

Abstract
Characterizing self-tolerance mechanisms and their failure is critical to understand immune homeostasis, cancer immunity, and autoimmunity. However, examination of self-tolerance mechanisms has relied primarily on transgenic mice expressing T-cell receptors (TCRs) targeting well-characterized, but non-physiologic model antigens, such as ovalbumin and hemagglutinin. Identifying TCRs directed against bona fide self-antigens is made difficult by the extraordinary diversity of TCRs and low prevalence of antigen-specific clones (<10–100 naïve cells/organism), limiting dissection of tolerance mechanisms restricting immunity to self-proteins. Here, we isolated and characterized TCRs recognizing the intestinal epithelial cell receptor and colorectal cancer antigen GUCY2C to establish a model to study self-antigen-specific tolerance mechanisms. GUCY2C-specific CD4+ effector T cells were isolated from immunized, non-tolerant Gucy2c−/− mice. Next-generation sequencing identified GUCY2C-specific TCRs, which were engineered into CD4+ T cells in vitro to confirm TCR recognition of GUCY2C. Further, the generation of “retrogenic” mice by reconstitution with TCR-transduced hematopoietic stem cells (HSCs) resulted in normal CD4+ T-cell development, responsiveness to immunization, and GUCY2C-induced tolerance in recipient mice, recapitulating observations in conventional models. This retrogenic model can be employed to define self-tolerance mechanisms restricting T and B cell responses to GUCY2C, to optimize colorectal cancer immunotherapy without autoimmunity.
INTRODUCTION
While cancer immunotherapies are emerging as important treatment options for some cancers, including melanoma (1), prostate (2), leukemia (3), and lung (4), effective applications for other malignancies remain elusive. Colorectal cancer (CRC), which falls into the latter category, is the second leading cause of cancer-related mortality, accounting for ~500,000 worldwide deaths annually (5). Currently, surgery has the greatest impact on CRC survival; however, it is most successful in the treatment of early-stage disease. With standard of care, ~50% of the patient population will develop metastatic disease and subsequently die (6), underscoring the critical need for novel therapies to improve patient outcomes.
The immune system requires a diverse repertoire of cells that can respond to invading foreign antigens while maintaining an appropriate balance with tolerance to self-antigens to prevent autoimmunity. Indeed, thymic selection plays a critical role in generating enormous diversity in the TCR repertoire (7), while limiting autoimmunity (8). Tolerance presents a major challenge to developing efficacious cancer immunotherapies, including vaccines targeting self-antigens. In that context, mechanistic studies examining self-tolerance opposing cancer immunotherapy are hampered by the low prevalence of antigen-specific clones (<10–100 cells per organism (9)) making identification of individual self/tumor antigen-specific TCRs difficult. Moreover, tolerance limits self-reactive TCR abundance further by eliminating clones that recognize self-proteins with high affinity (7). Because cancer cells often express self-antigens normally expressed by the healthy tissues from which they derive, tolerance also inhibits immunity against these malignant cells (10). Development of methods to overcome tolerance barriers has the potential to enhance cancer immunity and improve patient outcomes in cancers for which immunotherapeutics are limited by tolerance.
In the context of cancer immunity, previous tolerance studies have been conducted utilizing model antigens; however, the applicability of these studies are limited, as the physiological relevance of mechanistic tolerance determinations in such systems is questionable (11). In contrast, TCR “retrogenic” mice produced by bone marrow reconstitution with hematopoietic stem cells (HSCs) retrovirally engineered to express TCRs have yielded insights into mechanisms underlying autoimmune diseases (12–17), and may have similar utility in examining the convergence of tolerance, autoimmunity, and cancer immunity targeting self/cancer antigens. Here, developed a retrogenic model system to investigate mechanisms of tolerance driven by guanylate cyclase C (GUCY2C), a natural self-antigen and cancer immunotherapy target selectively expressed in intestinal epithelial cells (18). Importantly, GUCY2C is overexpressed in intestinal tumors of both mice and humans (18–21). Compartmentalization in the intestinal lumen coupled with universal expression by CRC, highlighted by its utility as an effective biomarker for metastatic disease (21), implicate GUCY2C as the index cancer mucosa antigen (22–25) and an immune target for treating CRC. Indeed, adenoviral vaccines expressing murine GUCY2C produce GUCY2C-specific CD8+ T-cell responses in mice (22, 26–29) that kill GUCY2C-expressing colon cancer cells in vitro (27, 28) and reduce or eliminate tumors and improve survival in vivo in mouse models of metastatic CRC in lung or liver (22, 27, 28). Of significance, vaccine-induced CD8+ T cells selectively target metastatic CRC, but not normal intestinal tissue endogenously expressing GUCY2C (22, 27, 28). These observations have been extended to patients which produce GUCY2C-specific immune responses without toxicity (30). Importantly, GUCY2C-specific immune responses in mice (22, 26, 27) and humans (30) are characterized by selective tolerance of GUCY2C-specific CD4+ T cells, but not CD8+ T cells or B cells. In turn, this selective CD4+ T cell tolerance corrupts GUCY2C-specific B and CD8+ T-cell responses and antitumor efficacy, representing the critical limitation to therapeutic GUCY2C vaccination in cancer.
Defining mechanisms underlying CD4+ T-cell tolerance, and potential benefits (antitumor immunity) and toxicities (autoimmunity) associated with their reversal, are critical to optimize next-generation vaccine therapies targeting GUCY2C in gastrointestinal (GI) cancers. Moreover, those approaches may be applicable to therapies targeting other self/cancer antigens to treat malignancies beyond colorectal cancer. Here, we employed next-generation sequencing to identify natural GUCY2C-specific CD4+ T-cell receptors (TCRs) and reconstitution with genetically engineered hematopoietic stem cells (HSCs) to establish TCR retrogenic models required to examine GUCY2C-specific CD4+ T-cell tolerance mechanisms, autoimmunity, and antitumor immunity.
MATERIALS AND METHODS
Mice and immunizations
All animal studies were performed in agreement with IACUC protocols and procedures. Male or female BALB/c Gucy2c−/− (27) and Guc2yc+/+ and Rag1−/− (The Jackson Laboratory, Bar Harbor, ME) mice ages 8–15 weeks were used in all experiments. Rag1−/− or Rag1−/−Gucy2c−/− BALB/c mice were used for all retrogenic experiments. Mice were immunized with Ad5-GUCY2CECD (1×108 IFU/mouse) i.m. and immune responses were quantified by ELISpot or intracellular cytokine staining (ICS) 11–14 days later (22, 26–28).
GUCY2C-specific CD4+ T-cell isolation and TCR sequencing
Bulk splenocytes from Ad5-GUCY2C-immunized Gucy2c−/− mice were plated at 1×107 cells/mL in a T75 tissue culture-treated flask. Cells were stimulated with a final concentration of 20 μg/mL GUCY2C153–167 and incubated for 4.5 h at 37°C. Cells were harvested into 50 mL conical tubes, centrifuged at 4°C and resuspended in 50 mL cold MACS buffer (1x PBS supplemented with 0.5% BSA and 2 mM EDTA) and washed two additional times. Cells were passed through a 40 μm strainer to remove cellular debris before a final spin. Cells were resuspended in cold CTL-TEST media (Cellular Technology Limited, Cleveland, OH) and labeled with a bispecific catch antibody (IFNγ Cytokine Secretion Assay, Miltenyi Biotec, Bergisch Gladbach, Germany) directed towards murine IFNγ and incubated on ice for 5 mins. Cells were resuspended in warm assay medium at 106 cells/mL in 50 mL conical tubes and incubated for 45 min at 37°C with continuous rotation to allow secretion and capture of the cytokines by the surface-bound capture reagent. The cells were then washed at 4°C and resuspended in cold MACS buffer and placed on ice for 10 min. The cells received PE-conjugated αIFNγ antibody and were incubated for 10 min on ice and washed in cold buffer. Anti-PE MicroBeads were added, and cells were incubated for 15 min at 4°C and subsequently washed in cold buffer. Cells were passed through two MS/LS columns, serially. Samples were taken pre- and post-MACs sorting, stained with Propidium Iodine (Millipore Sigma, Darmstadt, Germany), αCD4-FITC (clone RM4–5, BD Biosciences, San Jose, CA), αCD8α-Pacific Orange (Clone 5H10, Invitrogen, Carlsbad, CA) and αIFNγ-PE (IFNγ Cytokine Secretion Assay, Miltenyi Biotec), and analyzed by flow cytometry on a BD LSR II to determine purity of the sorted population. The remainder of eluted GUCY2C153–167-specific cells were pelleted, and RNA was purified using a PicoPure RNA Isolation Kit (Life Technologies, Carlsbad, CA) according to the manufacturers protocol. RNA was commercially sequenced on an Illumina MiSeq for individual TCR α and β sequences in the GUCY2C153–167-enriched population by iRepertoire, Inc. (Huntsville, AL).
TCR construction, retrovirus production, and T-cell transduction
Sequencing data produced ~200bp reads surrounding CDR3s, their frequencies, and V, D, and J usage. Generation of TCRα and TCRβ circos plots employed the open-source software suite VDJtools (31) and R. Hierarchical tree maps were generated using the iRepertiore software. Prevalent CDR3s (based on amino acid sequences) were identified and aligned to full-length TCR α and β sequences using IMGT (32). The corresponding full-length TCR α and β chains were synthesized (Life Technologies) and 12 TCRs composed of TCRα-Furin-V5-P2A-TCRβ constructs (33) were subcloned into the pMSCV-IRES-GFP (pMIG) retroviral vector (Addgene, Cambridge, MA, #2749). A DO11 TCR construct specific for OVA323–339 was similarly produced as a negative control (34). The Phoenix-Eco retroviral packaging cell line (Gary Nolan, Stanford University) was transfected with TCR-pMIG vectors and the pCL-Eco retroviral packaging vector (Novus Biologicals, Littleton, CO) using the Calcium Phosphate ProfectionR Mammalian Transfection System (Promega, Madison, WI). Retrovirus-containing supernatants were collected 48 h later, filtered through 0.45 μm filters, and aliquots were frozen at −80°C. Murine CD4+ T cells were purified from BALB/c splenocytes using a CD4+ T cell Isolation Kit (Miltenyi Biotec) and subsequently stimulated with αCD3/αCD28-coated beads (T Cell Activation/Expansion Kit, Miltenyi Biotec) at a 1:1 bead:cell ratio at 1×106 cells/mL in cRPMI (RMPI + 10% FBS, 10 μM HEPES, 0.05 μM 2-mercaptoethanol) under Th1 polarizing conditions: 100 U/mL recombinant human IL-2 (NCI Biological Resources Branch, Frederick, MD), 2 ng/mL recombinant mouse IL-12 (R&D Systems), and 5 μg/mL mouse αIL-4 antibody (R&D Systems, Minneapolis, MN). The day following stimulation, ½ of the culture media was replaced with an equal volume of thawed retroviral supernatant in the presence of polybrene (Millipore Sigma). Spinoculation was performed at room temperature for 90 min at 2,500 rpm followed by incubation at 37°C for 2.5 h at which point cells were pelleted and resuspended in fresh media supplemented with rh-IL-2, rm IL-12, and αIL-4 antibody. T cells were expanded for 7–10 d by daily dilution to 1×106 cells/mL and cytokines were replenished daily for 48 h, followed by 48 h of media supplemented with only rh-IL-2, and then 48–72 h of media supplemented with rh-IL-2 and rm-IL-12. T cells were harvested and used for functional assays on day 7–10. All cell counts employed a Muse Cell Analyzer and Count and Viability Assay (Millipore Sigma).
Surface markers and intracellular cytokine staining
TCR-transduced T cells were stimulated for 6 h with DMSO, GUCY2C153–167 or OVA323–339 peptides, or with Cell Stimulation Cocktail (PMA/Ionomycin, Thermo Fisher Scientific, Waltham, MA). Incubation included the Protein Transport Inhibitor Cocktail (Thermo Fisher Scientific) when assessing intracellular cytokines. Transduced T cells or blood obtained by retro-orbital eye bleeding were stained for surface markers using the following antibodies: αCD4-Pacific Blue (clone RM4–5, BD Biosciences), αCD8α-PerCP-Cy5.5 (clone 53.6–7, BD Biosciences), αGr1-APC (clone RB6–8C5, BioLegend, San Diego, CA), αB220-PerCP-Cy5.5 (clone RA3–6B2, BD Biosciences), αTer119-PE-Cy7 (clone TER-119, BD Biosciences), and αCD45.2-PE (clone 104, BD Biosciences) and αCD25-PE (clone PC61.5, eBioscience). ICS was performed using the BD Cytofix/Cytoperm Kit (BD Biosciences) and staining with the following antibodies: αGFP-Alexa-488 (polyclonal, Invitrogen) and αIFNγ-PE-CF594 (clone XMG1.2), αIL-2-APC (clone JES6–5H4) and αTNFα-PE-Cy7 (clone MP6-XT22) from BD Biosciences. Red blood cells were lysed using BD lysis buffer and cells were fixed in 2% PFA and analyzed on a BD LSR II or BD LSR Fortessa flow cytometer. Analyses were performed using FlowJo software (FlowJo, Ashland, Oregon).
Retrogenic models
Retrogenic mice were produced by reconstitution of irradiated recipients with retrovirally-transduced HSCs using established techniques (16, 17, 35, 36). Briefly, Rag1−/− mice were injected i.p. with 150 mg/kg of 5-fluorouracil (Millipore Sigma); bone marrow was harvested 72 h later and murine HSCs were purified using mouse HSC isolation kit and EasySep magnet (STEMCELL, Vancouver, BC, Canada). HSCs were cultured for 24 h in complete STEMSpan (STEMCELL) supplemented with 20 ng/ml IL-3, 50 ng/ml hIL-6, 50 ng/ml mSCF and 30 ng/ml hLDL (STEMCELL). Bone marrow cells were spinoculated with retroviral supernatant and 8 μg/ml polybrene for 90 min at 37°C at 2500 rpm at 24, 48, and 72 h. Fresh media and cytokines were added after each transduction. After 96 h, bone marrow cells were collected, resuspended in 1X PBS + 2% Heparin (Millipore Sigma), and injected i.v. at ~2×106 cells/mouse into 8.5 Gy-irradiated recipients (~1 donor/1 recipient). Mice were test-bled for TCR reconstitution ~8 weeks post-transplant for analysis.
Dual-color enzyme-linked ImmunoSpot assays
ELISpot assays were performed using a murine IL-2/IFNγ double-color enzymatic kit (CTL-mIFNgIL2–1M/2; Cellular Technology Limited), which contained plates, all antibodies, detection reagents, and substrate. Assays were performed according to the manufacturer’s instructions. Briefly, the PVDF membranes were pre-wet with 70% ethanol and plates were coated with murine IL-2/IFNγ Coating Antibodies overnight at 4°C. The next day, plates were washed once with PBS and then peptides were plated in CTL-Test Medium. After plating the splenocytes and antigen presenting cells (for retrogenic experiments), 96-well plates were placed in a humidified incubator at 37°C with 5% CO2. After 24 h of incubation, cells were removed, and the detection antibody and development reagents were added. Following completion of the ELISpot assay, plates were air dried prior to analysis. Plates were scanned and analyzed using an ImmunoSpot® S6 Universal Analyzer (Cellular Technology Limited). Spot Forming Cells (SFCs) were automatically calculated by the ImmunoSpot® Software for each antigen stimulation condition and the medium (negative) control using the SmartCount™ and Autogate™ functions. Antigen-specific responses were calculated by subtracting baseline (DMSO stimulation) responses from antigen-stimulated responses.
TCR avidity analysis
TCR avidity experiments were performed using the IL-2/IFNγ dual-color ELISpot assay described above. Splenocytes were stimulated with a range of GUCY2C peptide from 0 ug/mL to 200 ug/mL. Then, for each sample, responses were normalized (0 ug/mL = 0% and 200 ug/mL = 100%) to generate a peptide dose-response with responses ranging from 0 to 100%. TCR avidity measurements were then analyzed by nonlinear regression [log(peptide concentration) versus normalized response] using GraphPad Prism version 7 (La Jolla, CA) (37, 38).
Statistical analysis
Statistical significance was analyzed using Student’s t test and two-way ANOVA. When appropriate, p values were corrected for multiple comparisons using Bonferroni’s method. p < 0.05 was considered statistically significant. We chose sample sizes based on experience with previous similar experiments. All statistical analyses were performed using GraphPad Prism version 7. Analyses represent mean ± SEM, unless otherwise indicated, and * p < 0.05, ** p < 0.01, *** p < 0.001.
RESULTS
GUCY2C-specific CD4+ T-cell isolation
Gucy2c−/− mice that are deficient in GUCY2C do not exhibit self-tolerance to, and, consequently, produce immune responses against, this protein (22, 26, 27). To examine CD4+ T-cell tolerance mechanisms, we established a model system using GUCY2C-specific TCRs that develop in Gucy2c−/− mice in the absence of tolerance. To identify GUCY2C-specifc TCRs that are eliminated from the naïve T-cell repertoire in Gucy2c+/+ mice, we immunized non-tolerant Gucy2c−/− mice with Ad5-GUCY2C, producing robust GUCY2C-specific CD4+ T-cell responses directed towards the dominant GUCY2C153–167 epitope, measured by IFNγ ELISpot (27) (Fig. 1a–b). Bulk splenocytes were isolated from Ad5-GUCY2C-immunized Gucy2c−/− mice and GUCY2C153–167-specific CD4+ T cells were purified using magnetic-activated cell sorting (MACS) following a live-cell IFNγ Secretion Assay. FACS analysis of bulk CD4+ T cells labeled by this technique prior to magnetic separation confirmed the expected low frequency of GUCY2C-specific cells, while MACS separation enriched those cells >2000-fold (Fig. 1c). This enrichment was sufficient to identify GUCY2C-specific TCRs within the partially purified population of cells.
Figure 1. GUCY2C-specific TCRαβ identification in CD4+ T cells.

A) GUCY2C-specific TCR identification approach. B–C) Bulk splenocytes were isolated from 7 female Gucy2c−/− BALB/c mice 14 days after i.m. immunization with 108 IFU Ad5-GUCY2C and were stimulated with GUCY2C153–167 peptide and analyzed by IFNγ ELISpot (B) or stimulated with GUCY2C153–167 peptide for 4.5 h, stained with αCD4-FITC, αCD8-Pacific Orange and αIFNγ-PE, magnetically sorted for IFNγ+ cells and FACS analysis (C). D–F) Isolated IFNγ+ GUCY2C-specific CD4+ T cells were subjected to massively parallel sequencing. D) Circos plots showing frequencies of V–J pairs for TCRα (left) and TCRβ (right) chains. E) Hierarchical tree maps showing individual CDR3 diversity assessment for TCRα (left) and TCRβ (right) chains. Each bubble corresponds to a unique CDR3 (by amino acid sequence); bubble size indicates frequency of reads. F) CDR3s identified in (E) were rank ordered by read frequencies of individual CDR3s for TCRα and TCRβ chains, revealing predicted TCRαβ pairs.
Identifying GUCY2C-specific TCRs by massively parallel sequencing
RNA purified from GUCY2C-enriched CD4+ T cells isolated by MACS underwent massively parallel (next-generation) sequencing to identify individual TCR α and β sequences (Fig. 1a). We used an Illumina® platform to sequence all TCR α and β genes present in the isolated CD4+ T cells and clustered identical amino acid sequences to identify prevalent TCRs. During an immune response, a relatively small number of antigen-recognizing clones expand exponentially, producing an oligoclonal response. Nearly 3×106 individual sequences were generated and, as expected, the GUCY2C153–167-reactive immune repertoire lacked diversity, reflecting expansion of relatively few clones (Fig. 1d). For both TCR α and β, a small number of V(D)J pairs accounted for a large fraction of observed sequences. Examination of individual V(D)J junctions (CDR3s) further revealed the oligoclonal nature of the response (Fig. 1e). We identified the top TCRα and TCRβ CDR3s (Fig. 1e and Table S1) and used frequency-based matching to identify natural GUCY2C-specific pairs (39) (Fig. 1f).
GUCY2C TCR synthesis and screening
Given the possibility that dual-TCRα (40) T cells within the population may shift the TCRα and TCRβ frequency profiles (2 TCRα chains pair with 1 TCRβ chain), individual TCR α and β chains were synthesized for the top 4 TCRα sequences and top 3 TCRβ sequences and all combinations of TCRαβ pairs where generated from the synthesized chains. The 12 candidate TCRs were synthesized using a 2A ribosomal skip sequence between the TCR α and β sequences, permitting translation of both proteins from a single open reading frame, resulting in a near stoichiometric production of α and β chains (33). TCR constructs were cloned into a retroviral vector with an IRES-GFP marker (Fig. 2a). To examine the TCR constructs, naïve CD4+ T cells were isolated from Guc2yc+/+ mice and transduced with the candidate TCR constructs or a DO11 TCR construct specific for OVA323–339 (34). Following transduction and expansion in Th1-promoting conditions, transduced CD4+ T cells were stimulated with GUCY2C153–167 peptide and effector function was detected by TNFα and IFNγ ICS (Fig. 2b–c). Of 12 synthesized, TCRs 4A and 5B recognized GUCY2C153–167 peptide (Fig. 2b–c), but not vehicle control, with 55–60% of GFP+CD4+ T cells responding to GUCY2C153–167 peptide stimulation with cytokine production (sequences of TCR 4A and 5B are shown in Fig. S1).
Figure 2. GUCY2C recognition by engineered cells confirmed TCR specificity and activity.

A) Assembled TCRs were expressed as TCRαβ pairs linked by a 2A ribosomal skip sequence in engineered CD4+ T cells by retrovirus-mediated transduction of naïve CD4+ T cells under Th1-promoting conditions. The retroviral vector includes a GFP marker, allowing gating on transduced cells. B) Representative FACS analysis shows IFNγ and TNFα cytokine production following stimulation with GUCY2C153–167 peptide in engineered T cells identified TCR 4A and 5B as GUCY2C-specific. DMSO served as a vehicle control. DO11 served as a negative control for the TCRs. B) is representative and (C) is the mean of 2–4 experiments for each TCR construct. ** p < 0.01, Two-way ANOVA.
Hematopoietic stem cell (HSC) engineering
To establish a model to explore mechanisms underlying tolerance, GUCY2C-specific TCRs must be expressed in developing thymocytes in vivo, rather than in CD4+ T cells stimulated ex vivo. In that context, we inserted retroviral TCR constructs into purified mouse hematopoietic stem cells (HSCs) ex vivo which were then used to reconstitute bone marrow-depleted mice, producing retrovirally-transduced bone marrow chimeric (retrogenic) mice. In this model, transduced HSCs develop into all bone marrow-derived lineages, including T cells, permitting testing of tolerance directed by genetically engineered TCRs recognizing GUCY2C. For all retrogenic experiments, Rag1−/− HSCs were used to prevent donor cells from producing endogenous TCR rearrangements, ensuring that only engineered TCRs were expressed. Here, HSCs were purified from bulk bone marrow by magnetic separation and transduced serially 1, 2, or 3 times with TCR-IRES-GFP retrovirus (Fig. 3). HSCs transduced with control DO11 and GUCY2C TCRs 4A and 5B expanded ~4-fold (Fig. 3a) and remained undifferentiated (41) (Fig. 3b). Successive retroviral applications increased transduction efficiency (Fig. 3c).
Figure 3. HSC expansion and transduction.
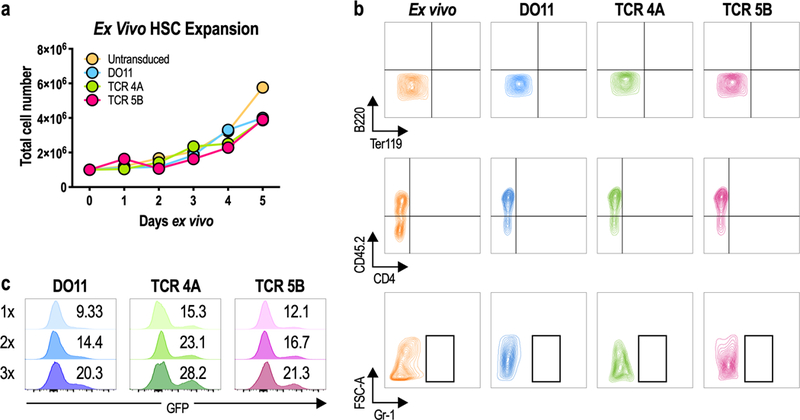
A–C) Bone marrow was isolated from donor Rag1−/− mice and HSCs were purified by negative selection of lineage-positive cells. Purified HSCs were left untransduced or transduced 1–3 times with DO11 or GUCY2C TCRs. A) HSC expansion ex vivo. B) Transduced HSCs remained lineage negative (mature lineage markers B220, Ter119, CD4, and Gr-1 shown here) following retroviral transduction and expansion, comparable to HSCs stained directly ex vivo. C) HSC transduction efficiency following 1, 2, or 3 applications of TCR retrovirus (1x, 2x or 3x). Values indicate percentage of GFP+ cells. Data are representative of 3 experiments.
Retrogenic mice
HSCs from Rag1−/− mice were transduced with TCR 4A, 5B, or DO11 retrovirus and transferred to lethally irradiated Rag1−/−Gucy2c−/− mice. The Rag1−/− genotype ensured that recipient mice were devoid of endogenous T cells. Retrogenic mice reconstituted with HSCs transduced with TCR 4A, 5B, or DO11 displayed CD4+ T-cell development that was comparable to control mice (Fig. 4). Moreover, mice reconstituted with HSCs transduced with TCR 4A and 5B exhibited minimal CD8+ T-cell maturation, compared to control mice (Fig. 4). These results confirm successful reconstitution of CD4+ T-cell development by delivery of exogenous TCRs to HSCs. Importantly, using the retrogenic system we can enrich for GUCY2C-specific CD4+ T cells (present at ~30–60% in peripheral blood of TCR4A or 5B retrogenic mice).
Figure 4. Reconstitution of retrogenic mice.

Transduced HSCs were adoptively transferred to lethally-irradiated Rag1−/−Gucy2c−/− mice. Reconstitution was determined nine weeks later by FACS on blood samples. % CD4+ T cells of total cells, % CD8+ T cells of total cells in blood. Reconstitution analysis of 3–5 mice/group.
Retrogenic mouse immune responses
Conventional Gucy2c−/− mice or TCR 4A or 5B retrogenic Rag1−/−Gucy2c−/− mice were immunized with Ad5-GUCY2C. Splenocytes were collected 14 days later and antigen-specific immune response quantified by IL-2/TNFα ICS by FACS (Fig. 5a). Importantly, TCRs 4A and 5B retrogenic mice produced antigen-specific responses, with ~30–50% of GFP+CD4+ T cells responding to GUCY2C153–167 peptide stimulation with cytokine production, compared to ~2% of CD4+ T cells in conventional Gucy2c−/− mice (Fig. 5b). Thus, in this retrogenic model, HSCs can reconstitute specific T-cell compartments (Fig. 4) and antigen-specific immune responses (Fig. 5). Moreover, GUCY2C-specific TCR 4A and 5B retrogenic mice performed similarly to DO11 TCR retrogenic mice which also demonstrated normal T-cell development (Fig. 4) and antigen-specificity (Fig. S2).
Figure 5. Retrogenic immune responses.

A–H) Rag1−/−Gucy2c−/− retrogenic mice were produced with TCR 4A or 5B and, along with conventional Gucy2c−/− mice, were immunized with Ad5-GUCY2C. Splenocytes were collected 14 days later and analyzed by IL-2/TNFα ICS after stimulation with DMSO (vehicle), GUCY2C153–167, or PMA/IONO (positive control). Data indicate responses by individual mice and are representative of two experiments with 3–5 mice/group. * p <0.05, ** p <0.01 t test.
Retrogenic TCR functional avidity
TCR avidity is a main factor controlling T-cell tolerance (42). Moreover, T cells with high functional avidity mediate superior T-cell responses and tumor immunity (43). Thus, the avidity of T cells from TCR retrogenic mice were compared to that of naturally-occurring GUCY2C-specific T cells found in conventional Gucy2c−/− mice. T cells were collected from immunized mice and functional avidity was quantified by GUCY2C153–167 peptide titration in an IL-2/IFNγ dual-color ELISpot assay (Fig. 6). Indeed, TCR 4A and 5B retrogenic Gucy2c−/− mice and conventional Gucy2c−/− mice produced GUCY2C-specific T cells with similar functional avidities (Fig. 6), reflecting similarities in TCRα and TCRβ usage between Gucy2c−/− mice and TCR 4A and 5B (Table S1). While transduction of HSCs with γ-retrovirus produces a continuum of TCR expression levels (Fig. S3a), only those retrogenic cells with surface TCR levels similar to conventional T cells produce cytokine following antigen stimulation (Fig. S3b). Together, these data confirm that this retrogenic model recapitulates endogenous T-cell responses and is suitable to study GUCY2C-specific CD4+ T-cell tolerance mechanisms and outcomes.
Figure 6. Retrogenic TCR Avidity.
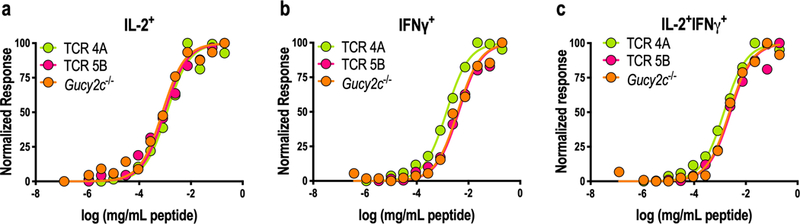
A–C) Rag1−/−Gucy2c−/− TCR 4A and 5B and conventional Gucy2c−/− mice were immunized with Ad5-GUCY2C. Splenocytes were collected 14 days later to measure TCR avidity by IL-2/IFNγ dual-color ELISpot using peptide titration. Normalized responses for each sample were calculated from 0% (no peptide) to 100% (highest peptide concentration) to determine the avidity of each sample. Avidity curves for IL-2+ (A), IFNγ+ (B), and IL-2+IFNγ+ double-positive (C) T cells were similar in TCR retrogenic and conventional Gucy2c−/− mice. Data points indicate the mean of 1–3 experiments containing 2–4 mice/group/experiment. Curves indicate non-linear regression computed from the average data points across experiments.
Retrogenic T-cell tolerance
This validated model was used to determine if GUCY2C-specific CD4+ T-cell tolerance is recapitulated in retrogenic mice. Lethally irradiated Rag1−/−Gucy2c−/− or Rag1−/−Gucy2c+/+ recipient mice were reconstituted with TCR 4A or 5B HSCs (Fig. 7). Endogenous GUCY2C-specific CD4+ T-cell responses eliminated in Gucy2c+/+ mice by tolerance were present in non-tolerant Gucy2c−/− mice (22, 26, 27) (Fig. 7a). Thus, we hypothesized that Gucy2c−/−, but not Gucy2c+/+, mice reconstituted with GUCY2C-specific TCRs 4A or 5B would produce GUCY2C-specific T-cell responses. Indeed, immunization produced GUCY2C-specific T-cell responses in Gucy2c−/−, but not Gucy2c+/+, TCR 4A or 5B mice (Fig. 7b–c). Together, these results confirm the utility of TCR retrogenic mice produced by ex vivo TCR repertoire sequencing as a model for exploring GUCY2C-specific tolerance mechanisms.
Figure 7. Retrogenic T-Cell Tolerance.
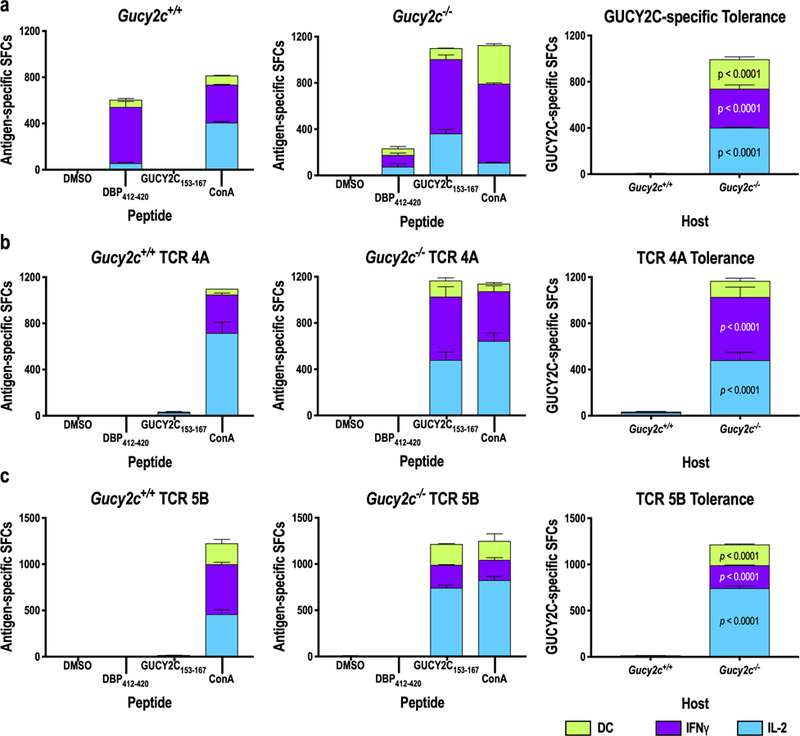
Gucy2c+/+ and Guc2yc−/− retrogenic hosts were established with TCR 5B or TCR 4A HSCs 6–9 weeks prior to immunization. GUCY2C-specific T-cell responses were quantified by IL-2/IFNγ double-color ELISpot assay 11–14 days after immunization with Ad5-GUCY2C. No GUCY2C-specific responses were detected in conventional Gucy2c+/+ mice, while robust responses were produced in Gucy2c−/− mice (A), recapitulated by TCR 4A (B) and 5B (C) retrogenic mice. Data indicate the means of two experiments with 2–4 mice/group/experiment. p values compare Gucy2c+/+ and Gucy2c−/− mice, determined by Two-way ANOVA. SFCs = spot-forming cells. Data indicate SFCs/106 splenocytes.
DISCUSSION
In this study, we identified a mouse model to explore mechanisms underlying T-cell tolerance targeting endogenous self-antigens, employing the mucosal self-antigen GUCY2C as the example. Indeed, GUCY2C is a self/tumor antigen undergoing clinical testing as a therapeutic target in metastatic colorectal cancer (30, 44, 45). Importantly, GUCY2C vaccination of mice and humans reveals conservation across species of selective elimination of GUCY2C-specific CD4⁺, but not CD8+, T-cell responses (22, 26, 27, 30). In turn, this conservation across species supports the utility of mice as a model to explore GUCY2C-specific tolerance mechanisms and outcomes. Thus, the retrogenic system developed here provides a unique platform to study the implications of tolerance on antitumor immunity and autoimmunity in the context of GUCY2C immunotherapy that has relevance to human therapies.
Previous studies of tolerance have utilized model antigens and are limited in their physiological applications and translational potential for human therapies (11). For example, ovalbumin (OVA) has been used as a model self-antigen expressed under the control of various tissue-specific promoters coupled with OVA-specific (OT-I) TCR transgene expression or CD8+ T cell transfer (46). Not surprisingly, when OT-I T cells are transferred to mice expressing OVA under the control of the promoter for intestinal fatty acid binding protein (IFABP-OVA), antigen-specific tolerance is observed (47). These results were recapitulated in a model in which OVA is expressed under the control of the rat insulin promoter (RIP-OVA). Importantly, both models express high, non-physiologic levels of OVA antigen that limit interpretations, as antigen levels are known to impact tolerance (48). Indeed, when OVA is expressed at lower levels, no tolerance is observed (49). Expressing the influenza virus membrane protein hemagglutinin (HA) under the control of the glial fibrillary acidic protein (GFAP) promoter as a model of Crohn’s disease revealed that adoptively transferred HA-specific (CL4) CD4+ T cells did not traffic to the intestine and were deleted in the periphery (50). This was similar to some findings in IFABP-OVA mice (51), but contrasts with studies in Vil-HA mice (52), which may reflect differences in patterns of expression of HA. Taken together, these differences in tolerance mechanisms achieved for the same exogenous antigens controlled by different promoters underscores the limits of using model antigens to predict tolerance mechanisms and outcomes directed by endogenous antigens.
Cre recombinase also has been employed as a model self-antigen under the control of different tissue-restricted promoters, including Clara Cell 10 kDa Secretory Protein (CC10-Cre) or Villin (Vil-Cre) for expression in lung epithelium or intestinal epithelium, respectively (53). Indeed, Cre-specific CD4+ T cells were preserved and Cre-specific Tregs were increased in CC10-Cre and Vil-Cre transgenic mice, suggesting that CD4+ T cells directed to mucosa-restricted antigens may be preferentially diverted to Treg development as a less durable form of tolerance than deletional mechanisms (53). Interestingly, CC10 and Villin genes are both AIRE-regulated and expressed by medullary thymic epithelial cells (54); however, thymic expression of Cre was not detectable in either CC10-Cre or Vil-Cre mice in this study (53), suggesting that artificial transgenic promoters may not accurately recapitulate AIRE-mediated regulation of endogenous genes. Thus, findings with exogenous antigens may not be applicable to natural self-antigens, necessitating the exploration of tolerance mechanisms to endogenous self-antigens.
In contrast to model antigens, carcinoembryonic antigen (CEA) is a cell-surface oncofetal glycoprotein expressed in human large intestine, and is over-expressed in most GI malignancies (55). However, developing experimental mouse models to study CEA as a potential immunotherapy target proved challenging as no mouse or rat homolog exists. Many transgenic mouse lines have been generated, but with limitations. There have been several reports of differences in patterns of CEA expression across cell types and mouse strains, with CEA levels higher than the concentration detected in humans by 20-fold in some cases (56), confounding findings of toxicity in these models (57). Although models improve as new technologies evolve, studies of CEA still require models with greater translational potential for the treatment of human disease.
In contrast to uncertainties of transgenic expression of exogenous antigens, GUCY2C is an endogenous self-antigen expressed in intestinal cells in mice and humans, and universally over-expressed in colorectal cancer cells in patients, where insights into tolerance mechanisms in murine models could be clinically translated. Indeed, we have previously demonstrated that nontolerant Gucy2c−/− mice have superior GUCY2C-specific tumor immunity compared to tolerant Gucy2c+/+ mice (22, 26, 27). Identifying mechanisms limiting immunity in Gucy2c+/+, but not Gucy2c−/−, mice may reveal cellular or molecular pathways that can be leveraged to maximize GUCY2C-targeted immunotherapy in patients. Moreover, selective tolerance of the CD4+ T-helper cell lineage of the GUCY2C immune repertoire limiting antitumor responses in humans (30) and mice (22, 26, 27) also has been observed with melanoma (Trp2) and breast cancer (Her2) antigens (27). Indeed, we have identified selective CD4+ T-cell tolerance that restricts vaccine efficacy across multiple self-antigens, tissues, MHC haplotypes and cancers, but preserves antigen-specific CD8+ T and B cells, highlighting the broad impact of characterizing mechanisms mediating GUCY2C self-tolerance to alter cancer immunotherapeutic outcomes.
Using a retrogenic system, we have demonstrated that immunization of Gucy2c−/− TCR 4A or 5B mice resulted in GUCY2C-specific T-cell responses, while no responses were detected in Gucy2c+/+ recipient mice, recapitulating findings in conventional models. This suggests that TCR 4A and 5B Gucy2c+/+ mice could be used to explore GUCY2C tolerance mechanisms selectively targeting CD4+ T cells, which are responsible for attenuation of Ad5-GUCY2C vaccine efficacy. Defining these mechanisms is critical to develop approaches that reverse or block tolerance and assess their associated antitumor and autoimmune outcomes. Elucidating targets to overcome these tolerance barriers provides a mechanism-based strategy for restoring GUCY2C-specific CD4+ T cell responses and maximizing antitumor efficacy with the potential for translation to life-long protection against metastatic disease in patients.
The importance of identifying GUCY2C-specific tolerance mechanisms is underscored in the context of our recently-completed first-in-man Phase I clinical trial (30) and planned Phase II clinical trials examining the efficacy of Ad5-GUCY2C vaccination in patients with colorectal cancer, as well as those with esophageal, gastric, and pancreatic cancers which ectopically express GUCY2C. Indeed, our studies of GUCY2C as a bona fide cancer mucosa antigen have been translated from murine discovery to human clinical trials, overcoming the limitations of model antigens previously employed in in tolerance studies. Understanding the mechanisms and consequences of GUCY2C-specific tolerance are vital to the development of second-generation GUCY2C immunotherapeutics to safely and effectively treat GUCY2C-expressing GI malignancies, diseases for which effective therapies are critically lacking. Moreover, the approach outlined in this study may be adopted to investigate tolerance mechanisms to other self-antigens implicated in autoimmunity and cancer.
Supplementary Material
Acknowledgments
Financial support was provided by: NIH (P30 CA56036 to the Kimmel Cancer Center of Thomas Jefferson University and R01 CA204881 and R01 CA206026 to S. Waldman); the Department of Defense Congressionally Directed Medical Research Programs (#W81XWH-17-1-0299 to A. Snook); Targeted Diagnostic & Therapeutics, Inc. (to S. Waldman); Courtney Ann Diacont Memorial Foundation (to S. Waldman); PhRMA Foundation (to A. Snook); the W.W. Smith Charitable Trust (to A. Snook); and Margaret Q. Landenberger Research Foundation (to A. Snook). S. Waldman is the Samuel M.V. Hamilton Professor of Medicine of Thomas Jefferson University. This project was funded, in part, by grants from the Pennsylvania Department of Health (SAP #4100059197, SAP #4100051723 to S. Waldman). The Department specifically disclaims responsibility for any analyses, interpretations, or conclusions. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, and Urba WJ. 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, and Investigators IS. 2010. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363: 411–422. [DOI] [PubMed] [Google Scholar]
- 3.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, and June CH. 2011. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science translational medicine 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, and Wigginton JM. 2012. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 366: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siegel RL, Miller KD, and Jemal A. 2018. Cancer statistics, 2018. CA Cancer J. Clin 68: 7–30. [DOI] [PubMed] [Google Scholar]
- 6.O’Connell JB, Maggard MA, and Ko CY. 2004. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst 96: 1420–1425. [DOI] [PubMed] [Google Scholar]
- 7.Hogquist KA, Baldwin TA, and Jameson SC. 2005. Central tolerance: learning self-control in the thymus. Nat. Rev. Immunol 5: 772–782. [DOI] [PubMed] [Google Scholar]
- 8.Cheng MH, Anderson MS, 2011. Monogenic Autoimmunity. Annu. Rev. Immunol [DOI] [PMC free article] [PubMed]
- 9.Malandro N, Budhu S, Kuhn NF, Liu C, Murphy JT, Cortez C, Zhong H, Yang X, Rizzuto G, Altan-Bonnet G, Merghoub T, and Wolchok JD. 2016. Clonal Abundance of Tumor-Specific CD4(+) T Cells Potentiates Efficacy and Alters Susceptibility to Exhaustion. Immunity 44: 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson SR, Yuan J, and Teague RM. 2014. Targeting CD8+ T-cell tolerance for cancer immunotherapy. Immunotherapy 6: 833–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCaughtry TM, and Hogquist KA. 2008. Central tolerance: what have we learned from mice? Semin. Immunopathol 30: 399–409. [DOI] [PubMed] [Google Scholar]
- 12.Lennon GP, Bettini M, Burton AR, Vincent E, Arnold PY, Santamaria P, and Vignali DA. 2009. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity 31: 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaparro RJ, Burton AR, Serreze DV, Vignali DA, and DiLorenzo TP. 2008. Rapid identification of MHC class I-restricted antigens relevant to autoimmune diabetes using retrogenic T cells. J Immunol Methods 335: 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton AR, Vincent E, Arnold PY, Lennon GP, Smeltzer M, Li CS, Haskins K, Hutton J, Tisch RM, Sercarz EE, Santamaria P, Workman CJ, and Vignali DA. 2008. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes 57: 1321–1330. [DOI] [PubMed] [Google Scholar]
- 15.Nakayama M, Castoe T, Sosinowski T, He X, Johnson K, Haskins K, Vignali DA, Gapin L, Pollock D, and Eisenbarth GS. 2012. Germline TRAV5D-4 T-cell receptor sequence targets a primary insulin peptide of NOD mice. Diabetes 61: 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bettini M, Blanchfield L, Castellaw A, Zhang Q, Nakayama M, Smeltzer MP, Zhang H, Hogquist KA, Evavold BD, and Vignali DA. 2014. TCR affinity and tolerance mechanisms converge to shape T cell diabetogenic potential. J Immunol 193: 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold PY, Burton AR, and Vignali DA. 2004. Diabetes incidence is unaltered in glutamate decarboxylase 65-specific TCR retrogenic nonobese diabetic mice: generation by retroviral-mediated stem cell gene transfer. J Immunol 173: 3103–3111. [DOI] [PubMed] [Google Scholar]
- 18.Carrithers SL, Parkinson SJ, Goldstein S, Park P, Robertson DC, and Waldman SA. 1994. Escherichia coli heat-stable toxin receptors in human colonic tumors. Gastroenterology 107: 1653–1661. [DOI] [PubMed] [Google Scholar]
- 19.Carrithers SL, Parkinson SJ, Goldstein SD, Park PK, Urbanski RW, and Waldman SA. 1996. Escherichia coli heat-stable enterotoxin receptors. A novel marker for colorectal tumors. Dis Colon Rectum 39: 171–181. [DOI] [PubMed] [Google Scholar]
- 20.Frick GS, Pitari GM, Weinberg DS, Hyslop T, Schulz S, and Waldman SA. 2005. Guanylyl cyclase C: a molecular marker for staging and postoperative surveillance of patients with colorectal cancer. Expert Rev Mol Diagn 5: 701–713. [DOI] [PubMed] [Google Scholar]
- 21.Waldman SA, Hyslop T, Schulz S, Barkun A, Nielsen K, Haaf J, Bonaccorso C, Li Y, and Weinberg DS. 2009. Association of GUCY2C expression in lymph nodes with time to recurrence and disease-free survival in pN0 colorectal cancer. JAMA 301: 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snook AE, Stafford BJ, Li P, Tan G, Huang L, Birbe R, Schulz S, Schnell MJ, Thakur M, Rothstein JL, Eisenlohr LC, and Waldman SA. 2008. Guanylyl cyclase C-induced immunotherapeutic responses opposing tumor metastases without autoimmunity. J Natl Cancer Inst 100: 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snook AE, Eisenlohr LC, Rothstein JL, and Waldman SA. 2007. Cancer mucosa antigens as a novel immunotherapeutic class of tumor-associated antigen. Clinical pharmacology and therapeutics 82: 734–739. [DOI] [PubMed] [Google Scholar]
- 24.Snook AE, Stafford BJ, Eisenlohr LC, Rothstein JL, and Waldman SA. 2007. Mucosally restricted antigens as novel immunological targets for antitumor therapy. Biomarkers in medicine 1: 187–202. [DOI] [PubMed] [Google Scholar]
- 25.Snook AE, and Waldman SA. 2009. Cancer Mucosa Antigens a Novel Paradigm in Cancer Immunotherapeutics. Bioforum Eur 3: 14–16. [PMC free article] [PubMed] [Google Scholar]
- 26.Snook AE, Li P, Stafford BJ, Faul EJ, Huang L, Birbe RC, Bombonati A, Schulz S, Schnell MJ, Eisenlohr LC, and Waldman SA. 2009. Lineage-specific T-cell responses to cancer mucosa antigen oppose systemic metastases without mucosal inflammatory disease. Cancer Res 69: 3537–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snook AE, Magee MS, Schulz S, and Waldman SA. 2014. Selective antigen-specific CD4(+) T-cell, but not CD8(+) T- or B-cell, tolerance corrupts cancer immunotherapy. Eur J Immunol 44: 1956–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snook AE, Magee MS, Marszalowicz GP, Schulz S, and Waldman SA. 2012. Epitope-targeted cytotoxic T cells mediate lineage-specific antitumor efficacy induced by the cancer mucosa antigen GUCY2C. Cancer Immunol Immunother 61: 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snook AE, Huang L, Schulz S, Eisenlohr LC, and Waldman SA. 2008. Cytokine adjuvanation of therapeutic anti-tumor immunity targeted to cancer mucosa antigens. Clinical and translational science 1: 263–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snook A, Baybutt T, Mastrangelo M, Lewis N, Goldstein S, Kraft W, Oppong Y, Hyslop T, Myers R, Alexeev V, Eisenlohr L, Sato T, and Waldman S. 2015. A Phase I study of AD5-GUCY2C-PADRE in stage I and II colon cancer patients. Journal for ImmunoTherapy of Cancer 3: P450. [Google Scholar]
- 31.Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI, Kirgizov KI, Skorobogatova EV, and Chudakov DM. 2015. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLoS computational biology 11: e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baum TP, Pasqual N, Thuderoz F, Hierle V, Chaume D, Lefranc MP, Jouvin-Marche E, Marche PN, and Demongeot J. 2004. IMGT/GeneInfo: enhancing V(D)J recombination database accessibility. Nucleic Acids Res 32: D51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang S, Cohen CJ, Peng PD, Zhao Y, Cassard L, Yu Z, Zheng Z, Jones S, Restifo NP, Rosenberg SA, and Morgan RA. 2008. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther 15: 1411–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee HM, Bautista JL, Scott-Browne J, Mohan JF, and Hsieh CS. 2012. A broad range of self-reactivity drives thymic regulatory T cell selection to limit responses to self. Immunity 37: 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holst J, Vignali KM, Burton AR, and Vignali DA. 2006. Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic mice. Nature methods 3: 191–197. [DOI] [PubMed] [Google Scholar]
- 36.Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, and Vignali DA. 2006. Generation of T-cell receptor retrogenic mice. Nat Protoc 1: 406–417. [DOI] [PubMed] [Google Scholar]
- 37.Xiang B, Baybutt TR, Berman-Booty L, Magee MS, Waldman SA, Alexeev VY, and Snook AE. 2017. Prime-Boost Immunization Eliminates Metastatic Colorectal Cancer by Producing High-Avidity Effector CD8(+) T Cells. J Immunol 198: 3507–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ioannidou K, Baumgaertner P, Gannon PO, Speiser MF, Allard M, Hebeisen M, Rufer N, and Speiser DE. 2017. Heterogeneity assessment of functional T cell avidity. Sci Rep 7: 44320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linnemann C, Heemskerk B, Kvistborg P, Kluin RJ, Bolotin DA, Chen X, Bresser K, Nieuwland M, Schotte R, Michels S, Gomez-Eerland R, Jahn L, Hombrink P, Legrand N, Shu CJ, Mamedov IZ, Velds A, Blank CU, Haanen JB, Turchaninova MA, Kerkhoven RM, Spits H, Hadrup SR, Heemskerk MH, Blankenstein T, Chudakov DM, Bendle GM, and Schumacher TN. 2013. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med 19: 1534–1541. [DOI] [PubMed] [Google Scholar]
- 40.Elliott JI, and Altmann DM. 1995. Dual T cell receptor alpha chain T cells in autoimmunity. J. Exp. Med 182: 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Challen GA, Boles N, Lin KK, and Goodell MA. 2009. Mouse hematopoietic stem cell identification and analysis. Cytometry. Part A : the journal of the International Society for Analytical Cytology 75: 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moon JJ, Dash P, Oguin TH 3rd, McClaren JL, Chu HH, Thomas PG, and Jenkins MK. 2011. Quantitative impact of thymic selection on Foxp3+ and Foxp3- subsets of self-peptide/MHC class II-specific CD4+ T cells. Proc. Natl. Acad. Sci. U. S. A 108: 14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vigano S, Utzschneider DT, Perreau M, Pantaleo G, Zehn D, and Harari A. 2012. Functional avidity: a measure to predict the efficacy of effector T cells? Clinical & developmental immunology 2012: 153863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baybutt TR, Aka AA, and Snook AE. 2017. The Heat-Stable Enterotoxin Receptor, Guanylyl Cyclase C, as a Pharmacological Target in Colorectal Cancer Immunotherapy: A Bench-to-Bedside Current Report. Toxins (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baybutt TR, Aka AA, and Snook AE. 2017. Immunotherapy in Colorectal Cancer: Where Are We Now? Current Colorectal Cancer Reports 13: 353–361. [Google Scholar]
- 46.Grabie N, Delfs MW, Westrich JR, Love VA, Stavrakis G, Ahmad F, Seidman CE, Seidman JG, and Lichtman AH. 2003. IL-12 is required for differentiation of pathogenic CD8+ T cell effectors that cause myocarditis. J. Clin. Invest 111: 671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vezys V, Olson S, and Lefrancois L. 2000. Expression of intestine-specific antigen reveals novel pathways of CD8 T cell tolerance induction. Immunity 12: 505–514. [DOI] [PubMed] [Google Scholar]
- 48.Tay SS, Wong YC, McDonald DM, Wood NA, Roediger B, Sierro F, McGuffog C, Alexander IE, Bishop GA, Gamble JR, Weninger W, McCaughan GW, Bertolino P, and Bowen DG. 2014. Antigen expression level threshold tunes the fate of CD8 T cells during primary hepatic immune responses. Proc. Natl. Acad. Sci. U. S. A 111: E2540–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Espinosa-Carrasco G, Le Saout C, Fontanaud P, Stratmann T, Mollard P, Schaeffer M, and Hernandez J. 2017. CD4(+) T Helper Cells Play a Key Role in Maintaining Diabetogenic CD8(+) T Cell Function in the Pancreas. Front. Immunol 8: 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Magnusson FC, Liblau RS, von Boehmer H, Pittet MJ, Lee JW, Turley SJ, and Khazaie K. 2008. Direct presentation of antigen by lymph node stromal cells protects against CD8 T-cell-mediated intestinal autoimmunity. Gastroenterology 134: 1028–1037. [DOI] [PubMed] [Google Scholar]
- 51.Lee JW, Epardaud M, Sun J, Becker JE, Cheng AC, Yonekura AR, Heath JK, and Turley SJ. 2007. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol 8: 181–190. [DOI] [PubMed] [Google Scholar]
- 52.Westendorf AM, Fleissner D, Deppenmeier S, Gruber AD, Bruder D, Hansen W, Liblau R, and Buer J. 2006. Autoimmune-mediated intestinal inflammation-impact and regulation of antigen-specific CD8+ T cells. Gastroenterology 131: 510–524. [DOI] [PubMed] [Google Scholar]
- 53.Legoux FP, Lim JB, Cauley AW, Dikiy S, Ertelt J, Mariani TJ, Sparwasser T, Way SS, and Moon JJ. 2015. CD4+ T Cell Tolerance to Tissue-Restricted Self Antigens Is Mediated by Antigen-Specific Regulatory T Cells Rather Than Deletion. Immunity 43: 896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, Heger A, Ponting CP, and Hollander GA. 2014. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res 24: 1918–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hance KW, Zeytin HE, and Greiner JW. 2005. Mouse models expressing human carcinoembryonic antigen (CEA) as a transgene: evaluation of CEA-based cancer vaccines. Mutat. Res 576: 132–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhattacharya-Chatterjee M, Saha A, Foon KA, and Chatterjee SK. 2008. Carcinoembryonic antigen transgenic mouse models for immunotherapy and development of cancer vaccines. Curr Protoc Immunol Chapter 20: Unit 20 28. [DOI] [PubMed]
- 57.Bos R, van Duikeren S, Morreau H, Franken K, Schumacher TN, Haanen JB, van der Burg SH, Melief CJ, and Offringa R. 2008. Balancing between antitumor efficacy and autoimmune pathology in T-cell-mediated targeting of carcinoembryonic antigen. Cancer Res 68: 8446–8455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.