

Abstract
Context:
Loss-of-function mutations in the coding region of MKRN3, a maternally imprinted gene at chromosome 15q11.2, are a common cause of familial central precocious puberty (CPP). Whether MKRN3 alterations in regulatory regions can cause CPP has not been explored to date. We aimed to investigate potential pathogenic variants in the promoter region of MKRN3 in patients with idiopathic CPP.
Patients/Methods:
A cohort of 110 patients with idiopathic CPP was studied. Family history of precocious sexual development was present in 25%. Mutations in the coding region of MKRN3 were excluded in all patients. Genomic DNA was extracted from peripheral blood leukocytes, and 1,100 nucleotides (nt) of the 5′-regulatory region of MKRN3 were amplified and sequenced. Luciferase assays were performed in GT1–7 cells transiently transfected with plasmids containing mutated and wild-type MKRN3 promoter.
Results:
We identified a rare heterozygous 4-nt deletion (c.−150_−147delTCAG; −38 to −41 nt upstream to the transcription start site) in the proximal promoter region of MKRN3 in a girl with CPP. In silico analysis predicted that this deletion would lead to the loss of a binding site for a downstream responsive element antagonist modulator (DREAM), a potential transcription factor for MKRN3 and GNRH1 expression. Luciferase assays demonstrated a significant reduction of MKRN3 promoter activity in transfected cells with a c.−150_−147delTCAG construct plasmid in both homozygous and heterozygous states when compared with cells transfected with the corresponding wild-type MKRN3 promoter region.
Conclusion:
A rare genetic alteration in the regulatory region of MKRN3 causes CPP.
Keywords: Central precocious puberty, MKRN3 promoter region, Genetic alteration, Regulatory region
Introduction
Distinct genetic abnormalities were recently recognized as a cause of familial central precocious puberty (CPP) in children of both sexes [1, 2]. Although family history of early or precocious puberty may be found in about one-third of CPP patients, most instances are sporadic or with unknown family history [3]. To date, loss-of-function mutations in MKRN3 represent the most common genetic etiology of CPP, accounting for up to 46% of familial cases and around 5% of sporadic cases [1, 2, 4]. MKRN3 is a ubiquitously expressed, highly conserved intronless gene, located on the long arm of chromosome 15, in a region that contains a cluster of maternally imprinted genes associated with Prader-Willi syndrome [5, 6]. Due to the maternal imprinting, MKRN3 is expressed exclusively from the paternally inherited allele [5, 7].
MKRN3 encodes a RING zinc-finger protein that belongs to the Makorin family, whose members are known to be E3 ubiquitin ligases [7]. MKRN3 protein structure has a ubiquitin ligase domain, and it has been postulated that MKRN3 might inhibit stimulators of GnRH secretion [8, 9]. Mkrn3 expression studies in the hypothalamic arcuate nucleus of mice have demonstrated a decrease in Mkrn3 mRNA levels just before puberty initiation, reinforcing the hypothesis that MKRN3 may act as an inhibitor of GnRH secretion during childhood [4].
Genetic variants in promoter regions have been shown to be associated with decreased gene expression [10, 11]. Although several studies have identified loss-of-function mutations in the coding region of MKRN3 in patients with CPP, abnormalities in its noncoding region leading to CPP have not yet been reported. Therefore, we aimed to investigate whether variants in the promoter region of MKRN3 could modulate gene expression and lead to CPP in humans.
Patients and Methods
We studied 110 patients (109 girls and 1 boy) with idiopathic CPP who were clinically evaluated at Hospital das Clínicas from São Paulo Medical School, São Paulo University, São Paulo, Brazil. Family history of precocious sexual development was identified in 25%. Abnormalities in the coding region of MKRN3 were excluded in all patients. Idiopathic CPP was characterized by clinical signs of pubertal development before age 8 years in girls and 9 years in boys, pubertal basal and/or GnRH-stimulated LH levels, advanced bone age (Greulich and Pyle method), and normal central nervous system magnetic resonance imaging (MRI) [12, 13]. A control group, consisting of 68 Brazilian individuals who had a history of spontaneous normal pubertal development, was also analyzed. Written informed consent was obtained from all participants and their parents.
Hormone Assays
Serum LH, FSH, testosterone, and estradiol concentrations were measured by immunofluorometric assay (IFMA) and electrochemiluminescence assay (ECLIA). The interassay coefficient of variation was 5% or less for all assays. For the acute GnRH stimulation test, serum LH and FSH were measured at −15, 0, 15, 30, 45, and 60 min after intravenous administration of 100 μg GnRH. A basal LH >0.6 U/L (IFMA) or >0.15 U/L (ECLIA) was considered to be a pubertal level for both sexes and a GnRH-stimulated LH peak >6.9 U/L for girls and >9.6 U/L for boys (IFMA) or >5.0 U/L for both sexes (ECLIA) was considered as a pubertal response [4, 14]. Basal estradiol levels >21 pg/mL in girls and basal testosterone levels >19 ng/dL in boys were considered as pubertal.
Genetic Analysis
Genomic DNA was isolated from peripheral blood leukocytes from all participants using standard procedures. The 5′-regulatory region (5′-UTR) of MKRN3 encompassing 1,100 nucleotides (−840 to +260 in relation to the transcription start site [TSS]) [5], including the proximal promoter, was amplified and sequenced. The TSS of MKRN3 was previously mapped by Jong et al. [5] using rapid amplification of cDNA ends, with 109 bp 5′-UTR. The promoter region of MKRN3 (GenBank accession number NC_000015.10) was amplified by polymerase chain reaction (PCR) and followed by sequencing of the products with the use of the conventional Sanger method (suppl. Appendix; for all online suppl. material, see https://www.karger.com/doi/10.1159/000490059). We used the 1000 Genomes and NHLBI EVS databases and gnomAD, as well as a database of 150 exomes from the University of Michigan and a control group of 68 Brazilian individuals, to exclude all common variants. Putative binding sites in the MKRN3 promoter region were predicted using the Genomatix software suite (https://www.genomatix.de/online_help/help_matinspector/matinspector_help.html) [15].
Plasmid Constructs
The pGL2 basic luciferase vector and pGL2hMKRN3p_WT vector were kindly provided by A. Lominiczi (Division of Neuroscience, Oregon National Primate Research Center, Beaverton, OR, USA). The pGL2hMKRN3p_WT construct contains 439 bp (−446 to −7 in relation to the ATG) of the promoter region of MKRN3 gene and was used as a control for heterozygous and homozygous variants. The 4-nt deletion (c.−150_−147delTCAG) was generated using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent, USA) as specified by the manufacturer. The mutagenic primers were designed using the web-based QuikChange Primer Design Program available online at www.agilent.com/genomics/qcpd. The site-directed mutagenesis was confirmed by automatic sequencing using the Sanger method (data not shown).
Luciferase Assays
Hypothalamic GnRH neuronal mouse GT1−7 cell line was used for the transfection studies. This cell line represents a good model for studying neuron-specific expression of the GNRH1 gene, as it retains many characteristics of in vivo GnRH neurons, including the pulsatile GnRH release [16, 17]. In addition, the GT1–7 cell line expresses the downstream responsive element antagonist modulator (DREAM) transcription factor, as demonstrated by Leclerc et al. [18]. These cells were purchased from Banco de Células do Rio de Janeiro (BCRJ) (Catalogue Number 0095, BCRJ, Duque de Caxias, RJ, Brazil) and cultured in DMEM (Life Technologies) with 10% fetal bovine serum (Life Technologies) and 1% antibiotics (Life Technologies) under a humid environment with 5% CO2 at 37 ° C overnight. The cells were plated in a 6-well plate at a density of 5 × 105 for 24 h before transfection. The GT1–7 cells were transiently transfected with increasing amounts (750, 1,500, and 3,000 ng/well) of pGL2hMKRN3p_WT or mutant MKRN3 expression vectors (pGL2hMKRN3p_delTCAG representing the homozygous condition, and 50% of pGL2hMKRN3p_WT + 50% of pGL2hMKRN3p_delTCAG representing the heterozygous condition) with co-transfection of pCMV_Renilla vector (100 ng/well; Promega, Madison, WI, USA). The transfection complex was prepared by incubation with 10 μL Lipofectamine 2000 (Invitrogen) and 250 μL OPTI-MEM (Life Technologies) for 30 min at room temperature, and then added to the cultured GT1–7 cells. Twenty-four hours later, the cells were lysed and luciferase assays were performed using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer’s instructions on a GloMAX®-Multi Detection System (Promega). All luciferase data were normalized for the Renilla internal control. The data were calculated as means ± standard deviations of 4 independent experiments, each performed in triplicate.
Statistical Analyses
Statistical analysis was performed using ANOVA and Brown-Forsythe and Bartlett’s post-tests. Tests with p values <0.05 were considered significant.
Results
DNA Sequencing
We identified a novel heterozygous 4-nt deletion, c.−150_−147delTCAG (−38 to −41 nt upstream to the TSS; [GRCh37/hg19] chr15:23,810,780–23,810,930delTCAG), in the proximal promoter region of MKRN3 in a girl with a nonfamilial idiopathic CPP. This variant was absent in several different databases (1000 Genomes, NHLBS EVS, gnomAD, and 150 exomes from Michigan University) and in the Brazilian control group. The affected girl had the onset of pubertal development around age 7 years. At the first medical visit (7.6 years), she had breast development pubertal stage Tanner 3 and advanced bone age (9 years). Hormonal evaluation revealed a pubertal basal LH level of 1.0 U/L and an LH level of 8.9 U/L after GnRH stimulation, both measured by IFMA, and a pubertal basal estrogen level (IFMA) of 70 pg/mL. She was treated with a GnRH analog (leuprorelin acetate) for 3 years with adequate clinical and hormonal control. Her mother had menarche at age 10 years, and she did not carry the 4-nt deletion. DNA from her father and 2 brothers were not available (Fig. 1).
Fig. 1.
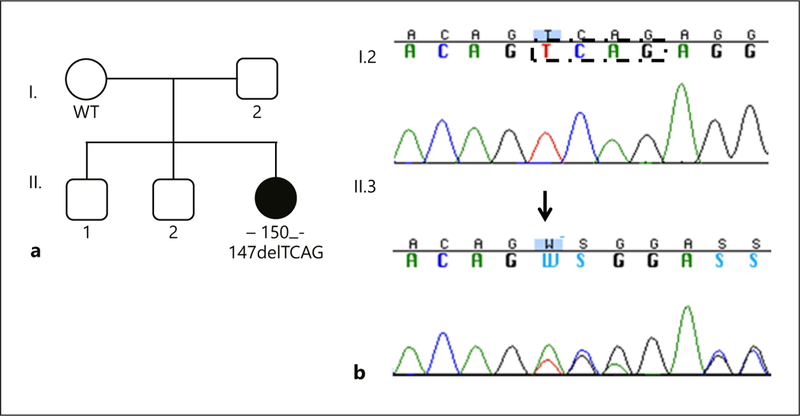
a Family pedigree showing the presence of the c.−150_−147delTCAG variant in the affected girl (individual II.3). The absence of the variant in the mother (individual I.2) suggests that this defect was likely inherited from her father (DNA not available) or it may represent a potential de novo MKRN3 defect. WT, wild type MKRN3 promoter region. b Chromatograms of the MKRN3 promoter showing the c.−150_−147delTCAG heterozygous variant in the genomic DNA of the affected girl (II.3) and the normal sequence of her unaffected mother (I.2). The heterozygous variant is indicated with an arrow. The black dashed box indicates the 4 bases that are deleted in the affected girl.
To understand the relevance of the c.−150_−147delTCAG variant, we performed in silico analysis using the Genomatix software suite. Comparison between deleted and reference sequences predicted that the c.−150_−147delTCAG variant would lead to the loss of a putative DREAM binding site.
Another rare variant, c.−274T>A ([GRCh37/hg19 Chr15:23810656 T/A; rs182933790), in the promoter region of MKRN3, was detected in a girl with pubertal onset at 6.6 years. The minor allele frequency of this variant was <0.03% in the databases (1000 Genomes, NHLBS EVS, and gnomAD), indicating that it is a rare nucleotide change. This variant was identified in 2 Brazilian control individuals (1 man and 1 woman) who had onset of pubertal development at adequate age and no history of CPP in their close relatives, suggesting a lack of genotype-phenotype correlation. However, the parents of the individuals were not studied; therefore, we cannot exclude that this rare promoter variant affected the maternal allele, characterizing asymptomatic carriers. The remaining participants did not have any detectable rare coding variants in the MKRN3 promoter.
Decreased Activity of the Mutant MKRN3 Promoter
The luciferase activity of pGL2hMKRN3p_delTCAG in homozygous state was significantly decreased (p < 0.0001 for 1,500 ng; p < 0.001 for 3,000 ng) compared with that of pGL2hMKRN3p_WT. Similarly, the transfection of pGL2hMKRN3p_WT/delTCAG mutant in heterozygous state also led to a significant reduction of the luciferase expression compared to the wild type (p < 0.01 for both 1,500 and 3,000 ng) (Fig. 2). Moreover, a dose-dependent regulation of MKRN3 promoter was verified with 3 different concentrations of DNA vectors (750, 1,500, and 3,000 ng in total) for each condition. As shown in Figure 2, a significant decreased regulation was demonstrated in all amounts analyzed (p < 0.0001).
Fig. 2.
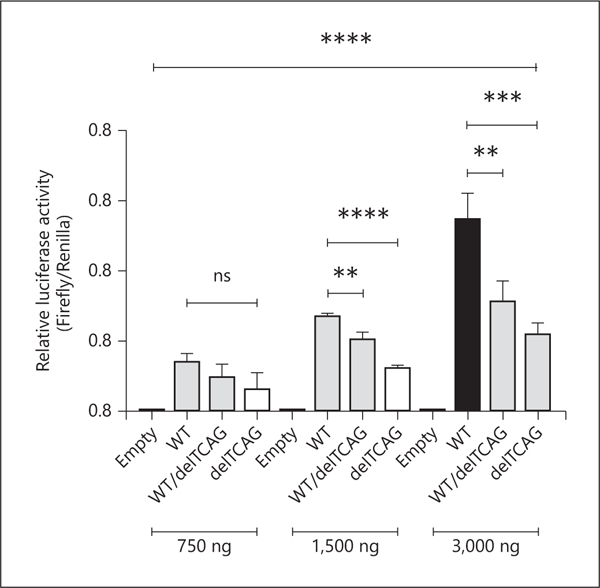
Functional study of MKRN3 promoter variant. On the x-axis, “Empty” indicates pGL2 vector; “WT” indicates pGL2hMKRN3p_WT; “wT/delTCAG” indicates 50% pGL2hMKRN3p_WT + 50% pGL2hMKRN3p_delTCAG; and “delTCAG” indicates pGL2hMKRN3p_delTCAG. Values represent means ± standard deviations of 4 independent experiments, each performed in triplicate. The dose-dependent ANOVA test is represented by **** p < 0.0001 with Brown-Forsythe test p = 0.0089 and Bartlett’s test p < 0.0001. ** p < 0.01; *** p < 0.001; **** p < 0.0001. pGL2 was excluded from ANOVA analysis. ANOVA 750 ng: ns. ANOVA 1,500 ng: <0.0001; WT × WT/delTCAG: **; WT × delTCAG: ****. ANOVA 3,000 ng: 0.0002; WT × WT/delTCAG: **; WT × delTCAG: ***. ns, not significant.
Discussion
Since the first description of MKRN3 loss-of-function mutations in children with idiopathic CPP in 2013 [3], more than 30 different variants affecting the MKRN3 have been associated with the CPP phenotype [1, 2, 19–21]. All previously identified mutations were located in the coding sequence of the only exon of MKRN3, with a hot-spot area between nucleotides 476–482, a cytosine homopolymer region [19, 20]. A recurrent mutation, c.482insC frame-shift, identified in this region, has been reported in several families with CPP [20, 21]. Here, we describe a rare heterozygous 4-nt deletion in the MKRN3 promoter region (c.−150_−147delTCAG) in a girl with typical features of CPP. She presented with progressive breast development (Tanner 3 at first medical visit) associated with advanced bone age and pubertal basal and GnRH-stimulated LH levels at age 7.5 years. There was no family history of premature sexual development among her close relatives, and the lack of MRI structural alterations suggested initially that she had an idiopathic form of CPP.
Notably, the 4-nt deletion c.−150_−147delTCAG is located at −38 to −41 nt upstream to the TSS, in the proximal promoter of MKRN3 (Fig. 3). This is an area typically very important for binding of the basal transcriptional machinery and for basal and regulated gene expression [22]. In addition, the 5′-UTR of MKRN3 is notable for potential transcription factor motifs [5] (Fig. 3). In silico analysis (Genomatix) predicted that the c.−150_−147delTCAG variant would lead to a loss of a DREAM binding site. DREAM, also known as KChIP-3 or calsenilin, is a transcription factor widely expressed in a variety of tissues, including the central nervous system and reproductive organs [16, 23]. In most cases, DREAM is a transcriptional repressor that acts in a Ca2+-regulated manner, which binds downstream response elements. Interestingly, in some promoters, DREAM may play a role as an activator, especially when the downstream response elements are located upstream, but not downstream, of the transcriptional initiation site [24]. Leclerc and Boock-for [16] demonstrated that the Ca2+-binding DREAM protein can also bind to a specific region in the GNRH1 promoter, increasing GNRH1 gene expression [16]. We speculate that DREAM could act as an activator in the MKRN3 promoter, by binding to a site upstream of the transcriptional start site; therefore, a loss of its binding site would lead to a decrease in MKRN3 expression resulting in premature activation of GnRH pulsatile secretion and, consequently, earlier onset of pubertal development (Fig. 3). Indeed, in vitro luciferase assay studies demonstrated a significant reduction of MKRN3 promoter activity in GT1–7 cells transfected with a plasmid encoding the deletion, indicating that this 4-nt deletion had a negative impact on MKRN3 transcription.
Fig. 3.
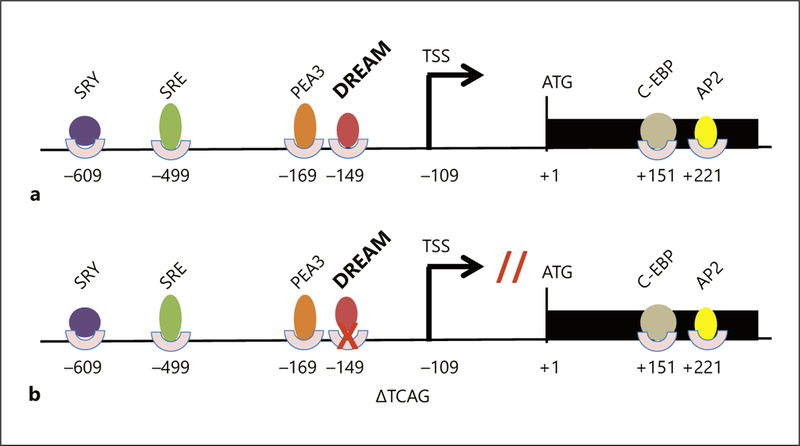
Schematic representation of the MKRN3 gene; the black rectangle indicates the coding DNA sequence of MKRN3; colorful circles and ovals represent putative transcription factor binding sites. a Wild-type MKRN3 promoter region with intact binding sites. b MKRN3 promoter region with the 4-nt deletion. TSS, transcription start site; SRY, sex-determining region Y (SRY) protein; PEA3, polyomavirus enhancer activator 3 homolog; SRE, serum response element; C-EBP, CCAAT/enhancer binding protein; AP2, activating protein-2.
DNA from the patient’s father was not available; therefore, the paternal inheritance of this novel 4-nt deletion located in the promoter region of the maternally imprinted MKRN3 was not confirmed. However, the patient’s mother, who had normal pubertal developmental, did not carry this variant, suggesting that this defect was likely inherited from her father. Alternatively, it may represent a potential de novo MKRN3 defect.
In summary, our current findings suggest that the 4-nt deletion in the MKRN3 proximal promoter resulted in MKRN3 deficiency and consequently CPP in a girl, indicating that potential abnormalities of the regulatory regions of MKRN3 can cause the CPP phenotype.
Supplementary Material
Acknowledgments
The authors thank Bruno Ferraz-de-Souza for support in the in vitro studies and Alexander Lominiczi for providing the plasmids for luciferase assays.
This work was supported by grant 302849/2015–7 (to A.C.L.), grant 303002/2016–6 (to B.B.M.) from Conselho Nacional de Desenvolvimento Científico e Tecnológico; grants 13/03236–5 (to A.C.L.) and 2013/06391–1 (to D.B.M.), and grant 2014/50137–5 (to B.B.M.) from the Fundação de Amparo à Pesquisa do Estado de Sao Paulo and National Institutes of Health R01 HD082314 (to U.B.K.).
Footnotes
Disclosure Statement
The authors have nothing to disclose.
References
- 1.Bulcao Macedo D, Nahime Brito V, Latronico AC: New causes of central precocious puberty: the role of genetic factors. Neuroendocrinology 2014;100:1–8. [DOI] [PubMed] [Google Scholar]
- 2.Simon D, Ba I, Mekhail N, Ecosse E, Paulsen A, Zenaty D, Houang M, Jesuran Perelroizen M, de Filippo GP, Salerno M, Simonin G, Reynaud R, Carel JC, Léger J, de Roux N: Mutations in the maternally imprinted gene MKRN3 are common in familial central precocious puberty. Eur J Endocrinol 2016; 174:1–8. [DOI] [PubMed] [Google Scholar]
- 3.de Vries L, Kauschansky A, Shohat M, Phillip M: Familial central precocious puberty suggests autosomal dominant inheritance. J Clin Endocrinol Metab 2004;89:1794–1800. [DOI] [PubMed] [Google Scholar]
- 4.Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, Cukier P, Thompson IR, Navarro VM, Gagliardi PC, Rodrigues T, Kochi C, Longui CA, Beckers D, de Zegher F, Montenegro LR, Mendonca BB, Carroll RS, Hirschhorn JN, Latronico AC, Kaiser UB: Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med 2013;368:2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jong MT, Gray TA, Ji Y, Glenn CC, Saitoh S, Driscoll DJ, Nicholls RD: A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum Mol Genet 1999;8:783–793. [DOI] [PubMed] [Google Scholar]
- 6.Nicholls RD, Saitoh S, Horsthemke B: Imprinting in Prader-Willi and Angelman syndromes. Trends Genet 1998;14:194–200. [DOI] [PubMed] [Google Scholar]
- 7.Gray TA, Hernandez L, Carey AH, Schaldach MA, Smithwick MJ, Rus K, Marshall Graves JA, Stewart CL, Nicholls RD: The ancient source of a distinct gene family encoding proteins featuring RING and C(3)H zinc-finger motifs with abundant expression in developing brain and nervous system. Genomics 2000;66:76–86. [DOI] [PubMed] [Google Scholar]
- 8.Abreu AP, Macedo DB, Brito VN, Kaiser UB, Latronico AC: A new pathway in the control of the initiation of puberty: the MKRN3 gene. J Mol Endocrinol 2015;54:R131–R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abreu AP, Kaiser UB: Pubertal development and regulation. Lancet Diabetes Endocrinol 2016;4:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newburger PE, Skalnik DG, Hopkins PJ, Eklund EA, Curnutte JT: Mutations in the promoter region of the gene for gp91-phox in X-linked chronic granulomatous disease with decreased expression of cytochrome b558. J Clin Invest 1994;94:1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spek CA, Greengard JS, Griffin JH, Bertina RM, Reitsma PH: Two mutations in the promoter region of the human protein C gene both cause type I protein C deficiency by disruption of two HNF-3 binding sites. J Biol Chem 1995;270:24216–24221. [DOI] [PubMed] [Google Scholar]
- 12.Carel JC, Lèger J: Precocious puberty. N Engl J Med 2008;358:2366–2377. [DOI] [PubMed] [Google Scholar]
- 13.Latronico AC, Brito VN, Carel JC: Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol 2016;4: 265–274. [DOI] [PubMed] [Google Scholar]
- 14.Neely EK, Hintz RL, Wilson DM, Lee PA, Gautier T, Argente J, Stene M: Normal ranges for immunochemiluminometric gonadotropin assays. J Pediatr 1995;127:40–46. [DOI] [PubMed] [Google Scholar]
- 15.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T: MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 2005; 21:2933–2942. [DOI] [PubMed] [Google Scholar]
- 16.Leclerc GM, Boockfor FR: Calcium influx and DREAM protein are required for GnRH gene expression pulse activity. Mol Cell Endocrinol 2007;267:70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI: Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 1990;5: 1–10. [DOI] [PubMed] [Google Scholar]
- 18.Leclerc GM, Leclerc GJ, Shorte SL, Stephen Frawley L, Boockfor FR: Cloning and mRNA expression of the Ca2+-binding DREAM protein in the pituitary. Gen Comp Endocrinol 2002;129:45–55. [DOI] [PubMed] [Google Scholar]
- 19.Macedo DB, Abreu AP, Reis AC, Montenegro LR, Dauber A, Beneduzzi D, Cukier P, Silveira LF, Teles MG, Carroll RS, Guerra Junior G, Guaragna Filho G, Gucev Z, Arnhold IJ, de Castro M, Moreira AC, Martinelli CE Jr, Hirschhorn JN, Mendonca BB, Brito VN, Antonini SR, Kaiser UB, Latronico AC: Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J Clin Endocrinol Metab 2014;99:E1097–E1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grandone A, Capristo C, Cirillo G, Sasso M, Umano GR, Mariani M, Miraglia Del Giudice E, Perrone L: Molecular screening of MKRN3, DLK1, and KCNK9 genes in girls with idiopathic central precocious puberty. Horm Res Paediatr 2017;88:194–200. [DOI] [PubMed] [Google Scholar]
- 21.Ortiz-Cabrera NV, Riveiro-Álvarez R, López-Martínez MA, Pérez-Segura P, Aragón-Gómez I, Trujillo-Tiebas MJ, Soriano-Guillén L: Clinical exome sequencing reveals MKRN3 pathogenic variants in familial and nonfamilial idiopathic central precocious puberty. Horm Res Paediatr 2017;87:88–94. [DOI] [PubMed] [Google Scholar]
- 22.Ohler U, Wassarman DA: Promoting developmental transcription. Development 2010; 137:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrión AM, Link WA, Ledo F, Mellström B, Naranjo JR: DREAM is a Ca2+-regulated transcriptional repressor. Nature 1999;398:80–84. [DOI] [PubMed] [Google Scholar]
- 24.Mellström B, Naranjo JR: Ca2+-dependent transcriptional repression and derepression: DREAM, a direct effector. Semin Cell Dev Biol 2001;12:59–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.