction: Titanocene-Catalyzed Reductive Opening of Epoxides. J. Am. Chem. Soc 1998, 120, 12849–12859. [Google Scholar]
- (18).Chakraborty TK; Dutta S Radical-Induced Opening of Trisubstituted Epoxides: Application in the Synthesis of the C1–C12 Segment of Epothilones. Tetrahedron Lett 1998, 39, 101–104. [Google Scholar]
- (19).Fernández-Mateos A; de la Nava E. M. n.; Coca GP; Silvo AR; González RR, Radicals from Epoxides: Intramolecular Addition to Aldehyde and Ketone Carbonyls. Org. Lett 1999, 1, 607–609. [Google Scholar]
- (20).Nakai K; Kamoshita M; Doi T; Yamada H; Takahashi T Stereo- and regio-selective Ti-mediated radical cyclization of epoxy-alkenes: synthesis of the A and C ring synthons of paclitaxel. Tetrahedron Lett 2001, 42, 7855–7857. [Google Scholar]
- (21).Bermejo F; Sandoval C Cp2TiCl-Promoted Isomerization of Trisubstituted Epoxides to exo-Methylene Allylic Alcohols on Carvone Derivatives. J. Org. Chem 2004, 69, 5275–5280. [DOI] [PubMed] [Google Scholar]
- (22).Justicia J; Jimenez T; Morcillo SP; Cuerva JM; Enrique Oltra J Mixed disproportionation versus radical trapping in titanocene(III)-promoted epoxide openings. Tetrahedron 2009, 65, 10837–10841. [Google Scholar]
- (23).(a) Perez Morales C; Catalan J; Domingo V; Gonzalez Delgado JA; Dobado JA; Mar Herrador M; Quilez del Moral JF; Barrero AF Protecting-Group-Free Synthesis of Chokols. J. Org. Chem 2011, 76, 2494–2501. [DOI] [PubMed] [Google Scholar]; (b) When 2 equiv of triethylamine was replaced with 2 equiv of 2,4,6-collidine, the yield of β-scission product in this study was diminished from 66% to 22% and the yield of hydrogen abstraction product increased from <15% to 45%. This is consistent with collidine acting as a hydrogen transfer reagent.
- (24).Barrero AF; Cuerva JM; Herrador MM; Valdivia MV A New Strategy for the Synthesis of Cyclic Terpenoids Based on the Radical Opening of Acyclic Epoxypolyenes. J. Org. Chem 2001, 66, 4074–4078. [DOI] [PubMed] [Google Scholar]
- (25).Barrero AF; Oltra JE; Cuerva JM; Rosales A Effects of Solvent and Water in Ti(III)-Mediated Radical Cyclizations of Epoxygermacrolides. Straightforward Synthesis and Absolute Stereochemistry of (+)-3α-Hydroxyreynosin and Related Eudesmanolides. J. Org. Chem 2002, 67, 2566–2571. [DOI] [PubMed] [Google Scholar]
- (26).Barrero AF; Rosales A; Cuerva JM; Oltra JE Unified Synthesis of Eudesmanolides, Combining Biomimetic Strategies with Homogeneous Catalysis and Free-Radical Chemistry. Org. Lett 2003, 5, 1935–1938. [DOI] [PubMed] [Google Scholar]
- (27).Justicia J; Alvarez de Cienfuegos L; Estevez RE; Paradas M; Lasanta AM; Oller JL; Rosales A; Cuerva JM; Enrique Oltra J Ti-catalyzed transannular cyclization of epoxygermacrolides. Synthesis of antifungal (+)-tuberiferine and (+)-dehydrobrachyaenolide. Tetrahedron 2008, 64, 11938–11943. [Google Scholar]
- (28).Filatov ES; Simanov EF; Orlova MA Reactivity of Hydrogen Atoms. Uspekhi Khimii 1981, 50, 2167–2187. [Google Scholar]
- (29).Gansäuer A; Behlendorf M; von Laufenberg D; Fleckhaus A; Kube C; Sadasivam DV; Flowers RA II. Catalytic Atom-Economical Radical Arylation of Epoxides. Angew. Chem. Int. Ed 2012, 51, 4739–4742. [DOI] [PubMed] [Google Scholar]
- (30).Gansäuer A; Kube C; Daasbjerg K; Sure R; Grimme S; Fianu GD; Sadasivam DV; Flowers RA II. Substituent Effects and Supramolecular Interactions of Titanocene(III) Chloride: Implications for Catalysis in Single Electron Steps. J. Am. Chem. Soc 2014, 136, 1663–1671. [DOI] [PubMed] [Google Scholar]
- (31).It cannot be completely ruled out that radical 7 could be trapped by Cp2TiCl affording a 3° alkyltitanium species which then undergoes protonolysis to afford saturated alcohols 13. We believe this is unlikely for reasons discussed in detail in the Supporting Information pp. S13-S14. Moreover, the results in Table 4 would require that this be a minor pathway. Nevertheless, it can be noted that Cp2Ti(R)Cl, R = 3° alkyl can be prepared for bridgehead alkyl derivative where β-hydride elimination would result in formation of an anti-Bredt alkene:Dimitrov V Preparation and Properties of Cyclopentadienyltitanium(IV)-1-Norbornyl Compounds. J. Organometal. Chem 1985, 282, 321–329.
- (32).Cuerva JM; Campaña AG; Justicia J; Rosales A; Oller-López JL; Robles R; Cárdenas DJ; Buñuel E; Oltra JE Water: The Ideal Hydrogen-Atom Source in Free-Radical Chemistry Mediated by TiIII and Other Single-Electron-Transfer Metals? Angew. Chem. Int. Ed 2006, 45, 5522–1526. [DOI] [PubMed] [Google Scholar]
- (33).Gansäuer A; Behlendorf M; Cangönül A; Kube C; Cuerva JM; Friedrich J; van Gastel M H2O Activation for Hydrogen-Atom Transfer: Correct Structures and Revised Mechanisms. Angew. Chem. Int. Ed 2012, 51, 3266–3270. [DOI] [PubMed] [Google Scholar]
- (34).Jin J; Newcomb M Rate Constants for Hydrogen Atom Transfer Reactions from Bis(cyclopentadienyl)titanium(III) Chloride-Complexed Water and Methanol to an Alkyl Radical. J. Org. Chem 2008, 73, 7901–7905. [DOI] [PubMed] [Google Scholar]
- (35).Zhang Y-Q; Jakoby V; Stainer K; Schmer A; Klare S; Bauer M; Grimme S; Cuerva JM; Gansäuer A Amide-Substituted Titanocenes in Hydrogen-Atom Transfer Catalysis. Angew. Chem., Int. Ed 2016, 55, 1523–1526. [DOI] [PubMed] [Google Scholar]
- (36).Tarantino KT, Miller DC; Callon TA; Knowles RR Bond-Weakening Catalysis: Conjugate Aminations Enabled by the Soft Homolysis of Strong N–H Bonds. J. Am. Chem. Soc 2015, 137, 6440–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Diethelm S; Schindler CS; Carreira EM Access to the Aeruginosin Serine Protease Inhibitors through the Nucleophilic Opening of an Oxabicyclo[2.2.1]heptane: Total Synthesis of Microcin SF608. Chem. Eur. J 2014, 20, 6071–6080. [DOI] [PubMed] [Google Scholar]
- (38).Laarhoven LLJ; Mulder P α-C–H Bond Strengths in Tetralin and THF: Application of Competition Experiments in Photoacoustic Calorimetry. J. Phys. Chem 1997, 101, 73–77. [Google Scholar]
- (39).Denisov ET; Tomanov VE Estimation of bond dissociation energies from the kinetic characteristics of liquid-phase radical reactions. Russ. Chem. Rev 2005, 74, 825–858. [Google Scholar]
- (40).O’Leary DJ; Allis DG; Hudson BS; James S; Morgera KB; Baldwin JE Vicinal Deuterium Perturbations on Hydrogen NMR Chemical Shifts in Cyclohexanes. J. Am. Chem. Soc 2008, 130, 13659–13663. [DOI] [PubMed] [Google Scholar]
- (41).Enemérke RJ; Larsen J; Skrydstrup T; Daasbjerg K Revelation of the Nature of the Reducing Species in Titanocene Halide-Promoted Reductions. J. Am. Chem. Soc 2004, 126, 7853–7864. [DOI] [PubMed] [Google Scholar]
- (42).Gansäuer A; Barchuk A; Keller F; Schmitt M; Grimme S; Gerenkamp M; Mueck-Lichtenfeld C; Daasbjerg K; Svith H Mechanism of Titaonocene-Mediated Epoxide Opening through Homolytic Substitution. J. Am. Chem. Soc 2007, 129, 1359–1371. [DOI] [PubMed] [Google Scholar]
- (43).Jaraiz M; Enriquez L; Pinacho R; Rubio JE; Lessari A; Lopez-Perez JL A DFT-Based Computational-Experimental Methodology for Synthetic Chemistry: Example of Application to the Catalytic Opening of Epoxides by Titanocene. J. Org. Chem 2017, 82, 3760–3766. [DOI] [PubMed] [Google Scholar]
- (44).Moreover, it has also been observed that ring-opening reactions of epoxides with Ti(III) form less deoxygenation side-products (reduction of epoxide to alkene) when run under protic catalysis as compared to stoichiometric conditions when the reactions are performed under comparable conditions (excess 1,4-cyclohexadiene).Gansäuer A; Pierobon M; Bluhm H Catalytic, Highly Regio- and Chemoselective Generation of Radicals from Epoxides: Titanocene Dichloride as an Electron Transfer Catalyst in Transition Metal Catalyzed Reactions. Angew. Chem. Int. Ed 1998, 37, 101–103.
- (45).Gansäuer A; Klatte M; Braendle GM; Friedrich J Catalytic Hydrogen Atom Transfer (HAT) for Sustainable and Diastereoselective Radical Reduction. Angew. Chem. Int. Ed 2012, 51, 8891–8894. [DOI] [PubMed] [Google Scholar]
- (46).Luinstra GA; Teuben JH; Brintzinger H-H IR studies at elevated pressures III. Kinetics of the CO induced disproportionation of (C5(CH3)5)2TiX (X = Cl, Br, I). J. Organometal. Chem 1989, 375, 183–190. [Google Scholar]
- (47).Castro-Rodriguez M; Rodriguez-Garcia I; Rodriguez-Maecker R; Pozo-Morales L; Oltra JE; Rosales Martinez A Cp2TiCl: An Ideal Reagent for Green Chemistry? Org. Process Res. Dev 2017, 21, 911–923. [Google Scholar]
- (48).Schomaker JM; Pulgam VR; Borhan B Synthesis of diastereomerically and enantiomerically pure 2,3-disubstituted tetrahydrofurans using a sulfoxonium ylide. J. Am. Chem. Soc 2004, 126, 13600–13601. [DOI] [PubMed] [Google Scholar]
- (49).Lhermet R; Durandetti M; Maddaluno J Intramolecular carbonickelation of alkenes. Beilstein J. Org. Chem 2013, 9, 710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wiitala KW; Al-Rashid FZ; Dvornikovs V; Hoye TR; Cramer CJ Evaluation of various DFT protocols for computing 1H and 13C chemical shifts to distingush stereoisomers: diastereomeric 2-, 3-, and 4-methylcyclohexanols as a test case. J. Phys. Org. Chem 2007, 20, 345–354. [Google Scholar]
- (51).Furche F; Ahlrichs R; Hättig C; Klopper W; Sierka M; Weigend F Turbomole. Wiley Interdisciplinary Reviews: Comput. Mol. Sci 2014, 4, 91–100. [Google Scholar]
- (52).Tao J; Perdew J; Staroverov V; Scuseria G Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett 2003, 91, 146401/1–146401/4. [DOI] [PubMed] [Google Scholar]
- (53).Grimme S; Antony J; Ehrlich S; Krieg H A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132, 154104/1–154104/19. [DOI] [PubMed] [Google Scholar]
- (54).Grimme S; Ehrlich S; Goerigt L Effect of damping function in dispersion corrected density functional theory. J. Comput. Chem 2011, 1456–1465. [DOI] [PubMed] [Google Scholar]
- (55).Weigend F; Ahlrichs R Balanced basis sets of split-valance, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (56).Zhao Y; Truhlar DG Design of Density Functions that Are Broadly Accurate for Thermochemistry, Thermochemical Kinetics, and Nonbonded Interactions. J. Phys. Chem. A 2005, 5656–5667. [DOI] [PubMed] [Google Scholar]
- (57).Becke AD, Density-functional thermochemistry III: The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (58).Stephens PJ; Devlin J; Chabalowski CF; Frisch MJ Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem 1994, 98, 11623–11627. [Google Scholar]
- (59).Klamt A Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitiative Calculation of Solvation Phenomena. J. Phys. Chem 1995, 99, 2224–2235. [Google Scholar]
- (60).Eckert F; Klamt A Fast solvent screening via quantum chemistry: COSMO-RS approach. AIChE J 2002, 48, 369–385. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.