

Abstract
Crystal structures exist for human, but not rodent, estrogen receptor-α ligand-binding domain (ERα-LBD). Consequently, rodent studies involving binding of compounds to ERα-LBD are limited in their molecular-level interpretation and extrapolation to humans. Because the sequences of rodent and human ERα-LBDs are > 95% identical, we expected their 3D structures and ligand binding to be highly similar. To test this hypothesis, we used the human ERα-LBD structure (PDB 3UUD) as a template to produce rat and mouse homology models. Employing the rodent models and human structure, we generated docking poses of 23 Group A ligands (17ß-estradiol, diethylstilbestrol, and 21 paraben analogs) in AutoDock Vina for interspecies comparisons. Ligand RMSDs (Å) (median, 95% CI) were 0.49 (0.21–1.82) (human-mouse) and 1.19 (0.22–1.82) (human-rat), well below the 2.0–2.5 Å range for equivalent docking poses. Numbers of interspecies ligand-receptor residue contacts were highly similar, with Sorensen Sc (%) = 96.8 (90.0–100) (human-mouse) and 97.7 (89.5–100) (human-rat). Likewise, numbers of interspecies ligand-receptor residue contacts were highly correlated: Pearson r = 0.913 (human-mouse) and 0.925 (human-rat). Numbers of interspecies ligand-receptor atom contacts were even more tightly correlated: r = 0.979 (human-mouse) and 0.986 (human-rat). Pyramid plots of numbers of ligand-receptor atom contacts by residue exhibited high interspecies symmetry and had Spearman rs = 0.977 (human-mouse) and 0.966 (human-rat). Group B ligands included 15 ring-substituted parabens recently shown experimentally to exhibit decreased binding to human ERα and to exert increased antimicrobial activity. Ligand efficiencies calculated from docking ligands into human ERα-LBD were well correlated with those derived from published experimental data (Pearson partial rp = 0.894 and 0.918; Groups A and B, respectively). Overall, the results indicate that our constructed rodent ERα-LBDs interact with ligands in like manner to the human receptor, thus providing a high level of confidence in extrapolations of rodent to human ligand-receptor interactions.
Keywords: estrogen receptor-α, homology model, in silico mutagenesis, ligand-receptor docking, paraben analogs, species comparisons
Graphical Abstract:
1. Introduction
Numerous toxicology studies have depended on rodents as sources of in vivo models [1, 2] or in vitro screening assays [3–5] to identify and characterize suspected endocrine disrupting compounds (EDCs). Traditional methods of generating toxicity data for risk assessment that rely on animal models or even in vitro assays can quickly become too costly or time-consuming to adequately screen and establish toxicological profiles for the tens of thousands of chemicals cataloged by the United States Environmental Protection Agency [6]. Efforts are underway to minimize the use of rodent models and establish new approach methodologies [7] that could be used more routinely to screen and identify suspected EDCs via in silico approaches [8, 9]. However, attempts to establish in silico protocols for identifying EDCs that act on estrogen signaling pathways are hampered due to the lack of reported structural data for mouse or rat estrogen receptor-α (ERα) within the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) [10, 11].
Interspecies sequence identities for the entire ERα receptor are 88.5% (human-mouse), 87.5% (human-rat), and 97.5% (mouse-rat). For the ligand binding domain (ERα-LBD) alone, the interspecies sequence identities are 95.5% (human-mouse), 95.1% (human-rat), and 99.2% (mouse-rat) [12] (Fig. 1). We therefore hypothesized that these receptor species should display similar 3D structures and exhibit comparable ligand binding interactions with known agonists. Although this contention appears logical and likely, subtle differences in the protein sequence could affect tertiary structure and/or ligand binding to a given receptor [13]. For example, several clinically relevant mutations resulting in single amino acid substitutions in the human ERα-LBD have been shown to confer a ligand-independent phenotype compared to the wild type ERα-LBD [14–16], which further demonstrates the apparent structural and functional sensitivity of the ERα-LBD to small differences in its primary sequence.
Fig. 1.

Multiple protein sequence alignment of human, mouse, and rat ERα. Residues differing from the human in any sequence are shaded gray. Domains: A/B (AF1) = magenta; C (DNA binding) = yellow; D (Hinge region) = light red; E (Ligand Binding Domain /AF2; region encompassed by PDB 3UUD) = green; F (C-terminal extension of Ligand Binding Domain/AF2) = light blue. The “X” residues 381 and 530 marked with orange annotations in the human sequence are hydroxyCys that were kept as Cys in the mouse and rat homology models. S537 marked with a red annotation in the human sequence is the Y537S mutation that was introduced to stabilize the agonist conformation in PDB 3UUD; the mouse and rat homology models were likewise mutated to serine residues at this site. Sequences were downloaded from Uniprot (www.uniprot.com). Alignment was carried out by Geneious 11.1.5 (Geneious, 2018; Kearse et al., 2012) using the Clustal-Omega algorithm. Domain assignments were adapted from Uniprot (2018a-c) and Sanchez et al. (2002).
To determine whether the differing residues among the rodent and human ERα-LBD receptors could be considered neutral substitutions, we created ERα-LBD structures for the unreported rodent species via in silico mutagenesis of a human ERα-LBD template using the YASARA molecular modeling suite [17]. The construction of these in silico receptors allowed us to address the lack of reported mouse and rat ERα-LBD structures and compare how each of these ERα-LBDs might interact with known or postulated ERα agonists using molecular docking simulations. Comparative docking into rodent and human ERα-LBDs was carried out with 23 compounds designated Group A ligands. These compounds included 17β-estradiol (E2), diethylstilbestrol (DES), 17 paraben analogs and 4 paraben metabolites (Fig. 2, Table 1).
Fig. 2.

Structures of Group A ligands used for docking studies. R-groups for parabens are listed in Table 1.
Table 1.
Group A ligands and their computed pKa and SASA values.
| Compound | Paraben R-groupa or Chemical Name |
pKab | SASA (Å2)c |
|---|---|---|---|
|
Parabens MP |
Methyl | 8.43 | 322.94 |
| EP | Ethyl | 8.54 | 353.49 |
| PrP | Propyl | 8.67 | 382.68 |
| BuP | Butyl | 8.78 | 412.14 |
| PeP | Pentyl | 8.88 | 441.39 |
| HxP | Hexyl | 8.96 | 471.17 |
| HpP | Heptyl | 9.02 | 499.83 |
| OcP | Octyl | 9.08 | 529.01 |
| NnP | Nonyl | 9.13 | 552.71 |
| DecP | Decyl | 9.18 | 581.77 |
| UnDecP | Undecyl | 9.22 | 611.46 |
| DoDecP | Dodecyl | 9.26 | 640.59 |
| iPrP | Iso-propyl | 8.61 | 378.45 |
| iBuP | Iso-butyl | 8.77 | 403.42 |
| iPeP | Iso-pentyl | 8.87 | 432.28 |
| PhP | Phenyl | 8.53 | 405.94 |
| BzP | Benzyl | 8.73 | 435.85 |
|
Parabens metabolites 4OH |
4-hydroxybenzoic acid | 4.01 | 280.28 |
| 2OH | 2-hydroxy-iso-butyl 4-hydroxybenzoate | 8.75 | 410.28 |
| 3OHR | (R)-3-hydroxy n-butyl 4-hydroxybenzoate | 8.77 | 416.90 |
| 3OHS | (S)-3-hydroxy n-butyl 4-hydroxybenzoate | 8.77 | 415.76 |
|
Established ERα agonists E2 |
17β-estradiol | 10.06 | 457.02 |
| DES | diethylstilbestrol | 10.31 | 489.45 |
Unless designated otherwise, all alkyl groups are normal (n) straight chains.
Most acidic pKa computed with ADMET_Predictor 9.0.
Solvent-accessible surface area computed with ADMET_Predictor 9.0.
Molecular docking of E2, the most potent endogenous estrogen in both humans and rodents [18, 19], with the human and rodent ERα-LBDs provided us with a point of comparison for analyzing ligand-residue interactions among different ERα-LBDs agonists. For further calibration, we included DES, a potent synthetic nonsteroidal ERα agonist [20] that has been used as a model compound in the characterization of EDCs with estrogenic activity [21].
Parabens (esters of p-hydroxybenzoic acid) constitute a class of chemicals that have received considerable [3, 22] and controversial [23, 24] attention as suspected EDCs. Although they are found in relatively low concentrations in human tissues and possess only weak estrogenic and antiandrogenic activity [25, 26], parabens are ubiquitous in the environment owing to their widespread use as preservatives in a variety of foods and personal care products [27]. However, apart from concerns about potential adverse health impacts of parabens, the main purpose of the present study was to use members of a homologous series of paraben compounds, paraben metabolites, E2, and DES as tools in molecular docking simulations to assess the degree of similarity of ligand binding between human and rodent ERα-LBDs. A set of experimental estrogenic activity data for the human ERα was also available for 13 of the parabens and E2 [28], enabling us to compare these results with corresponding potencies derived from docking.
Recently, it has been found that various substitutions in the benzene ring of n-butyl and n-octyl parabens, especially in the 3,5-position, result in decreased binding to human ERα with concomitant enhancement of antimicrobial activity [29]. This discovery opens up the possibility of replacing existing parabens with analogs that are more effective preservatives and even less likely to act as EDCs. Moreover, this publication provided us with an additional consistent set of experimental structure-activity data on paraben analogs for assessing the effectiveness of our docking protocols. Accordingly, we carried out docking of these 15 compounds (designated Group B ligands, Fig. 3, Table 2) into the human ERα-LBD to determine the extent to which computationally predicted affinities agreed with experimentally determined binding potencies.
Fig. 3.

Structures of Group B ligands used for docking studies. Compounds are parabens with various substituents in the benzene ring. Compound designations are the same as in (Bergquist et al., 2018) as listed in Table 2. R′ = butyl; R′′ = octyl.
Table 2.
Group B ligands and their computed pKa and SASA values.
| Compounda | Added Ring Substituentsb |
pKac | SASA (Å2)d |
|---|---|---|---|
|
n-Butyl parabens BuP |
None | 8.78 | 412.14 |
| 2a | 2,3,5,6-tetrafluoro | 3.73 | 434.90 |
| 2b | 3,5-dichloro | 5.28 | 465.74 |
| 2c | 3,5-dibromo | 5.30 | 483.57 |
| 2d | 3-bromo | 7.03 | 447.79 |
| 2e | 3,5-diiodo | 4.69 | 508.60 |
| 2f | 3-iodo | 6.68 | 460.43 |
| 2g | 3,5-dimethyl | 8.94 | 471.89 |
| 2h | 3,5-di-tert-butyl | 9.50 | 601.17 |
| 2i | 3,5-dihydroxy | 7.51 | 431.45 |
| 2j | 3,5-dimethoxy | 8.68 | 506.35 |
| 2k | 3,5-dinitro | 2.96 | 482.96 |
|
n-Octyl parabens OcP |
None | 9.08 | 529.01 |
| 3e | 3,5-diiodo | 5.08 | 625.49 |
| 3g | 3,5-dimethyl | 9.27 | 588.38 |
| 3i | 3,5-dihydroxy | 7.90 | 548.40 |
| 3k | 3,5-dinitro | 3.87 | 599.88 |
|
Established ERa agonist E2 |
None | 10.31 | 457.02 |
Parent n-butyl and n-octyl paraben and 17β-estradiol (E2) names from Group A (Fig. 2; Table 1). Ring-substituted paraben names from Fig. 3 (Bergquist et al., 2018).
4-position in each case occupied by a hydroxyl group.
Most acidic pKa computed with ADMET_Predictor 9.0.d
Solvent-accessible surface area computed with ADMET_Predictor 9.0.
A preliminary version of this work was presented at the 57th annual meeting of the Society of Toxicology [30].
2. Materials and Methods
2.1. Receptor preparation with YASARA
The crystal structure of the human ERα-LBD (Y537S) in complex with 17β-estradiol (PDB 3UUD) [31] was downloaded and prepared for docking with the YASARA molecular modelling suite (YASARA-Structure version 17.4.17 for Windows) [17]. This structure was selected as a template for creating homology models of mouse and rat ERα-LBD via in silico mutagenesis due to its high resolution (1.6 Å) and the presence of the Y537S mutated residue, which stabilizes the agonist-binding conformation of the receptor without compromising the overall structure or agonist ligand-binding properties of the protein [31]. The Y537S residue in the human ERα-LBD template receptor was retained for the mouse and rat structures created via in silico mutagenesis. All crystallographic waters were removed, hydrogen atoms were added, bond orders were corrected for S-hydroxycysteine residues, missing loops were repaired [32], and only chain-A in complex with its 17ß-estradiol ligand (E2) was retained. Differing residues between protein sequences for human [33], mouse [34], and rat [35] ERα-LBD were identified by protein sequence alignments (Fig. 1) performed with Geneious bioinformatics software (version 11.1.5 for Windows) [36, 37]. These residues in the human 3D structure were then mutated in silico using YASARA to create separate mouse and rat ERα-LBD receptors. The residue mutations for mouse were L306P, I326M, L327I, T334S, V368G, T371N, Q502R, and S527N. The residue mutations for rat were L306P, I326L, L327I, T334S, V368G, T371N, T483N, Q502R, and S527N. Side-chains of mutated residues were optimized with YASARA using the SCWALL method, which combines semi-empirical quantum mechanics, rotamer library, and steepest-descent algorithms [38].
For the human and rodent ERα-LBD receptors created via in silico mutagenesis, a cubic simulation cell was set to automatically encompass the entire receptor in YASARA plus an additional 2.5 Å margin in the x, y, and z directions. With the E2 ligand in the active site, each prepared receptor was separately subjected to energy minimization using the YASARA2 force field, which is derived from AMBER14 with the addition of knowledge-based dihedral and interaction potentials [38]. The energy-minimized structure was subjected to a 500 ps molecular dynamics refinement in explicit water solvent using YASARA and the YASARA2 force field [39]. Each receptor and refinement step were analyzed for structural errors and scored using MolProbity [40, 41]. Based on their MolProbity scores, the best ERα-LBD receptor for human, mouse, and rat was chosen for docking comparisons among all three receptor species as described in section 2.6 below.
2.2. Receptor preparation with UCSF Chimera
All crystallographic waters were removed, hydrogen atoms were added, and missing loops were repaired with UCSF Chimera (version 1.11 for Windows) [42] as a graphical user interface for MODELLER using default settings. Only chain-A with its native E2 ligand was retained for further refinement. Differing residues between human, mouse, and rat ERα-LBD were identified as described in the previous section. Residue differences observed in either mouse or rat ERα-LBD were swapped using the Dunbrack rotamer library via UCSF Chimera to create separate receptors using the prepared human ERα-LBD structure as a template. All observed clashes among swapped residues were optimized by subjecting them to energy minimization in UCSF Chimera using default settings for each ERα-LBD receptor. Energy minimization of the entire receptor was performed for comparison using 100 steepest descent minimization steps and 10 conjugate gradient steps. Molecular dynamics refinement in explicit solvent was performed with the YASARA molecular modeling suite with default settings using a 500 ps simulation as described in the previous section. Each receptor and refinement step were analyzed for structural errors and scored using MolProbity [40, 41].
2.3. Receptor preparation with I-TASSER
The protein sequence for mouse [34] and rat [35] ERα were uploaded to the I-TASSER online server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) for receptor assembly by iterative threading using default parameters [43]. I-TASSER does not produce a receptor with a ligand bound to it. Therefore, a cubic simulation cell was fitted around the entire mouse or rat ERα-LBD structure produced by I-TASSER plus an additional 5 Å margin in the x, y, and z directions. A docking simulation was performed with AutoDock Vina [44] for 100 runs using default parameters, and setup was conducted with YASARA as the graphical front-end [45] using the E2 ligand extracted from the crystal structure of human ERα-LBD (PDB 3UUD). The top pose for each docking run in the rodent receptors were selected for further structural refinement via energy minimization using the YASARA2 force field as described in section 2.1 above. The energy-minimized structure was subjected to 500 ps molecular dynamics refinement in explicit solvent using YASARA with the YASARA2 force field as described in section 2.1 [39]. Each receptor and refinement step were analyzed for structural errors and scored using MolProbity [40, 41].
2.4. Selection and preparation of Group A and Group B ligands
Group A ligands are shown in Fig. 2. These 23 compounds included E2, DES, and 21 paraben analogs. Many of the parabens in this group have been previously characterized in vitro in estrogen-dependent breast cancer cell lines or reporter assays, which have demonstrated relatively weak estrogenic behavior for these compounds [3, 5, 22, 28]. We also included (R)-and (S)-3-hydroxybutyl 4-hydroxybenzoate (3OHR and 3OHS) and 2-hydroxy iso-butyl 4-hydroxybenzoate (2OH) [46], metabolites of n-butylparaben (BuP) and iso-butylparaben (iBuP), which we recently characterized in vitro as weak estrogenic compounds in human ERα-expressing breast cancer cell lines [47].
Group B ligands are shown in Fig. 3. These 17 compounds consisted of BuP and E2 along with 15 parabens containing various substituents in the benzene ring [29].
Initial structures for Group A and Group B ligands were generated using ChemDraw Professional version 17.1.0.105 for Windows and saved as CDX structure files. Each CDX file was imported into Chem3D Ultra version 17.1.0.105 for Windows and energy-minimized using the MMFF94 functionality in Chem3D Ultra to ensure that the structures had the correct orders, lengths, and angles for all bonds. The minimized structures were saved as PDB files for use in the docking simulations.
Ligand PDB files were converted to SDF files using OpenBabel version 2.4.1 64-bit for Windows [48, 49] for calculation of pKa and solvent-accessible surface area (SASA) using SimulationsPlus ADMET_Predictor version 9.0.0.10 64-bit for Windows [50]. These values are listed in Table 1 (Group A ligands) and Table 2 (Group B ligands).
2.5. Docking simulations
Docking was performed with AutoDock Vina [44] using default parameters, and setup was conducted with YASARA as the graphical front-end [45, 51]. A cubic simulation cell (25 Å × 25 Å × 25 Å) was centered on the C9 carbon of E2 before removing the native ligand from each receptor and set as the search space for all test compounds, which were treated as flexible ligands. The best pose of 25 runs was selected based on two criteria: (1) as computed by the docking algorithm, the most favorable free energy of binding (ΔG); and (2) as determined by visual inspection, poses in which the 4-hydroxy group of the ligand benzene ring was oriented toward the R394 and E353 residues in the receptor binding site for potential hydrogen-bond formation, as seen in crystal structures of PrP, BuP, and E2 in complex with the human ERα-LBD[31, 52, 53]. Hydrogen-bond networks of the docked structures were optimized with YASARA [54], and protein structures of docking complexes were aligned with MUSTANG [55] before determining interspecies root-mean-square deviation (RMSD) values for docked ligands. Throughout this paper, ligand RMSDs are reported as heavy-atom (all atoms except hydrogen) values that have been symmetry-corrected to reduce false negative docking results [56]. Protein RMSDs are reported as CA backbone values [57, 58].
Because ΔG for docking tends to be biased in favor of larger molecules [59], we have expressed potency derived from docking in terms of ligand efficiency (LE). Thus, LEdock = -ΔG/Nh, where Nh = the number of heavy (non-hydrogen) atoms in the ligand) [60, 61].
In order to compare LEdock values with experimental data, we calculated experimental ligand efficiencies using the general relationship, LEexp = p(Activity)/Nh, where LEexp = experimental ligand efficiency, p(Activity) = -log(Activity), and Nh = the number of heavy (non-hydrogen) atoms in the ligand. This comparison requires self-consistent sets of experimental data obtained with a given method under the same assay conditions [60]. For Group A ligands, experimental activity data meeting these criteria were available for 14 of the 23 compounds in terms of EC20 values for transcriptional activation in an estrogen response element (ERE) luciferase reporter assay for human ERα [28]. For Group B ligands, experimental activity data were available for all 17 compounds (15 ring-substituted paraben analogs along with E2 and BuP) as IC50 values for binding to the human ERα receptor [29].
2.6. Receptor screening and assessment of unknown rodent structures
Prepared mouse and rat ERα-LBD receptors created with YASARA [17], UCSF Chimera [42], and I-TASSER [43] were scored and compared using MolProbity (see Supplementary Data, Tables S1 and S2). Side-chain optimization (SCWALL) followed by energy minimization (EM) of the entire ERα-LBD structure in YASARA produced the highest-scoring receptors with the fewest structural errors for ERα-LBD among all three receptor preparation methods. MD refinement resulted in the introduction of new errors and was therefore not used for preparation of the final models for subsequent docking simulations. The highest-scoring rodent receptors prepared with YASARA were selected to determine ligand-binding similarity among human and rodent ERα-LBD and to carry out the remainder of the docking simulations conducted in this study.
Structural similarity of the highest scoring ERα-LBD receptors were determined by the template-modeling score (TM-score) [58]. The TM-align web server (https://zhanglab.ccmb.med.umich.edu/TM-align/) was used to perform a structural alignment and generate TM-scores among the three species of ERα-LBD used for calibration. The TM-scores and receptor CA backbone RMSDs between human and mouse (TM-score: 0.99; RMSD: 0.16 Å) or rat (TM-score: 0.99; RMSD: 0.17 Å) were determined. The TM-score between mouse and rat (TM-score: 0.99; RMSD: 0.03 Å) was also calculated. A TM-score of 1.00 indicates a perfect match [58], and a CA backbone RMSD < 1 Å is within the generally accepted range for equivalent protein structures [62]. Thus, our three prepared in silico ERα-LBD models displayed a high degree of structural similarity.
The docking results for E2 were subjected to 3D protein alignment via MUSTANG in YASARA for human and mouse (Fig. 4A) or human and rat (Fig. 4B) ERα-LBD, which indicated a docking preference for the known active site of human ERα. Further receptor calibration was carried out by aligning the proteins associated with the DES docking poses in YASARA with MUSTANG for human and mouse (Fig. 4C) or human and rat (Fig. 4D) ERα-LBD. Protein-aligned mouse and rat docking poses for E2 (Fig. S1A) or DES (Fig. S1B) are likewise shown. Hydrogen-bonding interactions of E2 and DES with key side-chain residues in the ERα-LBD receptors are shown in Fig. 4. The ligand RMSD values for the aligned docking poses of E2 and DES in Fig. 4 are summarized in Table 3. Aligned mouse and rat ERα-LBD poses for docked E2 and DES were determined to have ligand RMSD values of 0.18 Å and 0.05 Å, respectively. Given that ligand RMSD values of 2.0 to 2.5 A are the traditional cutoff range for equivalent poses [63, 64], the extremely small values we obtained for E2 and DES to calibrate the prepared receptors indicate that our in silico mouse and rat ERα-LBD models produced virtually identical docking results among the human and rodent ERα-LBD structures. These calibrated structures were therefore selected for further analysis of ligand binding similarity.
Fig. 4.

Docking comparisons of known ERα agonist ligands in human and rodent ERα-LBD receptors. (A) E2 human and mouse. (B) E2 human and rat. (C) DES human and mouse. (D) DES human and rat. Ligand colors: gray = human, magenta = mouse, orange = rat. Hydrogen bonds are represented as yellow dashes. Oxygen and nitrogen atoms are colored red and blue, respectively. Active site helix labels (H5, H11, and H12) are displayed in bold face. Hydrogen-bonding residues are displayed and labeled.
Table 3.
Interspecies ligand RMSD values and Sc indices for ligand-receptor residue contacts of Group A ligands docked into human, mouse, and rat ERα-LBD receptors.
| Compound | RMSD (Å)a | Sc index (%)b | |||||
|---|---|---|---|---|---|---|---|
| Human/Mouse | Human/ Rat | Mouse/Rat | Human/Mouse | Human/ Rat | Mouse/Rat | ||
| Parabens | |||||||
| MeP | 1.82 | 1.83 | 0.08 | 84.8 | 84.8 | 100 | |
| EtP | 2.51 | 2.96 | 1.05 | 72.7 | 74.3 | 93.8 | |
| PrP | 1.20 | 1.33 | 0.47 | 84.2 | 89.5 | 95 | |
| BuP | 3.22 | 3.30 | 0.10 | 81.0 | 81.0 | 100 | |
| PeP | 0.31 | 0.21 | 0.19 | 100 | 100 | 100 | |
| HxP | 0.18 | 0.16 | 0.12 | 100 | 100 | 100 | |
| HpP | 0.21 | 0.13 | 0.13 | 100 | 100 | 100 | |
| OcP | 0.49 | 0.47 | 0.15 | 100 | 97.9 | 97.9 | |
| NnP | 0.23 | 0.27 | 0.23 | 100 | 100 | 100 | |
| DecP | 3.60 | 3.57 | 0.12 | 90.6 | 90.6 | 100 | |
| UnDecP | 3.45 | 1.19 | 3.52 | 96.0 | 98.0 | 93.9 | |
| DoDecP | 0.66 | 3.62 | 3.64 | 94.1 | 92.3 | 96.3 | |
| iPrP | 0.36 | 1.74 | 1.54 | 97.0 | 86.5 | 88.9 | |
| iBuP | 0.14 | 0.18 | 0.09 | 100 | 100 | 100 | |
| iPeP | 0.46 | 0.50 | 0.24 | 97.7 | 97.7 | 100 | |
| PhP | 1.79 | 1.72 | 0.13 | 95.5 | 97.7 | 97.8 | |
| BzP | 2.09 | 2.11 | 0.09 | 89.4 | 89.4 | 100 | |
| Parabens metabolites | |||||||
| 4OH | 0.15 | 0.27 | 0.34 | 96.8 | 89.7 | 93.3 | |
| 2OH | 2.09 | 2.08 | 1.02 | 88.4 | 88.4 | 100 | |
| 3OHR | 1.79 | 1.73 | 0.18 | 90.0 | 90.0 | 100 | |
| 3OHS | 0.15 | 0.13 | 0.16 | 97.7 | 97.7 | 100 | |
| Established ERα agonists | |||||||
| E2 | 0.08 | 0.22 | 0.18 | 100 | 100 | 100 | |
| DES | 0.11 | 0.14 | 0.05 | 100 | 100 | 100 | |
Symmetry-corrected heavy-atom interspecies ligand RMSD values for Group A ligands docked into mouse and rat ERα-LBD receptors. Overall group RMSD values (median, 95% CI, n = 23) were 0.49 (0.21, 1.82) Å (human-mouse), 1.19 (0.22, 1.83) Å (human-rat), and 0.18 (0.12, 0.34) Å (mouse-rat). By definition, RMSD (human-human) = 0.00 Å.
Sorenson similarity coefficient (Sc) = [2(NAB)/(NA + NB)] x 100, where NA = number of contacts in receptor A, NB = number of contacts in receptor B, and NAB = number of contacts shared by receptors A and B. In this case, contacts are ligand-receptor residue contacts. Sc values (median, 95% CI, n = 23) for all ligands were 96.8 (90.0, 100)% (human-mouse), 97.7 (89.5, 100)% (human-rat), and 100 (97.8, 100)% (mouse-rat). By definition, Sc (human-human) = 100%.
2.7. Statistical analyses
GraphPad Prism 7.04 for Windows was used to create correlation and pyramid plots of ligand-receptor residue or atom contacts obtained from the docking results; it was also used to determine Pearson (r) or Spearman (rs) correlation coefficients (GraphPad Software, La Jolla California USA, www.graphpad.com). The Pearson r was used for normally distributed data, and the Spearman rs was used for non-normally distributed data as determined by the D’Agostino & Pearson and Shapiro-Wilk normality tests in Prism. Data sets were treated as non-normally distributed if they failed to pass one or both normality tests (alpha = 0.05). Summary statistics of non-normally distributed data are presented as median values with 95% confidence intervals (CI). Correlations of LEexp vs. LEdock were obtained using Pearson partial correlation (rp) computed with OriginPro 2018b 64-bit for Windows to correct for the Nh covariate (OriginLab, Northampton, Massachusetts USA, www.originlab.com). The Sorensen similarity coefficient (Sc, expressed as a percentage; also known as the Dice or Sorensen-Dice coefficient) for each docking comparison of ligand-receptor atom or residue contacts was obtained with the following equation: Sc = [2(NAB)/(NA + NB)] × 100], where NAB = number of contacts for both species, NA = number of contacts for species A, and NB = number of contacts for species B [65, 66]. Sc was calculated using the Python “distance” package version 0.1.3 in Anaconda Python 3.6.5 for 64-bit Linux.
3. Results
3.1. Group A ligand RMSD values indicate overall agreement of docking poses between species
Upon calibration of the prepared ERα-LBD structures shown in Figs. 4 and S1, we used the Group A ligands shown in Fig. 2 and Table 1 to determine ligand-receptor binding similarities among the refined human structure and in silico rodent models of the ERα-LBD.
Our first comparison was an examination of the interspecies ligand RMSD values for each Group A compound (Table 3). Overall group RMSD values (median, 95% CI, n = 23) were 0.49 (0.21–1.82) Å (human-mouse), 1.19 (0.22–1.83) Å (human-rat), and 0.18 (0.12–0.34) Å (mouse-rat). By definition, RMSD (human-human) = 0.00 Å. Because the generally accepted RMSD range for equivalent docking poses is 2.0–2.5 Å [63, 64], the small values we obtained indicate excellent overall agreement in docking members of this set of ligands into human, mouse, and rat ERα-LBD receptors. In general, the calibration ligands, DES and E2, showed the most favorable ligand RMSD values among the compounds tested. Among the paraben compounds, n-pentyl through n-nonyl and iso-alkyl analogs generally displayed more favorable RMSD values than those with shorter or longer n-alkyl chains. This optimal behavior is likely due to a combination of factors, including an overall reduction and constriction of possible translational motion of the relatively larger compounds within the active site, as well as their ability to fully occupy the available space in this binding pocket in a manner similar to endogenous ERα agonists such as E2. Experimental data on relative estrogenic potency of paraben analogs also suggest that optimal activity reflects an ideal juxtaposition of molecular size and hydrophobicity [28].
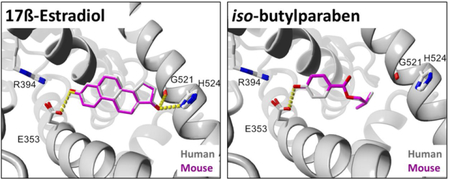
An example of a relatively poor docking result is shown with n-butylparaben (BuP) docked to human ERα-LBD and aligned with the top docking pose for mouse or rat (Fig. 5A, 5B). The rather large ligand RMSD values for the top docking poses of BuP in Fig. 5 indicate comparatively incongruent ligand alignment in human vs. rodent ERα-LBDs.
Fig. 5.

Docking comparisons of Group A paraben ligands in human and rodent ERα-LBD receptors. (A) BuP human and mouse. (B) BuP human and rat. (C) iBuP human and mouse. (D) iBuP human and rat. Ligand colors: gray = human, magenta = mouse, orange = rat. Hydrogen bonds are represented as yellow dashes. Oxygen and nitrogen atoms are colored red and blue, respectively. Active site helix labels (H5, H11, and H12) are displayed in bold face. Hydrogen-bonding residues are displayed and labeled.
In contrast, the top docking pose for iso-butylparaben (iBuP), a structural isomer of BuP, was found to display a nearly perfect alignment between human vs. mouse or rat receptors as evidenced by visual inspection (Fig. 5C, 5D, S2B) and by the ligand RMSDs (Table 3). The alignment for iBuP docked to mouse vs. rat was also found to have an exceptional ligand RMSD, as likewise observed in the case of BuP docked to mouse vs. rat receptors (Fig. S2A, Table 3).
3.2. Numbers of interspecies ligand-receptor contacts are highly correlated
Our next comparison was an exploration of the degree of interspecies correlations of the numbers of residue and atom receptor contacts for each compound in Group A. The numbers of ligand-receptor residue or atom contacts between human and mouse ERα-LBD among all Group A ligands tested were found to have Pearson r = 0.913 and 0.978, respectively (Fig 6A, 6B). Even higher correlations were obtained with the numbers of ligand-receptor receptor or atom contacts between human and rat ERα-LBD (r = 0.925 and 0.986, respectively, Fig. 6C, 6D). Finally, the numbers of ligand-receptor residue or atom contacts between mouse and rat ERα-LBD gave the highest correlations of all (r = 0.945 and 0.990, respectively, Fig. S3A, S3B). The extraordinary correspondence between the numbers of ligand-receptor interactions in mouse and rat ERα-LBDs agrees with the high level of sequence identity between the rodent species (Fig. 1). Overall, with respect to the numbers of residue or atom ligand-receptor contacts, the mouse and rat ERα-LBDs were found to interact with the series of paraben analogs and known ERα agonists from Group A ligands in a manner highly similar to that of the human ERα-LBD.
Fig. 6.

Correlations of numbers of contacts for Group A ligands docked into human and rodent ERα-LBD receptors. (A) Human-mouse residue contacts. (B) Human-mouse atom contacts. (C) Human-rat residue contacts. (D) Human-rat atom contacts. Residue contacts = filled circles, atom contacts = open circles. Each data point is labeled with the ligand name (See Fig. 2 and Table 1). The Pearson correlation coefficient (r) and associated p-value are shown in each panel.
3.3. Interspecies ligand-receptor contacts arising from specific residues are highly similar
To assess how similarly Group A compounds interacted with specific residues within the active site of ERα, we calculated the interspecies Sorensen similarity coefficients (Sc) for the numbers of residue contacts for each compound docked into human, rat, or mouse ERα-LBD (Table 3). Rather than simply correlating the number of ligand-receptor residue contacts for a given ligand between two receptors, the Sc takes into account the specific residues in each receptor making contacts with the ligand. This analysis yielded an interspecies Sc value for each compound. As can be seen in Table 3, low RMSD values for a given compound tended to be reflected by correspondingly high Sc values.
The group Sc coefficients (median, 95% CI) for all residue contacts among all Group A ligands for each pair of species were 96.8 (90.0–100)% (human-mouse), 97.7 (89.5–100)% (human-rat), and 100 (97.8–100)% (mouse-rat). These Sc values indicate an overall high degree of similarity between human and rodent ERα-LBDs as well as between mouse and rat ERα-LBDs with respect to the numbers of residue contacts between ligands and receptors arising from specific residues.
In order to gain a clearer picture of interspecies similarity of ligand-receptor atom contacts by residue for all Group A compounds, we displayed the data in the form of “pyramid plots”, in like manner to the classic “population pyramids” used to visually categorize demographic information by comparing the numbers of people in different age groups by gender [67]. Here, we replaced age groups with protein residue sequence numbers, and we replaced genders with species. As can be seen in Fig. 7 and Fig. S4, the remarkable interspecies similarity in ligand-receptor atom contacts by residue is readily apparent from the symmetry of the pyramid plots. Moreover, on a quantitative basis, Sc values were 96.9% (human-mouse), 93.5% (human-rat), and 96.9% (mouse-rat), and rs values were 0.977 (human-mouse), 0.966 (human-rat), and 0.991 (mouse-rat). These plots were also useful for assessing the relative prevalence of ligand-receptor interactions in a set of ligands. For example, among Group A ligands, there were highly frequent hydrophobic interactions with L346, L387, and F404 and less frequent hydrogen bonding interactions with R394, E353, and H524.
Fig. 7.

Numbers of ligand-receptor atom contacts by residue for all Group A ligands docked into human and rodent ERα-LBD receptors. (A) Human (blue bars on left) and mouse (green bars on right) receptors. Human (blue bars on left) and rat (red bars on right) receptors. The Sorensen similarity coefficient (Sc, expressed as a percentage) along with the Spearman correlation coefficient (rs) and associated p-value are shown in each panel. Vertical axis = ERα-LBD sequence number; horizontal axis = number of atom contacts by residue.
Overall, the Sc values for numbers of residue contacts along with the pyramid plots and their associated Sc values and rs coefficients demonstrate that the Group A ligands give rise to sets of specific active site contacts that are highly similar and consistent across human and rodent ERα-LBDs.
3.4. Group A LEdock values are highly correlated between species
Interspecies correlation plots of LEdock values for Group A ligands are shown in Fig. 8A,B (human-rodent) and Fig. S5A (mouse-rat). Each plot also shows the partial Pearson correlation coefficients (rp) to determine the degree of correlation corrected for the covariate, Nh (number of heavy atoms). These values were 0.958 (human-mouse), 0.981 (human-rat), and 0.960 (mouse-rat), demonstrating excellent interspecies agreement in the predicted strength of ligand interactions with the ERα-LBD receptors.
Fig. 8.

Human-rodent LEdock correlations for Group A and Group B ligands. (A) Human-mouse, Group A ligands. (B) Human-rat, Group A ligands. (C) Human-mouse, Group B ligands. (D) Human-rat, Group B ligands. LEdock = -ΔG/Nh, where LEdock = ligand efficiency for docking, ΔG = free energy of ligand-receptor binding from docking results, and Nh = number of heavy (non-hydrogen) atoms in the ligand. Each point is labeled with the name of the ligand (Group A ligands = closed circles, see Fig. 2 and Table 1; Group B ligands = open circles, see Fig. 3 and Table 2). The Pearson partial correlation coefficient (rp, to correct for the covariate, Nh) and associated p-value are shown in each panel.
It is also noteworthy that the three longest-chain parabens, DeP, UnDeP, and DoDeP, were clustered at the low end of the potency scale, below LEdock values of 0.35 kcal/mol/Nh. This result agrees with published in vitro data showing that these three compounds exhibited little or no estrogenic activity as assessed by transcriptional activation in an estrogen response element luciferase reporter assay [28]. However, the LEdock scores for 4OH and MeP calculated from our docking results were near the top end of the scale, whereas these compounds were negative in the ERE assay. Explanations of the apparently anomalous results for these low molecular weight compounds are provided below in the Discussion section.
3.5. Group B LEdock values are highly correlated between species and decreased by ring substitution
Interspecies correlation plots of LEdock values for Group B ligands are shown in Fig. 8C,D (human-rodent) and Fig. S5 (mouse-rat). In these cases, values for OcP from Group A ligands were included along with values for BuP that were already part of Group B in order to enable direct assessment of the effect of ring substitutions on predicted ligand binding of both n-butyl-and n-octylparabens. Values of rp were 0.957 (human-mouse), 0.964 (human-rat), and 0.990 (mouse-rat), indicating strong interspecies correlations.
Moreover, it was readily apparent from inspection of the plots in Fig. 8C,D and Fig. S5 that the predicted efficiencies of ligand binding of all ring-substituted n-butyl-and n-octylparabens (2a through 2k) were less than that of BuP. Furthermore, the LEdock values of all of the ring-substituted n-octylparabens (3e through 3k) were less than that of OcP. Thus, molecular docking predicts that adding ring substituents to n-butyl-or n-octylparaben as shown in Fig. 3 will decrease the avidity of binding of these ligands to human, mouse, and rat ERα-LBD receptors.
3.6. Human LEdock and LEexp values display good agreement for both Group A and Group B ligands
Fig. 9 shows correlation plots for human LEexp vs. LEdock for Group A and Group B ligands. Self-consistent experimental data sets were available for 14 of the 23 ligands in Group A [28] and all 17 of the ligands in Group B [29]. The rp values were 0.894 for Group A and 0.918 for Group B, indicating good agreement between ligand efficiencies computed from molecular docking and those derived from experimental data. In addition, Fig. 9B shows that both LEdock and LEexp values for all of the ring-substituted parabens fall below these values for BuP, which has no substituents in its benzene ring.
Fig. 9.

LEdock and LEexp correlations for Group A and B ligands with human ERa-LBD. (A) Group A ligands (filled circles) with names (Fig. 2, Table 1). (B) Group B ligands (open circles) with names according to Bergquist et al. (2018) (Fig. 3, Table 2). LEdock = -ΔG/Nh, where LEdock = ligand efficiency for docking, ΔG = free energy of ligand-receptor binding from docking results, and Nh = number of heavy (non-hydrogen) atoms in the ligand. The Pearson partial correlation coefficient (rp, to correct for the covariate, Nh) and associated p-value are shown in each panel.
4. Discussion
One of the primary goals of our present work was to address the current conspicuous absence of reported ERα-LBD crystal structures among the rodent species. Especially in view of the extensive use of rodent models for investigations of physiological and pathogenic processes involving the estrogen receptor [68–70], the lack of 3D structures for the mouse and rat ERα-LBDs constituted a highly significant knowledge gap that needed to be filled.
The frequently quoted “sequence-structure gap” between the prodigious amount of sequence data and the sparser quantity of 3D structural data can be bridged experimentally using X-ray crystallography, NMR, or cryo-EM [71]. Alternatively, this data disparity can be addressed computationally using iterative threading [43] or homology modeling [72, 73]. In our present work, we selected in silico mutagenesis [74] via YASARA Structure [17] as the means to create homology models after comparing results obtained with iterative threading using I TASSER [43] and homology modeling employing UCSF Chimera [42].
Owing to the high degree of sequence identity between the human and rodent ERα-LBD proteins, it has been tacitly assumed that the 3D structures and ligand-binding characteristics of these receptors are essentially identical across species [75, 76]. Indeed, in tests of estrogenic potential, a common scenario is to conduct ligand-binding assays using human receptors in vitro and to carry out in vivo assessments in rodents [77–80]. Thus, it was reasonable for us to hypothesize that the 3D structures of the human and rodent proteins would be highly similar and that the binding modes of agonists would likewise be highly similar. Although this predicted outcome seemed likely, in the absence of structural data, it was still necessary to test our contention.
We tested our hypothesis computationally by constructing homology models, assessing their quality and structural similarity to the human protein, and carrying out comparative docking studies of paraben analogs, E2, and DES. The results supported our hypothesis, which indicates that the differing residues among these receptors could be considered neutral substitutions with little or no effect on altering the tertiary structure of the ERα-LBD or its binding of the tested agonists. Thus, our results provide additional evidence to validate the use of rodent models in the assessment and characterization of the estrogenic activity of compounds arising from their interaction with the ERα-LBD.
We have shown via MolProbity analysis that our rodent homology models are of high structural quality and via docking studies that they can serve as viable docking targets. Accordingly, the models could be used to test other hypotheses concerning species differences among estrogen receptors. For example, it has been shown that there are significant differences in ligand selectivity between ERα and ERβ receptor among human, mouse, and rat species using ligands that we did not test in our models [76]. To our knowledge, these species differences remain unexplained. Our rodent homology models could be used to provide insight into these observations in future work.
Likewise, homology models of the estrogen receptor have been produced for a variety of other species, attesting to the relevance and value of computational models in the absence of experimentally determined structures. Indeed, in recent years, homology models in general have gained wide acceptance as a means of producing high-quality protein structures for research investigations and as docking targets [74, 81, 82]. With respect to estrogen receptors, homology models of ER receptors based on X-ray crystal structures of human ERα have been created for human (ERβ) [83], lizard (3 species) [84], medaka (3 varieties) [85], rainbow trout [86], rotifer [87], and zebrafish [88]. These estrogen receptor homology models have been used in evolutionary and physiological research and as docking targets with applications in ecological toxicology.
Furthermore, our use of in silico mutagenesis as a technique for creating homology models could easily be extended to the study of mutant receptors in breast cancer and other diseases [68, 70, 89–92]. In addition, the technique of in silico mutagenesis could be used for the rational design of estrogen receptors with substantially altered ligand affinities for applications in assays and biosensors [93]. Methodologically, it is noteworthy that in the case of the rodent ERα-LBD homology models, the simplest of the three approaches – using YASARA-Structure for in silico mutagenesis with optimization of side-chains and hydrogen-bonding networks followed by energy minimization – produced the highest-quality structures.
Another goal of the present work was to carry out comparative molecular docking simulations with the human, mouse, and rat ERα-LBD receptors using known ER agonists and a homologous series of parabens, considered toxicologically relevant as suspected endocrine disrupting compounds (EDCs). In this connection, the identification and characterization of suspected endocrine disrupting compounds EDCs using both in vitro and in vivo methods has been considered a major subject of toxicology research for several decades [94]. Furthermore, parabens represent one category of suspected EDCs that have been investigated for their potential action on ERα and related hormone signaling pathways [28, 47]. Although paraben compounds are generally considered weak agonists of ERα, they are still being investigated to determine whether current exposures may lead to adverse impacts on human health [24, 27, 95]. For example, an epidemiological study reported a possible association between increased BuP exposure and markers of sperm DNA damage in men [96]. Another study showed a possible dose-response relationship between higher paraben exposure and shorter self-reported menstrual cycle length among female Japanese university students [95]. In addition, the detection of parabens in numerous human tissues [27, 97–103] and their associations with possible endocrine disruption in humans further demonstrate the toxicological relevance of these compounds as test ligands in our study. These findings also highlight the need for the further development of in vitro and in silico screening methods for recognizing and categorizing EDCs.
Among computational approaches, there have been other reports on molecular docking of parabens. While these studies differed from our investigation in several respects (e.g., fewer paraben compounds and/or targets other than ERα), they also provided an important degree of corroboration and additional insight into our results. For example, molecular docking of parabens and other known or potentially estrogenic compounds using human crystal structures of ERα-LBD as receptors has been combined with other information from multiple studies to predict their estrogenic potential [104, 105]. These large-scale projects highlighted the utility of molecular docking in exploratory toxicology studies and demonstrated a high level of consistency across studies for highly active compounds. However, these studies also revealed a much lower level of consistency for weakly active compounds, indicating that further research is needed to improve the reliable classification of compounds that exhibit low potency against ERα, such as the paraben analogs used as ERα-LBD ligands in our study.
In a study of five n-alkyl parabens [106], docking of BuP into the human ERα-LBD was shown to have a comparatively high RMSD from the crystal structure owing to the relatively unconstrained n-butyl group being able to adopt multiple conformations, as noted in our present work. In other reports [107, 108], MeP, EtP, and BzP were found to exert estrogenic effects in a uterotrophic assay in rats, and docking was used to show that these compounds adopted apparent bioactive conformations in the human ERα-LBD, similar to our findings.
An investigation of interactions of parabens with the human androgen receptor [25] found that docking scores for n-alkyl parabens were found to be inversely correlated with chain length, as shown in our study with human and rodent estrogen receptors. Moreover, dividing their -ΔG values by Nh to yield LEdock scores resulted in changing their relative ranking of 4OH, PhP, and BzP from 7, 2, and 1 to 1, 4, and 6, respectively, compared to our relative ranking of the same nine compounds as 1, 5, and 6, respectively.
Finally, among other related docking studies, an investigation has been conducted on the inverse antagonistic activity of five parabens against the estrogen-related receptor gamma (ERR-γ) [26], which has recently been shown to function as a tumor suppressor in gastric cancer [109]. The binding pocket of ERR-γ is similar to that of ERα, and parabens were found to dock in the known agonist site in like manner to our docking poses of parabens in the human and rodent ERα-LBD structures.
Although ERβ might have been examined in the present study, ERα would be expected to contribute to a greater proportion of effects observed by binding of an ER agonist. Only the expression of ERα, and not ERβ, is currently used to make clinical decisions regarding estrogen-sensitive diseases, such as ER‑ positive breast cancer, primarily due to the relatively poor understanding of the role of ERß within ERα-expressing breast tumors [110]. Further study will be needed to determine the extent to which structural differences or differential expression of these estrogen receptor subtypes might elicit an estrogenic response from exposures to exogenous ER agonists.
It is also important to point out that there were some apparently anomalous findings among our results. In an experimental study comparing estrogenic activities of paraben analogs [28], the common metabolite, 4OH, failed to elicit an estrogenic response, yet we obtained a positive docking result with this compound in the three species of ERα-LBD.
The negative experimental result with 4OH is not surprising, given the fact that parabens are neutral esters, whereas 4OH is a carboxylic acid with an experimental pKa of 4.54 [111] and a calculated pKa of 4.01 (Table 1). Therefore, this compound would be ionized at a physiological pH of 7.4. Because of its negative charge, 4OH would be expected to encounter difficulty gaining access to the hydrophobic interior of the ERα; however, when docked in the active site, hydrogen bonds to the phenol group and hydrophobic interactions with the benzene ring would serve to stabilize the complex [106].
As shown in Fig. 10A, the mean LEdock values obtained from the docking results of all three species of ERα-LBD displayed a strong negative correlation with the solvent-accessible surface area (SASA) of the Group A parabens analogs, including the metabolite, 4OH. In this plot, 4OH aligns with the paraben analogs as the compound with the highest LEdock value and the lowest SASA. At the same time, a plot of mean LEdock vs. pKa (Fig. 10B) shows that 4OH clearly stands apart from the paraben analogs, which span the full range of LEdock values with little change in pKa. However, it is important to note that AutoDock Vina does not make use of partial atomic charges [112], and the same result was obtained whether 4OH was docked as a neutral molecule or as an ionized species (data not shown).
Fig. 10. Correlations for Group A and Group B ligands with physiochemical parameters.

(A) LEdock vs. SASA for Group A ligands. (B) LEdock vs. pKa for Group A ligands. (C) LEdock vs, SASA) for Group B ligands. (D) LEdock vs. pKa for Group B ligands. Group A = closed circles; see Fig. 2 and Table 1 for names. Group B = open circles, see Fig. 3 and Table 2 for names. Each point = mean ± SEM, n = 3 for human, mouse, and rat. In most cases, the error bars fall inside the diameter of the data markers. LEdock = -ΔG/Nh, where LEdock = ligand efficiency for docking, ΔG = free energy of ligand-receptor binding from docking results, and Nh = number of heavy (non-hydrogen) atoms in the ligand. SASA = solvent-accessible surface area of the ligand (Å2). The Pearson correlation coefficient (r) and associated p-value are shown in each panel. In panel (B), note that 4OH is a carboxylic acid, pKa = 4.01, that would be ionized at pH 7.4. The other Group A ligands are neutral esters with pKa values for their phenol groups within a narrow range, whereas their LEdock values span a wide range. In panel (D), note that some of the ring substitutions in the Group B ligands result in considerable lowering of the pKa of the phenol group, yet a strong correlation between LEdock and SASA is maintained (panel C).
In comparison, Fig. 10C shows a negative correlation between the mean LEdock values of the Group B paraben analogs with SASA, similar to what was observed with the Group A ligands. Note that in Fig. 10D, there is no statistically significant correlation between mean LEdock values and pKa, yet this representation and the data in Table 2 show that eight of the 17 compounds would be ionized to some extent at physiological pH. In the case of Group B compounds, the ionized group would be a phenolic oxygen. Here again, whereas docking scores were inversely related to molecular size, they were indifferent to the potential of a given compound to ionize.
Moreover, the discrepant relative ranking of 4OH potency came about from the simple calculation of dividing –ΔG by the number of heavy atoms in the molecule (Nh) to generate LEdock values. The motivation for this calculation arose from the fact that docking scores tend to favor larger molecules, and the LE metric provides an expedient way to correct this bias [60, 61]. If the Group A ligands were ranked by –ΔG rather than LEdock, 4OH would move from first to last place. Nevertheless, regardless of its relative ranking, 4OH would retain a docking score. Therefore, the compound would not be deemed completely inactive, in keeping with its weak estrogenic activity in mouse bioassays [113] and human breast cancer cell lines [114]. Moreover, the general notion of 4OH as a ligand in complex with proteins should not be surprising, given that it has been docked into human cyclooxygenase-2 (COX-2) [115], despite negative results in a COX-2 dependent human smooth muscle cell assay [116]. Furthermore, 4OH is found as a bound ligand in a variety of enzymes, including carbonic anhydrase [117], p-hydroxybenzoate hydroxylase [118], and 4-hydroxybenzoate octaprenyltransferase [119]. Lastly, 4OH was found to have intermediate antagonistic activity among paraben analogs against the human androgen receptor [25], and when its reported docking score was converted to an LEdock value, its relative rank increased from 7th to 1st place out of nine compounds, similar to the results we obtained in the present study.
Although LE values (LEdock and/or LEexp) should be used and interpreted with due caution [120, 121], their validity and utility have been well established and widely accepted [60, 61, 122]. Nevertheless, when LE values are employed to select optimally binding ligands for a given receptor, it is important to recognize that, in general, LE is not a linear function of Nh [59]. In particular, based on compilations from large databases of ligand-receptor complexes, LE values decrease markedly within an Nh range of 10 to 20 [121]. Thus, the use of LE values for compound selection can be problematic, especially for small molecules such as 4OH and paraben analogs with relatively short alkyl chains, such as MeP.
The problem of smaller ligands having disproportionately large LE values has given rise to a variety of compensatory methods with varying degrees of success [123]. The more effective methods for correcting LE according to molecular size depend on deriving parameters from curve-fitting plots of LE vs. Nh. Obtaining meaningful values for such parameters would require much larger data sets than the ones described in the present study. Moreover, apart from considerations of the receptor-binding capabilities of individual parabens and the implications this would have on assessing their potential human health impacts, the main purpose of our investigation to use these compounds as tools to evaluate the similarity of the human ERα-LBD structure to our homology models of mouse and rat ERα-LBDs. In this way, our use of LEexp-LEdock correlations along with those of other variables helped to demonstrate the strong structural and implied functional correspondence between human and rodent estrogen receptors.
5. Conclusions
We have demonstrated that in silico mutagenesis of the human ERα-LBD produced new high-quality homology models of mouse and rat ERα-LBD, as assessed by MolProbity analysis. Because there are currently no experimentally determined 3D structures of mouse or rat ERα-LBD in the PDB, our computational results serve to bridge this significant data gap by providing modeled 3D structures of the rodent receptors.
We compared three methods for producing the homology models: (1) in silico mutagenesis with YASARA-Structure; (2) in silico mutagenesis with UCSF Chimera; and (3) iterative threading with I-TASSER. Furthermore, each of the three methods included two additional refinement procedures consisting of energy minimization and a brief molecular dynamics simulation in explicit solvent. The best overall results were obtained with the simplest procedure: in silico mutagenesis and energy minimization with YASARA-Structure. This technique could be employed in future studies to gain insight into species differences in ligand binding [76], evolutionary or physiological investigations [84, 87, 88], studies of ERα-LBD mutations in breast cancer or other diseases [68–70, 89, 90, 92], ecological toxicology [85, 86, 124], and genetic engineering of mutant ERα-LBDs for use in bio-detection of ligands in assays or biosensors [93].
We have also shown that our in silico receptor structures are viable docking targets capable of reliably recognizing the known potent agonists E2 and DES as well as a homologous series of weak agonists consisting of parabens and metabolites. These results are in agreement with in vitro data regarding the ability of these compounds to bind to mammalian estrogen receptors and modulate estrogen signaling pathways [3, 5, 28, 47, 53, 125]. Moreover, our detailed comparisons of docking results consistently revealed high similarity between human and rodent ligand-receptor complexes in support of our working hypothesis and indicating that the interspecies sequence differences in these receptors could be regarded as neutral substitutions. Overall, the molecular docking results serve to bolster confidence in the application of computational techniques that have proved successful in drug discovery to the fields of computational and predictive toxicology.
In particular, our specific docking studies of the novel ring-substituted paraben analogs synthesized by Bergquist et al. [29] furnish computational corroboration of the decreased potencies of these compounds against the human ERα-LBD relative to their unsubstituted counterparts. These important new findings validate the use of ring-substituted parabens to decouple ERα agonist activity from their antimicrobial properties. In combination with data from Bergquist et al. [29], our results also have significant implications for improving product safety through the design and manufacture of consumer goods that would contain appropriately modified paraben analogs instead of the compounds currently employed.
Lastly, this paper has introduced from other fields ways of depicting and comparing data that to our knowledge are new to computational toxicology. For example, ligand efficiency, borrowed from medicinal chemistry, provides a simple way to compensate for the bias in docking scores favoring larger molecules [60]. The Sorensen similarity coefficient has been used in ecology, genetics, and other fields [126], but we are unaware of its previous use in computational or general toxicology. Moreover, we found that the Sorensen coefficient yielded scores that were more intuitively aligned with expectation than other similarity metrics, such as the Tanimoto coefficient, which is widely used in cheminformatics [127]. Finally, to our knowledge, our adaptation of population pyramid plots [67] for displaying docking information is novel. Furthermore, we found that our pyramid plots provided a visually appealing and pragmatic way to depict simultaneously the overall symmetry or asymmetry of ligand-receptor interactions between species along with quantitative information about the number of ligand contacts for each residue in the receptors.
Supplementary Material
Highlights:
Rodent ERα-LBDs were produced by in silico mutagenesis of the human receptor.
Docking poses of parabens were highly similar between rodent and human ERα-LBDs.
Sorenson coefficient proved useful as a new similarity metric for docking results.
Pyramid plots are effective graphical representations of ligand-receptor contacts.
3,5-disubstitution of parabens decreases predicted affinity toward ERα-LBD.
Acknowledgments
Research reported in this publication was supported in part by the National Institute of Environmental Health Sciences of the National Institutes of Health, under Award Numbers T32ES007062, R01ES028802 (to JAC) and P30ES017885. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary Data
Supplementary data associated with this article can be found in the online version at https://doi.org/xxx.
References
- [1].Oishi S, Effects of butylparaben on the male reproductive system in rats, Toxicol. Ind. Health 17 (2001) 31–39, 10.1191/0748233701th093oa. [DOI] [PubMed] [Google Scholar]
- [2].Schlumpf M, Cotton B, Conscience M, Haller V, Steinmann B, Lichtensteiger W, In vitro and in vivo Estrogenicity of UV Screens, Environ. Health Perspect 109 (2001) 239–244, 10.1289/ehp.01109239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Charles AK, Darbre PD, Combinations of parabens at concentrations measured in human breast tissue can increase proliferation of MCF-7 human breast cancer cells, J. Appl. Toxicol 33 (2013) 390–8, 10.1002/jat.2850. [DOI] [PubMed] [Google Scholar]
- [4].Miller D, Wheals BB, Beresford N, Sumpter JP, Estrogenic activity of phenolic additives determined by an in vitro yeast bioassay, Environ. Health Perspect 109 (2001) 133–8, 10.2307/3434765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Okubo T, Yokoyama Y, Kano K, Kano I, ER-dependent estrogenic activity of parabens assessed by proliferation of human breast cancer MCF-7 cells and expression of ERa and PR, Food. Chem. Toxicol 39 (2001) 1225–1232, 10.1016/S0278-6915(01)00073-4. [DOI] [PubMed] [Google Scholar]
- [6].Cote I, Andersen ME, Ankley GT, Barone S, Birnbaum LS, Boekelheide K, Bois FY, Burgoon LD, Chiu WA, Crawford-Brown D, Crofton KM, DeVito M, Devlin RB, Edwards SW, Guyton KZ, Hattis D, Judson RS, Knight D, Krewski D, Lambert J, Maull EA, Mendrick D, Paoli GM, Patel CJ, Perkins EJ, Poje G, Portier CJ, Rusyn I, Schulte PA, Simeonov A, Smith MT, Thayer KA, Thomas RS, Thomas R, Tice RR, Vandenberg JJ, Villeneuve DL, Wesselkamper S, Whelan M, Whittaker C, White R, Xia M, Yauk C, Zeise L, Zhao J, DeWoskin RS, The Next Generation of Risk Assessment Multi-Year Study-Highlights of Findings, Applications to Risk Assessment, and Future Directions, Environ. Health Perspect 124 (2016) 1671–1682, 10.1289/EHP233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kavlock RJ, Bahadori T, Barton-Maclaren TS, Gwinn MR, Rasenberg M, Thomas RS, Accelerating the Pace of Chemical Risk Assessment, Chem. Res. Toxicol 31 (2018) 287–290, 10.1021/acs.chemrestox.7b00339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Porta N, Roncaglioni A, Marzo M, Benfenati E, QSAR methods to screen endocrine disruptors, Nucl. Receptor Res 3 (2016) Article ID 101203, 10.11131/2016/101203 [DOI] [Google Scholar]
- [9].Vuorinen A, Odermatt A, Schuster D, In silico methods in the discovery of endocrine disrupting chemicals, J. Steroid. Biochem. Mol. Biol 137 (2013) 18–26, 10.1016/j.jsbmb.2013.04.009. [DOI] [PubMed] [Google Scholar]
- [10].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, The Protein Data Bank, Nucleic Acids Res 28 (2000) 235–242, 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].PDB, https://www.rcsb.org/ (Accessed 30 August 2018).
- [12].White R, Lees JA, Needham M, Ham J, Parker M, Structural Organization and Expression of the Mouse Estrogen Receptor, Mol. Endocrinol 1 (1987) 735–744, 10.1210/mend-1-10-735. [DOI] [PubMed] [Google Scholar]
- [13].Held M, Metzner P, Prinz JH, Noe F, Mechanisms of protein-ligand association and its modulation by protein mutations, Biophys. J 100 (2011) 701–10, 10.1016/j.bpj.2010.12.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, Roychowdhury S, Pienta KJ, Kim SY, Roberts JS, Rae JM, Van Poznak CH, Hayes DF, Chugh R, Kunju LP, Talpaz M, Schott AF, Chinnaiyan AM, Activating ESR1 mutations in hormone-resistant metastatic breast cancer, Nat. Genet 45 (2013) 1446–51, 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S, ESR1 ligand-binding domain mutations in hormone-resistant breast cancer, Nat. Genet 45 (2013) 1439–45, 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA, An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer, Cancer Res 57 (1997) 1244–1249, [PubMed] [Google Scholar]
- [17].Krieger E, Vriend G, YASARA View -molecular graphics for all devices -from smartphones to workstations, Bioinformatics 30 (2014) 2981–2, 10.1093/bioinformatics/btu426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dabrosin C, Increased extracellular local levels of estradiol in normal breast in vivo during the luteal phase of the menstrual cycle, J. Endocrinol 187 (2005) 103–8, 10.1677/joe.1.06163. [DOI] [PubMed] [Google Scholar]
- [19].Dhandapani KM, Brann DW, Estrogen-astrocyte interactions: implications for neuroprotection, BMC Neurosci 3 (2002) 6, 10.1186/1471-2202-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Warita K, Mitsuhashi T, Sugawara T, Tabuchi Y, Tanida T, Wang ZY, Matsumoto Y, Yokoyama T, Kitagawa H, Miki T, Takeuchi Y, Hoshi N, Direct effects of diethylstilbestrol on the gene expression of the cholesterol side-chain cleavage enzyme (P450scc) in testicular Leydig cells, Life Sci 87 (2010) 281–5, 10.1016/j.lfs.2010.06.020. [DOI] [PubMed] [Google Scholar]
- [21].Colborn T, vom Saal FS, Soto AM, Developmental effects of endocrine-disrupting chemicals in wildlife and humans., Environ. Health Perspect 101 (1993) 378–384, 10.1289/ehp.93101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wielogorska E, Elliott CT, Danaher M, Connolly L, Endocrine disruptor activity of multiple environmental food chain contaminants, Toxicol. In Vitro 29 (2015) 211–20, 10.1016/j.tiv.2014.10.014. [DOI] [PubMed] [Google Scholar]
- [23].Nohynek GJ, Borgert CJ, Dietrich D, Rozman KK, Endocrine disruption: Fact or urban legend?, Toxicol. Lett 223 (2013) 295–305, 10.1016/j.toxlet.2013.10.022. [DOI] [PubMed] [Google Scholar]
- [24].Sasseville D, Alfalah M, Lacroix JP, “Parabenoia” Debunked, or “Who’s Afraid of Parabens?”, Dermatitis 26 (2015) 254–9, 10.1097/DER.0000000000000147. [DOI] [PubMed] [Google Scholar]
- [25].Ding K, Kong X, Wang J, Lu L, Zhou W, Zhan T, Zhang C, Zhuang S, Side Chains of Parabens Modulate Antiandrogenic Activity: In Vitro and Molecular Docking Studies, Environ. Sci. Technol 51 (2017) 6452–6460, 10.1021/acs.est.7b00951. [DOI] [PubMed] [Google Scholar]
- [26].Zhang Z, Sun L, Hu Y, Jiao J, Hu J, Inverse antagonist activities of parabens on human oestrogen-related receptor γ (ERRγ): in vitro and in silico studies, Toxicol. Appl. Pharmacol 270 (2013) 16–22, 10.1016/j.taap.2013.03.030. [DOI] [PubMed] [Google Scholar]
- [27].Honda M, Robinson M, Kannan K, Parabens in human urine from several Asian countries, Greece, and the United States, Chemosphere 201 (2018) 13–19, 10.1016/j.chemosphere.2018.02.165. [DOI] [PubMed] [Google Scholar]
- [28].Watanabe Y, Kojima H, Takeuchi S, Uramaru N, Ohta S, Kitamura S, Comparative study on transcriptional activity of 17 parabens mediated by estrogen receptor alpha and beta and androgen receptor, Food Chem. Toxicol 57 (2013) 227–34, 10.1016/j.fct.2013.03.036. [DOI] [PubMed] [Google Scholar]
- [29].Bergquist BL, Jefferson KG, Kintz HN, Barber AE, Yeagley AA, Disconnecting the Estrogen Receptor Binding Properties and Antimicrobial Properties of Parabens through 3,5-Substitution, ACS Med. Chem. Lett 9 (2018) 51–55, 10.1021/acsmedchemlett.7b00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gonzalez TL, Richardson RJ, In silico mutagenesis and molecular docking of mouse and rat estrogen receptor a with paraben analogs., The Toxicologist, Supplement to Toxicol. Sci 162 (2018) Abstract 2109, https://www.toxicology.org/pubs/docs/Tox/2018Tox.pdf. [Google Scholar]
- [31].Delfosse V, Grimaldi M, Pons JL, Boulahtouf A, le Maire A, Cavailles V, Labesse G, Bourguet W, Balaguer P, Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes, Proc. Natl. Acad. Sci 109 (2012) 14930–14935, 10.1073/pnas.1203574109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Canutescu AA, Dunbrack RL Jr., Cyclic coordinate descent: A robotics algorithm for protein loop closure, Protein Sci 12 (2003) 963–972, 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Uniprot, UniProtKB -P03372 (ESR1_HUMAN), https://www.uniprot.org/uniprot/P03372, (2018),
- [34].Uniprot, UniProtKB -P19785 (ESR1_MOUSE), https://www.uniprot.org/uniprot/P19785, (2018),
- [35].Uniprot, UniProtKB -P06211 (ESR1_RAT), https://www.uniprot.org/uniprot/P06211, (2018),
- [36].Geneioius, https://www.geneious.com, (2018),
- [37].Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A, Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data, Bioinformatics 28 (2012) 1647–1649, 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K, Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8, Proteins 77 (2009) 114–122, 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Krieger E, Vriend G, New ways to boost molecular dynamics simulations, J. Comput. Chem 36 (2015) 996–1007, 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography, Acta. Crystallogr. D Biol. Crystallogr 66 (2010) 12–21, 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].MolProbity, http://molprobity.biochem.duke.edu/ (Accessed 30 August 2018).
- [42].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera—A visualization system for exploratory research and analysis., J. Comput. Chem 25 (2004) 1605–1612, 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- [43].Zhang Y, I-TASSER server for protein 3D structure prediction, BMC Bioinformatics 9 (2008) 40, 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Trott O, Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem 31 (2010) 455–61, 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Krieger E, Koraimann G, Vriend G, Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field, Proteins 47 (2002) 393–402, 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- [46].Moos RK, Angerer J, Dierkes G, Bruning T, Koch HM, Metabolism and elimination of methyl, iso-and n-butyl paraben in human urine after single oral dosage, Arch. Toxicol 90 (2016) 2699–2709, 10.1007/s00204-015-1636-0. [DOI] [PubMed] [Google Scholar]
- [47].Gonzalez TL, Moos RK, Gersch CL, Johnson MD, Richardson RJ, Koch HM, Rae JM, Metabolites of n-Butylparaben and iso-Butylparaben Exhibit Estrogenic Properties in MCF-7 and T47D Human Breast Cancer Cell Lines, Toxicol. Sci 164 (2018) 50–59, 10.1093/toxsci/kfy063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].OpenBabel, (2018), http://openbabel.org/wiki/Main_Page. (Accessed 12 August 2018).
- [49].O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR, Open Babel: An open chemical toolbox, J. Cheminf 3 (2011) 33, 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].SimulationsPlus, (2018), https://www.simulations-plus.com/software/admetpredictor/ (Accessed 12 August 2018).
- [51].Wijeyesakere SJ, Richardson RJ, In silico chemical–protein docking and molecular dynamics. In: Computational Systems Pharmacology and Toxicology (Johnson DE, and Richardson RJ, eds.), Royal Society of Chemistry, Cambridge (UK) (2017) 174–190, [Google Scholar]
- [52].Delfosse V, le Marie A, Balaguer P, Bourguet W, A structural perspective on nuclear receptors as targets of environmental compounds, Acta Pharmacol. Sin 36 (2015) 88–104, 10.1038/aps.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Delfosse V, Grimaldi M, Cavailles V, Balaguer P, Bourguet W, Structural and functional profiling of environmental ligands for estrogen receptors, Environ. Health Perspect 122 (2014) 1306–13, 10.1289/ehp.1408453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Krieger E, Dunbrack RL Jr., Hooft RW, Krieger B, Assignment of protonation states in proteins and ligands: combining pKa prediction with hydrogen bonding network optimization, Methods Mol. Biol 819 (2012) 405–21, 10.1007/978-1-61779-465-0_25. [DOI] [PubMed] [Google Scholar]
- [55].Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM, MUSTANG: A multiple structural alignment algorithm, Proteins Struct. Funct. Bioinf 64 (2006) 559–574, 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- [56].Allen WJ, Rizzo RC, Implementation of the Hungarian Algorithm to Account for Ligand Symmetry and Similarity in Structure-Based Design, J. Chem. Inf. Model 54 (2014) 518–529, 10.1021/ci400534h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kufareva I, Abagyan R, Methods of Protein Structure Comparison, in: Orry AJW, Abagyan R (Eds.), Homology Modeling: Methods and Protocols, Humana Press, Totowa, NJ, 2012, pp. 231–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang Y, Skolnick J, TM-align: a protein structure alignment algorithm based on the TM-score, Nucleic Acids Res 33 (2005) 2302–2309, 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Reynolds CH, Bembenek SD, Tounge BA, The role of molecular size in ligand efficiency, Bioorg. Med. Chem. Lett 17 (2007) 4258–4261, 10.1016/j.bmcl.2007.05.038. [DOI] [PubMed] [Google Scholar]
- [60].Hopkins AL, Keseru GM, Leeson PD, Rees DC, Reynolds CH, The role of ligand efficiency metrics in drug discovery, Nat. Rev. Drug Discov 13 (2014) 105, 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- [61].Murray CW, Erlanson DA, Hopkins AL, Keserü GM, Leeson PD, Rees DC, Reynolds CH, Richmond NJ, Validity of Ligand Efficiency Metrics, ACS Med. Chem. Lett 5 (2014) 616–618, 10.1021/ml500146d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wijeyesakere SJ, Richardson RJ, Stuckey JA, Crystal structure of patatin-17 in complex with aged and non-aged organophosphorus compounds, PLoS One 9 (2014) e108245, 10.1371/journal.pone.0108245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Damm-Ganamet KL, Smith RD, Dunbar JB Jr., Stuckey JA, Carlson HA, CSAR Benchmark Exercise 2011–2012: Evaluation of Results from Docking and Relative Ranking of Blinded Congeneric Series, J. Chem. Inf. Model 53 (2013) 1853–1870, 10.1021/ci400025f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ravindranath PA, Forli S, Goodsell DS, Olson AJ, Sanner MF, AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility, PLoS Comp. Biol 11 (2015) e1004586, 10.1371/journal.pcbi.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sorenson T, A method of establishing groups of equal amplitudes in plant sociology based on similarity of species content and its application to analyses of the vegetation on danish commons., K. dan Vidensk. Selsk. Biol. Skr 5 (1948) 1–34. [Google Scholar]
- [66].Maggiora G, Vogt M, Stumpfe D, Bajorath J, Molecular similarity in medicinal chemistry, J. Med. Chem 57 (2014) 3186–204, 10.1021/jm401411z. [DOI] [PubMed] [Google Scholar]
- [67].Pick JB, Computer display of population age structure, Demography 11 (1974) 673–682, 10.2307/2060477. [DOI] [PubMed] [Google Scholar]
- [68].Dabydeen SA, Furth PA, Genetically engineered ERalpha-positive breast cancer mouse models, Endocr. Relat. Cancer 21 (2014) R195–208, 10.1530/ERC-13-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hamilton KJ, Arao Y, Korach KS, Estrogen hormone physiology: reproductive findings from estrogen receptor mutant mice, Reprod. Biol 14 (2014) 3–8, 10.1016/j.repbio.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shull JD, Dennison KL, Chack AC, Trentham-Dietz A, Rat models of 17beta-estradiol-induced mammary cancer reveal novel insights into breast cancer etiology and prevention, Physiol. Genomics 50 (2018) 215–234, 10.1152/physiolgenomics.00105.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Schwede T, Protein modeling: what happened to the “protein structure gap”?, Structure 21 (2013) 1531–40, 10.1016/j.str.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sheehan D, O’Sullivan S, Online homology modelling as a means of bridging the sequence-structure gap, Bioeng. Bugs 2 (2011) 299–305, 10.4161/bbug.2.6.16116. [DOI] [PubMed] [Google Scholar]
- [73].Xiang Z, Advances in homology protein structure modeling, Curr. Protein Pept. Sci 7 (2006) 217–27, 10.2174/138920306777452312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Doering JA, Lee S, Kristiansen K, Evenseth L, Barron MG, Sylte I, LaLone CA, In silico site-directed mutagenesis informs species-specific predictions of chemical susceptibility derived from the Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS) tool, Toxicol. Sci (2018) kfy186–kfy186, 10.1093/toxsci/kfy186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chamkasem A, Toniti W, Sequence to structure approach of estrogen receptor alpha and ligand interactions, Asian Pac. J. Cancer Prev 16 (2015) 2161–6, 10.7314/apjcp.2015.16.6.2161. [DOI] [PubMed] [Google Scholar]
- [76].Harris HA, Bapat AR, Gonder DS, Frail DE, The ligand binding profiles of estrogen receptors alpha and beta are species dependent, Steroids 67 (2002) 379–84, 10.1016/S0039-128X(01)00194-5. [DOI] [PubMed] [Google Scholar]
- [77].Chang BY, Kim DS, Kim HS, Kim SY, Evaluation of estrogenic potential by herbal formula, HPC 03 for in vitro and in vivo, Reproduction 155 (2018) 105–115, 10.1530/REP-17-0530. [DOI] [PubMed] [Google Scholar]
- [78].Quinn AL, Regan JM, Tobin JM, Marinik BJ, McMahon JM, McNett DA, Sushynski CM, Crofoot SD, Jean PA, Plotzke KP, In vitro and in vivo evaluation of the estrogenic, androgenic, and progestagenic potential of two cyclic siloxanes, Toxicol. Sci 96 (2007) 145–53, 10.1093/toxsci/kfl185. [DOI] [PubMed] [Google Scholar]
- [79].Zhang Z, Jia C, Hu Y, Sun L, Jiao J, Zhao L, Zhu D, Li J, Tian Y, Bai H, Li R, Hu J, The estrogenic potential of salicylate esters and their possible risks in foods and cosmetics, Toxicol. Lett 209 (2012) 146–53, 10.1016/j.toxlet.2011.12.004. [DOI] [PubMed] [Google Scholar]
- [80].Zingue S, Michel T, Nde CBM, Njuh AN, Cisilotto J, Ndinteh DT, Clyne C, Fernandez X, Creczynski-Pasa TB, Njamen D, Estrogen-like and tissue-selective effects of 7-methoxycoumarin from Ficus umbellata (Moraceae): an in vitro and in vivo study, BMC Complement. Altern. Med 17 (2017) 383, 10.1186/s12906-017-1895-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bohnuud T, Luo L, Wodak SJ, Bonvin AM, Weng Z, Vajda S, Schueler-Furman O, Kozakov D, A benchmark testing ground for integrating homology modeling and protein docking, Proteins 85 (2017) 10–16, 10.1002/prot.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bordogna A, Pandini A, Bonati L, Predicting the accuracy of protein-ligand docking on homology models, J. Comput. Chem 32 (2011) 81–98, 10.1002/jcc.21601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].DeLisle RK, Yu SJ, Nair AC, Welsh WJ, Homology modeling of the estrogen receptor subtype beta (ER-beta) and calculation of ligand binding affinities, J. Mol. Graph. Model 20 (2001) 155–67, 10.1016/s1093-3263(01)00115-2. [DOI] [PubMed] [Google Scholar]
- [84].Yatsu R, Katsu Y, Kohno S, Mizutani T, Ogino Y, Ohta Y, Myburgh J, van Wyk JH, Guillette LJ Jr., Miyagawa S, Iguchi T, Characterization of evolutionary trend in squamate estrogen receptor sensitivity, Gen. Comp. Endocrinol 238 (2016) 88–95, 10.1016/j.ygcen.2016.04.005. [DOI] [PubMed] [Google Scholar]
- [85].Tohyama S, Miyagawa S, Lange A, Ogino Y, Mizutani T, Tatarazako N, Katsu Y, Ihara M, Tanaka H, Ishibashi H, Kobayashi T, Tyler CR, Iguchi T, Understanding the molecular basis for differences in responses of fish estrogen receptor subtypes to environmental estrogens, Environ. Sci. Technol 49 (2015) 7439–47, 10.1021/acs.est.5b00704. [DOI] [PubMed] [Google Scholar]
- [86].Marchand-Geneste N, Cazaunau M, Carpy AJ, Laguerre M, Porcher JM, Devillers J, Homology model of the rainbow trout estrogen receptor (rtERalpha) and docking of endocrine disrupting chemicals (EDCs), SAR QSAR Environ. Res 17 (2006) 93–105, 10.1080/10659360600562137. [DOI] [PubMed] [Google Scholar]
- [87].Jones BL, Walker C, Azizi B, Tolbert L, Williams LD, Snell TW, Conservation of estrogen receptor function in invertebrate reproduction, BMC Evol. Biol 17 (2017) 65, 10.1186/s12862-017-0909-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Costache AD, Pullela PK, Kasha P, Tomasiewicz H, Sem DS, Homology-modeled ligand-binding domains of zebrafish estrogen receptors alpha, beta1, and beta2: from in silico to in vivo studies of estrogen interactions in Danio rerio as a model system, Mol. Endocrinol 19 (2005) 2979–90, 10.1210/me.2004-0435. [DOI] [PubMed] [Google Scholar]
- [89].Alluri PG, Speers C, Chinnaiyan AM, Estrogen receptor mutations and their role in breast cancer progression, Breast Cancer Res 16 (2014) 494, 10.1186/s13058-014-0494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Herynk MH, Fuqua SA, Estrogen receptor mutations in human disease, Endocr. Rev 25 (2004) 869–98, 10.1210/er.2003-0010. [DOI] [PubMed] [Google Scholar]
- [91].Holen I, Speirs V, Morrissey B, Blyth K, In vivo models in breast cancer research: progress, challenges and future directions, Dis. Model. Mech 10 (2017) 359–371, 10.1242/dmm.028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Smits BM, Cotroneo MS, Haag JD, Gould MN, Genetically engineered rat models for breast cancer, Breast Dis 28 (2007) 53–61, [DOI] [PubMed] [Google Scholar]
- [93].Ferrero VE, Pedotti M, Chiado A, Simonelli L, Calzolai L, Varani L, Lettieri T, Rational modification of estrogen receptor by combination of computational and experimental analysis, PLoS One 9 (2014) e102658, 10.1371/journal.pone.0102658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Marty MS, Carney EW, Rowlands JC, Endocrine disruption: historical perspectives and its impact on the future of toxicology testing, Toxicol. Sci 120 Suppl 1 (2011) S93–108, 10.1093/toxsci/kfq329. [DOI] [PubMed] [Google Scholar]
- [95].Nishihama Y, Yoshinaga J, Iida A, Konishi S, Imai H, Yoneyama M, Nakajima D, Shiraishi H, Association between paraben exposure and menstrual cycle in female university students in Japan, Reprod. Toxicol 63 (2016) 107–13, 10.1016/j.reprotox.2016.05.010. [DOI] [PubMed] [Google Scholar]
- [96].Meeker JD, Yang T, Ye X, Calafat AM, Hauser R, Urinary concentrations of parabens and serum hormone levels, semen quality parameters, and sperm DNA damage, Environ. Health Perspect 119 (2011) 252–7, 10.1289/ehp.1002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Artacho-Cordon F, Arrebola JP, Nielsen O, Hernandez P, Skakkebaek NE, Fernandez MF, Andersson AM, Olea N, Frederiksen H, Assumed non-persistent environmental chemicals in human adipose tissue; matrix stability and correlation with levels measured in urine and serum, Environ. Res 156 (2017) 120–127, 10.1016/j.envres.2017.03.030. [DOI] [PubMed] [Google Scholar]
- [98].Azzouz A, Rascon AJ, Ballesteros E, Simultaneous determination of parabens, alkylphenols, phenylphenols, bisphenol A and triclosan in human urine, blood and breast milk by continuous solid-phase extraction and gas chromatography-mass spectrometry, J. Pharm. Biomed. Anal 119 (2016) 16–26, 10.1016/j.jpba.2015.11.024. [DOI] [PubMed] [Google Scholar]
- [99].Barr L, Metaxas G, Harbach CA, Savoy LA, Darbre PD, Measurement of paraben concentrations in human breast tissue at serial locations across the breast from axilla to sternum, J. Appl. Toxicol 32 (2012) 219–32, 10.1002/jat.1786. [DOI] [PubMed] [Google Scholar]
- [100].Frederiksen H, Jorgensen N, Andersson AM, Parabens in urine, serum and seminal plasma from healthy Danish men determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS), J. Expo. Sci. Environ. Epidemiol 21 (2011) 262–71, 10.1038/jes.2010.6. [DOI] [PubMed] [Google Scholar]
- [101].Hines EP, Mendola P, von Ehrenstein OS, Ye X, Calafat AM, Fenton SE, Concentrations of environmental phenols and parabens in milk, urine and serum of lactating North Carolina women, Reprod. Toxicol 54 (2015) 120–8, 10.1016/j.reprotox.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Valle-Sistac J, Molins-Delgado D, Diaz M, Ibanez L, Barcelo D, Silvia Diaz-Cruz M, Determination of parabens and benzophenone-type UV filters in human placenta. First description of the existence of benzyl paraben and benzophenone-4, Environ. Int 88 (2016) 243–9, 10.1016/j.envint.2015.12.034. [DOI] [PubMed] [Google Scholar]
- [103].Moos RK, Apel P, Schroter-Kermani C, Kolossa-Gehring M, Bruning T, Koch HM, Daily intake and hazard index of parabens based upon 24 h urine samples of the German Environmental Specimen Bank from 1995 to 2012, J. Expo. Sci. Environ. Epidemiol 27 (2016), 10.1038/jes.2016.65. [DOI] [PubMed] [Google Scholar]
- [104].Mansouri K, Abdelaziz A, Rybacka A, Roncaglioni A, Tropsha A, Varnek A, Zakharov A, Worth A, Richard AM, Grulke CM, Trisciuzzi D, Fourches D, Horvath D, Benfenati E, Muratov E, Wedebye EB, Grisoni F, Mangiatordi GF, Incisivo GM, Hong H, Ng HW, Tetko IV, Balabin I, Kancherla J, Shen J, Burton J, Nicklaus M, Cassotti M, Nikolov NG, Nicolotti O, Andersson PL, Zang Q, Politi R, Beger RD, Todeschini R, Huang R, Farag S, Rosenberg SA, Slavov S, Hu X, Judson RS, CERAPP: Collaborative Estrogen Receptor Activity Prediction Project, Environ. Health Perspect 124 (2016) 1023–33, 10.1289/ehp.1510267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Trisciuzzi D, Alberga D, Mansouri K, Judson R, Cellamare S, Catto M, Carotti A, Benfenati E, Novellino E, Mangiatordi GF, Nicolotti O, Docking-based classification models for exploratory toxicology studies on high-quality estrogenic experimental data, Future Med. Chem 7 (2015) 1921–36, 10.4155/fmc.15.103. [DOI] [PubMed] [Google Scholar]
- [106].Lee S, Barron MG, Structure-based understanding of binding affinity and mode of estrogen receptor α agonists and antagonists, PLoS One 12 (2017) e0169607, 10.1371/journal.pone.0169607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Sun L, Yu T, Guo J, Zhang Z, Hu Y, Xiao X, Sun Y, Xiao H, Li J, Zhu D, Sai L, Li J, The estrogenicity of methylparaben and ethylparaben at doses close to the acceptable daily intake in immature Sprague-Dawley rats, Sci Rep 6 (2016) 25173, 10.1038/srep25173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Hu Y, Zhang Z, Sun L, Zhu D, Liu Q, Jiao J, Li J, Qi M, The estrogenic effects of benzylparaben at low doses based on uterotrophic assay in immature SD rats, Food Chem. Toxicol 53 (2013) 69–74, 10.1016/j.fct.2012.11.043. [DOI] [PubMed] [Google Scholar]
- [109].Kang MH, Choi H, Oshima M, Cheong JH, Kim S, Lee JH, Park YS, Choi HS, Kweon MN, Pack CG, Lee JS, Mills GB, Myung SJ, Park YY, Estrogen-related receptor gamma functions as a tumor suppressor in gastric cancer, Nat. Commun 9 (2018) 1920, 10.1038/s41467-018-04244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Haldosen LA, Zhao C, Dahlman-Wright K, Estrogen receptor beta in breast cancer, Mol. Cell. Endocrinol 382 (2014) 665–672, 10.1016/j.mce.2013.08.005. [DOI] [PubMed] [Google Scholar]
- [111].PubChem, https://pubchem.ncbi.nlm.nih.gov/compound/4-hydroxybenzoic_acid#section=Chemical-and-Physical-Properties (Accessed 30 August 2018).
- [112].Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ, Computational protein-ligand docking and virtual drug screening with the AutoDock suite, Nat. Protoc 11 (2016) 905–19, 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Lemini C, Silva G, Timossi C, Luque D, Valverde A, Gonzalez-Martinez M, Hernandez A, Rubio-Poo C, Chavez Lara B, Valenzuela F, Estrogenic effects of p-hydroxybenzoic acid in CD1 mice, Environ. Res 75 (1997) 130–4, 10.1006/enrs.1997.3782. [DOI] [PubMed] [Google Scholar]
- [114].Pugazhendhi D, Pope GS, Darbre PD, Oestrogenic activity of p-hydroxybenzoic acid (common metabolite of paraben esters) and methylparaben in human breast cancer cell lines, J. Appl. Toxicol 25 (2005) 301–9, 10.1002/jat.1066. [DOI] [PubMed] [Google Scholar]
- [115].Finimundy TC, Barros L, Calhelha RC, Alves MJ, Prieto MA, Abreu RMV, Dillon AJP, Henriques JAP, Roesch-Ely M, Ferreira ICFR, Multifunctions of Pleurotus sajor-caju (Fr.) Singer: A highly nutritious food and a source for bioactive compounds, Food. Chem 245 (2018) 150–158, 10.1016/j.foodchem.2017.10.088. [DOI] [PubMed] [Google Scholar]
- [116].Schror K, Acetylsalicylic Acid, 2nd ed. Wiley-VCH, Weinheim, Germany: (2016), p. 139. [Google Scholar]
- [117].Martin DP, Cohen SM, Nucleophile recognition as an alternative inhibition mode for benzoic acid based carbonic anhydrase inhibitors, Chem. Commun. (Camb.) 48 (2012) 5259–61, 10.1039/c2cc32013d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Chen Z, Shen X, Wang J, Wang J, Yuan Q, Yan Y, Rational engineering of p-hydroxybenzoate hydroxylase to enable efficient gallic acid synthesis via a novel artificial biosynthetic pathway, Biotechnol. Bioeng 114 (2017) 2571–2580, 10.1002/bit.26364. [DOI] [PubMed] [Google Scholar]
- [119].Cheng W, Li W, Structural insights into ubiquinone biosynthesis in membranes, Science 343 (2014) 878–81, 10.1126/science.1246774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Cavalluzzi MM, Mangiatordi GF, Nicolotti O, Lentini G, Ligand efficiency metrics in drug discovery: the pros and cons from a practical perspective, Expert. Opin. Drug Discov 12 (2017) 1087–1104, 10.1080/17460441.2017.1365056. [DOI] [PubMed] [Google Scholar]
- [121].Polanski J, Tkocz A, Kucia U, Beware of ligand efficiency (LE): understanding LE data in modeling structure-activity and structure-economy relationships, J. Cheminform 9 (2017) 49, 10.1186/s13321-017-0236-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Velvadapu V, Farmer BT, Reitz AB, Fragment-based drug discovery, In: The Practice of Medicinal Chemistry 4th Edition (Wermouth CG, Aldous D, Raboisson P, Rognan D, eds.), Elsevier, Amsterdam: (2015) 161–179. [Google Scholar]
- [123].Orita M, Ohno K, Warizaya M, Amano Y, Niimi T, Chapter fifteen -Lead Generation and Examples: Opinion Regarding How to Follow Up Hits, in: Kuo LC (Ed.), Methods in Enzymology, Academic Press; 2011, pp. 383–419. [DOI] [PubMed] [Google Scholar]
- [124].Miyagawa S, Lange A, Hirakawa I, Tohyama S, Ogino Y, Mizutani T, Kagami Y, Kusano T, Ihara M, Tanaka H, Tatarazako N, Ohta Y, Katsu Y, Tyler CR, Iguchi T, Differing species responsiveness of estrogenic contaminants in fish is conferred by the ligand binding domain of the estrogen receptor, Environ. Sci. Technol 48 (2014) 5254–63, 10.1021/es5002659. [DOI] [PubMed] [Google Scholar]
- [125].Pan S, Yuan C, Tagmount A, Rudel RA, Ackerman JM, Yaswen P, Vulpe CD, Leitman DC, Parabens and Human Epidermal Growth Factor Receptor Ligands Cross-Talk in Breast Cancer Cells, Environ. Health Perspect 124 (2015) 563–569, 10.1289/ehp.1409200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Duarte JM, Santos JBD, Melo LC, Comparison of similarity coefficients based on RAPD markers in the common bean, Genet. Mol. Biol 22 (1999) 427–432, 10.1590/s1415-47571999000300024. [DOI] [Google Scholar]
- [127].Todeschini R, Consonni V, Xiang H, Holliday J, Buscema M, Willett P, Similarity coefficients for binary chemoinformatics data: overview and extended comparison using simulated and real data sets, J. Chem. Inf. Model 52 (2012) 2884–901, 10.1021/ci300261r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.