

Abstract
Clinical practice guidelines for Wilson's disease (WD) have been published by the American Association for the Study of Liver Diseases and European Association for the Study of the Liver in 2008 and 2012, respectively. Their focus was on the hepatic aspects of the disease. Recently, a position paper on pediatric WD was published by the European Society of Pediatric Gastroenterology Hepatology and Nutrition. A need was felt to harmonize guidelines for the hepatic, pediatric, and neurological aspects of the disease and contextualize them to the resource-constrained settings. Therefore, experts from national societies from India representing 3 disciplines, hepatology (Indian National Association for Study of the Liver), pediatric hepatology (Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition), and neurology (Movement Disorders Society of India) got together to evolve fresh guidelines. A literature search on retrospective and prospective studies of WD using MEDLINE (PubMed) was performed. Members voted on each recommendation, using the nominal voting technique. The Grades of Recommendation, Assessment, Development and Evaluation system was used to determine the quality of evidence. Questions related to diagnostic tests, scoring system, and its modification to a version suitable for resource-constrained settings were posed. While ceruloplasmin and 24-h urine copper continue to be important, there is little role of serum copper and penicillamine challenge test in the diagnostic algorithm. A new scoring system – Modified Leipzig score has been suggested with extra points being added for family history and serum ceruloplasmin lower than 5 mg/dl. Liver dry copper estimation and penicillamine challenge test have been removed from the scoring system. Differences in pharmacological approach to neurological and hepatic disease and global monitoring scales have been included. Rising bilirubin and worsening encephalopathy are suggested as indicators predicting need for liver transplant but need to be validated. The clinical practice guidelines provide recommendations for a comprehensive management of WD which will be of value to all specialties.
Keywords: Wilson's disease scoring, modified Leipzig scoring, rare disease, genetic disorder
Abbreviations: AASLD, American Association for the Study of Liver Diseases; ACLF, Acute on Chronic Liver Failure; ALF, Acute Liver Failure; ALT, Alanine Transaminase; AST, Aspartate Transaminase; Cu, Copper; DP, D-Penicillamine; EASL, European Association for the Study of the Liver; HCC, Hepatocellular Carcinoma; GAS for WD, Global Assessment Scale for Wilson's Disease; INR, International Normalized Ratio; KF, Kayser-Fleischer; LT, Liver Transplantation; MARS, Molecular Absorption Recirculating System; MELD, Model for End-Stage Liver Disease; MRI, Magnetic Resonance Imaging; NGS, Next-Generation Sequencing; NWI, New Wilson's Index; PELD, Pediatric end stage liver disease; TPE, Total Plasma Exchange; TTM, Tetrathiomolybdate; WD, Wilson's Disease
This document is the outcome of a consensus meeting in March 2017 of experts from the Indian National Association for Study of the Liver, the Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition, and the Movement Disorders Society of India, which represent the major subspecialties involved in the care of Wilson's disease (WD). The need for this was felt as most previous guidelines have either focused on the hepatic aspects (American Association for the Study of Liver Diseases [AASLD] and European Association for the Study of the Liver [EASL])1, 2 or pediatric aspects (European society of Pediatric Gastroenterology Hepatology and Nutrition)3 of the disease. Participants at the meeting presented recommendations on specific areas of the disease, and then, members voted on them using the nominal voting technique as per the standard guidelines.4 The quality of evidence was adapted from the Grades of Recommendation, Assessment, Development and Evaluation system.5 The final recommendations were then circulated to all the core group members and updated based on systematic literature search. These guidelines are thus comprehensive and cover all aspects of WD. They also reflect the challenges faced by clinicians in resource-limited settings.
Historical perspective
The first possible case of WD was described by Frerichs (1861) in a 9-year-old boy with movement and speech abnormalities, whose autopsy showed liver cirrhosis. Kayser (1902) followed by Fleischer (1903) reported a greenish-brown ring around cornea in patients of suspected multiple sclerosis. Wilson (1911), described the disease as familial “progressive lenticular degeneration” in association with cirrhosis of the liver and Hall (1921) coined the term “hepatolenticular degeneration,” while Umpel (1913) demonstrated increased copper (Cu) in the liver and basal ganglia in patients with WD. Subsequently, Mandelbrot (1948), Scheinberg-Gitlin (1952), and Cartwright (1954) reported increased urinary copper, decreased level of ceruloplasmin, and increased serum free copper, respectively in WD. Bearn (1957) first postulated that it was a genetically determined metabolic disease, and Walshe (1956)6 demonstrated treatment success with D-penicillamine (DP) (1956) and trientene (1969).7 Brewer first documented the use of zinc and tetrathiomolybdate (TTM) in neuro-WD.8, 9
Epidemiology
The World Health Organization estimates that the global prevalence of WD is 1/10,000 to 1/30,000.10 Clinical prevalence in West was estimated 5 decades ago as 5 per million.11 Since then, it has steadily increased to approximately 142 per million by modern genetic testing. Some parts of Europe such as Romania and Sardinia report highest prevalence (370–885 per million) with 6 mutations accounting for 85% of their cohort.12 Sequencing of ATP7B in 1000 control participants in the UK allowed the frequency of an individual carrying 2 mutant ATP7B alleles to be estimated at 1/7026. In China, the prevalence is estimated as 5.87 per 100,000. Newborn screening programs show disappointing results. Screening by serum ceruloplasmin in children aged 6 months to 9 years yield prevalence of 124 per million in Japan. Screening programs recommend 3 years and above as opportune time to detect the disease.13 In India, there are no community-based incidence and prevalence studies of WD. Among pediatric liver diseases, WD accounts for 7.6–19.7% in tertiary hepatobiliary centers. Fifteen to 20 new cases of WD are registered annually in referral neurology centers.14
Normal copper fluxes and pools
The body contains 110 mg of Cu, predominantly in the muscles (28 mg), bones (46 mg), and connective tissues.15, 16 The Cu pool in the musculoskeletal system is in constant exchange with plasma. Plasma contains approximately 1 mcg/mL, of which 60–95% bound to ceruloplasmin. Ceruloplasmin is a source of Cu for peripheral organs, where Cu is an essential cofactor for many enzymes.
The copper circulation is explained in Figure 1. Normal dietary Cu intake is 1.5–5 mg in 24 h, 50–60% of which is unabsorbed and excreted in feces; 25–40% is absorbed from the duodenum, stored by enterocytes and bound to metallothioneins in a nontoxic form. From this intestinal pool, 75% flows through the portal system with albumin or transcuprein and is taken up by the liver. The remaining 25% is bound to albumin in the circulation. In the liver, 20% of Cu is re-excreted back into the gastrointestinal tract through bile and 80% is transported to the periphery, bound to ceruloplasmin. The biliary excretion is approximately 2.5 mg/d. Near-similar amounts are excreted from other secretions (saliva, gastric, pancreatic, and intestinal fluid). These are the endogenous Cu excretions, and a large proportion (approximately 80%) is again reabsorbed by the intestinal mucosa. When copper is deficient in the diet, there is enhanced affinity of metallothioneins in enterocytes for copper, thus increasing its absorption and vice versa. Thus, the daily fecal losses are a combination of unabsorbed dietary Cu and small proportion of excreted endogenous Cu amounting to approximately 1.5–4 mg/d. Compared to fecal excretion, urinary Cu excretion is low (10–100 μg/d). The effect of dietary Cu level on urinary Cu excretion is inconsistent, with some studies reporting a small but significant positive relationship while others showing no effect.
Figure 1.

Circulation of copper in the body.
Pathogenesis
WD is an autosomal recessive disorder that affects the ATP7B gene (chromosome 13q) expressed in hepatocytes, kidneys, and placenta which encodes for the copper transporting P-type ATPase or copper translocase ATP7B.17, 18, 19 ATP7B helps in transport of copper to the trans-Golgi network and the biliary excretion of copper. Antioxidant protein 1, a copper chaperone, helps deliver copper to the 6 copper-binding domains in ATP7B. Binding of copper to ATP7B causes adenosine tri phosphate (ATP) hydrolysis, which supplies energy for the transport of copper to the lysosome. Here, copper is incorporated into ceruloplasmin, which is then released in the circulation. Levels of copper in the hepatocytes regulate the intracellular distribution and function of ATP7B. With normal copper levels, ATP7B helps in synthesis of cuproproteins such as ceruloplasmin. Apo-ceruloplasmin (copper-free ceruloplasmin) is less stable in circulation than holo-ceruloplasmin (copper-bound ceruloplasmin). When intracellular copper is in excess, ATP7B facilitates its excretion into bile by exocytosis. Mutations in ATP7B result in decreased synthesis of copper-bound ceruloplasmin, impaired excretion of copper, and increased cytosolic, mitochondrial, and nuclear levels of copper.
Copper-Induced Liver Injury
Although not yet fully understood, a few possible mechanisms have been postulated.18, 19 Increased intracellular copper induces oxidative stress leading to the production of hydroxyl radicals and reduced superoxide dismutase and glutathione which leads to damage of cellular lipids, proteins, and nucleic acids. Cardiolipin on the mitochondrial membrane has been shown to be fragmented by copper-induced oxidative stress. Reversible mitochondrial recovery by chelation therapy suggests that the initial damage was possibly due to multivalent interactions/multiprotein cross-linking in intermembrane space and condensations within mitochondria and various other organelles. The molecular reasons for variability of clinical symptoms and lack of genotype-phenotype relationship in WD are not yet fully elucidated. Genotype variations, gene–environment interactions, and altered activity of other modifier genes have been implicated.
Clinical features
Clinical features depend on the predominant organ involved (mainly liver and brain), and the disease has been reported from 3 to 85 years of age. Although copper starts accumulating soon after birth, the disease takes at least 3 years to manifest. Symptoms depend on the site of deposition of copper in the body. Walshe and Yealland20 demonstrated an age-phenotypic presentation. Hepatic presentation was seen in younger age groups (<10 years: 83%; 10–18 years: 52%; >18 years: 24%), while neuropsychiatric presentation increased as age advanced (<10 years: 17%; 10–18 years: 48%; >18 years: 74%). Symptom onset to diagnosis was shorter in hepatic (6 months) than neuropsychiatric presentation (18 months). Studies in children from India and Egypt have shown isolated hepatic involvement (20–54%), isolated neurological (8–22%), neurohepatic (11–36%), asymptomatic (15–35%), and other manifestations (0–22%).21, 22, 23
The clinical manifestations are summarized in Table 1, while the prevalence from different centers is shown in Table 2.
Table 1.
Clinical Manifestations of Wilson's Disease.
| Hepatic | Asymptomatic hepatomegaly, persistently elevated transaminases, acute hepatitis, chronic hepatitis, cirrhosis (compensated and decompensated), acute liver failure, acute on chronic liver failure, fatty liver, isolated splenomegaly, cholelithiasis |
| Neuropsychiatric | Tremors, dystonia, Parkinsonism, choreoathetosis, seizures, dysarthria, drooling, clumsiness, incoordination, gait disturbance, behavioral changes, deteriorating school performance, depression, anxiety, psychosis |
| Osseomuscular | Arthralgia, arthritis, fractures, osteoporosis, osteomalacia, chondromalacia |
| Hematological | Hemolytic anemia, thrombocytopenia, pancytopenia, coagulopathy |
| Ocular | Kayser-Fleischer rings, sunflower cataracts |
| Renal | Renal stones, renal tubular acidosis, Fanconi syndrome |
Table 2.
Studies With Prevalence of Various Clinical Manifestations of Wilson's Disease.
| Study | Hepatic | Neuropsychiatric | Presymptomatic | Other |
|---|---|---|---|---|
| Lee et al (n = 245)69 | 134 (54.8%) | 55 (22.4%) | 55 (22.4%) | 1 (0.4%) (osseomuscular) |
| Taly et al (n = 282)42 | 42 (15%) | 219 (77.6%) Neuro, 195; hepato-neuro, 10; psychiatric, 7; others, 7 |
15 (5.4%) | 6 (2%) (osseomuscular) |
| Walshe (n = 217)40 | 94 (43.1%) | 97 (44%) | 24 (11%) | 2 (0.9%) (osseomuscular) |
| Cheng et al (n = 1222)70 | 450 (37%) | 592 (48.5%) | 31 (2.5%) | 149 (12%) (neurovisceral) |
Hepatic Manifestation
Liver is the first organ to be involved because of the accumulation of copper in WD predominantly manifesting in childhood and is virtually involved in all cases of WD.24 The symptoms related to liver involvement are quite varied and include.
Asymptomatic WD
Asymptomatic WD has been documented in 3–40% patients in various studies.25, 26 These patients have incidentally detected hepatomegaly, raised transaminases, or are siblings of the index WD patients detected on screening. A majority of these cases are identified in first decade of life or in adolescence.
Acute Hepatitis
Acute hepatitis (10–25%)22, 27 mimics acute viral hepatitis, autoimmune hepatitis, and drug-induced liver injury. Jaundice, anorexia, nausea, malaise, fever, pale stools, and pain in the abdomen are often the predominant symptoms. The biochemical investigations show conjugated hyperbilirubinemia, raised transaminases, and normal or marginally low synthetic functions. It is important to consider WD in older children and adolescents with seronegative acute hepatitis.
Acute Liver Failure
Acute liver failure (ALF) (8–20%)22, 28 is predominantly seen in childhood and adolescents. It is usually associated with Coombs-negative non immune intravascular hemolysis.29 The presentation mimics acute hepatitis, but the condition deteriorates rapidly over days to weeks and is often fatal. It results in deep jaundice, hemolysis, coagulopathy, ascites, encephalopathy, and renal failure. The investigations show very high serum bilirubin, mild-to-moderate rise of liver enzymes, low serum alkaline phosphatase, low serum uric acid, and defective synthetic functions. This phenomenon has also been reported in patients who have stopped the chelation therapy abruptly. The combination of the ratio of serum alkaline phosphatase/total bilirubin <4 and aspartate transminase:alanine transaminase (AST:ALT) ratio >2.2 has been described to have a diagnostic sensitivity and specificity of 100%.30 However, this has not been validated in subsequent studies.31
Acute on Chronic Liver Failure
In 2 studies on acute on chronic liver failure (ACLF) (11–55%)25, 32 from India, WD was the underlying chronic liver disease in 42–43% of patients with a superadded acute viral hepatitis.33, 34 Viral hepatitis is often the acute event that deteriorates underlying hepatic WD and is the first manifestation of the disease. There is a prodrome followed by deep jaundice, early-onset ascites, encephalopathy, and coagulopathy rapidly progressing to a multiorgan dysfunction.
Chronic Hepatitis
Chronic hepatitis (10–30%)35 is seen especially in adolescents and young adults. Nonspecific and constitutional symptoms such as fatigue, anorexia, nausea, and malaise may present before onset of jaundice and hepatic dysfunction. Associated delayed puberty, amenorrhea, polyarthralgia, may be present.
Cirrhosis
Patients may present with complications of cirrhosis (35%–60%),22, 25, 28 including ascites (spontaneous bacterial peritonitis), encephalopathy, and renal failure (including hepatorenal syndrome) or portal hypertension (variceal bleeding).36 In adults, although neurological manifestations may dominate the underlying liver is usually cirrhotic. Any young patient more than 3 years of age presenting with cirrhosis should be evaluated for WD as an underlying cause.
Fatty Liver
WD should be thought of as differential diagnosis in a child less than 10 years presenting with a fatty liver where nonalcoholic fatty liver disease is less likely. Various histological and histological series report incidence from 28% to 35.7%.37, 38, 39
Cholelithiasis
Chronic hemolysis leads to mixed gall stones which are cholesterol predominant but also pigmented and patients may present with symptoms/complications of gall stones.35 Copper content of the gall stone is low as these patients have defective excretion of copper into the bile.
Malignancies
The development of hepatocellular carcinoma (HCC) in patients with WD is rare compared with other causes of cirrhosis. Retrospective analysis of 363 patients of WD diagnosed in the UK and Sweden has shown that 4.2–5.3% develop HCC or cholangiocarcinoma over a 10–29 years and 15% over a 39-year follow-up.39
Neurological Manifestations
Most patients who present with central nervous system (CNS) manifestations have liver disease at the time of presentation, though not symptomatic. Contrary to the initial belief that hepatic form is the predominant presentation of WD, over the years published literature has noted that neurological manifestations tend to be more common at presentation, accounting to as high as 60%.40, 41, 42 However, there could be a center-oriented bias in reporting depending on the specialty a patient has been referred to. Patients with neurological presentations tend to be older (second/third decade) and usually have a Kayser-Fleischer (KF) ring (Figure 2). Early or subtle neurological manifestations in young children include deterioration of handwriting and school performance, dysarthria, and drooling of saliva. The classical dystonia involving the facial and mandible muscles produce a characteristic “Wilson's facies”. Wilson's facies is scored on the presence of vacuous smile, open mouth, hypersalivation, and dull look, a unique feature which gives an estimate of severity43 (Figure 3). The presenting neurological symptoms tend to be wide and variable. In a series of 307 patients from India,44 the common presenting symptoms were tremors (31.6%), dysarthria (15.6%), jaundice (12.4%), abnormal gait (8.8%), abdominal distention (7.8%), musculoskeletal symptoms (5.2%), seizures (4.9%), behavioral problems (4.6%), dystonia (3.6%), clumsiness (2.6%), drooling of saliva (2.6%), generalized weakness (2.3%), decreased scholastic performance (1.9%), changed sensorium (1.3%), bleeding symptoms (1.3%), dysphagia (0.9%), chorea (0.3%), and poor vision (0.3%). Over the course of disease, patients tend to develop varied combination of these presenting features.
Figure 2.
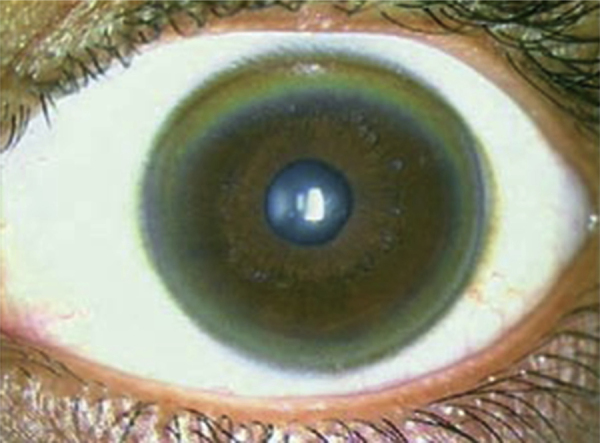
Golden-brown Kayser-Fleischer (KF) ring.
Figure 3.

Vacuous smile in Wilson's disease.
Primarily, the neurological form consists of extrapyramidal manifestations, but not exclusively limited to it.42 Broadly, the extrapyramidal neurological abnormalities can be classified as: (a) an akinetic-rigid syndrome similar to Parkinson's disease, (b) pseudosclerosis dominated by tremor, (c) ataxia, and (d) a dystonic syndrome. Other major neurological presentations include seizures and cognitive changes. Seizures are not uncommon and could occur at any stage of the disease with published frequency between 4.2% and 7.5%.42
Psycho-behavioral Manifestations
Psycho-behavioral issues are very common in WD and constitute almost for one-third of presenting symptoms. Almost all the patients will have some form of psychiatric symptoms during the course of their disease.45 Among the psycho-behavioral symptomatology, the following remain the core issues: organic dementia, psychosis, psychoneurosis, and behavioral disturbances characterized by impulsivity occasionally extending to unlawful behavior.46 Psychotic symptoms of WD are often undiagnosed and factors such as lack of awareness, failure to recognize coexisting neurological features, or misinterpretation of such symptoms as side effects of neuroleptic treatment are some of the possible cited reasons. The delay in diagnosis ranges from 1 to 5 years.47, 48 The reported incidence of psychiatric symptoms as the presenting manifestation varies from 2.4% to 20% and can be categorized into the following groups.49
Personality Changes
Personality changes include bizarre behavior, impulsivity occasionally extending to criminal behavior, disinhibition, irritability, emotionality, decreased threshold for anger, and aggression. Bizarre behavior and irritability are more often associated with bulbar and dystonic features rather than tremors.42, 49
Affective Disorders
Depression is the most common psychiatric manifestation and often co-occurs with neurological symptoms but not with hepatic disease. In the series by Shanmugiah et al,50 18% had bipolar affective disorder, 4% had major depression, and 2% had dysthymia. Other affective disorders such as hypomania and mania though rare have also been reported.
Psychosis
“Schizophrenia-like” and other forms of psychosis can rarely be the initial manifestation, but more commonly accompany other neurological features (16–51%).42, 45, 50, 51
Cognitive Impairment
Mental subnormality though not described as a feature of WD was found in 23% of patients with WD on neuropsychological and intellectual assessment.52
Others
Other psychiatric manifestations including substance abuse, catatonia, sexual preoccupation, and anxiety disorders have also been reported.53
Behavioral symptoms do respond to decoppering therapy, and few may require long-term symptomatic behavioral pharmacotherapies.
Ocular Manifestations
Copper accumulates in the Descemet's membrane of the cornea forming the KF ring, which is greenish-brown in color (Figure 2). They are always bilateral and are seen in 50–60% of hepatic and 95–100% cases of neurological WD.54 Although sometimes visible to the naked eye, a slit lamp examination is necessary for confirmation. They appear sequentially at the (upper > lower > medial > lateral) segment of limbus and on chelation therapy disappear in the reverse direction. KF rings may mimic bile pigment rings seen in the stromal layer of the cornea in advanced cholestasis and hence need to be confirmed by an experienced ophthalmologist especially if jaundice is present.55 In a study by Fenu et al,56 partial or total KF ring resolution was observed in 28%, deterioration in 6%, and static in the rest of the cohort over 1–3 years of therapy. After liver transplantation, a partial decrease in or complete disappearance of the KF ring has been documented.57
Sunflower cataract is uncommon (2–17%) and is due to copper deposition in the anterior capsule of the lens.58 These are always associated with KF rings, do not disturb vision, and disappear with chelation therapy.54 Abnormal oculomotor functions are frequently seen on electro-oculography in patients with WD but has doubtful clinical significance.
Renal Manifestations
Tubular injury (copper deposition in epithelium of proximal and distal convoluted tubules) occurs from WD (8%) but glomerular injury (10%) is usually a complication of chelation therapy.54 Tubular injury manifests as nephrocalcinosis (microscopic hematuria) and nephrolithiasis (renal colic). Although glomerular injury may be due to mesangial deposition of copper,59 it is more commonly related to the DP treatment. Hence, at presentation, all cases of WD should be screened for renal tubular dysfunction with a urine routine and microscopy, while on follow-up with chelators, proteinuria should be assessed to detect glomerular injury. In a study of 41 patients with WD (6–37 years of age) who were on DP treatment for 0–15 (mean 4.5) years, Sozeri and Feist60 have shown that 39% had significant proteinuria. Low-molecular-weight proteinuria was observed in the first 2 years of treatment, indicating early tubular damage, while high molecular proteinuria suggesting glomerular injury, persisted over longer periods. Children with tubular dysfunction need treatment with bicarbonate in addition to DP.61 DP needs to be discontinued (permanently) in those who develop glomerular injury especially if the proteinuria is in the nephrotic range.
Hematological Manifestations
These include Coombs-negative hemolytic anemia and thrombocytopenia with or without hemolysis.47, 54 The sudden release of excess free copper from the liver because of hepatocyte necrosis produce oxidative stress on red blood cells resulting in hemolysis. The hemolytic anemia may be may be mild with asymptomatic liver disease or an acute severe form heralding the development of ALF. Patients with advanced liver disease have abnormal coagulation profile and platelet dysfunction, while splenomegaly from portal hypertension can result in hypersplenism, but none of these are specific for WD.
Osseomuscular Manifestations
Osseomuscular symptoms may rarely be the presenting feature (2%)40, 42, or occur during the course of the disease in adults. Osteoporosis (24–88%), osteomalacia (14–35%), spontaneous fractures (9–35%), rickets, osteochondritis dissecans, chondromalacia patellae, premature osteopenia, and degenerative arthritis of knees and wrists have been reported in the second and third decade of life.62, 63 Although osteoporosis has been documented on densitometry in 43–67% of patients with WD,64 most patients are asymptomatic with only radiological abnormalities in the large joints. Except chelation, there is no other specific treatment available for osseomuscular disorders in WD. Paradoxically, DP itself can induce rheumatological disorders such as systemic lupus erythematosus, Goodpasture syndrome, myasthenia, dermatomyositis, and so forth.64 The cause for osseomuscular symptoms is unclear, although symptoms improve with chelation, and in vitro studies have documented abnormal copper deposition in cartilage and bones. Complete reversal of joint manifestations after liver transplantation has been documented in a case report from India.65
Other Manifestations
Asymptomatic cardiac arrhythmias are quite common in WD. Cardiomyopathy, autonomic dysfunction, and cardiac deaths due to copper accumulation in cardiac tissue have occasionally been reported in WD. Kuan66 reported electrocardiogram abnormalities in 34%, which include left ventricular hypertrophy, ST wave depression, and T wave inversion. Asymptomatic orthostatic hypotension was reported in 19%, abnormal response to Valsalva in 33%, and cardiac deaths in 2 cases (ventricular fibrillation and cardiomyopathy in one each).
Extensor hyperpigmentation due to increased melanin has been reported in 11% of patients with WD.67
Blue lunulae of nails (azure lunulae) and acanthosis nigricans have occasionally been reported in WD.54 Endocrine abnormalities reported include amenorrhea, gynecomastia, and testicular atrophy. Additional features include pancreatic insufficiency, diabetes mellitus, gigantism, and hypoparathyroidism.68
Consensus Statements on Clinical Features
Hepatic manifestation
-
1.
Isolated hepatic involvement is more common in childhood and adolescence than adulthood demonstrating an age-phenotypic nature of the disease (strength 1; level of evidence, A).
-
2.
Cirrhosis and portal hypertension is a common presentation in hepatic WD (strength 1; level of evidence, B).
-
3.
Acute liver failure, ACLF, acute hepatitis, asymptomatic hypertransaminasemia, fatty liver, cholelithiasis, and rarely hepatobiliary malignancies are the other manifestations of hepatic WD (strength 1; level of evidence, B).
-
4.
Concomitant hemolysis is invariably seen in patients with WD presenting with ALF (strength 1; level of evidence, C).
Neuropsychiatric manifestations
-
1.
Neurological/neuropsychiatric symptoms can be the sole presenting clinical symptom of WD (strength 1; level of evidence, A).
-
2.
Neuropsychiatric form of WD usually tends to present later than the hepatic form (strength 1; level of evidence, B).
-
3.
The clinical symptomatology varies significantly affecting various neurological domains, including from mild tremors, dystonia, seizures, Parkinsonism, ataxia, cognitive changes, and frank behavioral issues (strength 1; level of evidence, A).
-
4.
Any child/young adult presenting with neuropsychiatric features, screening for WD should be considered (strength 1; level of evidence, A).
Ocular manifestations
-
1.
KF rings are usually bilateral and present in almost all cases of neurological and more than half of hepatic WD. Slit lamp examination is mandatory for diagnosis (strength 1; level of evidence, B).
-
2.
Sunflower cataract is uncommon, even in neurological WD (strength 1; level of evidence, B).
-
3.
Disappearance of KF rings often correlates with adequate chelation but may take years (strength 1; level of evidence, B)
Renal manifestations
-
1.
Renal tubular dysfunction with nephrocalcinosis (manifesting as microscopic hematuria) is not uncommon in WD (strength 1; level of evidence, B).
-
2.
Children on DP should be periodically evaluated for proteinuria to detect drug-induced glomerular injury (strength 1; level of evidence, B).
Hematological manifestations
-
1.
Mild Coombs-negative hemolytic anemia can occur in asymptomatic WD (strength 2; level of evidence, C).
-
2.
Acute severe hemolysis can be an initial manifestation of ALF related to WD (strength 2; level of evidence, C).
-
3.
WD should be suspected in adolescents and young adults with Coombs-negative hemolytic anemia (strength 2; level of evidence, C).
Other system manifestations
-
1.
Joint manifestations may be the presenting symptoms of WD. Precocious onset degenerative joint disease (osteoarthritis, chondrocalcinosis) in the second and third decade should raise a suspicion of WD (strength 2; level of evidence, B)
-
2.
Asymptomatic arrhythmias are common, and therefore, a cardiac evaluation may be routinely performed in all adult patients (strength 1; level of evidence, B).
Diagnosis
A combination of serum ceruloplasmin, KF rings, and 24-hour urine copper is most commonly used to diagnose WD. Liver biopsy and estimation of dry copper has been traditionally thought to be of value in doubtful situations. Extrapyramidal symptoms associated with KF ring makes the diagnosis of WD certain. Genetic mutation studies are now available.
Serum Ceruloplasmin
Ceruloplasmin is a carrier protein produced mainly by the liver for the transport of copper in blood. A low level in a person with suspected disease may be suspicious of WD. The youngest age for testing serum ceruloplasmin to diagnose WD is 1 year.3 Enzymatic assays which measure copper-dependent oxidase activity and antibody-dependent immunologic assays such as radioimmunoassay, radial immunodiffusion, or nephelometry have been used to measure ceruloplasmin levels. Although the enzymatic method is superior and the preferred method for estimation, the immunological nephelometry method is the more commonly available method of measurement.70, 71, 72 Normal values range from 20 to 40 mg/dl. While values <10 mg/dl strongly favor the diagnosis of WD, those between 10 and 20 mg/dl may be seen in both patients and about 20% of heterozygotes.73 However, normal values may be seen in up to a third of patients with WD and may be falsely normal in acute inflammation (ceruloplasmin is an acute phase reactant). Serum ceruloplasmin is typically lower in neuropsychiatric disease compared with liver disease.74 Ceruloplasmin levels may be decreased in patients with other causes of cirrhosis, malabsorption, and renal disease. Low serum ceruloplasmin levels alone cannot be relied upon to make a diagnosis of WD, although very low levels below 5 mg/dl are highly suggestive of WD.
Twenty-four–Hour Urinary Copper Assay
This is a sensitive test that indirectly reflects the serum free copper level. Urine sample must be collected in a copper-free container and the test should be done before chelators are started. It is an excellent test in symptomatic patients, but may be false negative in those who are asymptomatic.75, 76 A level >100 mcg/24-hour is considered virtually diagnostic, but recent studies have shown that lowering the cutoff levels to 40 mcg/24-hour for asymptomatic patients increases the sensitivity.76 The assay is difficult to perform and should be performed only in reliable laboratories.
D-penicillamine Challenge Test
After a baseline 24-h collection for copper assay, a second 24-h sample of urine is collected while the patient is given two 500 mg doses of DP 12 h apart. Earlier considered to be a useful test in diagnosis, it is not recommended now in view of the high false-positive results.77 Lowering the threshold for basal urine copper excretion together with measurement of serum ceruloplasmin and KF ring may be more useful rather than D-penicillamine challenge test (PCT).78
Kayser-Fleischer Rings
KF ring should be sought in all patients suspected to have WD.76 This test may be negative in asymptomatic siblings or children less than 10 years of age. These are best seen on conventional slit lamp examination. A hand-held slit lamp apparatus may be used in patients who are not ambulatory.
Serum Copper
Serum copper measures the total serum copper (ceruloplasmin-bound copper + nonceruloplasmin copper or “free copper”). Ninety percent of the serum copper is bound to ceruloplasmin.
Total serum copper does not reflect tissue levels and therefore unreliable in diagnosis. Serum free copper may have a better correlation, especially in patients with ALF. However, the value of free copper is limited as it relies on the accuracy of tests measuring serum ceruloplasmin and serum copper.24
Serum Exchangeable Copper
This test corresponds to the copper which is bound to albumin and other peptides. A cutoff value of 15% of relative exchangeable copper (namely exchangeable copper to serum copper ratio) has been reported to have 100% sensitivity and 100% specificity for the diagnosis of WD in adults.79
Coombs-Negative Hemolytic Anemia
The presence of significant hemolysis on peripheral smear and a negative Coombs test in a patient with ALF makes the diagnosis of WD highly likely.24 Hemolysis contributes to the elevated serum bilirubin levels and a decrease in hemolysis reflects in a commensurate drop in serum bilirubin.2
Liver Biopsy and Liver Copper Estimation
Histopathological changes are not specific for WD and range from steatohepatitis, interface hepatitis, chronic hepatitis with Mallory's hyaline, bridging fibrosis, and cirrhosis, most of which are seen with other liver diseases as well.80
Although orcein, rhodamine and Timm's stain have been used to identify liver copper, the distribution is patchy, and interpretation is difficult. Positive staining may be seen in diseases associated with impaired bile secretion.81 These can be supportive evidence rather than of primary diagnostic importance.
Liver Copper Estimation
Often described as gold-standard test for WD, it is not easily available and may be fraught with logistic and quality issues. Biopsy specimens for estimation of copper should be sent in a dry condition in a copper-free container for atomic absorption analysis,81 although paraffin-embedded specimens can also be analyzed for copper. The normal copper content in liver tissue is <50 mcg/g of dry weight, while in WD a level >250 mcg/g is commonly encountered even in asymptomatic individuals. The copper level tends to be higher in those with liver dysfunction compared to those with neurological or asymptomatic disease. Liver copper content is physiologically increased in infancy.82 Uneven distribution of copper in the liver and low levels of copper in regenerating nodules limits its usefulness in those with cirrhosis and late stages of WD. In addition, patients with cholestatic liver disorders can have a high liver copper.83, 84
Magnetic Resonance Imaging Brain
The presence of neurocognitive features in conjunction with KF ring is often sufficient to make the diagnosis of WD. Accumulating experience has demonstrated magnetic resonance imaging (MRI) abnormalities to be almost universal in those with neurological disease. In one study, the MRI features with WD included “Face of giant panda” (14.3%), tectal plate hyperintensity (75%), central pontine myelinolysis–like abnormalities (62.5%), and concurrent signal changes in basal ganglia, thalamus, and brainstem (55.3%).85 These features when present are virtually pathognomonic of WD86(Figure 4).
Figure 4.

MRI changes in Wilson's disease. (A) Flair axial brain MRI showing hyperintensities in bilateral putamen and thalami with hypointensity of globus pallidi in a patient with neurological manifestations; (B) T1W axial brain MRI showing hyperintensity of bilateral globus pallidi in a patient with hepatic form of WD; (C) Flair axial brain MRI showing hyperintensities in dorsal midbrain and subcortical white matter signal changes in temporal region in a patient with neurological form of WD; (D) T2W axial MRI brain depicting “face of giant panda”; (E, F) Central pontine myelinolysis–like pontine signals changes in WD in flair axial brain MRI. MRI, Magnetic Resonance Imaging; WD, Wilson's Disease.
Family History
A positive family history of WD (including deaths from compatible liver or neurologic disease) is useful especially when parental consanguinity exists. A recent study from Vellore, India, identified consanguinity in 44% of patients with WD.87
Genetic Studies
WD is an autosomal recessive disease, and in 1993, 2 separate teams reported mutations in ATP7B gene on chromosome 13. Over 600 mutations have since been described and genetic studies have gradually become part of routine tests in diagnosis. The international consensus diagnostic score (Leipzig score 2003) gave maximum weightage to genetic testing.88 Within families, the risk of WD among siblings of an affected individual (“the proband” or index patient) is 25%, while among the progeny, the risk is 0.5%.89 If ATP7B mutations have been identified in a patient with WD, genetic testing to look for these mutations in his/her presymptomatic siblings is valuable to differentiate heterozygote carriers from homozygotes or compound heterozygotes for WD. Other copper-related liver disorders (Indian childhood cirrhosis, atypical copper cirrhosis)90 also can be differentiated with mutation studies. H1069Q mutation is the most common mutation seen in over 50% of patients in Western countries is almost nonexistent in other countries like India.91, 92 Despite the high rate of consanguinity noted in Indian families that are affected, there is a wide spectrum of mutations observed. This phenomenon can be considered as an “Indian paradox.”87 Various studies suggest that p.C271X may be the commonest mutation in patients from Western part of India94 and p.G1101R95 maybe the commonest mutation in patients from Eastern parts of India with WD. C813A was identified in 19% of patients from East India and 12% of patients from South India.96 Studies have not been able to establish a significant correlation between the ATP7B mutations and WD phenotype. The large number of ATP7B mutations adds to the complexity of this correlation. It is also recognized that affected siblings within a family (who share the same WD genotype) may have different phenotypes.93 It is still unclear what determines whether a patient with WD will develop hepatic or neurological disease or both. Looking for mutations in all 21 exons of ATP7B gene is an expensive and daunting challenge with conventional genetic tests. In addition, many patients with WD are compound heterozygotes. Earlier, mutation screening was performed to identify exons likely to harbor mutations as a preliminary step (using single-strand conformational polymorphism97 or conformation sensitive gel electrophoresis) and then sequencing only the selected exons as a second step. Technological advances now enable simultaneous analysis for mutations in all 21 exons of ATP7B gene using microarray-based tests98 or next-generation sequencing (NGS).96
Regional centers need to be established, where these tests can be performed at an affordable cost with a quick turn-around time. There is also a need for a nationwide network in various countries to determine the common mutations in each country.
Prenatal testing for WD is technically feasible.97 However, because it is an easily treatable disease, routine prenatal screening for WD does not appear justified.
Modified Leipzig score – A new scoring system
The Leipzig score of 199388 was modified by our consensus group members and the new "modified Leipzig score" (Table 3) was validated in 70 patients with proven WD. In this new score, additional points were given for family history suggestive of WD. In addition, weightage was also given to a serum ceruloplasmin value of <5 mg/dL versus a value of serum ceruloplasmin =>5 mg/dL. There are more than 600 mutations identified in the world although unlike the West, there are no common mutations identified in India. Mutational analysis was retained in the new score as genetic tests are now more accessible and may be performed in individuals in whom diagnosis is difficult to establish by clinical and biochemical testing. PCT was omitted from the modified Leipzig's score because of poor yield and inadequate validation. Also, liver copper estimation was excluded from this new score as has methodical flaws and is not easily available.
Table 3.
Modified Leipzig Scoring System for Diagnosis of Wilson's Disease.
| KF rings | |
| Present | 2 |
| Absent | 0 |
| Serum ceruloplasmin | |
| Normal (>20 mg/dl) | 0 |
| 0–5 mg/dl | 3 |
| 6–11 mg/dl | 2 |
| 11–20 mg/dl | 1 |
| 24-h urinary copper (in the absence of acute hepatitis) | |
| >100 mcg | 2 |
| 40–100 | 1 |
| <40 mcg | 0 |
| Coomb's-negative hemolytic anemia with liver disease | |
| Present | 1 |
| Absent | 0 |
| Mutational analysis | |
| On both chromosomes detected | 4 |
| On one chromosome detected | 1 |
| No mutation detected/test not done | 0 |
| Liver biopsy for histology S/O WD with | |
| Orcein- or rhodanine-positive granules | 1 |
| Neurobehavioral symptoms | 2 |
| Present | 2 |
| Absent | 0 |
| Typical features on MRI brain | 1 |
| Present | 1 |
| Absent | 0 |
| History of Wilson disease in a family member | |
| Sibling death from liver disease/neurological disease suggestive of WD | 1 |
| Total score | Evaluation |
| ≥4 or more | Diagnosis established |
| 3 | Diagnosis possible, more tests needed |
| ≤2 | Diagnosis very unlikely |
KF, Kayser-Fleischer; MRI, Magnetic Resonance Imaging; S/O, suggestive of; WD, Wilson's Disease.
Family screening
Family screening of first-degree relatives of WD has multiple advantages: it (1) allows early detection of disease in presymptomatic phase before a devastating course, (2) makes the family wiser and the physician more prepared, and (3) identifies a healthy family member or a heterozygote carrier who can be a potential donor for living related liver transplantation (LT) should a diseased member require LT in future. Ideally, family screening should include siblings, parents of affected children, and offspring of affected parents. There is a 25% chance of siblings carrying the homozygous disease gene. Screening is deferred till 2 years of age when presymptomatic patients can be started on chelation therapy. Screening can be initiated earlier if any stigmata of liver disease or occult warning signs are detected (eg, hepatomegaly or fatty or nodular liver sonologically in the absence of deranged liver function tests). Histological liver fibrosis as young as 4-month-old has been documented.99 Assessment should include good history, physical examination including slit lamp examination of KF ring, serum ceruloplasmin, liver function tests, and 24-h urinary copper. The role of testing for ATP7B mutation or haplotype studies assumes importance if the affected individual is alive or if the sample is stored before death where the specific mutation seen in the index case can be looked for in the family member being screened. Genetic testing is the only reliable method to separate heterozygote from homozygote siblings. There is, however, difficulty in diagnosing heterozygote carriers with certainty. Although NGS is promising in detecting both mutant alleles in 95%, the limitations include missing molecular defects outside the coding regions and detecting variants of unknown significance which may not be of clinical significance.3 More recently, relative exchangeable copper appears to be a promising tool for family screening in WD, especially for heterozygous ATP7B carriers who could present with slight biological abnormalities. However, further and larger studies are required to validate the same.100
Consensus Statements on Diagnosis of WD
Serum Ceruloplasmin
-
1.
Ceruloplasmin value of <10 mg/dl strongly favors the diagnosis of WD (strength 1; level of evidence, B).
-
2.
Borderline or normal levels do not exclude the diagnosis, and further tests are needed for confirmation WD (strength 1; level of evidence, A).
-
3.
Values above normal are unlikely in WD (strength 2; level of evidence, A).
Twenty-four–hour urine copper
-
1.
A basal 24-h level >100 mcg is a very useful diagnostic test in symptomatic patients (strength 1; level of evidence, A).
-
2.
Lower levels (>40 mcg) have been recommended especially for asymptomatic siblings but is less specific (strength 2; level of evidence, A).
-
3.
DP challenge test is not standardized, gives high false-positive results, and should not be used in diagnosis (strength 2, level of evidence, A).
KF rings
-
1.
KF ring is highly specific for WD, but its absence does not rule out the diagnosis (strength 1; level of evidence, A).
-
2.
While it is almost always present in patients with neuropsychiatric disease, only 50–60% of those with liver disease have a KF ring (strength 1, level of evidence, A).
-
3.
A slit lamp examination by an experienced ophthalmologist is necessary for confirming or ruling out a KF ring (strength 1; level of evidence, A).
Serum copper
-
1.
Serum total copper has no role in the diagnosis of WD (strength 1; level of evidence, A).
Hemolytic anemia
-
1.
Any degree of hemolysis in the setting of liver disease that is Coombs-negative heightens the suspicion of WD (strength 2; level of evidence, A).
-
2.
The presence of significant hemolysis in an individual with ALF is almost always due to WD (strength 2; level of evidence, A).
Liver biopsy and liver copper
-
1.
There are no histopathological features pathognomic for WD (strength 2; level of evidence, A).
-
2.
Liver copper estimation has limited usefulness due to nonavailability of the test as well as logistic and quality issues (strength 2; level of evidence, A).
-
3.
Histochemical stains for copper may only provide supportive evidence for diagnosis of WD (strength 2; level of evidence, B).
MRI brain
-
1.
MRI of brain with certain features such as tectal plate hyperintensity is pathognomonic of WD (strength 2; level of evidence, B).
Family history
-
1.
Positive family history is an indirect evidence of WD (strength 2; level of evidence, C).
-
2.
A history of sibling death in patient with features suggestive of WD features makes WD diagnosis likely although the presence of other associated features strengthens the diagnosis (strength 2; level of evidence, C).
Genetic studies
-
1.
ATP7B mutation analysis is recommended as a clinical diagnostic test to support the diagnosis of WD in a patient suspected to have WD (strength 1; level of evidence, B).
-
2.
Genetic testing for WD is recommended as a clinical diagnostic test in siblings of a proband with WD, especially if ATP7B mutation is identified in the proband (strength 1; level of evidence, B).
-
3.
Routine prenatal diagnosis of WD is not recommended (strength 1; level of evidence, C).
Family screening
-
1.
Family screening of patients diagnosed with WD is recommended (strength 1; level of evidence, A).
Drug therapy
D-penicillamine
DP is the preferred standard therapy for WD. It is rapidly absorbed from the intestine, bound to plasma proteins, and more than 80% is excreted in the urine.101 DP binds to copper through disulfide bonds, and every gram promotes urinary excretion of 200 mg of copper. It also induces hepatic metallothionein, a cytosolic metal-binding protein that sequesters copper, and renders it nontoxic. Clinical and biochemical improvement usually occurs within a year of treatment, but hepatic synthetic functions may take up to 10 years to normalize.102 Hepatic Cu levels fall during the first year but remain above normal for several years. DP chelates several heavy metals, not just copper, and has many adverse effects necessitating discontinuation in up to 30% of patients with WD (Table 4). Despite the serious adverse effects, DP is still the primary drug for management of hepatic WD in view of its time-tested efficacy, easy availability, and reasonable cost.
Table 4.
Adverse Effects of Drugs Used in Treatment of Wilson's Disease.
| Name of drug | Side effects |
|---|---|
| D-penicillamine |
Early: (1–3 weeks) hypersensitivity Fever, cutaneous eruptions, lymphadenopathy, neutropenia, thrombocytopenia, and proteinuria Late: (3 weeks–3 months) hypersensitivity
Nephrotoxicity, severe allergy on restarting drug, myasthenia gravis, polymyositis (<1%), loss of taste, immunoglobulin A depression, retinitis, hepatotoxicity (transaminitis), copper depletion leading to neutropenia, sideroblastic anemia, and hemosiderosis Direct dose dependent:
|
| Triethylenetetramine hydrochloride/trientine |
|
| Zinc |
|
ANA, antinuclear antibody.
In neurological WD, a paradoxical worsening of symptoms is occasionally observed after starting DP. While most centers report an incidence of 10%, one study by Brewer and Terry103 reported it in 50% of patients. DP is therefore started at the lowest possible dose in neurological WD. It may be initiated as 250 mg on alternate days. There is no definitive protocol on the rate of dose escalation. While some centers increase by 250 mg every 2 to 3 weeks, others do so every month, until the maximum of 1000–1500 mg/day in 2 or 3 divided doses is reached. The gradual increase in dose should be done under clinical and biochemical monitoring. Similarly rapid readministration of the drug in patients who have stopped therapy for a long time may lead to irreversible neurological changes.104, 105 The paradoxical worsening with treatment has been attributed to sudden mobilization of free copper from neurological tissues. Neurological improvement is much slower and usually reaches a plateau after around 2–3 years of treatment. A lack of improvement in neurological status (eg, dystonia) may be due to permanent neurological damage (putaminal necrosis).
The dose of DP is 20 mg/kg/day in children, while adults receive 750–1500 mg per day in 2–3 divided doses on an empty stomach. Food reduces its absorption by 50%, and so, food should not be given 1 h before and 2 h after the drug. Antacids and iron also significantly reduces absorption. Pyridoxine deficiency can occur with therapy because of the inhibition of pyridoxine kinase enzyme. Hence, vitamin B6 should be supplemented at a dose of 20–40 mg in children, pregnant women, and patients with malnutrition and intercurrent illness.106 Treatment with DP results in massive and rapid cupriuresis (>1000 μg/day) in the initial months of therapy, falling to 200–500 μg/day during the maintenance period.
Significant adverse effects are reported in 10–30% of patients on DP therapy (Table 4).
Children seem to tolerate the drug better than adults26, 107 In a series of 74 children from the UK, the reported incidence of side effects was 38%, and in 16%, the drug had to be switched over to trientine.78 Manolaki et al108 in their series of 54 children have reported a similar incidence of side effects at 16% where the initial treatment with DP had to be discontinued because of the adverse effects. If early hypersensitivity reactions occur, the drug should be stopped immediately. If the manifestations are only dermatological, DP can be introduced under cover of steroids. Once the skin lesions resolve, prednisolone is given initially at a dose of 0.5 mg/kg/day for 2–3 days. DP is then introduced at a low dose of 5 mg/kg/day and increased gradually while prednisolone is tapered and stopped. However, if the manifestations involve the bone marrow (agranulocytosis, aplastic anemia) or other organ systems, then DP should be stopped. Development of significant proteinuria or glomerulonephritis warrants stoppage of the drug. Direct dose-dependent adverse effects of DP is due to the interference with copper-dependent enzyme lysyl oxidase which mediates collagen cross linkage and elastin formation leading to progeria-like skin lesions, cutis laxa, and elastosis perforans serpiginosa109 (Figure 5). These usually occur when prolonged treatment is given in large doses.
Figure 5.
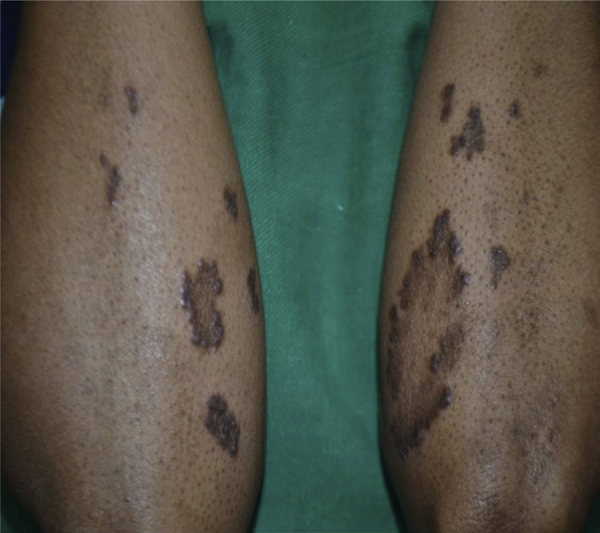
Elastosis perforans serpiginosa secondary to long-term D-penicillamine use. Multiple annular plaques seen over bilateral forearms with hyperpigmented papules arranged at the periphery, and few papules showing central small keratotic plugging.
Trientine
Trientine (triethylenetetramine-2-hydrochloride) is a chelator with a mechanism of action similar to DP, with fewer adverse reactions. The dosage is 750–1500 mg/day in 3 divided doses on empty stomach for adults (20 mg/kg/day for children). Although trientine has traditionally been used for patients intolerant to DP,110 recent studies suggest that it can be used as a first-line drug.111, 112 There are no clinical trials comparing the relative superiority of one over the other. In most developing countries including India, it is only imported for select patients because it is prohibitively expensive. It is heat sensitive and has to be stored in tightly closed containers between 2 and 8 °C. It chelates iron and other heavy metals as well; hence, treated subjects should be monitored for iron deficiency. Trientine can also cause paradoxical worsening in neurological WD, Hence, it should be started in low doses and increased slowly similar to DP.
Zinc
Zn acts by inducing metallothionein in enterocytes which preferentially binds absorbed Cu, sequesters it in the enterocytes, and prevents its entry into the portal circulation.113, 114, 115 As the enterocytes are naturally sloughed into the lumen, copper is excreted in the feces. Zinc also induces metallothionein in hepatocytes and protects against Cu toxicity. Unlike DP and trientine, Zn acts by increasing the fecal excretion of Cu. However, Zn has a slow action and takes much longer to achieve a negative Cu balance as compared with chelation therapy and hence may be less effective as first-line therapy in symptomatic liver disease. Although all 3 salts—acetate, sulfate and gluconate—are effective, acetate salts are preferred because of lesser incidence of gastric side effects.116 Adults require 150 mg/day of elemental zinc in 3 divided doses, whereas children and those under 50 kg are given only 75 mg/day. Zinc should be taken on empty stomach to ensure better absorption. Besides clinical and biochemical improvement, treatment efficacy is determined by a 24-h urinary copper excretion less than 100 μg. Urinary excretion of zinc should be more than 2000 μg/day to ensure compliance and also to determine the quality of the zinc preparation used. Zinc is used as a first-line drug in presymptomatic patients or symptomatic patients with neurological WD and for long-term maintenance therapy in others after optimal decoppering with chelators.117, 118, 119 In a recent study, hepatic treatment failure occurred more frequently on zinc therapy than on chelator therapy. Patients who did not respond to zinc therapy showed hepatic improvement after reintroduction of a chelating agent. This suggests that chelating agents are better first-line medications in symptomatic hepatic WD.
Ammonium Tetrathiomolybdate
Ammonium TTM, originally used to treat copper poisoning in veterinary practice, is a chelating drug with anti-angiogenic properties.120, 121 If ammonium TTM is taken after meals, it binds to the copper in the food, thus preventing its absorption. If taken on empty stomach, it is absorbed into the blood and forms a complex with circulating copper preventing cellular uptake, leading to its excretion in urine. The dosage used is 20 mg 3 times a day with meals and 20 mg 3 times a day in between the meals. Owing to its aggressive chelating effects, the reported side effects of ammonium TTM includes paradoxical worsening, bone marrow suppression, and hepatotoxicity. However, it has been stated that the potential neurological deterioration and side effects are lesser in comparison to trientine.122, 123 Currently, ammonium TTM is not available in India in many countries. Ongoing phase-2 multicenter trials with a more stable form of a chelator for copper (bischoline TTM [WTX101]) report that 57% patients show improvement in liver function test and 72% show improvement in free copper levels by week 24 of therapy.124 Further data on the use of this drug for various phenotypes of WD are awaited.
Treatment phases
Initial Phase
This phase aims to reduce the body copper levels to subtoxic threshold. The choice is between chelators (DP or trientine) alone, zinc alone, or a combination of both. There are no randomized controlled trials comparing the 3, and each center uses a protocol based on their experience and patient compliance.2, 125
DP has traditionally been the drug of choice unless the patients develop intolerance, in which case trientine is the preferred drug. In a study comparing DP versus zinc sulfate as a first-line therapy for neuro-WD, the neurological worsening on DP vs zinc within 180 days of starting the drug was 35% vs 19%.126 The authors concluded that DP and zinc sulfate were both effective in the majority of neuro-WD patients. Neither therapy appears to be clearly superior. Therefore, zinc may be considered a reasonable alternative to DP as a first-line therapy. Centers using both chelator and zinc together believe that DP has the most beneficial effect on the early decoppering phase, while zinc is inexpensive and assists by preventing copper absorption. There are insufficient data to prove the superiority of combination therapy but if it used, the 2 drugs must be given 6 h apart to prevent chelation of zinc by the chelator.
Maintenance Therapy
This is a lifelong therapy and prevents copper reaccumulation after the patient has been effectively decoppered. Zinc, in view of its good efficacy, low cost, and toxicity, is the drug of choice. DP in low dose is an alternative but patients should be monitored for side effects.
Presymptomatic patients
An asymptomatic sibling diagnosed to have WD by biochemical or genetic testing should be treated to prevent symptomatic disease. Zinc is the drug of choice. If a neonate is diagnosed to have WD based on genetic testing, it is unclear when treatment should be started. Because of the risk of body copper depletion, it should probably not be started in the first year.3 It may be suggested that because clinical presentation is rare below 3 years, zinc therapy could start at the age of 2 years.
WD presenting as ALF
Patients with encephalopathy should be considered for urgent liver transplantation. DP/trientine with or without zinc may be started as an ad hoc measure, but survival is unlikely without transplantation.
In patients without encephalopathy, the decision on transplantation should be individualized after discussion with the relatives. While delay carries the risk of sudden deterioration, encephalopathy, and death, unnecessary surgery may mean removing a native liver that may have recovered with medical treatment. The fulminant presentation with intense jaundice, hemolytic crisis, and rapid deterioration of hepatic encephalopathy rarely survive without a transplant. Rapid removal of free copper through molecular absorption recirculating system (MARS) or total plasma exchange (TPE) can benefit patients with fulminant presentation. TPE efficiently removes both ceruloplasmin- and albumin-bound copper, and the fresh-frozen plasma used for exchange can be helpful in treating the associated coagulopathy. MARS is also effective127, 128 but more expensive and less widely available than TPE.129
These modalities are seen as a bridge to LT, rather than a definitive treatment option.130 In a recent study, in 9 of 10 patients with WD, apheresis was a successful bridge to transplantation.
Liver transplantation
Liver Transplantation for Hepatic WD
Durand et al,102 in 2001, reported that early administration of DP may avoid LT for a vast majority of patients (90%) presenting with fulminant WD without hepatic encephalopathy (HE) at admission. In 2006, Nazer's score131 (serum bilirubin, international normalized ratio (INR), and serum albumin) was modified to add 2 parameters (AST and white blood cell count) to the score and renamed it as New Wilson's Index (NWI). An NWI score ≥11 (Table 5) was associated with nonsurvival without liver transplantation.77 In a study from South India,132 NWI and Pediatric End-Stage Liver Disease/Model for End-Stage Liver Disease (MELD) were found to have modest accuracy in predicting outcomes in WD. The authors derived a formula from regression analysis based on hepatic encephalopathy and bilirubin to predict the outcome in a fulminant presentation WD. Similarly, Fischer et al133 identified 3 of the 6 patients who had NWI scores predictive of death, and of these 3, 2 survived without a transplant. They cautioned about the scores not being accurate and this subgroup needing further study.
Table 5.
New Wilson's Index for Predicting Survival.77
| Score | Bilirubin (mg/dl) | INR | AST (IU/L) | WCC (109/L) | Albumin (g/dl) |
|---|---|---|---|---|---|
| 0 | 0–5.8 | 0–1.29 | 0–100 | 0–6.7 | =>4.5 |
| 1 | 5.9–8.7 | 1.3–1.6 | 101–150 | 6.8–8.3 | 3.4–4.4 |
| 2 | 8.8–11.6 | 1.7–1.9 | 151–200 | 8.4–10.3 | 2.5–3.3 |
| 3 | 11.7–17.5 | 2.0–2.4 | 201–300 | 10.4–15.3 | 2.1–2.4 |
| 4 | =>17.6 | =>2.5 | =>300 | =>15.4 | 0–2.0 |
AST, aspartate transaminase; INR, international normalized ratio; WCC, white cell count.
A recently published multicentric cohort (comprising 75 adults and 56 children) from France,134 the Kaplan-Meier survival analysis revealed good 5-year survival after LT (86%–96%) depending on whether transplants were performed before or after the year 2000. This reflects improvements in the selection of patients and perioperative management.
Liver Transplantation for Neurological WD
The indications for LT in neurologically affected patients are controversial. In a French study,134 only 6% received a LT for a purely neurological indication, and these patients were significantly worse after LT when compared with hepatic patients. All 3 patients with severe axial Parkinson's syndrome died (from infection) with a functional graft but without any neurological improvement. Although there is evidence that mild-to-moderate neurological involvement may improve after LT,135 neuropsychiatric disease is a predictor for poor outcome after LT.136 In a retrospective Italian study, neurological symptoms significantly improved after liver transplantation, but the survival of patients with hepatic and neuropsychiatric disease was significantly lower than those with liver disease alone.135
Sepsis was the main cause of death because neurological sequelae led to the lack of ambulation and bedridden patients. LT as a treatment option for those with only neuropsychiatric disease is not recommended.
Liver Grafts From Heterozygous Donors (Siblings/Parents)
These are safe for both recipient and donor, and disease recurrence risk is almost nonexistent. In developing countries such as India, deceased donor liver transplant program is still evolving, and most transplants are from heterozygous-related donors.
Treatment monitoring
Patients should be regularly monitored for ensuring compliance, efficacy of therapy, and early recognition of side effects. Effective decoppering (Table 6) is assessed on 24-hour urine copper and serum free copper value. Serum free copper is calculated by the formula: serum copper − 3 × serum ceruloplasmin. If serum ceruloplasmin is not measured by the enzymatic method, then this free copper calculation is not reliable. A 24-hour urine protein is estimated for renal toxicity of DP. This is initially done after a month, then 3 monthly, and subsequently 6–12 monthly.
Table 6.
Interpretation of Tests Used in Monitoring Drug Treatment of Wilson's Disease.
| Zinc | D-penicillamine/trientine | |
|---|---|---|
| Initial treatment | U Cu 100–500 μg/d S free Cu > 25 μg/dL U Zn > 2000 μg/d |
U Cu > 500 μg/d S free Cu > 25 μg/dL |
| Good control (Maintenance) | U Cu < 75 μg/d S free Cu 10–15 μg/dL |
U Cu 200–500 μg/d S free Cu 10–15 μg/dL |
| Non-compliance/Inadequate dose | U Zn < 2000 μg/d S free Cu > 15 μg/dL |
U Cu < 200 μg/d U Cu > 500 μg/d S free Cu > 15 μg/dL |
| Overtreatment | U Cu < 25 μg/d S. free Cu < 5 μg/dL |
U Cu < 200 μg/d S. free Cu < 5 μg/dL |
To document therapeutic efficiency, urinary copper excretion after 2 days of D-penicillamine cessation should be <50 μg in 24 h; If higher, it suggests poor compliance.138
U Cu, 24 h urinary copper, U Zn, 24 h urinary zinc, S. free Cu, Serum free copper, DP, D-penicillamine.
In hepatic WD, clinical improvement is characterized by decreasing jaundice, ascites, and portal hypertension. Complete blood counts and liver function tests are performed initially after a week, then at 2 and 4 weeks followed by 3 months, 6 months, and then yearly. Child-Pugh score (based on serum bilirubin, prothrombin time, serum albumin, presence of ascites, and encephalopathy) and MELD score (based on bilirubin, creatinine, and INR) should be documented in those with severe liver disease.
In neurological WD, symptoms on sequential evaluation remain the most critical outcome of therapeutic benefits. Scales are essential to objectively quantify the severity of the disease and its impact on patient's lifestyle. Over the years, of the many rating scales that have been used, the Global Assessment Scale for Wilson's Disease137 (GAS for WD) is the preferred scale because it assesses the neuropsychiatric, hepatic, and osseomuscular changes and their impact on quality of life over the observation period (Table 7).
Table 7.
Global Assessment Scale for Wilson's Disease.
| Tier | Areas of assessment | |
|---|---|---|
| Tier 1, domains scored from 0 to 5 in each domain based on impact on activities of daily living | Domain | |
| Liver* | Clinical, biochemical, and abdominal ultrasound evidence for liver disease | |
| Cognition and Behavior | Intellectual decline, depression, and psychosis | |
| Motor | Motor impairment on examination | |
| Osseomuscular | Joint or muscle involvement by clinical examination and radiographs | |
| Tier 2, items 1–13 scored from 0 to 4 based on clinical severity |
|
|
| Tier 2, item 14 is scored based on the presence or absence of uncommon manifestations (each scored as 1 point with a maximum of 4 points.) | Emotional lability Seizures over preceding 1 month Myoclonus Stereotypy Tics Pyramidal signs Eye movement abnormalities |
|
*In the liver domain, the scores range from no liver disease ever (L0) through active liver disease (L2), compensated (L3), decompensated liver disease (L4) to potentially life-threatening disease (L5).
Greater weightage is given to Wilson's facies and KF rings that are characteristic features of Wilson's disease.
Long-term outcomes
WD is well recognized as one of the treatable genetic disorders and early recognition and institution of therapy holds the key for good outcome. The response to therapy is dependent on various factors including drug compliance and duration/severity of symptoms at the time of institution of therapy. The Eurowilson Consortium in 2013139 analyzed the outcomes of patients on oral chelators in treatment of WD with a follow-up of 5–30 years. They noted that in 9 of 326 patients on penicillamine and in 3 of 141 patients on trientine, liver transplantation was needed. Overall improvement on therapy was 90% in hepatic presentation and >55% with neurological presentation. In studies that have observed the long-term outcomes in large cohorts of patients, good improvement (including near-normal quality of life, improvement in symptoms or stabilization) was noted in 85–93.5%.42, 104, 140, 141
Most patients tend to have improvements up to 18–30 months after initiating therapy with good compliance, following which there is a plateau effect.
Poor prognostic factors of patients of clinically severe neurological WD included strong family history and severe MRI brain changes.142 Despite severe neurological involvement, 50% of patients have good clinical improvement while on treatment. Nonresponders to therapy show progressive, MRI worsening over time.
Symptomatic management of neuro-WD
Dystonia
In addition to the decoppering therapy, patients with moderate to significant dystonia need medications such as trihexyphenidyl, tizanidine, baclofen, clonazepam, tetrabenazine for symptomatic treatment.143 Patients who do not respond to the aforementioned medications or have focal dystonia, botulinum toxin injection is the best option.142, 144 Deep brain stimulation and lesionectomies of globus pallidus interna has been performed with variable benefits, in those with refractory symptoms. Plasampharesis has been tried for patients with refractory generalized dystonia but is not part of the standard protocol.
Tremors
Mild tremors do not require any specific interventions or only simple physical therapies. In patients who have tremors affecting activities of daily living, drugs including propranolol, clonazepam, anticholinergics, topiramate, primidone, have been used.143 Pallidotomy or deep brain stimulation can be considered in resistant tremors, with some beneficial effects.
Parkinsonism
Levodopa should be tried in all patients with Parkinsonism, which might relieve symptoms. Other drugs of benefit include dopamine agonists, monoamine oxidase inhibitors, and amantadine.143
Seizures
Seizures occur in about 6–8% of patients with WD, either at initial presentation or during the course of the disease. Therapy is on standard lines with antiepileptic medications. In view of associated hepatic dysfunction, antiepileptics with first-pass metabolism in liver should be avoided.
Psychiatric Symptoms
While mild symptoms improve gradually with decoppering, those with severe symptoms require medical interventions. These include simple behavioral modifications to selective serotonin reuptake inhibitors, tricyclic antidepressants, serotonin-norepinephrine reuptake inhibitor, and atypical antipsychotics (typical antipsychotics can precipitate extrapyramidal symptoms). Patients with aggressive manic symptoms or significant psychosis have been treated with electroconvulsive therapies. There are no good randomized trials that have evaluated the efficacy of physical therapy. However, isolated case reports and small case series suggest that physical therapy can be useful in cases with dystonia, where stretching and splints can help prevent contractures and Parkinsonism where gait and balance techniques can be a benefit. There have been no randomized trials on the benefits of speech therapy for dysarthria. Neuromuscular electrical stimulation has been attempted in to improve dysphagia,145 and it is standard practice to use a percutaneous endoscopic gastrostomy (PEG) in patients where swallowing is markedly affected to improve nutrition until the patient is able to swallow well.
Dietary copper in the management
No prospective studies are available to define a cutoff level of copper in the diet of a patient with WD. Less than 1–2 mg/day of copper is widely acceptable and not specific for body weight or age (pediatric or adult). The AASLD and EASL recommend avoiding foods with high concentration of copper in the first year of treatment.1, 2 The rationale is justified as the body should not be overloaded with high dietary Cu during the critical first year of systemic chelation. However, this opinion is based on 2 case reports of WD on vegetarian diet and bioavailability of copper in healthy subjects with dietary changes.9 The evidence to restrict Cu is weak. In developing countries, this recommendation has practical difficulties especially in vegetarians because efforts to restrict dietary Cu to <2 mg/day results in protein intake being invariably reduced to 1–1.5 g/kg/d. This may be insufficient for a cirrhotic or advanced liver disease. Well-designed studies will be required to address this issue. It may be practical to restrict only very high copper containing foods such as nuts, soy, liver, and chocolates especially in the first year of chelation. Dairy, vegetables, and fruits can be allowed unrestricted. Spices need not be restricted as effective intake is <5 g/d. Contrary to the wide belief, mushroom, animal meat, fish, and poultry do not contain high copper. Table 8 shows food copper content in commonly used food items. The Cu content of foods is enhanced by drying, roasting, pickling, canning, and adding preservatives, while boiling, milling, polishing, and refining reduces the Cu content.146 If Cu pipe tubing systems is used for domestic supply, it is important to check whether the water supplied is soft or hard. Soft water is acidic and tends to solubilize the copper from the pipelines with considerable “leaching effect” in stored samples. Hard water is neutral to alkaline, and copper is unaffected. Drinking water should ideally contain <0.1 part per million (0.1 mg/L) of Cu. It is important to let the first 1–2 L of water be flushed away before using for drinking purposes. While there is no evidence that boiling, simple filtration, or reverse osmosis reduces the copper content, ion exchange methods used in ultrafiltration may be effective. Bottled mineral water is estimated to contain <0.05 mg/L Cu.147 However, further studies are required to confirm the same. Studies on Indian childhood cirrhosis have shown that copper content of boiled and 6 h stored buffalo milk (98.4–98.8 μmol/L) and water (5.1–5.4) in copper and brass vessels are higher than glass and aluminum vessels (milk: 1.8–2.0 and water 0.9–1.1 μmol/L).148
Table 8.
The Copper Content (mg) in Daily Dietary Items per 100 g Edible Portion.
| Low copper food items (copper content <1 mg/100 g edible portion) | High copper food items (copper content =>1 mg/100 g edible portion) |
|---|---|
| Rice (whole, puff, flakes) (0.23–0.27) | Red gram (1.14) |
| Wheat (whole grain, flour, semolina, vermicelli and noodles) (0.17–0.48) | Soybean (1.29) |
| Maize and products (0.11–0.45) | Lotus stem, water chestnut (1.2–1.3) |
| Barley and millet (0.43–0.67) | All nuts (1.1–2.2) |
| All legumes except red gram (0.6–0.97) | Cumin, coriander, black pepper, and mace (1.1–1.6) |
| All vegetables (0.1–0.4) | Liver (6.0) |
| Mushroom (0.09) | Oyster (3.4) |
| All fruits (0.1–0.6) | Duck meat (1.0) |
| All spices (except cumin, coriander, black pepper and mace) (0.1–0.6) | Cocoa (3.8) |
| Fish, prawns, chicken, red meat (0.1–0.5) | |
| Egg (0.07) | |
| Milk and dairy products (0.03–0.1) | |
| Coffee, tea (0.2–0.5) | |
| Jaggery (0.03) |
Adapted from the Indian Food Composition Table, National Institute of Nutrition 2017.
The authors cannot verify the Cu content of the following food items: gram flour, barley, oats, protein energy powder, soy milk, soy products, sauces, cheese and table salt. Gram flour and barley are likely to contain higher Cu content from their original pulses as the process requires drying and roasting. Most protein energy powders are derived from soy sources. All soy products, sauces and cheese undergo fermentation and contain preservatives thereby increasing the Cu content.
Additional treatment
All patients need to be vaccinated for hepatitis B and hepatitis A virus if they have not been previously exposed. Patients need to be managed for their complications of cirrhosis as per standard medical treatment. Alcohol and nonsteroidal anti-inflammatory drugs need to be avoided.
Young women with WD
Menstrual disturbances such as amenorrhea or oligomenorrhea are possibly related to disturbed hormonal activity. Impairment in conception and recurrent miscarriages in pregnancy have been reported. Women with undiagnosed and untreated WD have an increased rate of spontaneous abortions compared with women on treatment (odds ratio [OR]: 2.853, 95% confidence interval [CI]: 1.634–4.982]).149 Proposed mechanisms include ovarian dysfunction due to decreased levels of estradiol with increased testosterone concentrations secondary to liver dysfunction and placental abnormalities such as copper deposition. Higher spontaneous abortion noted in patients presented with neurologic manifestation compared with asymptomatic or hepatic or mixed manifestations (OR: 2.335, 95% CI: 1.323–4.118).150 Patients on DP and zinc showed lower spontaneous abortion rates compared with trientine (10% vs. 17% vs. 28%); but it was not statistically significant.150
Counseling the Family
Counseling also should address the risks of medication and risks of uncontrolled disease. Patients planning for pregnancy should have their copper status optimized before pregnancy. Also, the risk of 0.5% of the child being affected needs to be explained.
Maternal and Pregnancy Outcomes
Several small case series and case reports in the last decade demonstrated a favorable outcome of pregnancies in patients treated with DP. Recent case reports with trientine and a case series of 29 WD patients exclusively treated with zinc showed uneventful maternal and pregnancy outcomes.150, 151 A recently published largest study of 282 pregnancies in 136 women with WD showed a favorable course of pregnancies in vast majority of treated patients.152 In this study, presentation was 54% hepatic, 32% neurologic, and 9% were neurohepatic. Forty-one percent were on DP, 13% on trientine, 7% on zinc, 3% on combination treatment, and no therapy in 31%. Ninety-three percent had unchanged maternal outcome, 6% hepatic deterioration, and 1% neurological deterioration. Seventy-two percent had healthy babies and 26% had spontaneous abortion. Abortion rates were 20–29% in all presentations but 40% with neurological symptoms. Abortions were least with zinc (10%) and highest if undiagnosed without therapy (41%) or paused therapy (36%). Monitoring of liver functions and neurological symptoms throughout pregnancy is of utmost importance. The study reported slight alteration of biochemical values in 6% of cases, which subsided after delivery.152 Treatment interruption during pregnancy has been reported to result in ALF and appearance of neurologic symptoms. Safety of chelating agents in pregnancy has not been well established. Teratogenicity with high-dose DP has been reported in animal studies, and there are isolated case reports of connective tissue defects in humans.153, 154 Over zealous chelation and maternal copper deficiency can also result in impaired fetal development. Recent studies showed relatively low rate of birth defects (3%) with DP,152, 155 and the authors concluded that the overall benefits of continuing treatment outweigh the risks of treatment withdrawal. To avoid overchelation, the dose of DP and trientine may be reduced by 25–50% while zinc can be continued in the same dose.152, 156 Patients whose diagnosis was made only during pregnancy should be started on a standard dose of chelating agents considering the high copper burden. In women who are well controlled on chelation, there is a suggestion that switching to zinc before conception decreases the risk of miscarriage or teratogenicity. Available data support continuation of the previous chelator. Wound healing after caesarean section or episiotomy may be delayed, and therefore, the dose of DP should be reduced toward the later weeks of pregnancy. After delivery, the dose of chelating agents should be increased to prepregnancy levels. Breastfeeding is not recommended in women on DP because it is excreted into breast milk and might be potentially harmful to the infant. There are scant data on safety of trientine and zinc in breastfeeding. Detailed studies regarding contraception have not been performed so far. Spermicidal agents, barrier methods, and progesterone only preparations are advisable for contraception in patients with WD. Estrogen containing pills may interfere with biliary copper excretion and some of the intrauterine devices containing copper may be avoided.
Newer therapies
Studies on murine models recently reveal that genetic WD mice carrying a corrective adenoviral vector–containing human ATP7B DNA can bring long-term metabolic control and correction of copper metabolism.157 Significant improvement in liver injury (biochemical and histological), ceruloplasmin levels, urinary copper excretion, hepatic copper load, and increase in hepatic ATP7B expression by immunostaining has been documented. Cell therapy to repopulate the liver with healthy hepatocytes including the reconstitution of bile canalicular network has been tried.158
Consensus Statements on Treatment of WD
Drug Therapy
-
1.
A chelating agent should be used for symptomatic patients with hepatic WD. DP is preferred because of easy availability, cost, and efficacy (strength 1; level of evidence, B)
-
2.
Presymptomatic patients/those with hepatic disease on maintenance therapy/symptomatic patients with neuro-WD can be treated with zinc or chelating agent (strength 1; level of evidence, A).
-
3.
Lifelong treatment is necessary unless the patient has had a liver transplant (strength 1; level of evidence, A).
-
4.
MARS and TPE can be offered to patients with ALF especially in the presence of encephalopathy and is a good bridge therapy to LT, should the patient not show prompt recovery (strength 1; level of evidence, B).
-
5.
DP is an effective chelator although associated with several side effects (strength 1; level of evidence, A).
Liver transplantation
-
1.
Liver transplantation is indicated in patients with fulminant presentation with hepatic encephalopathy or hemolytic crises (strength 1; level of evidence, B).
-
2.
New Wilson's Index has been used as a predictor of LT (for both acute or chronic presentation). Rising bilirubin, advanced hepatic encephalopathy, and acute hemolysis have been suggested as better predictors for LT and need validation (strength 1; level of evidence, B).
-
3.
Living donor liver transplant from heterozygous sibling is effective and safe for both donor and recipient (strength 1; level of evidence, B).
-
4.
Liver transplantation is not indicated for isolated severe neurological WD. When the liver is also diseased, the decision should be individualized because significant neurological disease is a predictor of poor outcome (strength 1; level of evidence, B).
Monitoring on Treatment
-
1.
Careful clinical monitoring is necessary to determine benefit and adverse effects of the drugs. Complete blood counts, urine analyses, liver function tests, 24 h urinary copper and protein, and serum free copper should be monitored frequently at the initial phase of therapy and at least once every 6–12 months thereafter. KF ring should be evaluated annually (strength 1; level of evidence, B).
-
2.
GAS for WD is a comprehensive reliable and valid scale. It should be used to objectively assess the disability and to check the response to treatment on follow-up especially in patients with neuro-WD (strength 1; level of evidence, B).
Drug therapy for Neurological Symptoms
-
1.
For patients with dystonia not responding to decoppering therapy, anticholinergics and baclofen can be used. For those with refractory focal dystonia or residual dystonia, botulinum toxin injections can be considered (strength 2; level of evidence, C).
-
2.
Primidone and propranolol can be used in patients with residual postural and action tremors, levodopa in Parkinsonism, and tetrabenazine in hyperkinesias (strength 2; level of evidence, C).
-
3.
Physical therapy and speech therapy have some role to play in rehabilitation of patients with neurological WD (strength 2; level of evidence, C).
-
4.
In patients with severe dysphagia, PEG tubes are a temporary option till the dysphagia improves with chelation therapy (strength 2; level of evidence, C).
Maternal and Pregnancy Outcomes
-
1.
Pregnancy in patients with WD is safe, and most patients have successful outcomes (strength I; level of evidence, B).
-
2.
Patients with adequate copper control have better chances of successful pregnancies than untreated WD patients. Copper status of the patient should be optimized before pregnancy (strength I; level of evidence, B).
-
3.
Treatment should be continued with previous chelation medication during pregnancy with dose reduction of chelators in later part of pregnancy (strength 2, level of evidence, B).
-
4.
The risk of birth defects in WD is generally low, which also applies for patients treated with chelating agents (strength I, level of evidence, B).
Newer therapies
Newer therapies include hepatocyte transplant, stem cell transplant, and gene therapy, which attempt to restore hepatobiliary copper excretion. However, these seem to be still in experimental phase (strength 2; level of evidence, C).
Conflicts of interest
The authors have none to declare.
Nominal group technique of voting and writing of the guidelines
Hepatologists and neurologists manage WD, often with overlapping disease but no joint consensus exist till date. We felt the need for practice guidelines with holistic management. The process was primarily initiated by AN and RKD. Experts hepatologists (pediatric and adults) and neurologists were consulted to identify the gaps in literature and difficulties in management practices. Specific questions and areas of interest were identified and distributed to the core members. A two-day deliberation on WD was held in March 2017 in Mumbai. On the first day, various aspects of pathogenesis, management and recent literature review were presented and debated. On the second day, the core members met for formulating the guidelines. AN and RKD moderated the session. Presentations on specifically allotted topics were projected on the power-point by core members MSS, HD, SS (Sanjib Sinha), SA, AB, CEE. At the end of each presentation, different viewpoints were debated and finally voted. The statements were recorded. In addition to the presenters -MSS, HD, Sanjib Sinha, SA, AB, CEE, other authors PLK, RKD, VG, NM, RMK, MS,UP, AS, BRT, PMW, SKY contributed towards specific sections of the guidelines. The information was pooled. The draft was revised several times by AN,MSS and JM and reviewed by all contributors and AS, SS (Srinivas Sankaranaraynan) and AD. AN, HD, PLK and SS (Sanjib Sinha) additionally contributed in specific areas including the modified scoring system. The final manuscript was drafted by AN.
Acknowledgements
We would like to acknowledge Dr Priya Malde, Department of Gastroenterology, Apollo Hospital Navi Mumbai, India; Dr Smita Malhotra, Department of Pediatric Gastroenterology and Hepatology, Indraprastha Apollo Hospitals, New Delhi, India; Dr Deepak Goyal, Department of Pediatric Gastroenterology, Hepatology and Liver Transplantation, Medanta – The Medicity Hospital, Gurgaon, India and Jagadeesh Menon VR, Division of Pediatric Gastroenterology, Postgraduate Institute of Medical Education and Research, Chandigarh, India in contributing to the preparation of the manuscript.
References
- 1.Roberts E.A., Schilsky M.L. AASLD practice guidelines: a practice guideline on Wilson disease. Hepatology. 2008;47:2094–2108. [Google Scholar]
- 2.EASL Clinical Practice Guidelines Wilson's disease European association for the study of the liver. J Hepatol. 2012;56:671–685. doi: 10.1016/j.jhep.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Socha P., Janczyk W., Dhawan A. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018;66:334–344. doi: 10.1097/MPG.0000000000001787. [DOI] [PubMed] [Google Scholar]
- 4.Nominal Group Technique – Centre for Disease Control (CDC) https://www.cdc.gov/healthyyouth/evaluation/pdf/brief7.pdf.
- 5.Atkins D., Best D., Briss P.A. Grading quality of evidence and strength of recommendation. BMJ. 2004;328(7454):1490. doi: 10.1136/bmj.328.7454.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walshe J.M. Wilson's disease: new oral therapy. Lancet. 1956;270(6906):25–26. doi: 10.1016/s0140-6736(56)91859-1. [DOI] [PubMed] [Google Scholar]
- 7.Walshe J.M. Management of penicillamine nephropathy in Wilson's disease: a new chelating agent. Lancet. 1969;2(7635):1401–1402. doi: 10.1016/s0140-6736(69)90940-4. [DOI] [PubMed] [Google Scholar]
- 8.Brewer G.J., Hill G.M., Prasad A.S. Oral zinc therapy for Wilson's disease. Ann Intern Med. 1983;99:314–320. doi: 10.7326/0003-4819-99-3-314. [DOI] [PubMed] [Google Scholar]
- 9.Brewer G.J., Dick R., Gurkin V.Y. Initial therapy of patients with Wilson's disease with tetrathiomolybdate. Arch Neurol. 1991;48:42–47. doi: 10.1001/archneur.1991.00530130050019. [DOI] [PubMed] [Google Scholar]
- 10.Liu J., Luan J., Zhou X., Cui Y., Han J. Epidemiology, diagnosis, and treatment of Wilson's disease. Intractable Rare Dis Res. 2017 Nov;6(4):249–255. doi: 10.5582/irdr.2017.01057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sternlieb I., Scheinberg I.H. Prevention of Wilson's disease in asymptomatic patients. N Eng J Med. 1968;278:352–359. doi: 10.1056/NEJM196802152780702. [DOI] [PubMed] [Google Scholar]
- 12.Lo C., Bandmann O. Epidemiology and introduction to the clinical presentation of Wilson disease. Handb Clin Neurol. 2017;142:7–17. doi: 10.1016/B978-0-444-63625-6.00002-1. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi Y., Aoki T., Arashima S. Mass screening for Wilson's disease: results and recommendations. Pediatr Int. 1999;41:405–408. doi: 10.1046/j.1442-200x.1999.01096.x. [DOI] [PubMed] [Google Scholar]
- 14.Taly A.B., Prashanth L.K., Sinha S. Wilson's disease: an indian perspective. Neurol India. 2009;57:528–540. doi: 10.4103/0028-3886.57789. [DOI] [PubMed] [Google Scholar]
- 15.Bost M., Houdart S., Oberli M. Dietary copper and human health: current evidence and unresolved issues. J Trace Elem Med Biol. 2016;35:107–115. doi: 10.1016/j.jtemb.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Squitti R., Siotto M., Polimanti R. Low-copper diet as a preventive strategy for Alzheimer's disease. Neurobiol Aging. 2014;35:S40–S50. doi: 10.1016/j.neurobiolaging.2014.02.031. [DOI] [PubMed] [Google Scholar]
- 17.De Bie P., Muller P., Wijmenga C. Molecular pathogenesis of Wilson and Menke's disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44:673–688. doi: 10.1136/jmg.2007.052746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lalioti V., Sandoval I., Cassio D. Molecular pathology of Wilson's disease: a brief. J Hepatol. 2010;53:1151–1153. doi: 10.1016/j.jhep.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Wu F., Wang J., Pu C. Wilson's disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015;16:6419–6431. doi: 10.3390/ijms16036419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walshe J.M., Yealland M. Wilson's disease: the problem of delayed diagnosis. J Neurol Neurosurg Psychiatry. 1992 Aug;55(8):692–696. doi: 10.1136/jnnp.55.8.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Svetel M., Potrebić A., Pekmezović T. Neuropsychiatric aspects of treated Wilson's disease. Parkinsonism Relat Disord. 2009;15:772–775. doi: 10.1016/j.parkreldis.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 22.Pandit A.N., Bavdekar A., Bhave S. Wilson's disease. Indian J Pediatr. 2002;69:785–791. doi: 10.1007/BF02723693. [DOI] [PubMed] [Google Scholar]
- 23.Kalra V., Khurana D., Mittal R. Wilson's disease-Early onset and lessons from a pediatric cohort in India. Indian Pediatr. 2000;37:595–601. [PubMed] [Google Scholar]
- 24.Brewer G.J. Kluwer Academic Publishers; Boston: 2001. Wilson's Disease: A Clinician's Guide to Recognition, Diagnosis, and Management. [Google Scholar]
- 25.Lin L., Wang D., Ding N. Hepatic manifestations in Wilson disease: report of 110 cases. Hepatogastroenterol. 2015;62:657–660. [PubMed] [Google Scholar]
- 26.Medici V., Trevisan C.P., D'Inca R. Diagnosis and management of Wilson's disease: results of a single center experience. J Clin Gastroenterol. 2006;40:936–941. doi: 10.1097/01.mcg.0000225670.91722.59. [DOI] [PubMed] [Google Scholar]
- 27.Kleine, Truffa R., Mendes Wilson disease: an analysis of 28 Brazilian children. Clinics (Sao Paulo). 2012;67:231–235. doi: 10.6061/clinics/2012(03)05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayes F., Karim A.B., Helaly L. Spectrum of hepatic presentation of Wilsons disease in children attending a tertiary care centre of Dhaka city. Bangladesh J Child Health. 2014;38:86–93. [Google Scholar]
- 29.Walshe J.M. The acute haemolytic syndrome in Wilson's disease—a review of 22 patients. QJM. 2013;106:1003–1008. doi: 10.1093/qjmed/hct137. [DOI] [PubMed] [Google Scholar]
- 30.Korman J.D., Volenberg I., Balko J. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology. 2008;48:1167–1174. doi: 10.1002/hep.22446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eisenbach C., Sieg O., Stremmel W. Diagnostic criteria for acute liver failure due to Wilson disease. World J Gastroenterol. 2007;13:1711–1714. doi: 10.3748/wjg.v13.i11.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thanapirom K., Treeprasertsuk S., Komolmit P. Comparison of long-term outcome of patients with Wilson's disease presenting with acute liver failure versus acute-on- chronic liver failure. J Med Assoc Thai. 2013;96:150–156. [PubMed] [Google Scholar]
- 33.Lal J., Thapa B.R., Rawal P. Predictors of outcome in acute-on- chronic liver failure in children. Hepatol Int. 2011;5:693–697. doi: 10.1007/s12072-010-9217-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alam S., Lal B.B., Sood V. Pediatric acute-on- chronic liver failure in a specialized liver unit: prevalence, profile, outcome, and predictive factors. J Pediatr Gastroenterol Nutr. 2016;63:400–405. doi: 10.1097/MPG.0000000000001179. [DOI] [PubMed] [Google Scholar]
- 35.Sokol R.J. Copper metabolism and copper storage disorders. In: Suchy F., Sokol R., Balistreri W., editors. Liver Disease in Children. 4th ed. Cambridge University Press; Cambridge (UK): 2014. pp. 465–492. [Google Scholar]
- 36.Bem R.S., Muzzillo D.A., Degutti M.M. Wilson disease in southern Brazil: a 40-year follow-up study. Clinics (Sao Paulo). 2011;66:411–416. doi: 10.1590/S1807-59322011000300008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lecca S., Pilloni L., Ambu R. Multiple histochemical methods in the diagnosis of Wilson's disease. Presentation of 74 cases and review of the literature. Pathologica. 1998;90:771–775. [PubMed] [Google Scholar]
- 38.Akpinar E., Akhan O. Liver imaging findings of Wilson's disease. Eur J Radiol. 2007;61:25–32. doi: 10.1016/j.ejrad.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Walshe J.M., Waldenström E., Sams V. Abdominal malignancies in patients with Wilson's disease. QJM. 2003;96:657–662. doi: 10.1093/qjmed/hcg114. [DOI] [PubMed] [Google Scholar]
- 40.Walshe J.M. Wilson disease. In: Vinken P.J., Bruyn G.W., Klawans H.L., editors. vol. 49. Elsevier; 1986. pp. 223–238. (Handbook of Clinical Neurology). [Google Scholar]
- 41.Brewer G.J., Yuzbasiyan-Gurkan V. Wilson disease. Medicine. 1992;71:139–164. doi: 10.1097/00005792-199205000-00004. [DOI] [PubMed] [Google Scholar]
- 42.Taly A.B., Meenakshi-Sundaram S., Sinha S. Wilson disease: description of 282 patients evaluated over 3 decades. Medicine. 2007;86:112–121. doi: 10.1097/MD.0b013e318045a00e. [DOI] [PubMed] [Google Scholar]
- 43.Cetlin R.S., Rodrigues G.R., Pena-Pereira M.A. Teaching video neuroimages: excessive grinning in Wilson disease. Neurology. 2009;73(14):e73. doi: 10.1212/WNL.0b013e3181bacef0. [DOI] [PubMed] [Google Scholar]
- 44.Prashanth L.K., Taly A.B., Sinha S. Wilson's disease: diagnostic errors and clinical implications. J Neurol Neurosurg Psychiatry. 2004 Jun;75(6):907–909. doi: 10.1136/jnnp.2003.026310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akil M., Brewer G.J. Psychiatric and behavioral abnormalities in Wilson's disease. Adv Neurol. 1995;65:171–178. [PubMed] [Google Scholar]
- 46.Scheinberg I.H., Sternlieb I., editors. Wilson's Disease in: Major Problems in Internal Medicine. W B Saunders; Philadelphia: 1984. [Google Scholar]
- 47.Ala A., Walker A.P., Ashkan K. Wilson's disease. Lancet. 2007;369(9559):397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 48.Merle U., Schaefer M., Ferenci P. Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study. Gut. 2007;56(1):115–120. doi: 10.1136/gut.2005.087262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scwartz M., Feics S., Polak H. Psychiatric manifestations in Wilson's disease. Harefuah. 1993;124:75–77. [PubMed] [Google Scholar]
- 50.Shanmugiah A., Sinha S., Taly A.B. Psychiatric manifestations in Wilson's disease: a cross-sectional analysis. J Neuropsychiatry Clin Neurosci. 2008;20(1):81–85. doi: 10.1176/jnp.2008.20.1.81. [DOI] [PubMed] [Google Scholar]
- 51.Dening T.R., Berrios G.E. Wilson's disease: a prospective study of psychopathology in 31 cases. Br J Psychiatry. 1989;155:206–213. doi: 10.1192/bjp.155.2.206. [DOI] [PubMed] [Google Scholar]
- 52.Dening T.R., Berrios G.E. Wilson's disease. Psychiatric symptoms in 195 cases. Arch Gen Psychiatry. 1989;46(12):1126–1134. doi: 10.1001/archpsyc.1989.01810120068011. [DOI] [PubMed] [Google Scholar]
- 53.Dening T.R., Berrios G.E. Wilson's disease: a longitudinal study of psychiatric symptoms. Biol Psychiatry. 1990;28(3):255–265. doi: 10.1016/0006-3223(90)90581-l. [DOI] [PubMed] [Google Scholar]
- 54.Pfeiffer R.F. Wilson's disease. Hand Clin Neurol. 2011;100:681–709. doi: 10.1016/B978-0-444-52014-2.00049-5. [DOI] [PubMed] [Google Scholar]
- 55.Nagral A., Jhaveri A., Nalawade S. Kayser–Fleischer rings or bile pigment rings? Indian J Gastroenterol. 2015;34:410–412. doi: 10.1007/s12664-015-0603-2. [DOI] [PubMed] [Google Scholar]
- 56.Fenu M., Liggi M., Demelia E. Kayser-Fleischer ring in Wilson's disease: a cohort study. Eur J Intern Med. 2012;23:e150–e156. doi: 10.1016/j.ejim.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 57.Schilsky M.L., Scheinberg I.H., Sternlieb I. Liver transplantation for Wilson's disease: indications and outcome. Hepatology. 1994;19:583–587. doi: 10.1002/hep.1840190307. [DOI] [PubMed] [Google Scholar]
- 58.Langwinska-Wosko E., Litwin T., Dziezyc K. The sunflower cataract in Wilson disease: pathognomonic sign or rare finding? Acta Neurol Belg. 2016;116:325–328. doi: 10.1007/s13760-015-0566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sarles J., Durand J.M., Scheiner C., Picon G. Wilson disease, IgA glomerulonephritis and vascular purpura: an incidental association? Arch Fr Pediatr. 1993;50:501–504. [PubMed] [Google Scholar]
- 60.Sozeri E., Feist D., Ruder H. Proteinuria and other renal functions in Wilson's disease. Pediatr Nephrol. 1997;11:307–311. doi: 10.1007/s004670050282. [DOI] [PubMed] [Google Scholar]
- 61.Sinha R., Akhtar S. Gross hematuria in a case of Wilson disease: Answers. Pediatr Nephrol. 2012;27:919–920. doi: 10.1007/s00467-011-2011-x. [DOI] [PubMed] [Google Scholar]
- 62.Aggarwal A., Bhatt M. Update on Wilson disease. Int Rev Neurobiol. 2013;110:313–348. doi: 10.1016/B978-0-12-410502-7.00014-4. [DOI] [PubMed] [Google Scholar]
- 63.Selimoglu M.A., Ertekin V., Doneray H. Bone mineral density of children with Wilson disease: efficacy of penicillamine and zinc therapy. J Clin Gastroenterol. 2008;42:194–198. doi: 10.1097/MCG.0b013e318032388d. [DOI] [PubMed] [Google Scholar]
- 64.Hegedus D., Ferencz V., Lakatos P.L. Decreased bone density, elevated serum osteoprotegerin, and beta-cross-laps in Wilson disease. J Bone Miner Res. 2002;17:1961–1967. doi: 10.1359/jbmr.2002.17.11.1961. [DOI] [PubMed] [Google Scholar]
- 65.Nagral A., Sathe K. Reversal of severe Wilson arthropathy by liver transplantation. Indian Pediatr. 2011;48:406–407. [PubMed] [Google Scholar]
- 66.Kuan P. Cardiac Wilson's disease. Chest. 1987;91:579–583. doi: 10.1378/chest.91.4.579. [DOI] [PubMed] [Google Scholar]
- 67.Leu M.L., Strickland T., Wang C.C. Skin pigmentation in Wilson's disease. J Am Med Assoc. 1970;211:1542–1543. [PubMed] [Google Scholar]
- 68.Krysiak R., Handzlik-Orlik G., Okopien B. Endocrine symptoms as the initial manifestation of Wilson's disease. Yale J Biol Med. 2012;85:249–254. [PMC free article] [PubMed] [Google Scholar]
- 69.Lee B.H., Kim J.H., Lee S.Y. Distinct clinical courses according to presenting phenotypes and their correlation to ATP7B mutations in a large Wilson's disease cohort. Liver Int. 2011;31:831–839. doi: 10.1111/j.1478-3231.2011.02503.x. [DOI] [PubMed] [Google Scholar]
- 70.Cheng N., Wang H., Wu W. Spectrum of ATP7B mutations and genotype-phenotype correlation in large-scale Chinese patients with Wilson disease. Clin Genet. 2017;92:69–79. doi: 10.1111/cge.12951. [DOI] [PubMed] [Google Scholar]
- 71.Gromadzka G., Schmidt H.H., Genschel J. Frameshift and non- sense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson's disease. Clin Genet. 2005;68:524–532. doi: 10.1111/j.1399-0004.2005.00528.x. [DOI] [PubMed] [Google Scholar]
- 72.Merle U., Eisenbach C., Weiss K.H. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson's disease. J Hepatol. 2009;51:925–930. doi: 10.1016/j.jhep.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 73.Seo Jeong Kee. Diagnosis of Wilson disease in young children: molecular genetic testing and a paradigm shift from the laboratory diagnosis. Pediatr Gastroenterol Hepatol Nutr. 2012;15(4):197–209. doi: 10.5223/pghn.2012.15.4.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steindl P., Ferenci P., Dienes H.P. Wilson's disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology. 1997;113:212–218. doi: 10.1016/s0016-5085(97)70097-0. [DOI] [PubMed] [Google Scholar]
- 75.Rosencrantz R.A., Schilsky M.L. Mining for a diagnosis of Wilson's disease in children: genetics, score and ore. Hepatology. 2010;52:1872–1874. doi: 10.1002/hep.24054. [DOI] [PubMed] [Google Scholar]
- 76.Nicastro E., Ranucci G., Vajro P. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology. 2010;52:1948–1956. doi: 10.1002/hep.23910. [DOI] [PubMed] [Google Scholar]
- 77.Dhawan A., Taylor R.M., Cheeseman P. Wilson's disease in children: 37-year experience and revised King's score for liver transplantation. Liver Transpl. 2005;11:441–448. doi: 10.1002/lt.20352. [DOI] [PubMed] [Google Scholar]
- 78.Schilsky M.L. Non-invasive testing for Wilson disease: revisiting the d-penicillamine challenge test. J Hepatol. 2007;47:172–173. doi: 10.1016/j.jhep.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 79.El Balkhi S., Trocello J.M., Poupon J. Relative exchangeable copper: a new highly sensitive and highly specific biomarker for Wilson's disease diagnosis. Clin Chim Acta. 2011;412:2254–2260. doi: 10.1016/j.cca.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 80.Pilloni L., Lecca S., Van Eyken P. Value of histochemical stains for copper in the diagnosis of Wilson's disease. Histopathology. 1998;33:28–33. doi: 10.1046/j.1365-2559.1998.00455.x. [DOI] [PubMed] [Google Scholar]
- 81.Ludwig J., Moyer T.P., Rakela J. The liver biopsy diagnosis of Wilson's disease. Methods in pathology. Am J Clin Pathol. 1994;102:443–446. doi: 10.1093/ajcp/102.4.443. [DOI] [PubMed] [Google Scholar]
- 82.Araya M., Koletzko B., Uauy R. Copper deficiency and excess in infancy: developing a research agenda. J Pediatr Gastroenterol Nutr. 2003;37:422–429. doi: 10.1097/00005176-200310000-00005. [DOI] [PubMed] [Google Scholar]
- 83.Sato C., Koyama H., Satoh H. Concentrations of copper and zinc in liver and serum samples in biliary atresia patients at different stages of traditional surgeries. Tohoku J Exp Med. 2005;207:271–277. doi: 10.1620/tjem.207.271. [DOI] [PubMed] [Google Scholar]
- 84.Bayliss E.A., Hambidge K.M., Sokol R.J. Hepatic concentrations of zinc, copper and manganese in infants with extrahepatic biliary atresia. J Trace Elem Med Biol. 1995;9:40–43. doi: 10.1016/S0946-672X(11)80007-6. [DOI] [PubMed] [Google Scholar]
- 85.Sinha S., Taly A.B., Ravishankar S. Wilson's disease: cranial MRI observations and clinical correlation. Neuroradiology. 2006;48:613–621. doi: 10.1007/s00234-006-0101-4. [DOI] [PubMed] [Google Scholar]
- 86.Prashanth L.K., Sinha S., Taly A.B. Do MRI features distinguish Wilson's disease from other early onset extrapyramidal disorders? An analysis of 100 cases. Mov Disord. 2010;25:672–678. doi: 10.1002/mds.22689. [DOI] [PubMed] [Google Scholar]
- 87.Kumar S.S.1, Kurian G., Eapen C.E. Genetics of Wilson's disease: a clinical perspective. Indian J Gastroenterol. 2012;31:285–293. doi: 10.1007/s12664-012-0237-6. [DOI] [PubMed] [Google Scholar]
- 88.Ferenci P., Caca K., Loudianos G. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 89.Nayak N.C., Chitale A.R. Indian childhood cirrhosis(ICC) and ICC- like diseases: the changing scenario of facts versus notions. Indian J Med Res. 2013;137:1029–1042. [PMC free article] [PubMed] [Google Scholar]
- 90.Ramakrishna B., Date A., Kirubakaran C. Atypical copper cirrhosis in Indian children. Ann Trop Paediatr. 1995;15:237–242. doi: 10.1080/02724936.1995.11747778. [DOI] [PubMed] [Google Scholar]
- 91.Møller L.B., Horn N., Jeppesen T.D. Clinical presentation and mutations in Danish patients with Wilson disease. Eur J Hum Genet. 2011;19:935–941. doi: 10.1038/ejhg.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gromadzka G., Schmidt H.H., Genschel J. p.H1069Q mutation in ATP7B and biochemical parameters of copper metabolism and clinical manifestation of Wilson's disease. Mov Disord. 2006;21:245–248. doi: 10.1002/mds.20671. [DOI] [PubMed] [Google Scholar]
- 93.Gupta A., Aikath D., Neogi R. Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype-phenotype correlation in Indian patients. Hum Genet. 2005;118:49–57. doi: 10.1007/s00439-005-0007-y. [DOI] [PubMed] [Google Scholar]
- 94.Aggarwal A., Chandhok G., Todorov T. Wilson disease mutation pattern with genotype-phenotype correlations from Western India: confirmation of p.C271* as a common Indian mutation and identification of 14 novel mutations. Ann Hum Genet. 2013;77:299–307. doi: 10.1111/ahg.12024. [DOI] [PubMed] [Google Scholar]
- 95.Mukherjee S., Dutta S., Majumdar S. Genetic defects in Indian Wilson disease patients and genotype-phenotype correlation. Parkinsonism Relat Disord. 2014;20:75–81. doi: 10.1016/j.parkreldis.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 96.Santhosh S., Shaji R.V., Eapen C.E. ATP7B mutations in families in a predominantly Southern Indian cohort of Wilson's disease patients. Indian J Gastroenterol. 2006;25:277–282. [PubMed] [Google Scholar]
- 97.Cossu P., Pirastu M., Nucaro A. Prenatal diagnosis of Wilson's disease by analysis of DNA polymorphism. N Engl J Med. 1992;327:57. doi: 10.1056/NEJM199207023270116. [DOI] [PubMed] [Google Scholar]
- 98.Kumar S., Thapa B.R., Kaur G., Prasad R. Familial gene analysis for Wilson disease from north-west Indian patients. Ann Hum Biol. 2006;33:177–186. doi: 10.1080/03014460500503275. [DOI] [PubMed] [Google Scholar]
- 99.Rosencrantz R., Schilsky M. Wilson disease: pathogenesis and clinical considerations in diagnosis and treatment. Semin Liver Dis. 2011;31:245–259. doi: 10.1055/s-0031-1286056. [DOI] [PubMed] [Google Scholar]
- 100.Trocello J.M., El Balkhi S., Woimant F. Relative exchangeable copper: a promising tool for family screening in Wilson disease. Mov Disord. 2014;29:558–562. doi: 10.1002/mds.25763. [DOI] [PubMed] [Google Scholar]
- 101.Kukovertz W.R., Beubler E., Kreuzig F. Bioavailability and pharmacokinetics of D-penicillamine. J Rheumatol. 1983;10:90–94. [PubMed] [Google Scholar]
- 102.Durand F., Bernuau J., Giostra E. Wilson's disease with severe hepatic insufficiency: beneficial effects of early administration of D-penicillamine. Gut. 2001;48:849–852. doi: 10.1136/gut.48.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brewer G.J., Terry C.A., Aisen A.M. Worsening of neurologic syndrome in patients with Wilson's disease with initial penicillamine therapy. Arch Neurol. 1987;44:490–493. doi: 10.1001/archneur.1987.00520170020016. [DOI] [PubMed] [Google Scholar]
- 104.Walshe J.M., Yealland M. Chelation treatment of neurological Wilson's disease. QJM. 1993;86:197–204. [PubMed] [Google Scholar]
- 105.Kalita J., Kumar V., Chandra S. Worsening of Wilson disease following penicillamine therapy. Eur Neurol. 2014;71:126–131. doi: 10.1159/000355276. [DOI] [PubMed] [Google Scholar]
- 106.Gibbs K.R., Walshe J.M. Penicillamine and pyridoxine requirements in man. Lancet. 1966;309:175–179. doi: 10.1016/s0140-6736(66)90700-8. [DOI] [PubMed] [Google Scholar]
- 107.Walshe J.M. Wilson's disease presenting with features of hepatic dysfunction: a clinical analysis of eighty-seven patients. QJM. 1989;70:253–263. [PubMed] [Google Scholar]
- 108.Manolaki N., Nikolopoulou G., Daikos G.L. Wilson disease in children: analysis of 57 cases. J Pediatr Gastroenterol Nutr. 2008;48:72–77. doi: 10.1097/MPG.0b013e31817d80b8. [DOI] [PubMed] [Google Scholar]
- 109.Pavithra S., Rao S., Vishal B. D-penicillamine induced elastosis perforans serpiginosa mimicking acne keloidalis nuchae. Indian J Dermatol. 2011;56:449–450. doi: 10.4103/0019-5154.84730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Scheinberg I.H., Jaffe M.E., Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson's disease. N Engl J Med. 1987;317:209–213. doi: 10.1056/NEJM198707233170405. [DOI] [PubMed] [Google Scholar]
- 111.Saito H., Watanbe K., Sahara M. Triethylene-tetramine (trien) therapy for Wilson's disease. Tohoku J Exp Med. 1991;164:29–35. doi: 10.1620/tjem.164.29. [DOI] [PubMed] [Google Scholar]
- 112.Santos Silva E.E., Sarles J., Buts J.P. Successful medical treatment of severely decompensated Wilson disease. J Pediatr. 1996;128:285–287. doi: 10.1016/s0022-3476(96)70412-2. [DOI] [PubMed] [Google Scholar]
- 113.Cousins R.J. Absorption, transport and hepatic metabolism of copper and zinc: special reference to metallothionein and ceruloplasmin. Physiol Rev. 1985;65:238–309. doi: 10.1152/physrev.1985.65.2.238. [DOI] [PubMed] [Google Scholar]
- 114.Schilsky M., Blank R.R., Czaja M.J. Hepatocellular copper toxicity and its attenuation by zinc. J Clin Invest. 1989;84:1562–1568. doi: 10.1172/JCI114333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hill G.M., Brewer G.J., Prasad A.S. Treatment of Wilson's disease with zinc I. Oral zinc therapy regimens. Hepatology. 1987;7:522–528. doi: 10.1002/hep.1840070318. [DOI] [PubMed] [Google Scholar]
- 116.Wiernicka A., Janczyk W., Dądalski M. Gastrointestinal side effects in children with Wilson's disease treated with zinc sulphate. World J Gastroenterol. 2013;19:4356–4362. doi: 10.3748/wjg.v19.i27.4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Czlonkowska A., Gajda J., Rodo M. Effects of long term treatment in Wilson's disease with D-penicillamine and zinc sulphate. J Neurol. 1996;243:269–273. doi: 10.1007/BF00868525. [DOI] [PubMed] [Google Scholar]
- 118.Hoogenraad T.U., Van Hattum J., Van den Hamer C.J. Management of Wilson's disease with zinc sulphate. Experience in a series of 27 patients. J Neurol Sci. 1987;77:137–146. doi: 10.1016/0022-510x(87)90116-x. [DOI] [PubMed] [Google Scholar]
- 119.Brewer G.J., Dick R.D., Johnson V.D. Treatment of Wilson's disease with zinc: XV Long-term follow-up studies. J Lab Clin Med. 1998;132:264–278. doi: 10.1016/s0022-2143(98)90039-7. [DOI] [PubMed] [Google Scholar]
- 120.Pan Q., Kleer C.G., van Golen K.L. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62:4854–4859. [PubMed] [Google Scholar]
- 121.Brewer G.J. Copper lowering therapy with tetrathiomolybdate as an antiangiogenic strategy in cancer. Curr Cancer Drug Targets. 2005;5:195–202. doi: 10.2174/1568009053765807. [DOI] [PubMed] [Google Scholar]
- 122.Brewer G.J., Hedera P., Kluin K.J. Treatment of Wilson disease with ammonium tetrathiomolybdate: III. Initial therapy in a total of 55 neurologically affected patients and follow-up with zinc therapy. Arch Neurol. 2003;60:379–385. doi: 10.1001/archneur.60.3.379. [DOI] [PubMed] [Google Scholar]
- 123.Brewer G.J., Askari F., Lorincz M.T. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63:521–527. doi: 10.1001/archneur.63.4.521. [DOI] [PubMed] [Google Scholar]
- 124.Weiss K.H., Askari F.K., Czlonkowska A. Bis-choline tetrathiomolybdate in patients with Wilson's disease: an open-label, multicenter, phase 2 study. Lancet Gastroenterol Hepatol. 2017;2:869–876. doi: 10.1016/S2468-1253(17)30293-5. [DOI] [PubMed] [Google Scholar]
- 125.Wiggelinkhuizen M., Tilanus M.E., Bollen C.W., Houwen R.H. Systematic review: clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease. Aliment Pharmacol Ther. 2009;29:947–958. doi: 10.1111/j.1365-2036.2009.03959.x. [DOI] [PubMed] [Google Scholar]
- 126.Członkowska A., Litwin T., Karliński M. D-penicillamine versus zinc sulfate as first-line therapy for Wilson's disease. Eur J Neurol. 2014;21:599–606. doi: 10.1111/ene.12348. [DOI] [PubMed] [Google Scholar]
- 127.Stange J., Mitzner S.R., Risler T. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs. 1999;23:319–330. doi: 10.1046/j.1525-1594.1999.06122.x. [DOI] [PubMed] [Google Scholar]
- 128.Sen S., Felldin M., Steiner C. Albumin dialysis and Molecular Adsorbents Recirculating System (MARS) for acute Wilson's disease. Liver Transpl. 2002;8:962–967. doi: 10.1053/jlts.2002.35546. [DOI] [PubMed] [Google Scholar]
- 129.Sarles J., Lefevre P., Picon G. Plasma exchange for fulminant Wilson disease. Eur J Pediatr. 1992;151:310. doi: 10.1007/BF02072237. [DOI] [PubMed] [Google Scholar]
- 130.Pham H.P., Schwartz J., Cooling L. Report of the ASFA apheresis registry study on Wilson's disease. J Clin Apher. 2016;31:11–15. doi: 10.1002/jca.21396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nazer H., Ede R.J., Mowat A.P. Wilson's disease: clinical presentation and use of prognostic index. Gut. 1986;27:1377. doi: 10.1136/gut.27.11.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Devarbhavi H., Singh R., Adarsh C.K. Factors that predict mortality in children with Wilson disease associated acute liver failure and comparison of Wilson disease specific prognostic indices. J Gastroenterol Hepatol. 2014;29:380–386. doi: 10.1111/jgh.12356. [DOI] [PubMed] [Google Scholar]
- 133.Fischer R.T., Soltys K.A., Squires R.H. Prognostic scoring indices in Wilson disease: a case series and cautionary tale. J Pediatr Gastroenterol Nutr. 2011;52:466–469. doi: 10.1097/MPG.0b013e31820b0211. [DOI] [PubMed] [Google Scholar]
- 134.Guillaud O., Dumortier J., Sobesky R. Long term results of liver transplantation for Wilson's disease: experience in France. J Hepatol. 2014;60:579–589. doi: 10.1016/j.jhep.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 135.Stracciari A., Tempestini A., Borghi A. Effect of liver transplantation on neurological manifestations in Wilson disease. Arch Neurol. 2000;57:384–386. doi: 10.1001/archneur.57.3.384. [DOI] [PubMed] [Google Scholar]
- 136.Medici V., Mirante V.G., Fassati L.R. Liver transplantation for Wilson's disease: the burden of neurological and psychiatric disorders. Liver Transpl. 2005;11:1056–1063. doi: 10.1002/lt.20486. [DOI] [PubMed] [Google Scholar]
- 137.Aggarwal A., Aggarwal N., Nagral A. A novel global assessment scale for Wilson's disease (GAS for WD) Mov Disord. 2009 15;24:509–518. doi: 10.1002/mds.22231. [DOI] [PubMed] [Google Scholar]
- 138.Dzieżyc K., Litwin T., Chabik G. Measurement of urinary copper excretion after 48-h d-penicillamine cessation as a compliance assessment in Wilson's disease. Funct Neurol. 2015;30:264–268. doi: 10.11138/FNeur/2015.30.4.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weiss K.H., Thurik F., Gotthardt D.N., EUROWILSON Consortium Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11:1028–1035. doi: 10.1016/j.cgh.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 140.Beinhardt S., Leiss W., Stättermayer A.F. Long-term outcomes of patients with Wilson disease in a large Austrian cohort. Clin Gastroenterol Hepatol. 2014;12:683–689. doi: 10.1016/j.cgh.2013.09.025. [DOI] [PubMed] [Google Scholar]
- 141.Kumar N., Joshi D., Ansari A.Z. Clinical, biochemical and radiological profile of Wilson's disease from a tertiary care referral center in India. Arch Med. 2015;7:1–7. [Google Scholar]
- 142.Prashanth L.K., Taly A.B., Sinha S. Prognostic factors in patients presenting with severe neurological forms of Wilson's disease. QJM. 2005;98:557–563. doi: 10.1093/qjmed/hci095. [DOI] [PubMed] [Google Scholar]
- 143.Holscher S., Leinweber B., Hefter H. Evaluation of the symptomatic treatment of residual neurological symptoms in Wilson disease. Eur Neurol. 2010;64:83–87. doi: 10.1159/000316066. [DOI] [PubMed] [Google Scholar]
- 144.Teive H.A., Kluppel L.E., Munhoz R.P. Jaw-opening oromandibular dystonia secondary to Wilson's disease treated with botulinum toxin type A. Arq Neuropsiquiatr. 2012;70:407–409. doi: 10.1590/s0004-282x2012000600005. [DOI] [PubMed] [Google Scholar]
- 145.Lee S.Y., Yang H.E., Yang H.S. Neuromuscular electrical stimulation therapy for dysphagia caused by Wilson's disease. Ann Rehabil Med. 2012;36:409–413. doi: 10.5535/arm.2012.36.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brewer G.J., Gurkan V.Y., Dick R., Wang Y., Johnson V. Does a vegetarian diet control Wilson's disease? J Am Coll Nutr. 1993;12:527–530. doi: 10.1080/07315724.1993.10718347. [DOI] [PubMed] [Google Scholar]
- 147.Babaji P., Shashikiran N., Reddy S. Comparative evaluation of trace elements and residual bacterial content of different brands of bottled waters. J Indian Soc Pedod Prev Dent. 2004;22:201–204. [PubMed] [Google Scholar]
- 148.Pandit A., Bhave S. Present interpretation of the role of copper in Indian childhood cirrhosis. Am J Clin Nutr. 1996;63:830S–835S. doi: 10.1093/ajcn/63.5.830. [DOI] [PubMed] [Google Scholar]
- 149.Klee J.G. Undiagnosed Wilson's disease as cause of unexplained miscarriage. Lancet. 1979;2:423. doi: 10.1016/s0140-6736(79)90440-9. [DOI] [PubMed] [Google Scholar]
- 150.Scheinberg I.H., Sternlieb I. Pregnancy in penicillamine-treated patients with Wilson's disease. N Engl J Med. 1975;293:1300–1302. doi: 10.1056/NEJM197512182932507. [DOI] [PubMed] [Google Scholar]
- 151.Brewer G.J., Johnson V.D., Dick R.D., Hedera P., Fink J.K., Kluin K.J. Treatment of Wilson's disease with zinc. XVII: treatment during pregnancy. Hepatology. 2000;31:364–370. doi: 10.1002/hep.510310216. [DOI] [PubMed] [Google Scholar]
- 152.Pfeiffenberger J., Beinhardt S., Gotthardt D.N. Pregnancy in Wilson disease:management and outcome. Hepatology. 2018;67:1261–1269. doi: 10.1002/hep.29490. [DOI] [PubMed] [Google Scholar]
- 153.Keen C.L., Mark-Savage P., Lonnerdal B. Teratogenic effects of D penicillamine in rats: relation to copper deficiency. Drug-Nutr Interact. 1983;2:17–34. [PubMed] [Google Scholar]
- 154.Pinter R., Hogge W.A., McPherson E. Infant with severe penicillamine embryopathy born to a woman with Wilson disease. Am J Med Genet. 2004;128a:294–298. doi: 10.1002/ajmg.a.10871. [DOI] [PubMed] [Google Scholar]
- 155.Walshe J.M. Pregnancy in Wilson's disease. QJM. 1977;46:73–83. [PubMed] [Google Scholar]
- 156.Kaushansky A., Frydman M., Kaufman H., Homburg R. Endocrine studies of the ovulatory disturbances in Wilson's disease (hepatolenticular degeneration) Fertil Steril. 1987;47:270–273. doi: 10.1016/s0015-0282(16)50004-1. [DOI] [PubMed] [Google Scholar]
- 157.Murillo O., Luqui D.M., Gazquez C. Long-term metabolic correction of Wilson's disease in a murine model by gene therapy. J Hepatol. 2016;64:419–426. doi: 10.1016/j.jhep.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 158.Filippi C., Dhawan A. Current status of human hepatocyte transplantation and its potential for Wilson's disease. Ann N Y Acad Sci. 2014;1315:50–55. doi: 10.1111/nyas.12386. [DOI] [PubMed] [Google Scholar]