

Abstract
In the past two decades, mutations in multiple genes have been linked to autosomal dominant or recessive forms of monogenic Parkinson’s disease (PD). Collectively, these monogenic (often familial) cases account for less than 5% of all PD, the majority being apparently sporadic cases. More recently, large-scale genome-wide association studies have identified over 40 loci that increase risk of PD. Importantly, there is overlap between monogenic and sporadic PD genes, particularly for the loci that contain the genes SNCA and LRRK2, which are mutated in monogenic dominant PD. There have also been reports of idiopathic PD cases with heterozygous variants in autosomal recessive genes suggesting that these mutations may increase risk of PD. These observations suggest that monogenic and idiopathic PD may have shared pathogenic mechanisms. Here, we focus mainly on the role of monogenic PD genes that represent pleomorphic risk loci for idiopathic PD. We also discuss the functional mechanisms that may play a role in increasing risk of disease in both monogenic and idiopathic forms.
Keywords: Idiopathic Parkinson’s disease, pleomorphic risk loci, genetic risk factors, functional genomics
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects multiple brain regions, resulting in a syndrome that includes symptoms related to neurological control of movement as well as other brain functions including cognition (Langston, 2006). PD is both common and age-related, being rare before the age of 50, affecting about 1% of the population worldwide over the age of 65 years and about 4–5% of the population over 85 years old (de Lau and Breteler, 2006). Since aging remains the largest risk factor for developing PD, the economic and social impact resulting from PD will continue to rise with the overall longevity of many populations (Collier et al., 2011; Driver et al., 2009).
Historically, other than aging there have been relatively few widely confirmed causal factors that influence lifetime risk of PD, making this a classic sporadic disorder. However, genetic linkage analysis in families, has identified several underlying rare but penetrant pathogenic mutations. To date, 19 PARK loci have been designated for different genetic forms of PD and the underlying gene mutation has been identified in 11 of them, with some uncertainty about the accuracy of assignment of several genes in four loci (Hernandez et al., 2016). Although these discoveries have provided important insights into the pathogenesis of PD, the cumulative set of genes only explain up to 5% of all PD cases (Klein and Westenberger, 2012). Therefore, the remaining 95% of PD remain apparently sporadic.
Large genome-wide association studies (GWAS) of PD cases have identified common risk variants that have modest influence on lifetime risk of PD. The first reasonably well-powered PD GWAS identified several loci that contain common variation associated with PD risk (Simón-Sánchez et al., 2009; Satake et al., 2009). Subsequent meta-analyses have confirmed these associations (International Parkinson Disease Genomics Consortium et al., 2011; Nalls et al., 2014) and the latest GWAS, which consists of 37,688 PD cases, 18,618 PD proxies and over 1,400,000 controls, has robustly identified association signals in 92 loci (Nalls et al., 2018). What is particularly interesting in PD, but not generally true in other neurodegenerative diseases, is that the genes that cause disease in families are also represented in the GWAS loci. There are multiple examples of these pleomorphic risk loci, so called because they harbor variants that, likely through slightly different mechanisms, impact both inherited and sporadic PD.
Here, we provide an overview and interpretation of how monogenic genes may play roles in sporadic PD. We will focus mainly on genes that contain deleterious and highly penetrant causal mutations, but also harbor risk variants for idiopathic disease. These genes are particularly important because their presence implies that there are functional pathogenic links between monogenic and idiopathic PD, which in turn has implications for understanding and treating this disorder.
SNCA links protein deposition and genetic risk of PD
The first definitive genetic cause of PD was the discovery of a missense mutation (p.A53T) in SNCA (PARK1) that was linked to disease in a large family with an autosomal dominant pattern of inheritance (Polymeropoulos et al., 1997). Soon after being linked to monogenic forms of PD, α-synuclein was also identified as the primary component of Lewy Bodies and the major pathological hallmark of PD (Spillantini et al., 1997). Since its initial discovery, several other SNCA missense point mutations have been described (p.A30P, p.E46K, p.G51D, p.A53E), all of which are located in the N-terminal region of the protein that normally folds into a helical conformation to bind to neuronal synaptic membranes (Krüger et al., 1998; Zarranz et al., 2004; Lesage et al., 2013; Pasanen et al., 2014). In addition to these point mutations, duplications and triplications of the SNCA locus also cause inherited PD (Ibáñez et al., 2004; Chartier-Harlin et al., 2004; Singleton, 2003). Interestingly, individuals carrying triplications present with a more severe and aggressive phenotype than cases with duplications, which are more similar to idiopathic PD (Fuchs et al., 2007), suggesting that SNCA dosage is important in disease pathogenesis.
The SNCA locus was first implicated as a common genetic risk factor when polymorphisms in REP1, a variable repeat microsatellite sequence located upstream of the SNCA promoter, were associated with idiopathic PD (Maraganore et al., 2006). Subsequently, at least three independent single nucleotide polymorphisms (SNPs) across the SNCA locus have now been associated with increased risk for PD by GWAS (Pihlstrøm et al. 2018; Simón-Sánchez et al., 2009; Nalls et al., 2014; Chang et al., 2017; Nalls et al., 2018). Additionally, a recent GWAS in Dementia with Lewy Bodies (DLB), a synucleinopathy with overlapping symptoms, identified only one of the three independent signals at the 5’ end of SNCA as contributing to disease risk (Guerreiro et al., 2018). These distinct patterns of associations with PD and DLB at the SNCA locus suggest that these variants have different effects on SNCA gene regulation.
While the SNCA locus harbors multiple types of genetic variation associated with PD risk, an important question is whether there is convergence of these variants on disease processes or whether each type of variation causes disease by different mechanisms (Nalls et al., 2014). Several non-coding risk variants have been demonstrated to play a role in regulating SNCA expression levels in various model systems. For example, the SNCA-REP1 allele has been shown to increase human SNCA mRNA and protein levels in a transgenic mouse model (Cronin et al., 2009). Recently, a study employing a CRISPR/Cas9 strategy in human induced pluripotent stem cells (iPSCs), found that an intronic SNP in SNCA associated with PD by GWAS is located in an enhancer that contributes to the regulation of SNCA expression (Soldner et al., 2016). More recently, it was suggested that one of the lead SNPs from the PD GWAS is a major functional SNP and is predicted to act by increasing SNCA expression in the brain (Pihlstrøm et al. 2018). If we consider that inherited PD can be influenced by the number of copies of SNCA without coding variation, then we might expect that higher expression level of SNCA controlled by common genetic variants would influence sporadic PD risk. If this is true, then sporadic disease caused by common non-coding variants may be a subtler form of the multiplication cases.
As opposed to variants that influence expression level, coding missense point mutations in SNCA have a variety of structural effects on the protein that include changes in the ratio of tetrameric to monomeric species, formation of oligomeric aggregates and loss of membrane binding. Which of these activities is critically important for neuronal damage in PD is not resolved, as each have been shown to cause cellular damage. However, as the main neuropathological and clinical phenotypes in point mutations and multiplication mutations overlap, it seems likely that there are some common mechanisms that underlie disease pathogenesis. However, the likelihood of cognitive impairment, psychosis and related phenotypes in SNCA mutation carriers correlates with the type of the mutation. Missense mutation carriers are least likely to display these non-motor phenotypes while individuals with a locus triplication are most likely to exhibit severe forms of disease, and phenotypes in duplication carriers often lie in between these two ends of this range (Tambasco et al. 2016). Another, more specific example of common mechanisms between different types of PD is given by the LRRK2 locus that we will discuss below.
Variation at the LRRK2 polymorphic risk locus nominates common mechanisms in sporadic and monogenic PD
Mutations in LRRK2 were first identified as the cause of PARK8-linked autosomal dominant PD in multiple families in 2004 (Paisán-Ruíz et al., 2004; Zimprich et al., 2004). By 2008, 46 point mutations, excluding those commonly found in controls, had been identified in LRRK2 (Biskup and West, 2009) and by 2010 the total number of exonic variants had expanded to 121 (Ross et al., 2011). Of these variants, only six, p.R1441C/G/H, p.Y1699C, p.G2019S and p.I2020T, have reliably been shown to segregate with disease in extended pedigrees (Paisán-Ruíz et al., 2004; Zimprich et al., 2004; Di Fonzo et al., 2005; Nichols et al., 2005). LRRK2 p.G2019S, the most common disease associated variant, causes monogenic PD with an age-and population-dependent incomplete penetrance. Penetrance estimates range from a lower bound of 16.7% in the Ashkenazi population to an upper estimate of 85% by the age of 80 (Lee et al., 2017; Kachergus et al., 2005). This lack of complete penetrance explains the relatively high number of apparently idiopathic cases that carry the p.G2019S allele, with particularly high frequencies in Ashkenazi Jewish and North African populations (Gilks et al., 2005; Ozelius et al., 2006; Lesage et al., 2005). Other LRRK2 mutations also show incomplete penetrance (Gosal et al., 2007; Ruiz-Martínez et al., 2010), suggesting that while all of the variants initially found in families increase risk substantially, they do not invariably lead to disease.
Additional risk variants have been identified in other populations, with p.R1628P and p.G2385R being the most common in Asian populations (Funayama et al., 2007; Farrer et al., 2007; Tan et al., 2010; Gopalai et al., 2014). Interestingly, a protective variant of LRRK2, p.R1398H, has also been identified in multiple populations (Chen et al., 2011; Heckman et al., 2014). The effect size of these variants is quantitatively less than p.G2019S, having a less than two-fold effect on PD risk.
LRRK2 encodes a large multi-domain protein consisting of 2527 amino acids. Interestingly, all proven monogenic pathogenic mutations are clustered in the ROC (ras of complex proteins), COR (C-terminal of Roc) and kinase domains. Pathogenic mutations either work by decreasing the GTPase activity encoded by the ROC-COR tandem bidomain (West et al., 2005; Lewis et al., 2007; Berwick et al., 2017) or by increasing the activity of kinase domain (West et al., 2005; Greggio et al., 2006; West et al., 2007). The protective allele p.R1398H is also located in the COR domain and has been shown to decrease kinase activity of the protein (Tan et al., 2010; Nixon-Abell et al., 2016). An exception to these general observations, is the risk variant p.G2385R which is located in a C-terminal WD40 domain and shows lower steady state protein levels and altered protein binding due to changes in protein structure (Rudenko et al., 2012; Ho et al., 2016; Rudenko et al., 2017; Carrion et al., 2017). Speculatively, the lower steady state levels may help negate the pathogenic effects and explain why this variant is only risk factor rather than a more penetrant allele like p.G2019S.
Perhaps counter-intuitively, the lower GTPase activity of several pathogenic mutations is likely to enhance overall LRRK2 function as hydrolysis of GTP to form GDP is typically an inactivation event for GTPases. Thus, it has been postulated that while different mutations have slightly different biochemical mechanisms, they rapidly converge with consistent direction of effect on immediate downstream biology (Cookson, 2010). This contention has received support from experiments arising from an understanding that LRRK2 interacts with several small RAB family GTPases. First, LRRK2 interacts with a specific RAB at the trans-Golgi network (TGN) and all pathogenic mutations enhance the recruitment of LRRK2 to the TGN relative to WT, with the risk factor variant p.G2385R having an intermediate effect (Beilina et al., 2014). Second, LRRK2 can phosphorylate a series of RAB proteins and in cells (but not in vitro) all mutations enhance RAB phosphorylation (Steger et al., 2016). Therefore, all pathogenic coding mutations appear to have consistent effects on cellular events that are likely linked to intracellular membrane sorting, a well-defined function of RABs.
In early GWAS studies, the LRRK2 locus was noted to have potential association signal in both European and Japanese populations. However, the association did not pass correction for genome-wide significance in the European population and so was labeled as a suggestive association (Simón-Sánchez et al., 2009; Satake et al., 2009). As GWAS study sizes have significantly increased, it has become evident that there is a common non-coding risk variant at the LRRK2 locus (Nalls et al., 2014). The most recent meta-GWAS identified rs76904798 as the most significantly associated SNP in the LRRK2 region with a p-value of 1.52×10−28 (Nalls et al., 2018). It has been suggested that this specific PD risk variant is associated with higher expression of LRRK2 mRNA, being an example of an expression quantitative trait locus (eQTL) (Ryan et al., 2017). Although this result needs to be confirmed in additional sample series, it suggests that non-coding risk factor variants act in the same direction as pathogenic alleles, i.e., by increasing overall LRRK2 activity. Thus, as for SNCA, the pleomorphic risk locus containing LRRK2 likely has several genetic variants that lead to disease by similar mechanisms.
Heterozygous mutations in recessive genes may increase PD risk
Pathogenic mutations in PRKN, PINK1, DJ1, ATP13A2, PLA2G6, FBXO7, and DNAJC6 are causes of recessive, predominantly early-onset PD (EOPD) (Hauser et al., 2017). In each case, disease is associated with homozygous or compound heterozygous loss of function mutations in the same gene. In many ways, the phenotypes associated with recessive gene mutations are distinct from sporadic PD. Recessively inherited forms of PD are rare and often found in consanguineous pedigrees that may have other symptoms in addition to those characteristic of typical PD. Furthermore, unlike most PD cases, mutations in these genes result in early onset disease, sometimes as early as teenage years. EOPD cases tend to progress more slowly than typical sporadic PD or dominant gene mutations. Finally, autopsy examination of brains from EOPD suggests that α-synuclein deposition does not always occur, unlike sporadic PD where Lewy pathology is required for a definitive diagnosis (Mori et al., 1998; Hayashi et al., 2000; van de Warrenburg et al., 2001; Farrer et al., 2001; Sasaki et al., 2004; Samaranch et al., 2010). However, this data is complicated to interpret as LRRK2 PD cases also show variable protein deposition pathology despite high clinical overlap (Kalia et al., 2015; Poulopoulos et al., 2012).
Although the classical definition of recessive disease genes is that carriers of one risk allele are not affected, it has been reported that heterozygous mutations in some of these genes may act as risk factors for sporadic PD (Klein et al., 2007). One possible mechanism for this proposal is that heterozygous nonsense mutations predispose an individual to PD through partial loss of function. However, it is more likely that some individuals have a second undiscovered mutation or structural genetic variant that might explain their disease and be consistent with compound heterozygosity. None of the published PD GWAS, including the largest and most recent meta-analysis (Nalls et al., 2018), have identified a recessive PD gene as a risk locus. Most likely these variants are not detected because they are too rare for identification by GWAS or on their own they do not act as risk variants for PD. Most of the EOPD mutations are too rare to have been studied in the heterozygous state however for two of the most commonly mutated autosomal recessive PD genes (PRKN and PINK1) there are some reports that heterozygous mutations have a potential role in development of PD, and each will be discussed separately below.
PRKN (PARK2)
Mutations in PRKN are diverse in nature, owing to its large genomic size of 1.3Mb on chromosome 6 and recognition as a common fragile site in the genome (Smith et al., 2006). PD-linked PRKN mutations consist of homozygous or compound heterozygous point mutations as well as partial deletions or duplications (Abbas et al., 1999). Parkin mutations are the most common cause of EOPD with frequency estimations ranging from 4.6% to 10.5%, depending on the population (Abbas et al., 1999; Leroy et al., 1998; Taghavi et al., 2017). PRKN encodes the cytosolic E3 ubiquitin ligase parkin which is recruited to the mitochondrial membrane when phosphorylated by PINK1 to induce mitophagy (Kane et al., 2014).
Several studies have suggested that PRKN variants increase risk for sporadic PD (Lincoln et al., 2003; Lücking et al., 2000; Lesage et al., 2008; Clark et al., 2006; Wang et al., 1999; Hedrich et al., 2002) and/or influence age at onset (Foroud et al., 2003; Sun et al., 2006). However, others have shown that heterozygous mutations and structural genetic variants are observed with the same frequency in cases and healthy controls (Kay et al., 2007; Lincoln et al., 2003; Lücking et al., 2000; Kay et al., 2007). These conflicting studies make the role of heterozygous PRKN mutations in disease development uncertain. A meta-analysis of 4,538 cases and 4,213 controls that screened for PRKN copy number variants (CNVs) supported the idea that heterozygous carriers of CNVs containing coding exons had increased risk of developing PD compared to non-carriers (Huttenlocher et al., 2015). Additionally, although neuroimaging and electrophysiological findings associated with PD have shown some premorbid changes in heterozygous mutation carriers, such as reduced fluorodopa uptake in the striatum, these individuals have not been reported to be clinically diagnosed with PD (Khan et al., 2002; Hilker et al., 2001; Khan et al., 2005; Inzelberg et al., 2005).
One argument that has been advanced to explain the presence of heterozygous mutations in PRKN in apparently sporadic disease is that these variants might be associated with dominant inheritance but with diminished penetrance, suggesting that partial loss of function mutations would lead to milder forms of PD. Several studies have been performed in families, with the expectation that heterozygous carriers would also have PD, but these studies have yielded conflicting results. Some have reported that heterozygous relatives of PRKN-linked cases suffer from mild parkinsonism (Klein et al., 2000; Farrer et al., 2001) but not a full PD-like phenotype. However, others have not replicated any observations of parkinsonism in heterozygous carriers (Wang et al., 2013). Due to its large genomic size and diversity of mutations it is possible that some mutations in the second allele remain undetected in apparently heterozygous individuals.
PINK1 (PARK6)
The PARK6 locus was initially mapped to chromosome 1 in three different consanguineous families (Valente et al., 2001; Valente et al., 2002). Upon sequencing candidate genes in the region, PINK1 was confirmed to contain homozygous missense mutations (Valente et al., 2004a). Additional missense mutations have since been identified in several other consanguineous pedigrees (Hatano et al., 2004). It has been estimated that PINK1 mutations are found in 3.7% of EOPD cases worldwide, with frequencies ranging from 0.6% in European descent cases to 13.5% in Asian populations (Kilarski et al., 2012).
Similarly to PRKN, several lines of evidence suggest that heterozygous PINK1 mutations can act as risk factors for idiopathic PD (Rogaeva et al., 2004; Bonifati et al., 2005; Abou-Sleiman et al., 2006; Valente et al., 2004b). A recent study reported that carrying one copy of the rare p.G411S mutation in PINK1 increases risk of PD to a greater degree than other disease-associated variants (Puschmann et al., 2017). The p.G411S variant significantly decreases PINK1 kinase activity in neurons and the average age at disease onset is significantly younger in p.G411S mutation carriers than in non-carriers. Some clinical examinations of heterozygous relatives of homozygous PINK1 carriers have shown signs of mild parkinsonism (Criscuolo et al., 2006; Hedrich et al., 2006; Hiller et al., 2007; Djarmati et al., 2006). However, not all heterozygous relatives present with such symptoms. Similar to PRKN mutations, therefore, whether PINK1 alleles cause disease by haploinsufficiency or a low-penetrance dominant mechanism is uncertain. However, a meta-analysis of approximately 1,000 cases and 400 controls for heterozygous PINK1 variants found no significant difference in frequencies between the populations (Marongiu et al., 2008). The conflicting evidence at this locus suggests a role for PINK1 in idiopathic PD but more data is needed to validate this correlation.
Deciphering whether heterozygous variants in recessive genes are risk factors for idiopathic PD is important for the understanding the etiology of disease. Among individuals with PD, the number of carriers of heterozygous mutations in recessive genes surpasses the number of homozygous or compound heterozygous carriers, suggesting that they could be susceptibility factors or disease modifiers. These genes might also contribute to the heritability of idiopathic PD in a subset of carriers making them possible drug targets. Conversely, it is also possible that other mutations have been missed in PRKN or PINK1 or that there is a secondary mutation in an unknown modifier gene. Theoretically, non-coding variation at either of these loci that reduce expression on the unaffected allele may result in a PD phenotype if only the mutant allele is expressed. In the coming years, well powered human genetic studies will be decisive in robustly uncovering the role of heterozygous variants in recessive genes and their effect in PD.
Genome-wide association studies link sporadic and monogenic parkinsonism
There is growing evidence that the multiple pathways identified in monogenic PD also play a role in sporadic PD, showing that they are not separate entities and several genes might interact to regulate downstream common targets. This type of pleomorphism can be extrapolated to several PD related loci identified by GWAS including ACMSD, CSMD1, GCH1, and VPS13C. These candidate loci contain common variants linked to sporadic forms of PD, and putative rare pathogenic variants have also been described in monogenic cases with either PD or a parkinsonism syndrome.
Supporting the link between monogenic and sporadic etiologies, mutations in GCH1 have been found to segregate in families with a combination of members with adult-onset parkinsonism or dopa-responsive dystonia following an autosomal dominant pattern of inheritance with incomplete penetrance (Hagenah et al., 2005). Prompted by this observation, a follow-up large exome sequencing study showed that known GCH1 pathogenic mutations are more frequent in sporadic PD cases than in controls and are associated to a 7-fold increase in the risk for developing PD (Mencacci et al., 2014). These results were also supported by the latest PD meta-analysis which also nominated this locus at a significant level.
Another example of possible shared etiologies comes from a recent screening of individuals in a three generation pedigree affected with familial cortical myoclonic tremor and epilepsy, which pointed to p.Trp26Stop in ACMSD as a disease-segregating and predicted pathogenic mutation (Martí-Massó et al., 2013). Interestingly, one family member also exhibited parkinsonism and ACMSD is in a region associated with sporadic PD by GWAS (International Parkinson Disease Genomics Consortium et al., 2011). Subsequently, the ACMSD p.Glu298Lys mutation was detected in a single individual with late onset sporadic PD (Vilas et al., 2017) suggesting that rare variants within ACMSD may cause PD. Another genetic study performed in two unrelated families with PD identified two novel, heterozygous variants in the CSMD1, each resulting in mutation of a highly conserved amino acid, suggesting that they may cause PD (Ruiz-Martínez et al., 2017). The most convincing example, VPS13C was first reported as a susceptibility risk locus for PD (Nalls et al., 2014). Later, homozygous and compound heterozygous truncating mutations were found to cause a very severe type of autosomal recessive PD (Lesage et al., 2016). The shared role of these genes in monogenic and sporadic PD requires further validation in large well-powered studies but indicates that loci associated with sporadic forms of PD may also contain very rare variants that can cause monogenic PD.
Mutations in MAPT can cause parkinsonism and are risk factors for Parkinson’s disease
Although MAPT, which encodes the neuronal structure protein tau, is not considered a PARK gene there are several lines of evidence that link this gene to PD. Rare pathogenic variants in MAPT have been identified in several neurodegenerative diseases including tauopathies such frontotemporal dementia (FTD) (Hutton et al., 1998) and PSP (Spillantini et al., 1998; Clark et al., 1998; Haussmann et al., 2017; Poorkaj et al., 2002). Individuals carrying these MAPT mutations often present with a typical behavioral FTD phenotype as well as motor symptoms resembling parkinsonism (Wszolek et al., 2006).
The MAPT gene is found within a region of high linkage disequilibrium (LD) that covers ~1 Mb of chromosome 17. Two major MAPT haplotypes have been identified, H1 and H2, that are inverted relative to each other and each have several sub-haplotypes (Steinberg et al., 2012). Common variants within the H1 haplotype have been associated with PD (Nalls et al., 2014) and several other neurodegenerative diseases, including FTD (Verpillat et al., 2002), PSP (Höglinger et al., 2011) and AD (Jun et al., 2016; Desikan et al., 2015). It is noteworthy that MAPT is the only risk locus that is shared between Alzheimer’s disease (AD) and PD. A recent study has shown that PD patients who are homozygous for the H1 haplotype have a significantly increased burden of Lewy bodies in the neocortex compared to cases with the H2 haplotype (Robakis et al., 2016).
There are several transcripts of MAPT expressed in the CNS and multiple eQTLs have been identified that are associated with differences in alternate transcript levels (Blauwendraat et al., 2016; Ramasamy et al., 2014; Myers et al., 2007). A specific SNP within the H1 haplotype has been suggested to be involved in the regulation of exon 3 retention and thus there may be splicing quantitative traits as well eQTLs at this locus (Lai et al., 2017). This is a potential disease mechanism as exon 3 retention may change the interaction partners of tau protein. Overall, these findings show that the MAPT locus is highly pleomorphic, although the genetic and molecular underpinnings of its association with PD remain to be determined.
Mutations in GBA increase risk of PD
Mutations in GBA, encoding the glucocerebrosidase enzyme, were first identified as the cause of the autosomal recessive lysosomal storage disorder Gaucher disease in the 1980s (Tsuji et al. 1987). Currently over 300 GBA mutations have been identified which typically result in a reduced enzyme activity (Montfort et al. 2004). Clinically, Gaucher disease patients can display parkinsonian symptoms, and many studies have identified an increased frequency of heterozygous GBA mutations in PD cohorts (Aharon-Peretz et al., 2004; Clark et al., 2007). Subsequently, large multicenter studies identified a significant increase of GBA coding variants in both PD and DLB cases compared to controls and that the genetic influence of GBA is higher in DLB than PD (Sidransky et al. 2009; Nalls et al., 2013). Others have shown that PD patients who are GBA mutation carriers are more likely to develop cognitive impairment and dementia (Cilia et al. 2016) that is independent of Alzheimer disease pathology (Tsuang et al., 2012). Additionally, mutations in GBA are associated with an earlier age of onset of PD compared to non-carriers (Clark et al. 2007; Alcalay et al., 2012, Blauwendraat et al., 2018)
Heterozygous coding variants in GBA are therefore a common genetic cause of PD and they have also been associated with sporadic PD by GWAS (Nalls et al., 2014; Chang et al., 2017; Nalls et al. 2018). Although GBA coding variants explain the majority of the GBA GWAS signal there also appears to be independent non-coding signal (Blauwendraat et al., 2018; Berge-Seidl et al., 2017; Nalls et al., 2018). Interestingly, some coding variants like p.E365K are associated with PD and have a significant effect on glucocerebrosidase activity (Alcalay et al., 2015) but do not cause Gaucher disease in homozygous state. Reduction of functional glucocerebrosidase has been shown to result in an accumulation of SNCA protein in neurons (Cullen et al. 2011; Du et al. 2015) highlighting the importance of functional lysosomes in healthy aging (Robak et al. 2017). All of this evidence points to GBA as a significant, but low penetrant, risk factor for PD with alleles that may or may not cause Gaucher’s disease.
Future directions
Remarkable progress has been achieved in the understanding of the genetic architecture underlying monogenic and idiopathic PD in the past twenty years. Over 15 genes now have been identified to cause monogenic forms of PD and over 40 independent loci are associated with increased risk of sporadic PD. It is becoming clear that some genes exist that contain both deleterious and highly penetrant coding mutations as well as coding and non-coding variants that increase risk for idiopathic disease. This data is prima facie evidence suggesting a pathophysiological link between monogenic and idiopathic forms of PD. Monogenic and sporadic cases of PD are often clinically indistinguishable and it is clear that both forms share common genetic determinants. It is likely that the monogenic and sporadic dichotomy will break down in the coming years, when stratifying and redefining disease subtypes improves.
Despite the considerable success in identifying the genetic components associated with disease risk, a major challenge remains to understand the mechanisms by which pleomorphism affects biological function to contribute to PD risk. Observations at both the LRRK2 and SNCA loci suggest that risk factors act in a similar manner to more penetrant mutations, in both cases by providing an enhancement of function but in a quantitatively smaller manner. This has important implications for disease-modifying treatments as it suggests that strategies to limit toxicity of dominant PD gene products might be helpful for sporadic PD.
Figure 1.
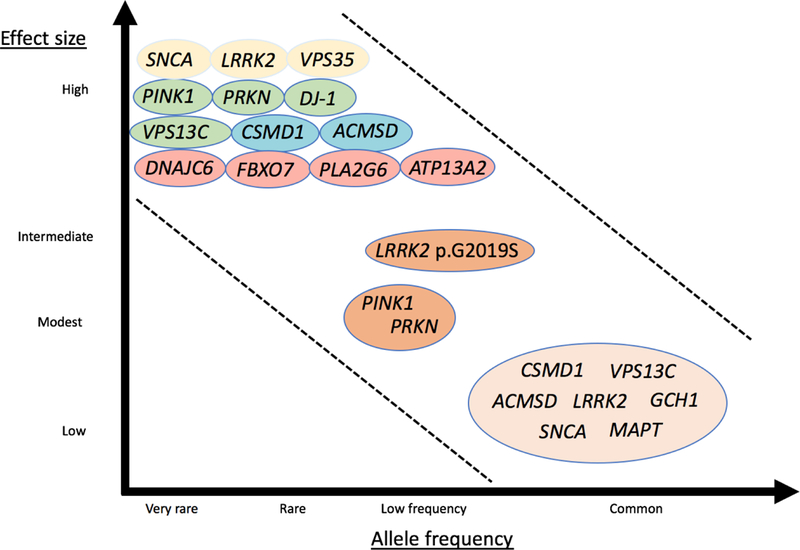
Continuum of genes of different phenotypic effect sizes and allele frequencies. Colors symbolize modes of inheritance: dominant (yellow), recessive (green), recessive atypical parkinsonism (pink), possibly disease-causing genes (blue), dominant with incomplete penetrance (orange), risk loci (light orange). Modified from McCarthy et al., 2008 (McCarthy et al. 2008).
Figure 2.
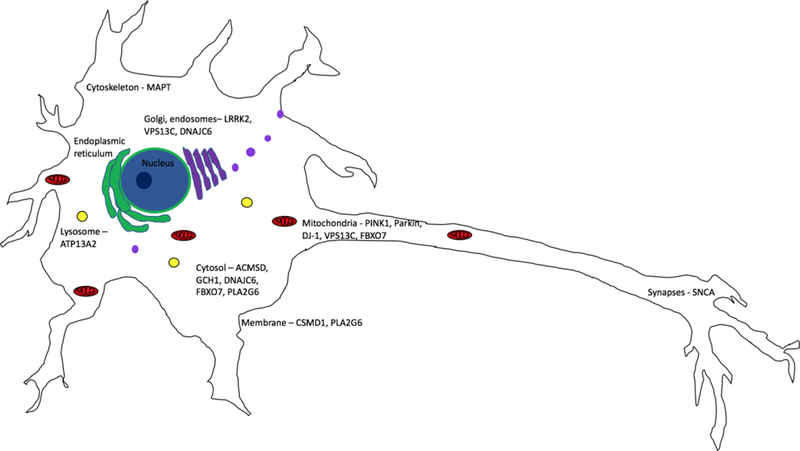
Subcellular localization of genes predicted to be involved in sporadic Parkinson’s disease. The most common subcellular localization for genes associated with sporadic PD is in the cytosol, mitochondria, and in organelles involved in vesicular trafficking, Golgi Network and endosomes.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institutes of Health (National Institute of Neurological Disorders and Stroke, National Institute on Aging; projects 1ZIA-NS003154-2 and Z01-AG000949)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas N, Lücking CB, Ricard S, Dürr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Böhme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A, 1999. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum. Mol. Genet 8, 567–574. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW, 2003. The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann. Neurol 54, 283–286. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Muqit MMK, McDonald NQ, Yang YX, Gandhi S, Healy DG, Harvey K, Harvey RJ, Deas E, Bhatia K, Quinn N, Lees A, Latchman DS, Wood NW, 2006. A heterozygous effect for PINK1 mutations in Parkinson’s disease? Ann. Neurol 60, 414–419. [DOI] [PubMed] [Google Scholar]
- Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R, 2004. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med 351, 1972–1977. [DOI] [PubMed] [Google Scholar]
- Alcalay RN, Caccappolo E, Mejia-Santana H, Tang M-X, Rosado L, Orbe Reilly M., Ruiz D, Ross B, Verbitsky M, Kisselev S, Louis E, Comella C, Colcher A, Jennings D, Nance M, Bressman S, Scott WK, Tanner C, Mickel S, Andrews H, Waters C, Fahn S, Cote L, Frucht S, Ford B, Rezak M, Novak K, Friedman JH, Pfeiffer R, Marsh L, Hiner B, Siderowf A, Payami H, Molho E, Factor S, Ottman R, Clark LN, Marder K, 2012. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology 78, 1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo S-H, Mazzoni P, Pauciulo MW, Nichols WC, Gan-Or Z, Rouleau GA, Chung WK, Wolf P, Oliva P, Keutzer J, Marder K, Zhang X, 2015. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138, 2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, Ding J, Nalls MA, International Parkinson’s Disease Genomics Consortium, North American Brain Expression Consortium, Olszewski M, Hauser DN, Kumaran R, Lozano AM, Baekelandt V, Greene LE, Taymans J-M, Greggio E, Cookson MR, 2014. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. U. S. A 111, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge-Seidl V, Pihlstrøm L, Maple-Grødem J, Forsgren L, Linder J, Larsen JP, Tysnes O-B, Toft M, 2017. The GBA variant E326K is associated with Parkinson’s disease and explains a genome-wide association signal. Neurosci. Lett 658, 48–52. [DOI] [PubMed] [Google Scholar]
- Berwick DC, Javaheri B, Wetzel A, Hopkinson M, Nixon-Abell J, Grannò S, Pitsillides AA, Harvey K, 2017. Pathogenic LRRK2 variants are gain-of-function mutations that enhance LRRK2-mediated repression of β-catenin signaling. Mol. Neurodegener 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, West AB, 2009. Zeroing in on LRRK2-linked pathogenic mechanisms in Parkinson’s disease. Biochim. Biophys. Acta 1792, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat C, Francescatto M, Gibbs JR, Jansen IE, Simón-Sánchez J, Hernandez DG, Dillman AA, Singleton AB, Cookson MR, Rizzu P, Heutink P, 2016. Comprehensive promoter level expression quantitative trait loci analysis of the human frontal lobe. Genome Med 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat C, Heilbron K, Vallerga CL, Bandres-Ciga S, von Coelln R, Pihlstrom L, Simon-Sanchez J, Schulte C, Sharma M, Krohn L, Siitonen A, Iwaki H, Leonard H, Noyce AJ, Tan M, Raphael Gibbs J., Hernandez DG, Scholz SW, Jankovic J, Shulman LM, Lesage S, Corvol J-C, Brice A, van Hilten JJ, Marinus J, Tienari P, Majamaa K, Toft M, Grosset DG, Gasser T, Heutink P, Shulman JM, Wood N, Hardy J, Morris HR, Hinds DA, Gratten J, Visscher PM, Gan-Or Z, Nalls M, Singleton A, The 23andMe Research Team, International Parkinsons Disease Genomics Consortium (IPDGC), 2018. Parkinson disease age of onset GWAS: defining heritability, genetic loci and a-synuclein mechanisms 10.1101/424010 [DOI]
- Blauwendraat C, Bras JM, Nalls MA, Lewis PA, Hernandez DG, Singleton AB, International Parkinson’s Disease Genomics Consortium, 2018. Coding variation in GBA explains the majority of the SYT11-GBA Parkinson’s disease GWAS locus. Mov. Disord 10.1002/mds.103 [DOI] [PMC free article] [PubMed]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MCJ, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P, 2003. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rohé CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, Tavella A, Marconi R, Nicholl DJ, Chien HF, Fincati E, Abbruzzese G, Marini P, De Gaetano A, Horstink MW, Maat-Kievit JA, Sampaio C, Antonini A, Stocchi F, Montagna P, Toni V, Guidi M, Dalla Libera A., Tinazzi M, De Pandis F, Fabbrini G, Goldwurm S, de Klein A, Barbosa E, Lopiano L, Martignoni E, Lamberti P, Vanacore N, Meco G, Oostra BA, Italian Parkinson Genetics Network, 2005. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65, 87–95. [DOI] [PubMed] [Google Scholar]
- Carrion MDP, Marsicano S, Daniele F, Marte A, Pischedda F, Di Cairano E, Piovesana E, von Zweydorf F, Kremmer E, Gloeckner CJ, Onofri F, Perego C, Piccoli G, 2017. The LRRK2 G2385R variant is a partial loss-of-function mutation that affects synaptic vesicle trafficking through altered protein interactions. Sci. Rep 7, 5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, International Parkinson’s Disease Genomics Consortium, 23andMe Research Team, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR, 2017. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin M-C, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A, 2004. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Chen L, Zhang S, Liu Y, Hong H, Wang H, Zheng Y, Zhou H, Chen J, Xian W, He Y, Li J, Liu Z, Pei Z, Zeng J, 2011. LRRK2 R1398H polymorphism is associated with decreased risk of Parkinson’s disease in a Han Chinese population. Parkinsonism Relat. Disord 17, 291–292. [DOI] [PubMed] [Google Scholar]
- Clark LN, Afridi S, Karlins E, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K, 2006. Case-control study of the parkin gene in early-onset Parkinson disease. Arch. Neurol 63, 548–552. [DOI] [PubMed] [Google Scholar]
- Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K, 2007. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69, 1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D’Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC, 1998. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc. Natl. Acad. Sci. U. S. A 95, 13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Kanaan NM, Kordower JH, 2011. Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat. Rev. Neurosci 12, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR, 2010. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci 11, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo C, Volpe G, De Rosa A, Varrone A, Marongiu R, Mancini P, Salvatore E, Dallapiccola B, Filla A, Valente EM, De Michele G, 2006. PINK1 homozygous W437X mutation in a patient with apparent dominant transmission of parkinsonism. Mov. Disord 21, 1265–1267. [DOI] [PubMed] [Google Scholar]
- Cronin KD, Ge D, Manninger P, Linnertz C, Rossoshek A, Orrison BM, Bernard DJ, El-Agnaf OMA, Schlossmacher MG, Nussbaum RL, Chiba-Falek O, 2009. Expansion of the Parkinson disease-associated SNCA-Rep1 allele upregulates human alpha-synuclein in transgenic mouse brain. Hum. Mol. Genet 18, 3274–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen V, Sardi SP, Ng J, Xu Y-H, Sun Y, Tomlinson JJ, Kolodziej P, Kahn I, Saftig P, Woulfe J, Rochet J-C, Glicksman MA, Cheng SH, Grabowski GA, Shihabuddin LS, Schlossmacher MG, 2011. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann. Neurol 69, 940–953. [DOI] [PubMed] [Google Scholar]
- Dekker MCJ, Eshuis SA, Maguire RP, Veenma-van der Duijn L, Pruim J, Snijders PJLM, Oostra BA, van Duijn CM, Leenders KL, 2004. PET neuroimaging and mutations in the DJ-1 gene. J. Neural Transm 111, 1575–1581. [DOI] [PubMed] [Google Scholar]
- de Lau LML, Breteler MMB, 2006. Epidemiology of Parkinson’s disease. Lancet Neurol 5, 525–535. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Schork AJ, Wang Y, Witoelar A, Sharma M, McEvoy LK, Holland D, Brewer JB, Chen C-H, Thompson WK, Harold D, Williams J, Owen MJ, O’Donovan MC, Pericak-Vance MA, Mayeux R, Haines JL, Farrer LA, Schellenberg GD, Heutink P, Singleton AB, Brice A, Wood NW, Hardy J, Martinez M, Choi SH, DeStefano A, Ikram MA, Bis JC, Smith A, Fitzpatrick AL, Launer L, van Duijn C, Seshadri S, Ulstein ID, Aarsland D, Fladby T, Djurovic S, Hyman BT, Snaedal J, Stefansson H, Stefansson K, Gasser T, Andreassen OA, Dale AM, ADNI, ADGC, GERAD, CHARGE and IPDGC Investigators, 2015. Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus. Mol. Psychiatry 20, 1588–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, Rohé CF, Ferreira J, Chien HF, Vacca L, Stocchi F, Guedes L, Fabrizio E, Manfredi M, Vanacore N, Goldwurm S, Breedveld G, Sampaio C, Meco G, Barbosa E, Oostra BA, Bonifati V, Italian Parkinson Genetics Network, 2005. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365, 412–415. [DOI] [PubMed] [Google Scholar]
- Djarmati A, Hedrich K, Svetel M, Lohnau T, Schwinger E, Romac S, Pramstaller PP, Kostić V, Klein C, 2006. Heterozygous PINK1 mutations: a susceptibility factor for Parkinson disease? Mov. Disord 21, 1526–1530. [DOI] [PubMed] [Google Scholar]
- Driver JA, Logroscino G, Gaziano JM, Kurth T, 2009. Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology 72, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T-T, Wang L, Duan C-L, Lu L-L, Zhang J-L, Gao G, Qiu X-B, Wang X-M, Yang H, 2015. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 11, 1803–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston JW, 2001. Lewy bodies and parkinsonism in families with parkin mutations. Ann. Neurol 50, 293–300. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, Stone JT, Lin C-H, Dächsel JC, Hulihan MM, Haugarvoll K, Ross OA, Wu R-M, 2007. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat. Disord 13, 89–92. [DOI] [PubMed] [Google Scholar]
- Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC, Parkinson Study Group, 2003. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 60, 796–801. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson E-M, Schüle B, Langston JW, Middleton FA, Ross OA, Hulihan M, Gasser T, Farrer MJ, 2007. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68, 916–922. [DOI] [PubMed] [Google Scholar]
- Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, Murata M, Toda T, Mizuno Y, Hattori N, 2007. Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport 18, 273–275. [DOI] [PubMed] [Google Scholar]
- Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T, Wood NW, 2005. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365, 415–416. [DOI] [PubMed] [Google Scholar]
- Gopalai AA, Lim S-Y, Chua JY, Tey S, Lim TT, Mohamed Ibrahim N., Tan AH, Eow GB, Abdul Aziz Z., Puvanarajah SD, Viswanathan S, Looi I, Lim SK, Tan LP, Chong YB, Tan CT, Zhao Y, Tan EK, Ahmad-Annuar A, 2014. LRRK2 G2385R and R1628P mutations are associated with an increased risk of Parkinson’s disease in the Malaysian population. Biomed Res. Int 2014, 867321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosal D, Lynch T, Ross OA, Haugarvoll K, Farrer MJ, Gibson JM, 2007. Global distribution and reduced penetrance: Lrrk2 R1441C in an Irish Parkinson’s disease kindred. Mov. Disord 22, 291–292. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR, 2006. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis 23, 329–341. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD, Shepherd CE, Parkkinen L, Darwent L, Heckman MG, Scholz SW, Troncoso JC, Pletnikova O, Ansorge O, Clarimon J, Lleo A, Morenas-Rodriguez E, Clark L, Honig LS, Marder K, Lemstra A, Rogaeva E, St George-Hyslop P, Londos E, Zetterberg H, Barber I, Braae A, Brown K, Morgan K, Troakes C, Al-Sarraj S, Lashley T, Holton J, Compta Y, Van Deerlin V, Serrano GE, Beach TG, Lesage S, Galasko D, Masliah E, Santana I, Pastor P, Diez-Fairen M, Aguilar M, Tienari PJ, Myllykangas L, Oinas M, Revesz T, Lees A, Boeve BF, Petersen RC, Ferman TJ, Escott-Price V, Graff-Radford N, Cairns NJ, Morris JC, Pickering-Brown S, Mann D, Halliday GM, Hardy J, Trojanowski JQ, Dickson DW, Singleton A, Stone DJ, Bras J, 2018. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 17, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenah J, Saunders-Pullman R, Hedrich K, Kabakci K, Habermann K, Wiegers K, Mohrmann K, Lohnau T, Raymond D, Vieregge P, Nygaard T, Ozelius LJ, Bressman SB, Klein C, 2005. High mutation rate in dopa-responsive dystonia: detection with comprehensive GCHI screening. Neurology 64, 908–911. [DOI] [PubMed] [Google Scholar]
- Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu C-S, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N, 2004. Novel PINK1 mutations in early-onset parkinsonism. Ann. Neurol 56, 424–427. [DOI] [PubMed] [Google Scholar]
- Hauser DN, Primiani CT, Cookson MR, 2017. The Effects of Variants in the Parkin, PINK1, and DJ-1 Genes along with Evidence for their Pathogenicity. Curr. Protein Pept. Sci 18, 702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussmann R, Wysocki M, Brandt MD, Hermann A, Donix M, 2017. MAPT mutation associated with frontotemporal dementia and parkinsonism (FTDP-17). Int. Psychogeriatr 29, 869–871. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Wakabayashi K, Ishikawa A, Nagai H, Saito M, Maruyama M, Takahashi T, Ozawa T, Tsuji S, Takahashi H, 2000. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov. Disord 15, 884–888. [DOI] [PubMed] [Google Scholar]
- Heckman MG, Elbaz A, Soto-Ortolaza AI, Serie DJ, Aasly JO, Annesi G, Auburger G, Bacon JA, Boczarska-Jedynak M, Bozi M, Brighina L, Chartier-Harlin M-C, Dardiotis E, Destée A, Ferrarese C, Ferraris A, Fiske B, Gispert S, Hadjigeorgiou GM, Hattori N, Ioannidis JPA, Jasinska-Myga B, Jeon BS, Kim YJ, Klein C, Kruger R, Kyratzi E, Lin C-H, Lohmann K, Loriot M-A, Lynch T, Mellick GD, Mutez E, Opala G, Park SS, Petrucci S, Quattrone A, Sharma M, Silburn PA, Sohn YH, Stefanis L, Tadic V, Tomiyama H, Uitti RJ, Valente EM, Vassilatis DK, Vilariño-Güell C, White LR, Wirdefeldt K, Wszolek ZK, Wu R-M, Xiromerisiou G, Maraganore DM, Farrer MJ, Ross OA, Genetic Epidemiology Of Parkinson’s Disease (GEO-PD) Consortium, 2014. Protective effect of LRRK2 p.R1398H on risk of Parkinson’s disease is independent of MAPT and SNCA variants. Neurobiol. Aging 35, 266 e5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich K, Djarmati A, Schäfer N, Hering R, Wellenbrock C, Weiss PH, Hilker R, Vieregge P, Ozelius LJ, Heutink P, Bonifati V, Schwinger E, Lang AE, Noth J, Bressman SB, Pramstaller PP, Riess O, Klein C, 2004. DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology 62, 389–394. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Hagenah J, Djarmati A, Hiller A, Lohnau T, Lasek K, Grünewald A, Hilker R, Steinlechner S, Boston H, Kock N, Schneider-Gold C, Kress W, Siebner H, Binkofski F, Lencer R, Münchau A, Klein C, 2006. Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch. Neurol 63, 833–838. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Marder K, Harris J, Kann M, Lynch T, Meija-Santana H, Pramstaller PP, Schwinger E, Bressman SB, Fahn S, Klein C, 2002. Evaluation of 50 probands with early-onset Parkinson’s disease for Parkin mutations. Neurology 58, 1239–1246. [DOI] [PubMed] [Google Scholar]
- Hering R, Strauss KM, Tao X, Bauer A, Woitalla D, Mietz E-M, Petrovic S, Bauer P, Schaible W, Müller T, Schöls L, Klein C, Berg D, Meyer PT, Schulz JB, Wollnik B, Tong L, Krüger R, Riess O, 2004. Novel homozygous p.E64D mutation in DJ1 in early onset Parkinson disease (PARK7). Hum. Mutat 24, 321–329. [DOI] [PubMed] [Google Scholar]
- Hernandez DG, Reed X, Singleton AB, 2016. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem 139 Suppl 1, 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilker R, Klein C, Ghaemi M, Kis B, Strotmann T, Ozelius LJ, Lenz O, Vieregge P, Herholz K, Heiss WD, Pramstaller PP, 2001. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann. Neurol 49, 367–376. [PubMed] [Google Scholar]
- Hiller A, Hagenah JM, Djarmati A, Hedrich K, Reetz K, Schneider-Gold C, Kress W, Münchau A, Klein C, 2007. Phenotypic spectrum of PINK1-associated parkinsonism in 15 mutation carriers from 1 family. Mov. Disord 22, 145–147. [DOI] [PubMed] [Google Scholar]
- Ho DH, Jang J, Joe E-H, Son I, Seo H, Seol W, 2016. G2385R and I2020T Mutations Increase LRRK2 GTPase Activity. Biomed Res. Int 2016, 7917128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglinger GU, Melhem NM, Dickson DW, Sleiman PMA, Wang L-S, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, PSP Genetics Study Group, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu C-E, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Müller U, Schellenberg GD, 2011. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet 43, 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher J, Stefansson H, Steinberg S, Helgadottir HT, Sveinbjörnsdóttir S, Riess O, Bauer P, Stefansson K, 2015. Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. Hum. Mol. Genet 24, 5637–5643. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P, 1998. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. [DOI] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet A-M, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A, 2004. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171. [DOI] [PubMed] [Google Scholar]
- International Parkinson Disease Genomics Consortium,Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin U-M, Saad M, Simón-Sánchez J, Schulte C, Lesage S, Sveinbjörnsdóttir S, Stefánsson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW, 2011. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 377, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inzelberg R, Hattori N, Mizuno Y, 2005. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology 65, 1843. [DOI] [PubMed] [Google Scholar]
- Jun G, Ibrahim-Verbaas CA, Vronskaya M, Lambert J-C, Chung J, Naj AC, Kunkle BW, Wang L-S, Bis JC, Bellenguez C, Harold D, Lunetta KL, Destefano AL, Grenier-Boley B, Sims R, Beecham GW, Smith AV, Chouraki V, Hamilton-Nelson KL, Ikram MA, Fievet N, Denning N, Martin ER, Schmidt H, Kamatani Y, Dunstan ML, Valladares O, Laza AR, Zelenika D, Ramirez A, Foroud TM, Choi S-H, Boland A, Becker T, Kukull WA, van der Lee SJ, Pasquier F, Cruchaga C, Beekly D, Fitzpatrick AL, Hanon O, Gill M, Barber R, Gudnason V, Campion D, Love S, Bennett DA, Amin N, Berr C, Tsolaki M, Buxbaum JD, Lopez OL, Deramecourt V, Fox NC, Cantwell LB, Tárraga L, Dufouil C, Hardy J, Crane PK, Eiriksdottir G, Hannequin D, Clarke R, Evans D, Mosley TH Jr, Letenneur L, Brayne C, Maier W, De Jager P, Emilsson V, Dartigues J-F, Hampel H, Kamboh MI, de Bruijn RFAG, Tzourio C, Pastor P, Larson EB, Rotter JI, O’Donovan MC, Montine TJ, Nalls MA, Mead S, Reiman EM, Jonsson PV, Holmes C, St George-Hyslop PH, Boada M, Passmore P, Wendland JR, Schmidt R, Morgan K, Winslow AR, Powell JF, Carasquillo M, Younkin SG, Jakobsdóttir J, Kauwe JSK, Wilhelmsen KC, Rujescu D, Nöthen MM, Hofman A, Jones L, IGAP Consortium, Haines JL, Psaty BM, Van Broeckhoven C, Holmans P, Launer LJ, Mayeux R, Lathrop M, Goate AM, Escott-Price V, Seshadri S, Pericak-Vance MA, Amouyel P, Williams J, van Duijn CM, Schellenberg GD, Farrer LA, 2016. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 21, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M, 2005. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am. J. Hum. Genet 76, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Lang AE, Hazrati L-N, Fujioka S, Wszolek ZK, Dickson DW, Ross OA, Van Deerlin VM, Trojanowski JQ, Hurtig HI, Alcalay RN, Marder KS, Clark LN, Gaig C, Tolosa E, Ruiz-Martínez J, Marti-Masso JF, Ferrer I, López de Munain A, Goldman SM, Schüle B, Langston JW, Aasly JO, Giordana MT, Bonifati V, Puschmann A, Canesi M, Pezzoli G, Maues De Paula A, Hasegawa K, Duyckaerts C, Brice A, Stoessl AJ, Marras C, 2015. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 72, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ, 2014. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol 205, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay DM, Moran D, Moses L, Poorkaj P, Zabetian CP, Nutt J, Factor SA, Yu C-E, Montimurro JS, Keefe RG, Schellenberg GD, Payami H, 2007. Heterozygous parkin point mutations are as common in control subjects as in Parkinson’s patients. Ann. Neurol 61, 47–54. [DOI] [PubMed] [Google Scholar]
- Khan NL, Brooks DJ, Pavese N, Sweeney MG, Wood NW, Lees AJ, Piccini P, 2002. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain 125, 2248–2256. [DOI] [PubMed] [Google Scholar]
- Khan NL, Scherfler C, Graham E, Bhatia KP, Quinn N, Lees AJ, Brooks DJ, Wood NW, Piccini P, 2005. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology 64, 134–136. [DOI] [PubMed] [Google Scholar]
- Kilarski LL, Pearson JP, Newsway V, Majounie E, Knipe MDW, Misbahuddin A, Chinnery PF, Burn DJ, Clarke CE, Marion M-H, Lewthwaite AJ, Nicholl DJ, Wood NW, Morrison KE, Williams-Gray CH, Evans JR, Sawcer SJ, Barker RA, Wickremaratchi MM, Ben-Shlomo Y, Williams NM, Morris HR, 2012. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Mov. Disord 27, 1522–1529. [DOI] [PubMed] [Google Scholar]
- Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE, 2007. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol 6, 652–662. [DOI] [PubMed] [Google Scholar]
- Klein C, Pramstaller PP, Kis B, Page CC, Kann M, Leung J, Woodward H, Castellan CC, Scherer M, Vieregge P, Breakefield XO, Kramer PL, Ozelius LJ, 2000. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann. Neurol 48, 65–71. [PubMed] [Google Scholar]
- Klein C, Westenberger A, 2012. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med 2, a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O, 1998. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Lai MC, Bechy A-L, Denk F, Collins E, Gavriliouk M, Zaugg JB, Ryan BJ, Wade-Martins R, Caffrey TM, 2017. Haplotype-specific MAPT exon 3 expression regulated by common intronic polymorphisms associated with Parkinsonian disorders. Mol. Neurodegener 12, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, 2006. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann. Neurol 59, 591–596. [DOI] [PubMed] [Google Scholar]
- Lee AJ, Wang Y, Alcalay RN, Mejia-Santana H, Saunders-Pullman R, Bressman S, Corvol J-C, Brice A, Lesage S, Mangone G, Tolosa E, Pont-Sunyer C, Vilas D, Schüle B, Kausar F, Foroud T, Berg D, Brockmann K, Goldwurm S, Siri C, Asselta R, Ruiz-Martinez J, Mondragón E, Marras C, Ghate T, Giladi N, Mirelman A, Marder K, Michael J Fox LRRK2 Cohort Consortium, 2017. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov. Disord 32, 1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy E, Anastasopoulos D, Konitsiotis S, Lavedan C, Polymeropoulos MH, 1998. Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson’s disease. Hum. Genet 103, 424–427. [DOI] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, Verny C, Brice A, French Parkinson’s Disease Genetics Study Group, 2013. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol 73, 459–471. [DOI] [PubMed] [Google Scholar]
- Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S, Erpapazoglou Z, Usenko T, Maurage C-A, Sahbatou M, Liebau S, Ding J, Bilgic B, Emre M, Erginel-Unaltuna N, Guven G, Tison F, Tranchant C, Vidailhet M, Corvol J-C, Krack P, Leutenegger A-L, Nalls MA, Hernandez DG, Heutink P, Gibbs JR, Hardy J, Wood NW, Gasser T, Durr A, Deleuze J-F, Tazir M, Destée A, Lohmann E, Kabashi E, Singleton A, Corti O, Brice A, French Parkinson’s Disease Genetics Study (PDG), International Parkinson’s Disease Genomics Consortium (IPDGC), 2016. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet 98, 500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M, Leutenegger A-L, Guimaraes J, Bonnet A-M, Agid Y, Dürr A, Brice A, French Parkinson’s Disease Genetics Study Group, 2005. G2019S LRRK2 mutation in French and North African families with Parkinson’s disease. Ann. Neurol 58, 784–787. [DOI] [PubMed] [Google Scholar]
- Lesage S, Lohmann E, Tison F, Durif F, Dürr A, Brice A, French Parkinson’s Disease Genetics Study Group, 2008. Rare heterozygous parkin variants in French early-onset Parkinson disease patients and controls. J. Med. Genet 45, 43–46. [DOI] [PubMed] [Google Scholar]
- Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR, 2007. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem. Biophys. Res. Commun 357, 668–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln SJ, Maraganore DM, Lesnick TG, Bounds R, de Andrade M, Bower JH, Hardy JA, Farrer MJ, 2003. Parkin variants in North American Parkinson’s disease: cases and controls. Mov. Disord 18, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denèfle P, Wood NW, Agid Y, Brice A, French Parkinson’s Disease Genetics Study Group, European Consortium on Genetic Susceptibility in Parkinson’s Disease, 2000. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med 342, 1560–1567. [DOI] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin M-C, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert J-C, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan E-K, Van Broeckhoven C, Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium, 2006. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 296, 661–670. [DOI] [PubMed] [Google Scholar]
- Marongiu R, Ferraris A, Ialongo T, Michiorri S, Soleti F, Ferrari F, Elia AE, Ghezzi D, Albanese A, Altavista MC, Antonini A, Barone P, Brusa L, Cortelli P, Martinelli P, Pellecchia MT, Pezzoli G, Scaglione C, Stanzione P, Tinazzi M, Zecchinelli A, Zeviani M, Cassetta E, Garavaglia B, Dallapiccola B, Bentivoglio AR, Valente EM, Italian PD Study Group, 2008. PINK1 heterozygous rare variants: prevalence, significance and phenotypic spectrum. Hum. Mutat 29, 565. [DOI] [PubMed] [Google Scholar]
- Martí-Massó JF, Bergareche A, Makarov V, Ruiz-Martinez J, Gorostidi A, López de Munain A, Poza JJ, Striano P, Buxbaum JD, Paisán-Ruiz C, 2013. The ACMSD gene, involved in tryptophan metabolism, is mutated in a family with cortical myoclonus, epilepsy, and parkinsonism. J. Mol. Med 91, 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, Hirschhorn JN, 2008. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet 9, 356–369. [DOI] [PubMed] [Google Scholar]
- Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, Bras J, Hersheson J, Stamelou M, Pittman AM, Noyce AJ, Mok KY, Opladen T, Kunstmann E, Hodecker S, Münchau A, Volkmann J, Samnick S, Sidle K, Nanji T, Sweeney MG, Houlden H, Batla A, Zecchinelli AL, Pezzoli G, Marotta G, Lees A, Alegria P, Krack P, Cormier-Dequaire F, Lesage S, Brice A, Heutink P, Gasser T, Lubbe SJ, Morris HR, Taba P, Koks S, Majounie E, Raphael Gibbs J., Singleton A, Hardy J, Klebe S, Bhatia KP, Wood NW, International Parkinson’s Disease Genomics Consortium and UCL-exomes consortium, 2014. Parkinson’s disease in GTP cyclohydrolase 1 mutation carriers. Brain 137, 2480–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montfort M, Chabás A, Vilageliu L, Grinberg D, 2004. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: Pathogenic changes and “modifier” polymorphisms. Hum. Mutat 23, 567–575. [DOI] [PubMed] [Google Scholar]
- Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y, 1998. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51, 890–892. [DOI] [PubMed] [Google Scholar]
- Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, Lees A, Leung D, McKeith IG, Perry RH, Morris CM, Trojanowski JQ, Clark C, Karlawish J, Arnold S, Forman MS, Van Deerlin V, de Silva R, Hardy J, 2007. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol. Dis 25, 561–570. [DOI] [PubMed] [Google Scholar]
- Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, Morris CM, Theuns J, Crosiers D, Cras P, Engelborghs S, De Deyn PP, Van Broeckhoven C, Mann DMA, Snowden J, Pickering-Brown S, Halliwell N, Davidson Y, Gibbons L, Harris J, Sheerin U-M, Bras J, Hardy J, Clark L, Marder K, Honig LS, Berg D, Maetzler W, Brockmann K, Gasser T, Novellino F, Quattrone A, Annesi G, De Marco EV, Rogaeva E, Masellis M, Black SE, Bilbao JM, Foroud T, Ghetti B, Nichols WC, Pankratz N, Halliday G, Lesage S, Klebe S, Durr A, Duyckaerts C, Brice A, Giasson BI, Trojanowski JQ, Hurtig HI, Tayebi N, Landazabal C, Knight MA, Keller M, Singleton AB, Wolfsberg TG, Sidransky E, 2013. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, International Parkinson’s Disease Genomics Consortium (IPDGC), Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI), 23andMe, GenePD, NeuroGenetics Research Consortium (NGRC), Hussman Institute of Human Genomics (HIHG), Ashkenazi Jewish Dataset Investigator, Cohorts for Health and Aging Research in Genetic Epidemiology (CHARGE), North American Brain Expression Consortium (NABEC), United Kingdom Brain Expression Consortium (UKBEC), Greek Parkinson’s Disease Consortium, Alzheimer Genetic Analysis Group, Ikram MA, Ioannidis JPA, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB, 2014. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simon-Sanchez J, Schulte C, Sharma M, Krohn L, Pihlstrom L, Siitonen A, Iwaki H, Leonard H, Faghri F, Raphael Gibbs J., Hernandez DG, Scholz SW, Botia JA, Martinez M, Corvol J-C, Lesage S, Jankovic J, Shulman LM, Sutherland M, Tienari P, Majamaa K, Toft M, Brice A, Yang J, Gan-Orr Z, Gasser TM, Heutink PM, Shulman JM, Wood NA, Hinds DA, Hardy JR, Morris HR, Gratten JM, Visscher PM, Graham RR, Singleton AB, The 23andMe Research Team, System Genomics of Parkinson’s Disease (SGPD) Consortium, International Parkinson’s Disease Genomics Consortium, 2018. Parkinson’s disease genetics: identifying novel risk loci, providing causal insights and improving estimates of heritable risk 10.1101/388165 [DOI]
- Nichols WC, Pankratz N, Hernandez D, Paisán-Ruíz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, Singleton A, Foroud T, Parkinson Study Group-PROGENI investigators, 2005. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 365, 410–412. [DOI] [PubMed] [Google Scholar]
- Nixon-Abell J, Berwick DC, Grannó S, Spain VA, Blackstone C, Harvey K, 2016. Protective LRRK2 R1398H Variant Enhances GTPase and Wnt Signaling Activity. Front. Mol. Neurosci 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB, 2006. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med 354, 424–425. [DOI] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB, 2004. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, Paetau A, 2014. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35, 2180 e1–5. [DOI] [PubMed] [Google Scholar]
- Pihlstrøm L, Blauwendraat C, Cappelletti C, Berge-Seidl V, Langmyhr M, Henriksen SP, van de Berg WDJ, Gibbs JR, Cookson MR, International Parkinson Disease Genomics Consortium, North American Brain Expression Consortium, Singleton AB, Nalls MA, Toft M, 2018. A comprehensive analysis of SNCA-related genetic risk in sporadic parkinson disease. Ann. Neurol 84, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL, 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, Koller WC, Bird TD, Trojanowski JQ, Lee VM-Y, Schellenberg GD, 2002. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann. Neurol 52, 511–516. [DOI] [PubMed] [Google Scholar]
- Poulopoulos M, Levy OA, Alcalay RN, 2012. The neuropathology of genetic Parkinson’s disease. Mov. Disord 27, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puschmann A, Fiesel FC, Caulfield TR, Hudec R, Ando M, Truban D, Hou X, Ogaki K, Heckman MG, James ED, Swanberg M, Jimenez-Ferrer I, Hansson O, Opala G, Siuda J, Boczarska-Jedynak M, Friedman A, Koziorowski D, Aasly JO, Lynch T, Mellick GD, Mohan M, Silburn PA, Sanotsky Y, Vilariño-Güell C, Farrer MJ, Chen L, Dawson VL, Dawson TM, Wszolek ZK, Ross OA, Springer W, 2017. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 140, 98–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, De T, UK Brain Expression Consortium, North American Brain Expression Consortium, Coin L, de Silva R, Cookson MR, Singleton AB, Hardy J, Ryten M, Weale ME, 2014. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci 17, 1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, International Parkinson’s Disease Genomics Consortium (IPDGC), Heutink P, Shulman JM, 2017. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robakis D, Cortes E, Clark LN, Vonsattel JPG, Virmani T, Alcalay RN, Crary JF, Levy OA, 2016. The effect of MAPT haplotype on neocortical Lewy body pathology in Parkinson disease. J. Neural Transm 123, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, Johnson J, Lang AE, Gulick C, Gwinn-Hardy K, Kawarai T, Sato C, Morgan A, Werner J, Nussbaum R, Petit A, Okun MS, McInerney A, Mandel R, Groen JL, Fernandez HH, Postuma R, Foote KD, Salehi-Rad S, Liang Y, Reimsnider S, Tandon A, Hardy J, St George-Hyslop P, Singleton AB, 2004. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch. Neurol 61, 1898–1904. [DOI] [PubMed] [Google Scholar]
- Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, Bacon JA, Bardien S, Bozi M, Brice A, Brighina L, Van Broeckhoven C, Carr J, Chartier-Harlin M-C, Dardiotis E, Dickson DW, Diehl NN, Elbaz A, Ferrarese C, Ferraris A, Fiske B, Gibson JM, Gibson R, Hadjigeorgiou GM, Hattori N, Ioannidis JPA, Jasinska-Myga B, Jeon BS, Kim YJ, Klein C, Kruger R, Kyratzi E, Lesage S, Lin C-H, Lynch T, Maraganore DM, Mellick GD, Mutez E, Nilsson C, Opala G, Park SS, Puschmann A, Quattrone A, Sharma M, Silburn PA, Sohn YH, Stefanis L, Tadic V, Theuns J, Tomiyama H, Uitti RJ, Valente EM, van de Loo S, Vassilatis DK, Vilariño-Güell C, White LR, Wirdefeldt K, Wszolek ZK, Wu R-M, Farrer MJ, Genetic Epidemiology Of Parkinson’s Disease (GEO-PD) Consortium, 2011. Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case-control study. Lancet Neurol 10, 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko IN, Kaganovich A, Hauser DN, Beylina A, Chia R, Ding J, Maric D, Jaffe H, Cookson MR, 2012. The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem. J 446, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko IN, Kaganovich A, Langston RG, Beilina A, Ndukwe K, Kumaran R, Dillman AA, Chia R, Cookson MR, 2017. The G2385R risk factor for Parkinson’s disease enhances CHIP-dependent intracellular degradation of LRRK2. Biochem. J 474, 1547–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Martínez J, Azcona LJ, Bergareche A, Martí-Massó JF, Paisán-Ruiz C, 2017. Whole-exome sequencing associates novel gene mutations with familial Parkinson disease. Neurol Genet 3, e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Martínez J, Gorostidi A, Ibañez B, Alzualde A, Otaegui D, Moreno F, López de Munain A, Bergareche A, Gómez-Esteban JC, Martí Massó JF, 2010. Penetrance in Parkinson’s disease related to the LRRK2 R1441G mutation in the Basque country (Spain). Mov. Disord 25, 2340–2345. [DOI] [PubMed] [Google Scholar]
- Ryan KJ, White CC, Patel K, Xu J, Olah M, Replogle JM, Frangieh M, Cimpean M, Winn P, McHenry A, Kaskow BJ, Chan G, Cuerdon N, Bennett DA, Boyd JD, Imitola J, Elyaman W, De Jager PL, Bradshaw EM, 2017. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl. Med 9 10.1126/scitranslmed.aai7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J, Pastor P, 2010. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133, 1128–1142. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shirata A, Yamane K, Iwata M, 2004. Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 63, 678–682. [DOI] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T, 2009. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet 41, 1303–1307. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen C-M, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung H-C, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen G-J, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan E-K, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu Y-R, Zabetian CP, Zhao Y, Ziegler SG, 2009. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med 361, 1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T, 2009. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, 2003. -Synuclein Locus Triplication Causes Parkinson’s Disease. Science 302, 841–841. [DOI] [PubMed] [Google Scholar]
- Smith DI, Zhu Y, McAvoy S, Kuhn R, 2006. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett 232, 48–57. [DOI] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R, 2016. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B, 1998. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U. S. A 95, 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, 1997. Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M, 2016. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5 10.7554/eLife.12813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L, Malig M, Scheinfeldt L, Beggs W, Ibrahim M, Lema G, Nyambo TB, Omar SA, Bodo J-M, Froment A, Donnelly MP, Kidd KK, Tishkoff SA, Eichler EE, 2012. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat. Genet 44, 872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Parsian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, Labelle N, Growdon JH, Singer C, Watts RL, Goldwurm S, Pezzoli G, Baker KB, Pramstaller PP, Burn DJ, Chinnery PF, Sherman S, Vieregge P, Litvan I, Gillis T, MacDonald ME, Myers RH, Gusella JF, 2006. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch. Neurol 63, 826–832. [DOI] [PubMed] [Google Scholar]
- Taghavi S, Chaouni R, Tafakhori A, Azcona LJ, Firouzabadi SG, Omrani MD, Jamshidi J, Emamalizadeh B, Shahidi GA, Ahmadi M, Habibi SAH, Ahmadifard A, Fazeli A, Motallebi M, Petramfar P, Askarpour S, Askarpour S, Shahmohammadibeni HA, Shahmohammadibeni N, Eftekhari H, Shafiei Zarneh A.E., Mohammadihosseinabad S, Khorrami M, Najmi S, Chitsaz A, Shokraeian P, Ehsanbakhsh H, Rezaeidian J, Ebrahimi Rad R, Madadi F, Andarva M, Alehabib E, Atakhorrami M, Mortazavi SE, Azimzadeh Z, Bayat M, Besharati AM, Harati-Ghavi MA, Omidvari S, Dehghani-Tafti Z, Mohammadi F, Mohammad Hossein Pour B, Noorollahi Moghaddam H., Esmaili Shandiz E., Habibi A, Taherian-Esfahani Z, Darvish H, Paisán-Ruiz C, 2017. A Clinical and Molecular Genetic Study of 50 Families with Autosomal Recessive Parkinsonism Revealed Known and Novel Gene Mutations. Mol. Neurobiol 10.1007/s12035-017-0535-1 [DOI] [PMC free article] [PubMed]
- Tambasco N, Nigro P, Romoli M, Prontera P, Simoni S, Calabresi P, 2016. A53T in a parkinsonian family: a clinical update of the SNCA phenotypes. J. Neural Transm 123, 1301–1307. [DOI] [PubMed] [Google Scholar]
- Tan E-K, Peng R, Teo Y-Y, Tan LC, Angeles D, Ho P, Chen M-L, Lin C-H, Mao X-Y, Chang X-L, Prakash KM, Liu J-J, Au W-L, Le W-D, Jankovic J, Burgunder J-M, Zhao Y, Wu R-M, 2010. Multiple LRRK2 variants modulate risk of Parkinson disease: a Chinese multicenter study. Hum. Mutat 31, 561–568. [DOI] [PubMed] [Google Scholar]
- Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, Buchman AS, Larson EB, Crane PK, Kaye JA, Kramer P, Woltjer R, Kukull W, Nelson PT, Jicha GA, Neltner JH, Galasko D, Masliah E, Trojanowski JQ, Schellenberg GD, Yearout D, Huston H, Fritts-Penniman A, Mata IF, Wan JY, Edwards KL, Montine TJ, Zabetian CP, 2012. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 79, 1944–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA, Ginns EI, 1987. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N. Engl. J. Med 316, 570–575. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW, 2004a. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW, 2001. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet 68, 895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Ferraris A, Graham EA, Davis MB, Breteler MMB, Gasser T, Bonifati V, Bentivoglio AR, De Michele G, Dürr A, Cortelli P, Wassilowsky D, Harhangi BS, Rawal N, Caputo V, Filla A, Meco G, Oostra BA, Brice A, Albanese A, Dallapiccola B, Wood NW, European Consortium on Genetic Susceptibility in Parkinson’s Disease, 2002. PARK6-linked parkinsonism occurs in several European families. Ann. Neurol 51, 14–18. [PubMed] [Google Scholar]
- Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR, 2004b. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol 56, 336–341. [DOI] [PubMed] [Google Scholar]
- van de Warrenburg BP, Lammens M, Lücking CB, Denèfle P, Wesseling P, Booij J, Praamstra P, Quinn N, Brice A, Horstink MW, 2001. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 56, 555–557. [DOI] [PubMed] [Google Scholar]
- Verpillat P, Camuzat A, Hannequin D, Thomas-Anterion C, Puel M, Belliard S, Dubois B, Didic M, Michel B-F, Lacomblez L, Moreaud O, Sellal F, Golfier V, Campion D, Clerget-Darpoux F, Brice A, 2002. Association between the extended tau haplotype and frontotemporal dementia. Arch. Neurol 59, 935–939. [DOI] [PubMed] [Google Scholar]
- Vilas D, Fernández-Santiago R, Sanchez E, Azcona LJ, Santos-Montes M, Casquero P, Argandoña L, Tolosa E, Paisán-Ruiz C, 2017. A Novel p.Glu298Lys Mutation in the ACMSD Gene in Sporadic Parkinson’s Disease. J. Parkinsons. Dis 7, 459–463. [DOI] [PubMed] [Google Scholar]
- Wang L, Nuytemans K, Bademci G, Jauregui C, Martin ER, Scott WK, Vance JM, Zuchner S, 2013. High-resolution survey in familial Parkinson disease genes reveals multiple independent copy number variation events in PARK2. Hum. Mutat 34, 1071–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Hattori N, Matsumine H, Kobayashi T, Yoshino H, Morioka A, Kitada T, Asakawa S, Minoshima S, Shimizu N, Mizuno Y, 1999. Polymorphism in the parkin gene in sporadic Parkinson’s disease. Ann. Neurol 45, 655–658. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM, 2005. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U. S. A 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim K-L, Dawson VL, Dawson TM, 2007. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet 16, 223–232. [DOI] [PubMed] [Google Scholar]
- Wszolek ZK, Tsuboi Y, Ghetti B, Pickering-Brown S, Baba Y, Cheshire WP, 2006. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet J. Rare Dis 1, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E., del Ser T, Muñoz DG, de Yebenes JG, 2004. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol 55, 164–173. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T, 2004. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]