

Summary
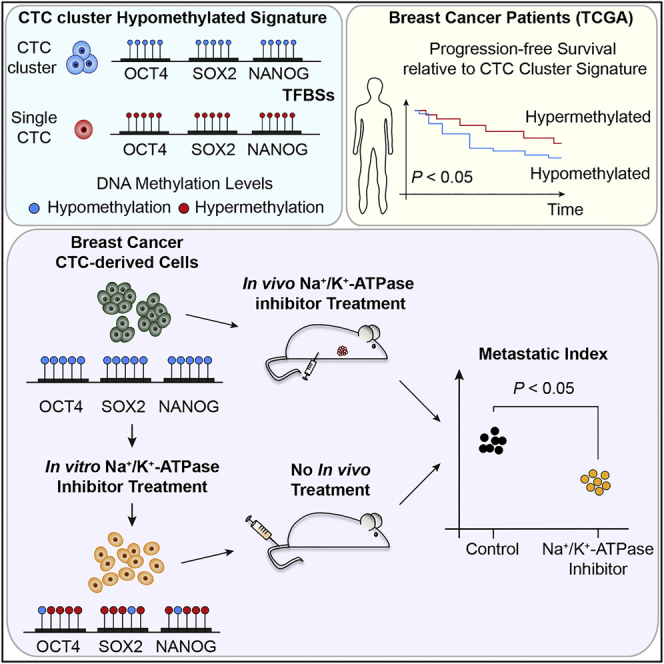
The ability of circulating tumor cells (CTCs) to form clusters has been linked to increased metastatic potential. Yet biological features and vulnerabilities of CTC clusters remain largely unknown. Here, we profile the DNA methylation landscape of single CTCs and CTC clusters from breast cancer patients and mouse models on a genome-wide scale. We find that binding sites for stemness- and proliferation-associated transcription factors are specifically hypomethylated in CTC clusters, including binding sites for OCT4, NANOG, SOX2, and SIN3A, paralleling embryonic stem cell biology. Among 2,486 FDA-approved compounds, we identify Na+/K+ ATPase inhibitors that enable the dissociation of CTC clusters into single cells, leading to DNA methylation remodeling at critical sites and metastasis suppression. Thus, our results link CTC clustering to specific changes in DNA methylation that promote stemness and metastasis and point to cluster-targeting compounds to suppress the spread of cancer.
Keywords: circulating tumor cells, circulating tumor cell clusters, single cell sequencing, bisulfite sequencing, RNA sequencing, stemness-associated transcription factors, proliferation-associated transcription factors, drug screen
Graphical Abstract
Highlights
-
•
Binding sites for OCT4, SOX2, NANOG, and SIN3A are hypomethylated in CTC clusters
-
•
CTC cluster hypomethylation profile correlates with a poor prognosis in breast cancer
-
•
Treatment with FDA-approved Na+/K+-ATPase inhibitors dissociates CTC clusters
-
•
Dissociation reverts the methylation profile of CTC clusters and suppresses metastasis
A comparative analysis of the methylation landscape of single and clusters of circulating tumor cells reveals patterns of similarity to embryonic stem cells and identifies pharmacological agents that can target clustering, suppress stemness, and blunt metastatic spreading.
Introduction
Circulating tumor cells (CTCs) are those cells that depart from cancerous lesions and enter the bloodstream (Alix-Panabières and Pantel, 2013). Although extraordinarily rare compared with blood cells and forced to strive for survival in circulation, CTCs are considered to be precursors of metastasis in various cancer types, including breast cancer (Aceto et al., 2015, Alix-Panabières and Pantel, 2014). CTCs are found in the blood of cancer patients as single CTCs and CTC clusters (Fidler, 1973, Liotta et al., 1976), with the latter featuring a higher ability to seed metastasis (Aceto et al., 2014). However, it is unknown what drives their enhanced metastatic potential and what are the vulnerabilities of clustered CTCs.
Abnormal DNA methylation patterns, including both genome-wide hypomethylation and hypermethylation, have been associated with several human cancers (Ehrlich, 2002, Ehrlich, 2009, Feinberg et al., 2006, Klutstein et al., 2016, Lee et al., 2006). Generally, these cancer-associated epigenetic modifications appear to affect distinct genomic areas, with hypomethylation favoring regulatory and repetitive elements versus hypermethylation, which is more frequent in CpG islands (Ehrlich, 2002). Both modifications have the ability to alter the expression of neighboring genes and contribute to the cancer phenotype (Ehrlich, 2009, Klutstein et al., 2016). For regulatory elements, loss of DNA methylation at transcription factor binding sites (TFBSs) can designate active transcription factor networks or networks primed for activation at later stages, e.g., during processes such as the derivation of induced pluripotent stem cells from differentiated cells (Lee et al., 2014) or cancer progression (Feinberg and Vogelstein, 1983). Although DNA methylation analysis of primary tumors is extensively investigated (Feinberg and Vogelstein, 1983, Klutstein et al., 2016), the forces that shape the DNA methylome during metastatic dissemination are largely uncharacterized.
Here, we combine microfluidic-based CTC capture from breast cancer patients and mouse models, single-cell resolution DNA methylation and RNA expression analysis, a drug screen with 2,486 FDA-approved compounds, and functional validation studies in mouse models to gain insights into the biology and vulnerabilities of CTC clusters. Our study provides a genome-wide DNA methylation landscape of single and clustered CTCs in breast cancer, highlighting fundamental differences that affect metastasis and enabling the identification of cluster-targeting compounds with immediate clinical applicability.
Results
Identification of Differentially Methylated Regions in CTC Clusters and Single CTCs
We first sought to identify active transcription factor networks by means of accessible TFBSs in single and clustered human breast CTCs, matched within individual liquid biopsies, through a genome-wide single-cell resolution DNA methylation analysis. To this end, blood samples were drawn from 43 patients with progressive breast cancer and processed with the Parsortix device (Xu et al., 2015), a microfluidic technology that allows a size-based, antigen-agnostic enrichment of CTCs from unprocessed blood samples, specifically adapted to achieve a capture rate of >97.2% for single CTCs and >99.3% for CTC clusters, and no artificial cluster formation during sample processing (Figures S1A and S1B). Upon capture, live CTCs were stained for cell surface expression of EpCAM, HER2, and EGFR, and counterstained with antibodies against CD45 to identify contaminant leukocytes (Figure S1C). Upon staining verification, we identified matched single and clustered CTCs in 19% of the analyzed samples (8/43 patients), and a total of 18 marker-positive single CTCs and 29 marker-positive CTC clusters from four patients were individually micromanipulated and deposited in lysis buffer for single-cell resolution whole-genome bisulfite sequencing (Table S1) (Farlik et al., 2015, Farlik et al., 2016). In parallel, we isolated spontaneously generated GFP-labeled single CTCs and CTC clusters from three mouse xenograft models, including two human breast CTC-derived cell lines (BR16 and BRx50) and the human breast cancer cell line MDA-MB 231 (lung metastatic variant, referred to as LM2) (Minn et al., 2005, Yu et al., 2014). In this setting, we individually micromanipulated 71 single CTCs and 48 CTC clusters (Table S1) and also processed them for single-cell resolution whole-genome bisulfite sequencing (Farlik et al., 2015, Farlik et al., 2016). Samples with a low coverage (< 1,000 unique CpGs) or a low bisulfite conversion efficiency (CG/CHG/CHH < 97%)—corresponding to 10.7% of patient-derived samples and 0.8% of xenograft-derived samples—were excluded from the analysis, resulting in a total of 89 single CTCs and 71 CTC clusters from patients and xenografts. On average, we achieved 3.68% CpG coverage for single CTCs and 5.86% CpG coverage for CTC clusters, in line with recent single-cell whole-genome bisulfite sequencing studies (Farlik et al., 2015, Farlik et al., 2016) (Figures S1D and S1E; Table S2). As expected, principal component analysis (PCA) mainly segregated CTCs based on the patient of origin or the specific xenograft model (Figures S1F and S1G). Metagene plot of CpG methylation revealed comparable methylation levels between single CTCs and CTC clusters across CpG islands, gene bodies, upstream (promoters) and downstream regions, including a drop of CpG methylation around the transcriptional start site, as expected (Figures S1H and S1I). We then specifically investigated differentially methylated regions (DMRs) between single CTCs and CTC clusters, evaluating average methylation levels in overlapping 5-kb windows, as previously established for single-cell DMR analysis (Farlik et al., 2015, Farlik et al., 2016). For patient-derived CTCs, with this approach we identified 3,347 DMRs with a ≥ 80% methylation difference between single CTCs and CTC clusters. Of these, 1,305 regions were hypomethylated in CTC clusters and 2,042 were hypomethylated in single CTCs (Figure 1A). We then looked at xenograft-derived CTCs, and to evaluate a comparable number of DMRs as found in patients, we assessed overlapping regions with a ≥ 70% methylation difference between single CTCs and CTC clusters. We found a total of 1,430 DMRs, of which 909 hypomethylated in CTC clusters and 521 hypomethylated in single CTCs (Figure 1B). We then analyzed DMRs from both patient- and xenograft-derived CTCs using i-cisTarget (Herrmann et al., 2012). With this analysis, among hypomethylated regions that are specific to either single CTCs or CTC clusters, we found a significant enrichment for several TFBSs, many of which overlapped between patient- and xenograft-derived CTCs (Figures 1C and 1D), thus allowing us to define specific hypomethylated TFBSs that globally characterize either single CTCs or CTC clusters in both patients and xenografts. Integrated gene ontology (GO) and pathway analysis of global CTC cluster hypomethylated TFBSs revealed a remarkable enrichment for stemness-related transcription factors that coordinately regulate proliferation and pluripotency, including OCT4, NANOG, SOX2, and SIN3A, paralleling embryonic stem cell (ESCs) biology (Figure 1E) (Kim et al., 2008, McDonel et al., 2012, Niwa, 2007, Saunders et al., 2017, van den Berg et al., 2010). Differently, single CTCs featured hypomethylation of other TFBSs, including those that are occupied by MEF2C, JUN, MIXL1, and SHOX2, commonly enriched in various cancers (Hong et al., 2014, Jiao et al., 2010, Laszlo et al., 2015, Raymond et al., 2014), yet independent of a core pluripotency network (Figure 1F) (Kim et al., 2008, Kim et al., 2010). To gain insights into more subtle changes in DNA methylation occurring specifically within promoters, gene bodies, and super enhancer regions, we carried out hypergeometric-based gene set enrichment analysis of genomic features in xenograft-derived CTCs (displaying a higher homogeneity compared to patient-derived CTCs). Consistently, this analysis revealed hypermethylation and H3K27me3 repression of Polycomb-repressive complex 2 (PRC2) target gene promoters and gene bodies (including those for SUZ12 and EED) in CTC clusters (Figures S1J–S1L), as previously alluded to in cancer specimens with stem-like and proliferative features (Avissar-Whiting et al., 2011, Kron et al., 2013, Lauss et al., 2012, Reddington et al., 2014, Wolff et al., 2010) and mirroring ESCs biology (Lee et al., 2006).
Figure S1.

Whole-Genome Bisulfite Sequencing of Single CTCs and CTC Clusters, Related to Figure 1
(A) CTC capture efficiency from blood spiked with BR16 or BRx50 single CTCs and CTC clusters, using the Parsortix device (n = 2 per cell line with 500 single CTCs and 150 CTC clusters). Error bars represent SEM.
(B) 250 single BR16-GFP+ and 250 single BR16-RFP+ cells are spiked in blood and CTCs are enriched using the Parsortix device. Captured CTCs are of single color, revealing no artificial cluster formation during processing (n = 2). Error bars represent SEM.
(C) Representative pictures of single CTCs and CTC clusters from breast cancer patients, enriched with the Parsortix microfluidic device and stained for EpCAM, HER2 and EGFR (green). White blood cells (WBCs) are counterstained with CD45 (red).
(D and E) Bar graph showing the percent of CpG sites that are covered in individual CTC clusters and single CTCs from patients (D) and xenografts (E).
(F and G) Principal component analysis of patient-derived (F) and xenograft-derived (G) single CTCs and CTC clusters, based on all features with p ≤ 0.05.
(H and I) Metaplots showing the percentage (%) of CpG methylation at CpG islands (H) and reference genes (I) in CTC clusters (blue line) and single CTCs (dotted red line). TSS: Transcription Start Site; TES: Transcription End Site.
(J–L) Hypergeometric gene set enrichment analysis of promoters (J), gene bodies (K) and super-enhancers (L) displaying ≥ 20% methylation difference (p value ≤ 0.01) in xenograft-derived CTC clusters compared to single CTCs. Gene sets with adjusted p value ≤ 0.05 are shown for promoters (J) and gene bodies (K). For super-enhancers (L), the top-20 significant gene sets with adjusted p value ≤ 0.05 are shown. Gene sets related to PRC2 activity are highlighted in red.
(M) Histogram showing mapped reads in patient CTCs corresponding to a methylated cytosine (C) (red) or a thymine (T) (blue; corresponding to a bisulfite-converted,
unmethylated cytosine) in representative regions that include binding sites for OCT4, SOX2, NANOG and SIN3A (shaded-orange box). n = number of CpGs covered.
Figure 1.

Whole-Genome Bisulfite Sequencing Analysis of CTCs from Breast Cancer Patients and Xenografts
(A) Heatmap showing methylation variable regions with ≥ 80% methylation difference between patient-derived CTC clusters and single CTCs (false discovery rate [FDR] < 0.05).
(B) Heatmap showing methylation variable regions with ≥ 70% methylation difference between xenograft-derived CTC clusters and single CTCs (FDR < 0.05).
(C and D) Normalized enrichment score (NES) representing enrichment (NES ≥ 3.4) of transcription factor binding sites (TFBSs) in CTC cluster hypomethylated regions (blue) and single CTC hypomethylated regions (red) of patients (C) or xenografts (D), identified using i-cisTarget.
(E and F) Integrated gene ontology (GO) and pathway enrichment analysis of TFBSs identified using i-cisTarget in hypomethylated regions of both patient- and xenograft-derived CTC clusters (E) or single CTCs (F). The bars represent the percentage of genes detected per GO and pathway term with p ≤ 0.05.
Thus, CTC clusters are clearly distinguishable from single CTCs based on their DNA methylation status at DMRs, where they mainly feature hypomethylation of bindings sites for stemness- and proliferation-associated TFs, such as OCT4, NANOG, SOX2, and SIN3A (Figure S1M, shown for patient CTCs), accompanied by a subtler hypermethylation of PRC2 target genes in promoters and gene bodies. This indicates that phenotypic differences in circulation affect DNA methylation dynamics.
Cluster-Associated Hypomethylated Regions Correlate with Poor Prognosis in Patients with Breast Cancer
We then tested whether the regions that are globally hypomethylated in CTC clusters are also hypomethylated in primary breast cancer. When analyzing bisulfite-sequencing data from The Cancer Genome Atlas (TCGA), we found 673/2,214 (30.39%) overlapping probes in 789 breast cancer patients, with 198 and 197 patients displaying either low (quantile Q1) or high (quantile Q4) methylation levels, respectively, in addition to a high correlation with genome-wide methylation levels (Figures 2A, S2A, and S2B). Progression-free survival (PFS) analysis on this subset of patients showed that low methylation levels within the regions that are hypomethylated in CTC clusters significantly correlate with a poor prognosis (p < 0.05) (Figure 2B). In contrast, no correlation with PFS is observed for those regions that are hypomethylated in single CTCs (Figures S2C–S2F). Of note, while the majority of breast cancer patients in both quantile Q1 and Q4 belong to the hormone receptor-positive subtype (ER+/PR+), the proportion of patients with the HER2-positive subtype is significantly lower in quantile Q1 compared to quantile Q4 for both CTC cluster- and single CTC-associated hypomethylated regions (Figures S2G–S2J).
Figure 2.

CTC Cluster Hypomethylated Regions Are Associated with a Poor Prognosis in Breast Cancer Patients
(A) Percentage of DNA methylation (mean beta values) of overlapping probes identified in the TCGA breast cancer patient dataset. Patients are grouped in four quantiles Q1-Q4, depending on the mean DNA methylation percentage of CTC cluster hypomethylated regions.
(B) Kaplan-Meier curve showing progression-free survival of breast cancer patients with overlapping probes in quantiles Q1 versus Q4 (top). The number of patients that progressed at each time point is shown (bottom).
See also Figure S2.
Figure S2.

TCGA Progression-free Survival Analysis of Breast Cancer Patients, Related to Figure 2
(A) Overlap of patient- and xenograft-derived CTC cluster hypomethylated regions with TCGA probes in breast cancer patients.
(B) Correlation plot of methylation levels across CTC cluster hypomethylated regions that are overlapping with TCGA probes and genome-wide methylation levels detected in the same samples.
(C) Overlap of patient- and xenograft-derived single CTC hypomethylated regions with TCGA probes in breast cancer patients.
(D) Percent of DNA methylation (mean beta values) of overlapping single CTC hypomethylated probes identified in the TCGA breast cancer patient dataset.
Patients are grouped in four quantiles Q1-Q4, depending on the mean DNA methylation percentage of single CTC hypomethylated regions.
(E) Correlation plot of methylation levels across single CTC hypomethylated regions that are overlapping with TCGA probes and genome-wide methylation levels detected in the same samples.
(F) Kaplan-Meier curve showing progression-free survival of breast cancer patients with single CTC hypomethylated overlapping probes in quantiles Q1 versus Q4 (top panel). The number of patients that progressed at each time point of the progression-free survival analysis is shown (bottom panel).
(G) Pie chart showing the percent of ER+/PR+, HER2+ and Triple Negative (TN) breast cancer patients with CTC cluster hypomethylated overlapping probes in quantiles Q1 versus Q4.
(H) Bar graphs showing the percent of ER+/PR+, HER2+ and Triple Negative breast cancer patients in Q1 and Q4 quantiles for CTC cluster hypomethylated overlapping probes. ns, not significant. p = by Student’s t test.
(I) Pie chart showing the percent of ER+/PR+, HER2+ and Triple Negative (TN) breast cancer patients with single CTC hypomethylated overlapping probes in quantiles Q1 versus Q4.
(J) Bar graphs showing the percent of ER+/PR+, HER2+ and Triple Negative breast cancer patients in Q1 and Q4 quantiles for single CTC hypomethylated overlapping probes. ns, not significant. p = by Student’s t test.
Together, our patient PFS analysis revealed that CTC cluster-associated hypomethylated regions are also hypomethylated at the level of the primary tumor in a subset of breast cancer patients characterized by a poor prognosis, compared to patients that display higher methylation levels in the same regions.
Proliferation-Related Genes Are Enriched in CTC Clusters
Our DNA methylation analysis led us to hypothesize that CTC clusters are characterized by active TF networks that support both stemness and proliferation. To identify whether the stemness- and proliferation-related TF networks are also transcriptionally active in CTC clusters compared to single CTCs, we performed single-cell resolution RNA sequencing analysis of 48 single CTCs and 24 CTC clusters, matched within individual liquid biopsies and isolated from six breast cancer patients with progressive metastatic disease, and of 49 single CTCs and 54 CTC clusters isolated from three xenograft models (Figures S3A and S3B; Tables S1 and S3). First, we performed a transcriptome-wide weighted gene co-expression network analysis (WGCNA) and identified 32 co-expression modules, revealing gene groups that are co-enriched in either single CTCs or CTC clusters (Figure 3A). Particularly, with this approach we identified two co-expression modules, containing total of 1,976 genes, enriched in patient-derived CTC clusters (p < 0.02; n = 1,544 for red; n = 432 for pink expression modules), while no co-expression modules were found to be significantly enriched in single CTCs (Figure 3B; Table S4). Gene ontology (GO) network analysis of the red and pink modules revealed an enrichment of gene groups related to cell-cell junctions, cellular proliferation, and platelet activation among others (Figure 3C). We then repeated the same analysis with xenograft-derived CTCs. Transcriptome-wide WGCNA identified 21 co-expression modules (Figure 3D), of which four significantly enriched in xenograft-derived CTC clusters and containing a total of 8,332 genes (p < 0.003; n = 159 for magenta, n = 337 for green, n = 753 for yellow, and n = 7,083 for turquoise expression modules), and three significantly enriched in xenograft-derived single CTCs and containing a total of 294 genes (p < 0.005; n = 202 for gray, n = 41 for dark turquoise, and n = 51 for dark red) (Figure 3E; Table S4). GO network analysis of the xenograft-derived CTC cluster networks revealed an enrichment of genes related to cell proliferation, DNA replication, cell-cell adhesion, and metabolic processes, among others (Figure 3F). In contrast, GO network analysis of the xenograft-derived single CTCs networks pointed to different processes that included RNA splicing, ATP metabolism, and ER stress response (Figure S3C). When specifically asking which processes were commonly found enriched in CTC clusters from both patients and xenografts, we identified genes related to both cellular proliferation and cell-cell adhesion (Figure 3G). While we previously reported upregulation of cell-cell adhesion components in CTC clusters (Aceto et al., 2014), whether cells within CTC clusters are also more proliferative compared to single CTCs is poorly understood. To directly address this point and validate our RNA sequencing findings, we stained CTCs from both patients and xenografts with the proliferation marker Ki67 and found that the percentage of Ki67-positive cells is indeed greatly increased in CTC clusters compared to matched single CTCs (Figures S3D–S3G).
Figure S3.

RNA Sequencing of Patient- and Xenograft-Derived Single CTCs and CTC Clusters, Related to Figure 3
(A and B) The plots show the number of detected features by number of cells in patient-derived (A) and xenograft-derived (B) CTCs processed with RNA sequencing.
(C) Gene Ontology (GO) enrichment analysis of genes in the Dark Red, Dark Turquoise and Grey expression modules, significantly enriched in xenograft-derived single CTCs.
(D–F) Dot plots showing the percent of Ki67-positive single CTCs and Ki67-positive CTCs within CTC clusters, detected in BR16 xenograft-derived CTCs (D), LM2 xenograft-derived CTCs (E), and patient 3-derived CTCs (F). ∗p < 0.05 by Student’s t test. Error bars represent SEM. ID = Internal ID.
(G) Representative pictures of BR16 xenograft-derived single CTCs and CTC clusters, stained with Pan Cytokeratin (PanCK) (green), Ki67 (red) and DAPI (blue).
Figure 3.

RNA Sequencing Analysis of Single CTCs and CTC Clusters from Breast Cancer Patients and Xenografts
(A) Weighted gene co-expression network analysis (WGCNA) in patient-derived single CTCs and CTC clusters, showing a hierarchical clustering tree of co-expression modules. Each module corresponds to a branch, which is labeled by a distinct color shown underneath.
(B) WGCNA identifies 32 modules with highly correlated gene expression patterns in patient-derived single CTCs and CTC clusters. Correlations between each module and CTC clusters or single CTCs are indicated by the intensity of red or green color, respectively. p value for each module is shown in brackets.
(C) GO term analysis of transcripts enriched in the red and pink expression modules of patient-derived CTC clusters.
(D) WGCNA in xenograft-derived single CTCs and CTC clusters, showing a hierarchical clustering tree of co-expression modules. Each module corresponds to a branch, labeled by a distinct color shown underneath.
(E) WGCNA identifies 21 modules with highly correlated gene expression patterns in xenograft-derived single CTCs and CTC clusters. Correlations between each module and CTC clusters or single CTCs are indicated by the intensity of red or green color, respectively. p value for each module is shown in brackets.
(F) GO term analysis of transcripts enriched in the magenta, green, yellow, and turquoise expression modules of xenograft-derived CTC clusters.
(G) Venn diagram showing the overlap of GO terms enriched in CTC clusters from patients and xenografts.
In summary, our transcriptome-wide WGCNA revealed that CTC clusters—additionally to upregulating cell-cell junction components—are also characterized by a higher proliferation rate compared to single CTCs, in line with our DNA methylation results.
Stem-Cell-Related Genes Are Enriched in CTC Clusters
Our transcriptome-wide analysis revealed that the most significant differences between single and clustered CTCs involve the expression of cell-cell junction components and cell cycle progression. Yet our DNA methylation analysis pointed to both proliferation- and stemness-related TF networks being accessible in CTC clusters. To specifically ask whether CTC clusters would differ from single CTCs also in regard to the expression of stemness-related genes (which may be regulated with more subtle expression changes compared to cell cycle- and cell-cell junction-related genes), we focused our analysis on 335 genes that were previously shown to be consistently upregulated in mouse and human embryonic stem cells and embryonal carcinoma cells, as opposed to their differentiated counterparts (Wong et al., 2008), and asked whether they are co-enriched in CTC clusters. First, a subset of 302 of these 335 genes were found to be expressed in our CTC samples from breast cancer patients (cutoff ≥ 3 transcripts per million). WGCNA with these genes identified four expression modules co-enriched in either single CTCs or CTC clusters. Particularly, we identified two co-expression modules, containing a total of 85 genes, enriched in patient-derived CTC clusters (n = 66 for blue; n = 19 for gray expression module) and in two co-expression modules, containing 217 genes, enriched in patient-derived single CTCs (n = 156 for turquoise; n = 61 for brown) (Figures 4A and 4B; Table S4). GO network analysis of these modules confirmed that CTC clusters co-express stemness-related genes interconnected to cellular proliferation, while single CTCs co-express genes more related to metabolic processes (Figures 4C, 4D, and S4A; Table S4). Importantly, TF target gene analysis (Janky et al., 2014) of CTC cluster-enriched genes confirmed, among others, activity of SIN3A and OCT4, in line with our DNA methylation findings (Figure 4E; Table S5). We then extended our analysis of the 302 stemness-related genes to xenograft-derived CTCs. In this case, WGCNA revealed four expression modules, of which two were found to be enriched in xenograft-derived CTC clusters and containing a total of 153 genes (n = 151 for green; n = 2 for yellow expression module) (Figures 4F and 4G; Table S4), in high concordance (85% overlap) with patient-derived modules enriched in CTC clusters (Figure 4H; Table S4). TF target gene analysis of those genes that are enriched in xenograft CTC clusters also revealed the activity of several stemness- and proliferation-related TFs, including SIN3A, OCT4, NANOG, and SOX2 (Figure 4I; Table S5). Interestingly, the majority of these genes also displays hypomethylated promoter regions in CTC clusters compared to single CTCs (Figures S4B and S4C). Co-expression modules found in single CTCs from patients and xenografts also displayed significant overlap between each other (56.7%) (Figure S4D; Table S4), and TF analysis revealed the activity of MYF6 and ASCL1, also displaying hypomethylated binding sites in single CTCs (Figure S4E; Table S5).
Figure 4.

Stemness-Related Gene Expression Analysis of Single CTCs and CTC Clusters from Breast Cancer Patients and Xenografts
(A) WGCNA of 302 stemness-related transcripts in patient-derived single CTCs and CTC clusters, showing a hierarchical clustering tree of co-expression modules. Each module corresponds to a branch, which is labeled by a distinct color shown underneath.
(B) WGCNA identifies four modules with highly correlated gene expression patterns in patient-derived single CTCs and CTC clusters. Correlations between each module and CTC clusters or single CTCs are indicated by the intensity of red or green color, respectively. p value for each module is shown in brackets.
(C) GO network analysis of patient-derived transcripts identified in the CTC cluster-associated blue and gray modules using iRegulon. The node size indicates significance (p < 0.05), and color intensity corresponds to the percentage of genes that are associated to each GO category. Indicative GO categories are shown.
(D) GO term analysis of transcripts enriched in the blue and gray modules of patient-derived CTC clusters.
(E) Patient-derived CTC cluster-associated blue and gray module gene regulatory network analysis showing putative transcription factor dependence on SIN3A, OCT4, and CBFB (green octagons).
(F) WGCNA of 302 stemness-related transcripts in xenograft-derived single CTCs and CTC clusters, showing a hierarchical clustering tree of co-expression modules. Each module corresponds to a branch, labeled by a distinct color shown underneath.
(G) WGCNA identifies four modules with highly correlated gene expression patterns in xenograft-derived single CTCs and CTC clusters. Correlations between each module and CTC clusters or single CTCs are indicated by the intensity of red or green color, respectively. p value for each module is shown in brackets.
(H) Venn diagram showing the overlap of CTC cluster-enriched transcripts between patients (blue and gray modules) and xenograft (yellow and green modules).
(I) Xenograft-derived CTC cluster-associated green and yellow module gene regulatory network analysis showing putative transcription factor dependence on SIN3A, OCT4, NANOG, BHLHE40, RORA, and FOXO1 (green octagons) and on SOX2 (orange circle).
Figure S4.

Stemness-Related Gene Expression Analysis of Single CTCs and CTC Clusters, Related to Figure 4
(A) GO term analysis of transcripts identified in patient-derived single CTC-associated turquoise and brown modules.
(B and C) Whisker plots showing the average methylation difference in CTC clusters relative to single CTCs, detected on the 5kb region upstream of the transcription start site of each target gene, in patient- (A) and xenograft-derived (B) CTCs. Hypomethylated genes in CTC clusters are represented with blue color, hypomethylated genes in single CTCs are represented with red color. Transcription factors relative to target genes are shown within gray boxes.
(D) Venn diagram showing the overlap between genes enriched in single CTCs of patient-derived (turquoise and brown) and xenograft-derived (orange and purple) expression modules.
(E) Regulatory network analysis of the 129 stemness-related genes that are commonly enriched in single CTCs in patient-derived (turquoise and brown) and xenograft-derived (orange and purple) expression modules, showing TF dependence on MYF6 and ASCL1, which also display hypomethylated binding sites based on the DMR analysis (green octagons).
Altogether, our gene expression data both at the transcriptome-wide level and also focused on stem cell-related genes strongly supports the model proposed with the DNA methylation analysis, suggesting that when compared to single CTCs, CTC clusters are endowed with a stemness- and proliferation-related network centered on the activity of key transcription factors including OCT4, SOX2, NANOG, and SIN3A. Activation of these programs may play a pivotal role in determining the metastasis-seeding ability of CTC clusters.
Identification of FDA-Approved Cluster-Targeting Agents
Next, we sought to identify actionable vulnerabilities of CTC clusters, and to test whether the epigenetic and transcriptional features of clustered CTCs are reversible upon cluster dissociation into single cells. To this end, we evaluated 2,486 FDA-approved compounds (Table S6) for their ability to dissociate clusters of human breast CTC-derived cells obtained from two patients with breast cancer (BR16 and BRx50), without affecting cellular viability. Cluster dissociation was assessed using an image-based high-content screening system and comparing CTCs treated with individual compounds to untreated CTCs as well as 40-μm-filtered single-cell suspension as negative and positive controls, respectively (Figures 5A, S5A, and S5B). For the majority of the 2,486 FDA-approved compounds, we observed no detectable reduction in cell viability (>70% viability) or in the mean CTC cluster size (> 450 μm2) of CTC-derived cells upon treatment (gray circles; Figure 5B). Yet we identified 39 compounds that significantly (p < 0.0001) reduced mean CTC cluster size without compromising viability (orange circles; Figure 5B). These compounds include inhibitors of Na+/K+ ATPase (n = 6), histone deacetylase (HDAC) (n = 2), nucleotide biosynthesis (n = 5), kinases (n = 3), GPCRs (n = 2), cholesterol biosynthesis (n = 1), nuclear export (n = 1), tubulin (n = 10), DNA binding compounds (n = 8), and antibiotics (n = 1) (Figures 5B and S5C). Importantly, reducing compound concentration resulted in a gradual increase in mean CTC cluster size in both CTC-derived cell lines, followed by an increase in the number of nuclei detected, and a slight improvement in mitochondrial membrane potential and overall viability (Figures 5C and S5D). Under these conditions, six compounds consistently led to a significant decrease in mean CTC cluster size for both CTC-derived cell lines, even at the lowest concentrations tested. These compounds display similar functions and can be grouped into two families based on their mechanism of action, namely the Na+/K+-ATPase inhibitors digitoxin and ouabain octahydrate and the tubulin binding agents rigosertib, podofilox, colchicine, and vincristine sulfate (Figures 5C, S5C, and S5D).
Figure 5.

Screen for FDA-Approved Compounds that Dissociate CTC Clusters
(A) Representative images of unfiltered and filtered BR16 CTC-derived cells stained with Hoechst (blue) and TMRM (orange) (left). Representative images of single and clustered CTCs outline based on nuclei proximity as determined using the Colombus Image Analysis System (right). The plots show the mean CTC cluster size (area in micrometers squared) and percentage (%) of viability of unfiltered versus filtered BR16 cells (n = 4; ∗∗∗p < 0.001 by Student’s t test; ns, not significant) (bottom).
(B) Effect of a 2-day treatment of BR16 cells with 2,486 FDA-approved compounds at 5 μM concentration, plotted as mean CTC cluster size (area in micrometers squared) versus percentage (%) of viability (n = 2). Thirty-nine FDA-approved compounds (orange circles) result in significant decrease in mean CTC cluster size (p < 0.0001, F value = 7.71; DF = 38 using one-way ANOVA test; horizontal dashed red line, < 450 μm2) and > 70% detectable viability (vertical-dashed red line). BR16 cells that were untreated (red) or 40 μm filtered (green) are shown as controls.
(C) The plot shows the mean CTC cluster size of BR16 cells treated with each of the 39 cluster-targeting compounds at four different concentrations. BR16 cells that were untreated (red) or 40 μm filtered (green) are shown as controls (top panel). The heatmap shows the number of nuclei, mean TMRM intensity, and percentage (%) of viability of BR16 cells treated with cluster-targeting compounds at the indicated concentrations (bottom). n = 2. Error bars represent SEM.
Figure S5.

Screen for CTC Cluster-Targeting FDA-Approved Compounds, Related to Figure 5
(A) Average TMRM intensity of unfiltered and filtered BR16 CTC-derived cells. n = 4; Error bars represent S.E.M; ∗∗∗p < 0.001 by Student’s t test.
(B) Assessment of mean CTC cluster size (area in μm2), percent (%) of viability and TMRM intensity of unfiltered and filtered BRx50 CTC-derived cells. n = 4; Error bars represent SEM; ∗∗∗p < 0.001 ∗∗∗∗p < 0.0001 by Student’s t test; ns, not significant.
(C) Mean CTC cluster size and viability distribution of BR16 cells treated with cluster-targeting FDA-approved compounds at 5 μM concentration for 2 days (p < 0.0001). BR16 cells that were not treated with FDA-approved compounds (red) or that were filtered (green) are shown for comparison. p = by one-way ANOVA test. n = 2; Error bars represent SEM.
(D) Mean CTC cluster size (area in μm2) of BRx50 cells treated with 3 different concentrations (0.1, 0.5 and 1 μM) of CTC cluster-targeting FDA-approved compounds (n = 2; Error bars represent SEM). BRx50 cells that were not treated with FDA-approved compounds (red) or that were filtered (green) are shown for comparison (top). Heatmaps showing the number of nuclei, the average TMRM intensity and the percent (%) of viability of BRx50 cells treated with CTC cluster-targeting FDA-approved compounds at the indicated concentrations (bottom). Compounds that consistently dissociate CTC clusters in both BR16 and Brx-50 cells are highlighted in red.
Dissociation of CTC Clusters Leads to DNA Methylation Remodeling of Key Sites
To assess whether CTC cluster dissociation into single cells would lead to DNA methylation remodeling, we treated four CTC-derived cell lines with the six CTC cluster-dissociating compounds individually. We found that a prolonged treatment (17 days) with 20 nM digitoxin or ouabain was unique in ensuring the highest degree of CTC cluster dissociation while allowing high viability and optimal proliferation rates comparable to untreated control cells (Figure S6A). Of note, the treatment period of 17 days was chosen to allow all cells (treated and untreated) to double at least 5 times and therefore enabling DNA methylation remodeling events to occur. Upon treatment, we processed BR16 and Brx50 cells for WGBS and RNA sequencing analysis to assess the molecular consequences of clusters dissociation. First, WGBS analysis revealed that the treatment did not affect global DNA methylation levels (data not shown). Yet a number of DMRs that were hypomethylated in CTC clusters gained methylation upon treatment (Figures S6B and S6C). Within the CTC cluster regions that gained methylation, i-cisTarget analysis showed enrichment for binding sites of stemness-related TFs, such as OCT4, SOX2, and NANOG, and for SIN3A, among others (Figure 6A). Simultaneously, RNA expression analysis upon treatment with digitoxin or ouabain revealed a high concordance between the expression changes of stemness-related genes that were enriched in patient- and xenograft-derived CTC clusters (r = 0.69; p < 2.22−16; Figures 6B), with the majority of these genes being downregulated as a consequence of the treatment (p < 0.002; Figure 6B). TF analysis revealed that downregulated genes were targets of OCT4, NANOG, SOX2, and SIN3A transcription factors, among others (Figure 6C). In contrast, TF analysis of those genes that were upregulated as a consequence to CTC cluster dissociation highlighted target genes of other TFs, independent from a core pluripotency network (Kim et al., 2008) (Figure S6D). Thus, CTC cluster dissociation into single cells with ouabain and digitoxin leads to DNA methylation remodeling and gain in methylation of critical binding sites for OCT4, SOX2, NANOG, and SIN3A, paralleled by downregulation of their corresponding target genes.
Figure S6.

Treatment of CTC-Derived Cell Lines with Digitoxin and Ouabain, Related to Figure 6
(A) Mean CTC cluster size (area in μm2) of BR16, BRx50, BRx07 and BRx68 CTC-derived cells treated with five different concentrations (1, 5, 10, 20 and 50 nM) of digitoxin or ouabain (n = 4; Error bars represent SEM). Cells that were not treated with FDA-approved compounds (red) or that were filtered (green) are shown for comparison (top). Heatmaps showing the number of nuclei, the average TMRM intensity and the percent (%) of viability of CTC-derived cells treated with digitoxin or ouabain at the indicated concentrations (bottom).
(B and C) The histograms show the number of CTC cluster hypomethylated regions that become differentially methylated upon treatment of BR16 (B) and BRx50 (C) cells with 20 nM dixitoxin or ouabain (q ≤ 0.05). Regions that gain >40% methylation are shown in red.
(D) Regulatory network analysis of the genes (orange) that are commonly upregulated (log2 fold-change ≥ 0.5) in BR16 and BRx50 CTC-derived cell lines upon 17-day treatment with digitoxin or ouabain, showing TF dependence on PBX3, MAX, NFYA, ELF2, PARP1 and HOXA10. Upregulated genes that do not show dependence on the above TFs are shown below in red.
(E) The plot shows the mean cell intensity of Ca2+-bound Fluo-3 after 30 min treatment of BR16 or BRx50 cells with 0.1, 0.5, 1 or 5 μM digitoxin or ouabain, respectively, relative to the untreated control (red) (n = 4; Error bars represent SEM).
(F) Western blot for CLDN3, CLDN4 and GAPDH on BR16 cells with double knockout (2KO) of CLDN3 and CLDN4. The dashed line represents the point where irrelevant lanes were spliced out from the original scan.
(G) Plot showing the reduction in mean cluster size (area in μm2) of the CLDN3/4 double KO BR16 cells, relative to control BR16 cells. ∗p < 0.05; ∗∗p < 0.01 by Student’s t test. Error bars represent SEM.
Figure 6.

Treatment with Na+/K+ ATPase Inhibitors and Tight Junction Dissociation Restores Methylation at Key Sites
(A) i-cisTarget analysis of CTC clusters hypomethylated regions, showing ≥ 40% methylation increase upon in vitro treatment with 20 nM of digitoxin or ouabain. Shown are enriched TFBSs in each CTC-derived cell line (NES ≥ 3).
(B) The plot shows expression changes in stemness-related transcripts that are enriched in patient- and xenograft-derived CTC clusters (n = 168) upon treatment of BR16 and BRx50 cells with 20 nM digitoxin or ouabain. Pearson’s correlation coefficient (r) and p values are shown.
(C) Gene regulatory network analysis of downregulated genes upon treatment with 20 nM digitoxin or ouabain in both BR16 and BRx50 (log FC ≤ −0.5), showing putative dependence on OCT4, SOX2, NANOG, and SIN3A, among other TFs (green octagons). Downregulated genes that are not regulated by the TFs mentioned above are shown in red.
(D) The plot shows the mean CTC cluster size of BR16 and BRx50 CTC-derived cells treated with FCCP or CCCP at different concentrations (n = 4). BR16 and BRx50 cells that were untreated (red) or 40 μm filtered (green) are shown as controls (top). The heatmap shows the percentage (%) of viability of treated cells at the indicated concentrations (middle). The plot shows the mean cell intensity of Ca2+-bound Fluo-3 after treatment with FCCP or CCCP, relative to the untreated control (red) (n = 4) (bottom). Error bars represent SEM.
(E) Heatmap showing methylation variable regions among CTC cluster hypomethylated regions that gain ≥ 40% methylation upon CLDN3/4 double knockout in BR16 cells.
(F) i-cisTarget analysis of CTC cluster hypomethylated regions, showing ≥ 40% methylation increase upon CLDN3/4 double knockout in BR16 cells. Shown are enriched TFBSs (NES ≥ 3).
Inhibition of the Na+/K+ ATPase using ouabain and intracellular increase of calcium levels have been previously shown to negatively affect the formation of tight junctions and desmosomes in epithelial cells (Nigam et al., 1992, Rajasekaran et al., 2001, Stuart et al., 1994). Here, to gain more insights into the mechanism of action of Na+/K+ ATPase inhibitors in the context of CTC clusters disruption, we tested whether the suppression of ATP production and consequently, an increase in intracellular calcium levels—using the proton uncouplers FCCP and CCCP (Demaurex et al., 2009)—would also dissociate CTC clusters. Our results show that treatment of CTC-derived cells with increasing concentrations of FCCP and CCCP for 18 hr leads to a gradual decrease of mean cluster size (Figure 6D), paralleled by a significant increase in intracellular Ca2+ levels (Figure 6D). Interestingly, a similar gradual increase in intracellular Ca2+ is also observed when treating with increasing concentrations of digitoxin and ouabain (Figure S6E). These results support a model whereby intracellular Ca2+ increase leads to the inability of cancer cells to properly form cell-cell junctions.
To further confirm this model, we assessed whether cell-cell junction disruption in CTC-derived cells would lead to clusters dissociation as well as DNA methylation remodeling at CTC cluster-associated DMRs. To this end, we employed the CRISPR technology to simultaneously knockout both claudin 3 (CLDN3) and claudin 4 (CLDN4) in BR16 CTC-derived cells, two of the highest-expressed tight junction proteins in CTC clusters (Table S4). Using two independent sgRNAs for each gene, we generated three BR16 lines with double CLDN3/4 knockout, which also displayed a significant reduction of mean CTC cluster size (Figures S6F and S6G). Whole-genome bisulfite sequencing of the CLDN3/4 double-knockout cells showed that, upon dissociation into single cells and similarly to the events that occurred upon Na+/K+ ATPase inhibition, a number of CTC cluster-associated hypomethylated regions gained methylation (Figure 6E). Interestingly, i-cisTarget analysis of the regions that gained higher levels of methylation revealed an enrichment of binding sites for OCT4, SOX2, NANOG, and SIN3A (Figure 6F), further indicating that CTC clustering directly affects DNA methylation dynamics at bindings sites for stemness- and proliferation-associated TFs.
Together, our results indicate that Na+/K+ ATPase inhibition leads to CTC clusters dissociation through the increase of the intracellular Ca2+ concentration and the consequent inhibition of cell-cell junction formation, resulting in DNA methylation remodeling at critical stemness- and proliferation-related binding sites.
Treatment with Na+/K+-ATPase Inhibitors Suppresses Spontaneous Metastasis Formation
To test whether ouabain and digitoxin would also enable CTC clusters disruption in vivo, we took a dual approach. First, we tested whether a 17-day in vitro treatment with ouabain and digitoxin would translate into a reduced ability of the treated cells to efficiently seed metastasis in untreated mice (Figure 7A). To this end, upon treatment, BR16 cells stably expressing GFP-luciferase were injected into the tail vein of NSG mice and noninvasively monitored through luminescence imaging for their ability to seed and propagate metastatic lesions. We found that while the treatment with digitoxin or ouabain did not affect the ability of BR16 cells to lodge in the lung tissue immediately after injection (see “day 0”; Figure 7B), it led to a reduced ability to survive during the first day upon arrival, as confirmed by a significant increase in the expression of cleaved caspase 3 compared to control cells (see “day 1”; Figures 7B, S7A, and S7B). Overall, this difference in the ability to survive during the very early steps of metastasis seeding resulted in a delayed metastatic outgrowth despite the absence of further treatment in vivo, as measured during the course of 72 days upon injection (Figure 7C).
Figure 7.

Treatment with Na+/K+ ATPase Inhibitors Suppresses Spontaneous Metastasis Formation
(A) Schematic representation of the experiment.
(B) The plots show the total bioluminescence flux at day 0 (left) and day 1 (right) upon tail vein injection of BR16 cells pre-treated with 20 nM digitoxin or ouabain. n = 5 for controls and ouabain, n = 4 for digitoxin; ∗p < 0.05 by Student’s t test. ns, not significant. Error bars represent SEM.
(C) Metastasis growth curve over 72 days upon tail vein injection of BR16 cells pre-treated with 20 nM digitoxin or ouabain. n = 5; ∗p < 0.05; ∗∗p < 0.01 by Student’s t test. Error bars represent SEM.
(D) Schematic representation of the experiment.
(E) The plots show the percentage (%) of spontaneously generated single CTCs and CTC clusters detected in the blood of BR16 xenografts treated with ouabain. n = 5; ∗∗∗p < 0.001 by Student’s t test. error bars represent SEM.
(F) The plot shows the metastatic index of BR16 xenografts treated with ouabain. n = 11 for controls, n = 5 for ouabain; ∗∗p < 0.01 by Student’s t test. Error bars represent SEM.
(G) Representative images of the bioluminescence signal measured in brain and in liver of control and ouabain-treated NSG mice.
See also Figure S7.
Figure S7.

Treatment with Digitoxin and Ouabain Reduces Metastasis Formation, Related to Figure 7
(A) The plots show the percent of Ki67-positive cancer cells detected in the lungs of NSG mice at Day 0 or Day 1 upon injection with BR16 CTC-derived cells, treated in vitro with digitoxin or ouabain. Cancer cells are identified through Pan Cytokeratin staining; n = 4 mice for each condition. Error bars represent SEM; ns, not significant.
(B) The plots show the percent of Caspase 3-positive cancer cells detected in the lungs of NSG mice at Day 0 or Day 1 upon injection with BR16 CTC-derived cells, treated in vitro with digitoxin or ouabain. Cancer cells are identified through Pan Cytokeratin staining; n = 4 mice for each condition. ∗p < 0.05 by Student’s t test; Error bars represent SEM; ns, not significant.
(C) The plot shows the total bioluminescence flux emitted from the primary tumor of BR16 xenografts treated with vehicle (control) or ouabain. Error bars represent SEM; ns, not significant.
(D) The plot shows the total number of CTCs, including both single CTCs and CTC clusters, detected per mL of blood in BR16 xenografts treated with vehicle
(control) or ouabain. n = 6 for controls and n = 5 ouabain; Error bars represent SEM; ns, not significant.
(E) The plots show the percent (%) of spontaneously generated single CTCs and CTC clusters detected in the blood of LM2 xenografts treated with vehicle (control) or ouabain. n = 11 for controls, n = 8 for ouabain; ∗∗p < 0.01.
(F) The plot shows the total bioluminescence flux emitted from the primary tumor of LM2 xenografts treated with vehicle (control) or ouabain. Error bars represent SEM; ns, not significant.
(G) The plot shows the total number of CTCs, including both single CTCs and CTC clusters, detected per mL of blood in LM2 xenografts treated with vehicle (control) or ouabain. n = 11 for controls and n = 8 ouabain; Error bars represent SEM; ns, not significant.
(H) The plot shows the metastatic index of LM2 xenografts treated with vehicle (control) or ouabain. n = 18 for controls, n = 8 for oubain. ∗p < 0.01 by Student’s t test; Error bars represent SEM.
Second, mimicking more closely the clinical setting, to assess the effect of our CTC cluster-dissociation strategy for the spontaneous formation of CTC clusters and metastasis from a primary tumor, we injected BR16 cells in the mammary fat pad of NSG mice. Fourteen weeks after primary tumor formation we administered ouabain daily for three weeks and assessed CTC composition and the occurrence of spontaneous metastatic lesions (Figure 7D). Importantly, we observed that ouabain treatment reduced the frequency of spontaneously generated CTC clusters while increasing the frequency of single CTCs (Figure 7E), without altering the size of the primary tumor nor overall CTC numbers (Figures S7C and S7D). Along with a reduction in the frequency of CTC clusters, ouabain treatment also resulted in a remarkable suppression (80.7-fold) of the total metastatic burden (Figures 7F and 7G). In a similar fashion, when administering ouabain treatment to NSG mice carrying spontaneously metastasizing LM2 tumors, we also observed an increase in the proportion of single CTCs and a decrease in CTC clusters (Figure S7E), without any change in the primary tumor size nor overall CTC numbers (Figures S7F and S7G), leading to a reduced metastatic burden compared to control (Figure S7H).
Together, these results demonstrate that Na+/K+ ATPase inhibition in vivo suppresses the ability of a cancerous lesion to spontaneously shed CTC clusters, leading to a remarkable reduction in metastasis seeding ability.
Discussion
Our study provides a comprehensive genome-wide analysis of the DNA-methylation events that characterize CTCs in patients and xenografts. Surprisingly, we find that phenotypic differences—such as the ability of CTCs to navigate through the bloodstream as single cells or multicellular clusters—shape the DNA methylome. Clustering of CTCs results in hypomethylation of binding sites that are typically occupied by master stemness and proliferation regulators, including OCT4, NANOG, SOX2, and SIN3A, and hypermethylation of Polycomb target genes. More globally, we also find that the DNA methylation profile of CTC clusters is detected at the level of the primary tumor in a subset of breast cancers that are characterized by a poor prognosis. CTC clusters dissociation into single cells with Na+/K+ ATPase inhibitors or through cell-cell junction knockdown enables DNA methylation remodeling at critical sites, highlighting a direct connection between clustering and methylation status. As a result, Na+/K+ ATPase inhibitors treatment emerges as a new strategy to significantly reduce the spread of cancer, providing a rationale for using these compounds in clinical studies.
Our results suggest that CTC clusters may share several properties that commonly feature stem cell biology. For instance, OCT4, NANOG, SOX2, and SIN3A are predominantly active in embryonic stem cells (ESCs), simultaneously regulating self-renewal and proliferation (Kim et al., 2008, McDonel et al., 2012, Niwa, 2007, van den Berg et al., 2010). In addition, ESCs rely on Polycomb-mediated repression of differentiation genes and chromatin remodeling to maintain their active pluripotency network (Lee et al., 2006). Cell-cell junction activity has been shown in several instances to safeguard pluripotency and to be required for a complete reprogramming of somatic cells into stem cells, and disruption of cell-cell junctions (e.g., through targeting of E-cadherin) in human ESC results into OCT4, NANOG, and SOX2 downregulation along the loss of stemness features (Li et al., 2010a, Li et al., 2010b, Li et al., 2012, Pieters and van Roy, 2014). Thus, by analogy with stem cell biology, elevated expression of cell-cell junction components in cancer cells may not only enable their intravasation in the bloodstream as multicellular clusters but also their ability to retain stem-like features that facilitate metastasis initiation.
Identifying FDA-approved compounds that dissociate CTC clusters provides a new tool to reduce the spread of cancer. Our results demonstrate that CTC clusters disruption into single cells via the inhibition of the Na+/K+ ATPase has a dual, yet interconnected effect. The fact that clusters dissociation leads to DNA methylation remodeling at critical sites provides a direct link between a phenotypic state of the cells (i.e., clustered versus single) and DNA methylation dynamics. Second, CTC clusters disruption increases the proportion of single CTCs in the bloodstream but suppresses overall metastasis formation, indicating that targeting CTC clusters could be a valuable therapeutic strategy. In this regard, while we cannot exclude that an anti-cluster treatment might be beneficial also at the late disease stages in those patients whose metastasis may seed other metastases (McPherson et al., 2016, Reiter et al., 2017, Zhang et al., 2017), in vivo data suggest that treatment with ouabain and digitoxin should be administered early, ideally at the time of localized disease and before dissemination to distant sites, with the objective to prevent CTC cluster formation.
Ouabain and digitoxin are cardiac glycosides with a similar chemical structure, used in low doses for the treatment of hypotension and cardiac arrhythmias, acting through nonselective inhibition of the Na+/K+ ATPase (Altamirano et al., 2006, Schwartz, 1976). Upon inhibition of the Na+/K+ ATPase, cellular uptake of Na+ occurs, leading to a simultaneous increase in intracellular Ca2+ and impaired translocation of desmosomal and tight junction proteins to the cellular membrane (Altamirano et al., 2006, Arispe et al., 2008). Based on this, our experimental evidences support a model whereby pharmacological inhibition of the Na+/K+ ATPase either directly or indirectly (i.e., by depletion of available ATP) leads to a concomitant increase in intracellular Ca2+ levels and CTC cluster disruption through suppression of functional cell-cell junction assembly (Cavey and Lecuit, 2009, Kim et al., 2011).
Together, our study provides key insights into the biology of CTCs and highlights a fundamental connection between phenotypic features of CTCs (such as their ability to circulate as multicellular clusters) and DNA methylation dynamics at critical stemness- and proliferation-related sites. Further, we identify the Na+/K+ ATPase inhibitors ouabain and digitoxin as FDA-approved agents capable to dissociate CTC clusters into single cells and to suppress spontaneous metastasis formation in xenograft models, providing a rationale for applying these compounds for the treatment of patients with breast cancer.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EpCAM-AF488 | Cell Signaling | Cat# CST5198; RRID: AB_10692105 |
| HER2-AF488 | BioLegend | Cat# 324410; RRID: AB_2099256 |
| EGFR-FITC | GeneTex | Cat# GTX11400; RRID: AB_368217 |
| CD45-BV605 (anti human) | Biolegend | Cat# 304042; RRID: AB_2562106 |
| CD45-BV605 (anti mouse) | Biolegend | Cat# 103140; RRID: AB_2562342 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2,486 FDA-approved molecules | NEXUS platform, ETH Zurich | Refer to Table S6 for individual chemical abstracts service (CAS) registry # |
| Deposited Data | ||
| RNA sequencing | This paper | GEO: GSE111065 |
| Bisulfite sequencing | This paper | ENA: PRJEB25101 |
| Experimental Models: Cell Lines | ||
| BR16 CTC-derived | Aceto lab, University of Basel | n/a |
| Brx50, Brx07 and Brx68 CTC-derived | Haber lab, MGH Cancer Center | n/a |
| MDA-MB-231 LM2 | Massague lab, MSKCC | n/a |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nicola Aceto (Nicola.Aceto@unibas.ch).
Experimental Model and Subject Details
Human blood samples collection
Blood specimens for CTC analysis were obtained from the University Hospital Basel after informed patient consent, according to protocols EKNZ BASEC 2016-00067 and EK 321/10, which received ethical approval from the Swiss authorities (EKNZ, Ethics Committee northwest/central Switzerland).
Mouse blood samples collection
For mouse studies, blood was retrieved via cardiac puncture and up to 1 mL of blood was collected. All mouse experiments were carried out in compliance with institutional and cantonal guidelines (approved mouse protocol #2781, cantonal veterinary office of Basel-City).
Cell lines
BR16 CTC-derived cells were generated from a patient with metastatic breast cancer at the University Hospital Basel as previously described (Yu et al., 2014). Brx50, Brx07 and Brx68 CTC-derived cells were obtained from the Haber and Maheswaran lab (MGH Cancer Center, Harvard Medical School, Boston, MA). MDA-MB-231 (LM2) cells were donated from Joan Massague’s lab (MSKCC, New York, NY, USA).
Method Details
Cell Culture
CTC-derived cells were maintained under hypoxic conditions (5% oxygen) on ultra-low attachment (ULA) 6-well plates (Corning, Cat# 3471-COR). CTC growth medium containing 20 ng/ml recombinant human Epidermal Growth Factor (GIBCO, Cat# PHG0313), 20 ng/ml recombinant human Fibroblast Growth Factor (GIBCO, Cat#100-18B), 1x B27 supplement (Invitrogen, Cat#17504-044) and 1x Antibiotic-Antimycotic (Invitrogen, Cat# 15240062) in RPMI 1640 Medium (Invitrogen, Cat# 52400-025) was added every third day. For passaging, cells were spun down at 800 g for 5 min using a Heraeus Multifuge X3R centrifuge (Invitrogen, Cat#75004515). The supernatant was subsequently aspirated and cells were resuspended in 2 ml/well CTC medium and plated in 6-well ULA plates. LM2 cells were passaged in DMEM/F-12 medium (Invitrogen, Cat#11330057) supplemented with 10% FBS (Invitrogen, Cat# 10500064) and Antibiotic-Antimycotic (Invitrogen, Cat# 15240062). For passaging, LM2 cells were washed once with D-PBS (Invitrogen, Cat#14190169) and dissociated using 0.25% Trypsin (Invitrogen, Cat#25200056).
CTC Capture and Identification
An average of 7.5 mL of blood per patient or up to 1 mL per mouse was drawn in EDTA vacutainers. Within 1 hr from blood draw, the blood was processed through a Parsortix GEN3D6.5 Cell Separation Cassette (Angle Europe) according to the manufacturer’s instructions. Captured CTCs were further stained on the Parsortix cassette with EpCAM-AF488 (CellSignaling, Cat# CST5198), HER2-AF488 (#324410, BioLegend), EGFR-FITC (GeneTex, Cat# GTX11400) and CD45-BV605 (Biolegend, Cat# 304042 (anti-human); Cat# 103140 (anti-mouse)) antibodies or imaged directly for GFP.
Tumorigenesis Assays
For tail vein experiments, NOD SCID Gamma (NSG) mice (Jackson Labs) were injected with 7x105 BR16-mCherry cells resuspended in 100 μL D-PBS and monitored with IVIS Lumina II (Perkin Elmer). For xenografts, 1x106 LM2-GFP, 1x106 BRx50-GFP or 1x106 BR16-GFP cells were resuspended in 100 μL of 50% Cultrex PathClear Reduced Growth Factor Basement Membrane Extract (R&D Biosystems, Cat# 3533-010-02) in D-PBS and injected orthotopically in NSG mice. Blood draw was performed 4-5 weeks after tumor onset for LM2 cells, 5 months after tumor onset for BR16 and 6-7 months after tumor onset for BRx50 cells. For in vivo treatment with ouabain, NSG mice were injected IP daily with 100 μL containing 0.67 mg/Kg ouabain diluted in PBS.
Immunofluorescence Staining
For immunofluorescence staining of CTCs, after Parsortix enrichment, CTCs were released in PBS and cytospinned for 3 min at 500 rpm on coated microscope slides (Thermo Scientific, Cat# 5991056), left to air dry for 5 min and then fixed with 4% PFA (Sigma; Cat# 58127) for 12 min. For lung tissue, immunoflourescence was performed on 4 μm paraffin sections. Paraffin sections were deparaffinized prior to immersion in Tris-EDTA buffer (10 mM Tris base/1 mM EDTA/0.05% Tween20, pH 9.0) at 95°C for 40 minutes for antigen retrieval. Slides were then washed twice in PBS and premeabilized with 5% Triton X-100 (Sigma) in PBS for 20 min, washed once with 0.2% Tween in PBS and twice with PBS. Blocking was performed for 1 hour at room temperature in 10% horse serum (Sigma, Cat# 2-05F00-I) followed by 2 times wash in PBS. Primary antibodies for Pan CytoKeratins (Abcam, Cat# ab190707), Ki67 (Abcam; Cat# ab15580) and Cleaved Caspase-3 (Cell Signaling, Cat# 9664S) were added at 1:50, 1:60 and 1:100 dilutions, respectively, in 0.5% BSA in PBS and incubated overnight at 4°C. The following day the slides were washed once with 5% horse serum, 0.2% tween in PBS followed by one wash with PBS. Secondary antibodies anti mouse-AF488 (Invitrogen; Cat# A21202) and anti-rabbit-AF647 (Invitrogen; Cat# A31573) were added at 1:200 dilution in 0.5% BSA in PBS for 30 min followed by three washes with PBS. Finally, slides were incubated for 5 min with 10 μM DAPI in PBS (Sigma, Cat# D9542) and mounted using ProLong Gold Antifade (Invitrogen; Cat# P36934).
FCCP, CCCP treatment and Intracellular Ca2+ measurement
FCCP (Abcam, Cat# ab120081) and CCCP (Abcam, Cat# ab141229) were reconstituted in DMSO at 100 mM concentration and further diluted in CTC medium to the indicated concentrations. For Ca2+ measurements, Fluo-3 AM (cell permeant, Invitrogen; Cat# F1241) was added to the 20’000 cells/well into a 96 well Black/clear Tissue culture treated plate (BD Falcon, Cat#353219) at 2 μM concentration together with 4 μM Hoechst 34580 (Invitrogen, Cat# H21486) and 4 μM TOTO-3 (Invitrogen, Cat# T3604) and cells were incubated for 30 min at 37°C. Plates were scanned using Operetta High Content Imaging System (Perkin Elmer) and Ca2+ bound Fluo-3 intensity per cell was calculated using Columbus Image Data Storage and Analysis System (Perkin Elmer).
CRISPR-CAS9 CLDN3/4 Double knock out in BR16
We used lentiviral delivery of pLenti-Cas9-EGFP vector (Addgene) to generate a BR16 CTC-derived cell line that stably expresses the Cas9 protein together with GFP. In BR16-Cas9-GFP line we then introduced sgRNA sequences that target either CLDN3 or CLDN4. In detail, sgRNA sequences were designed using the GPP Web Portal (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). Two sgRNAs targeting CLDN3 ((sense) 5′-CACGTCGCAGAACATCTGGG-3′ and (sense) 5′-ACGTCGCAGAACATCTGGGA-3′) were cloned in vector pLentiGuide-Puro (Addgene) and 2 sgRNAs targeting CLDN4 ((sense) 5′-CAAGGCCAAGACCATGATCG-3′ and (sense) 5′-ATGGGTGCCTCGCTCTACGT-3′) were cloned in vector pLentiGuide-Blast. Vector pLentiGuide-Blast was generated by replacing puromycin resistance gene on plasmid pLentiGuide-Puro with the blasticidin resistance gene using the MluI and BsiWI restriction enzyme sites. Double positive-clones were selected based on puromycin (1 μg/ml) and blasticidin (10 μg/ml) antibiotic selection for 2 weeks and CLDN3/CLDN4 knockout was verified by western blot.
Western Blot
Cells were pelleted at 800 rpm for 5 min and lysed in 1% SDS, 50 mM Tris pH7.8, 10 mM EDTA pH 8.0 and 0.1 M DTT in PBS with the addition of protease inhibitors according to manufacturer’s instructions (Complete EDTA-free Protease Inhibitor Cocktail Tablets - Roche; Cat#11873580001). Cell lysates were passed through a 27-G syringe to achieve homogeneous disruption and centrifuged at 10’000 rpm for 5 min at 4°C. The protein containing supernatant was transferred to a fresh tube. Protein concentration was determined using the DC Protein Assay Detection System (Biorad, Cat# 500-0114). The proteins were separated on a 12% SDS gel using the Bolt system (Invitrogen) and transferred to a nitrocellulose membrane using the iBlot2 system (Invitrogen) according to manufacturer’s instructions. Membranes were blocked in 5% milk in PBS for 1 hour at room temperature and primary antibodies for the detection of CLDN3 (1:1000 dilution; Novus; Cat#NB100-91711), CLDN4 (1:200 dilution; Invitrogen; Cat# 32-9400) and GAPDH (1:1000 dilution; Cell Signaling; Cat# 2118S) were added for incubation overnight at 4°C. The next day membranes were washed 3 times for 5 min in 0.2% tween in PBS. Secondary antibodies were added at 1:5000 dilution in 5% Milk in PBS and detection was performed using the Odyssey imaging system (Li-Cor) all according to manufacturer’s instructions.
Single-Cell Micromanipulation
CTCs were harvested from the Parsortix cassette in 1 mL D-PBS solution (Invitrogen, Cat#14190169) in a 6-well ultra-low attachment plate (Corning, Cat# 3471-COR) and visualized using a CKX41 Olympus inverted fluorescent microscope (part of the AVISO CellCelector Micromanipulator –ALS). Single CTCs and CTC clusters were identified based on intact cellular morphology, AF488/FITC-positive staining and lack of BV605 staining. Target cells were individually micromanipulated with a 30 μm glass capillary on the AVISO CellCelector micromanipulator (ALS) and deposited into individual PCR tubes (Axygen, Cat#321-032-501) containing 10 μL of 2x Digestion Buffer (EZ DNA Methylation Direct Kit -Zymo, Cat# D5020) for WGBS or 2 μL of RLT lysis buffer (QIAGEN, Cat#79216) supplemented with 1U/μl SUPERase In RNase inhibitor (Invitrogen, Cat# AM2694) for RNA sequencing, and immediately flash frozen in liquid nitrogen.
Single Cell Whole-genome Bisulfite Sequencing
Proteinase K digestion and bisulfite treatment was performed according to the manufacturer’s instructions for EZ DNA Methylation Direct Kit (Zymo, Cat# D5020). Bisulfite-treated DNA was eluted using 9 μL of Elution Buffer and used for library generation with TruSeq DNA methylation kit (Illumina, Cat# EGMK91396) according to manufacturer’s instructions. For amplification, 18 cycles were performed using Failsafe Enzyme (Illumina, Cat# FSE51100) and indexes were introduced with Index Primers’ Kit (Illumina, Cat# EGIDX81312). Library purification was performed using Agencourt AMPure XP beads at a ratio of 1:1 according to manufacturer’s instructions. To avoid DNA loss during pipetting steps, Corning DeckWork low binding barrier pipet tips were used (Sigma, Cat# CLS4135-4X960EA). Library concentration was estimated using Qubit DS DNA HS Assay Kit according to manufacturer’s instructions (Invitrogen, Cat#Q32854).
RNA-Seq Library generation
RNA was captured on beads conjugated with oligo-dT primer according to a published protocol (Macaulay et al., 2016). cDNA was generated according to the Smart-Seq 2 protocol (Picelli et al., 2014). Sequencing libraries were generated and indexed from 0.25 ng of cDNA per sample using the Nextera XT DNA Library Preparation Kit (Illumina, Cat# FC-131-2001) according to manufacturer’s instructions.
FDA-Approved Compound Screen
A library containing 2,486 FDA-approved compounds was obtained from the Nexus Platform – ETH Zurich. Each compound was resuspended using CTC medium at a 15 μM concentration and 20 μL were aliquoted in duplicate in a total of 64 Flat Bottom Clear Ultra Low attachment 96-well plates (Corning, Cat#3474). To obtain a single cell suspension from CTC-derived cells, a 40 μm cell strainer was used (Corning, Cat# 431750). 40 μL containing 15,000 CTC-derived cells were seeded per well in 96-well ultra-low attachment plates that contained 20 μL of pre-aliquoted FDA-approved compounds at 15 μM concentration, so that the final compound concentration was 5 μM. Plates were incubated in hypoxic conditions (5% oxygen) for 2 days and then 20 μL were transferred into a 96 well Black/clear Tissue culture treated plate (BD Falcon, Cat#353219) containing 40 μL of D-PBS (Invitrogen, Cat#14190169) and stained for 1hr at 37°C with a final concentration of 4 μM Hoechst 34580 (Invitrogen, Cat# H21486), 2 μM TMRM (Invitrogen, Cat#T668) and 4 μM TOTO-3 (Invitrogen, Cat# T3604). For each plate, two negative controls (non-treated cells) and two positive controls (40 μm-filtered cells) were included. Z-factors were calculated per individual plate using the following formula: Z′ = 1-3(σs + σc)/|μs-μc| (σ: standard deviation, μ: mean, s: positive control and c: negative control) (Martin et al., 2014) and ranged between 0.62-0.937. Plates were scanned using Operetta High Content Imaging System (Perkin Elmer) and CTC cluster analysis was performed using Columbus Image Data Storage and Analysis System (Perkin Elmer).
Quantification and Statistical Analysis
Whole Genome Bisulfite Data Processing
After sequencing, initial quality assessment of data was performed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). Adaptor sequences, low quality ends, first 9 base pairs from 5′ end and 3 base pairs from 3′ were removed with Trim Galore (v0.4.2, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/; parameters:–paired–retain_unpaired -r1 36 -r2 36–clip_r1 9–three_prime_clip_r1 3–clip_r2 9–three_prime_clip_r2 3–quality 20–phred33). Trimmed reads were aligned to GRCh38 human genome using Bismark Bisulfite Mapper (v0.17.0; paramters:–non_directional–bowtie2) (Krueger and Andrews, 2011) with the alignment tool Bowtie2 (v2.2.9) (Langmead and Salzberg, 2012). Reads with a conversion rate lower than 90% or less than three non-converted cytosines in non-CpG context were removed. Finally, methylation calls were extracted after deduplication using Bismark. Resulting data was processed using MethylKit (Akalin et al., 2012) (v1.4.0) to aggregate methylation values into genomic tiling regions of 5 kilobases using a step size of 1kilobase normalizing coverage between samples. Annotation used for metaplots of genes and CpG islands was performed using genomation toolkit in R/bioconductor package (Akalin et al., 2015).
Differential Methylation Analysis and Enrichment Analysis
Differential methylation at genomic tiles was assessed using Methylkit Chisq method to compare the fraction of methylated CpGs in test and control conditions, and correcting for overdispersion. Tiles with at least 2 samples covered per group and with a Q-value ≤ 0.05 using the SLIM (Wang et al., 2011) method were considered as differentially methylated. The region set enrichment analysis was performed following the example of Farlik et al. (2016). Normalized enrichment scores (NESs) of TBFSs in DMRs were calculated using i-cisTarget (https://gbiomed.kuleuven.be/apps/lcb/i-cisTarget/) (Herrmann et al., 2012) using full motif analysis and NES score threshold ≥ 3. Gene ontology network analysis was generated using ClueGO (Vocci and London, 1997). LOLA (v1.8; http://databio.org/regiondb) (Sheffield and Bock, 2016) was used to annotate the methylation data with a catalog of regulatory region sets from Codex database and ENCODE.
Enrichment Analysis Based on Genomic Features
Individual CpGs were mapped to genes and their promoters using RefSeq genes annotation from the UCSC genome browser (https://genome.ucsc.edu; date: 01/06/2018). Promoters were defined as ± 2 kb region around the transcription start site (TSS). Mapping to super-enhancer regions was based on dbSUPER (http://asntech.org/dbsuper/), an integrated database of super-enhancers that provides a list of genes associated to each region. Genomic coordinates from dbSUPER were converted from GRCh37 to GRCh38 via LiftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Each genomic feature was interrogated for differential methylation in the same fashion as we did for genomic tiles. Genes corresponding to genomic features with a nominal P value ≤ 0.01 and an absolute methylation difference ≥ 20% were considered for enrichment analysis using the gene set collection C2 from the Molecular Signatures Database v6.1 (MSigDB). Gene set enrichment analysis was performed using hypergeometric test implemented in the clusterProfiler R/bioconductor package. Gene-sets with an adjusted P value ≤ 0.05 were considered significant.
Survival Analysis using TCGA DNA Methylation Data
A total of 789 primary breast cancer samples from TCGA were available from the Illumina HumanMethylation 450 (HM450K) array. Level 3 data was downloaded from The National Cancer Institute (NCI) Genomic Data Commons (GDC) using the TCGAbiolinks bioconductor package (Colaprico et al., 2016). Downloaded data are β-values ranging from 0 to 1 and reflecting the fraction of methylated alleles at each CpG. Probes overlapping regions of the CTC cluster-associated DNA methylation signature were selected for further analysis. A curated set of clinical parameters was obtained from the TCGA Pan-Cancer Clinical Data Resource (TCGA-CDR) (Liu et al., 2018). For survival analysis, participants were divided into quartiles based on the average methylation across the CTC cluster-associated DNA methylation signature loci and progression free survival (PFS) was used as an endpoint. The Kaplan-Meier survival curves were drawn comparing the lowest (25%, Q1) versus the highest (75%, Q4) quartile and log-rank test P value was calculated using the survival R package.
RNA-Seq Data Processing
Initial quality assessment for RNA-seq data was performed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc), FastQ Screen (https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen), and visualized with MultiQC (v0.8). Adaptor sequences, first 9 bp and low quality ends were removed with Trim Galore (v0.4.2, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/; parameters:–phred33–length 36–clip_R1 9). Trimmed reads were aligned to a combined human (GRCh38) and mouse (GRCm38) genome reference using STAR (v 2.5.2a; parameters:–runMode alignReads–genomeLoad LoadAndExit) (Dobin et al., 2013) in order to exclude potential contaminations in the xenograft models. Quality control of resulting BAM files was performed with RSeQC (v2.6.4) (Wang et al., 2012). The gene-level expression counts were computed with featureCounts (v1.5.1) (Liao et al., 2014) using the gene annotations obtained from RefSeq (release 70). Samples with less than 800 features detected (threshold ≥ 1 mapped read) or showing more than 5% of contamination from the other species were removed from further analysis. To normalize gene counts for cell-specific biases in single-cell data, we used size factors computed using the deconvolution strategy developed by Lun et al. (2016) and implemented in the scran (v1.6.5) package available on R/bioconductor (Huber et al., 2015). RNA-seq data processing, quality control and visualization was performed with the help the R/bioconductor package scater (v1.6.0). Gene regulatory network analysis to identify putative transcription factor dependence was generated using iRegulon (Janky et al., 2014).
Weighted Gene Co-Expression Analysis
A signed co-expression network was constructed using Weighted Gene Correlation Network Analysis (WGCNA) R package (v1.61) based on 335 genes from a well-established stem cell gene signature (Wong et al., 2008). The analysis was performed only on those genes with a Fragments Per Kilobase Million (FPKM) value ≥ 1 in at least three samples (n = 301). A soft-threshold power used to create the pairwise distance matrix was defined as the lowest power for which the scale-free topology fit index reaches 0.80. Dynamic tree cut was (dynamicTreeCut R-package, v1.63) used to defined modules of genes. The association between the modules eigenvalues and CTC groups was evaluated using Wilcoxon rank sum. Gene ontology network analysis was generated using ClueGO (Vocci and London, 1997).
Data and Software Availability
Data Analysis and Data Availability
Data analysis, statistical test and visualization were conducted in R (version 3.4.0; R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism (v7.0). RNA sequencing data have been deposited to Gene Expression Omnibus (Edgar et al., 2002). The accession number for the RNA sequencing data reported in this paper is GEO: GSE111065. Bisulfite sequencing data have been deposited to the European Nucleotide Archive. The accession number for the bisulfite sequencing data reported in this paper is ENA: PRJEB25101.
Acknowledgments
We thank all patients that participated in our study, all involved clinicians and study nurses, and all members of the Aceto lab for feedback and discussions. We thank Dr. Joan Massagué (MSKCC, New York); Dr. Daniel Haber and Dr. Shyamala Maheswaran (MGH and Harvard Medical School, Boston, MA) for donating cell lines; and Dr. Gerhard Christofori (University of Basel, Switzerland) for helpful comments on the manuscript. We thank Katja Eschbach and Elodie Burcklen from the Genomics Facility Basel (ETH Zürich) for sequencing support; Falco Kilchmann and Eva Maria Riegler from NEXUS PHT (ETH Zurich) for help with the drug screening; and Dr. Robert Ivanek (University of Basel) for support with data analysis. Calculations were performed at sciCORE scientific computing center at the University of Basel. Research in the Aceto lab is supported by the European Research Council (678834), the Swiss National Science Foundation (PP0P3_163938), the Swiss Cancer League (KFS-3811-02-2016 and KLS-4222-08-2017), the Basel Cancer League (KLbB-4173-03-2017), the two Cantons of Basel through the ETH Zürich (PMB-01-16), and the University of Basel.
Author Contributions
S.G. and N.A. conceived the study, generated and interpreted the results, and wrote the manuscript. F.C.-G. conceived and performed the bioinformatics analyses. B.M.S. generated RNA sequencing (RNA-seq) libraries from CTCs. M.V., J.L., C.K. C.R., V.H.-S., and W.P.W. provided clinical input, consented breast cancer patients, and provided blood samples for CTC isolation. R.S. and I.K. performed cell culture experiments and contributed to the HTS compound screen. M.C.S. performed the CRISPR-related experiments. C.B. performed the WGBS and the RNA-seq library sequencing. C.U.S. provided the FDA-approved library for the drug screen. All authors approved the manuscript.
Declaration of Interests
N.A., S.G., and I.K. are listed as inventors in patent applications related to cancer treatment through CTC clusters inhibition. All other authors declare no competing interests.
Published: January 10, 2019
Footnotes
Supplemental Information includes seven figures and seven tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.11.046.
Supplemental Information
References
- Aceto N., Bardia A., Miyamoto D.T., Donaldson M.C., Wittner B.S., Spencer J.A., Yu M., Pely A., Engstrom A., Zhu H. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–1122. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aceto N., Toner M., Maheswaran S., Haber D.A. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer. 2015;1:44–52. doi: 10.1016/j.trecan.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Akalin A., Kormaksson M., Li S., Garrett-Bakelman F.E., Figueroa M.E., Melnick A., Mason C.E. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A., Franke V., Vlahoviček K., Mason C.E., Schübeler D. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics. 2015;31:1127–1129. doi: 10.1093/bioinformatics/btu775. [DOI] [PubMed] [Google Scholar]
- Alix-Panabières C., Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin. Chem. 2013;59:110–118. doi: 10.1373/clinchem.2012.194258. [DOI] [PubMed] [Google Scholar]
- Alix-Panabières C., Pantel K. Challenges in circulating tumour cell research. Nat. Rev. Cancer. 2014;14:623–631. doi: 10.1038/nrc3820. [DOI] [PubMed] [Google Scholar]
- Altamirano J., Li Y., DeSantiago J., Piacentino V., 3rd, Houser S.R., Bers D.M. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J. Physiol. 2006;575:845–854. doi: 10.1113/jphysiol.2006.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N., Diaz J.C., Simakova O., Pollard H.B. Heart failure drug digitoxin induces calcium uptake into cells by forming transmembrane calcium channels. Proc. Natl. Acad. Sci. USA. 2008;105:2610–2615. doi: 10.1073/pnas.0712270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avissar-Whiting M., Koestler D.C., Houseman E.A., Christensen B.C., Kelsey K.T., Marsit C.J. Polycomb group genes are targets of aberrant DNA methylation in renal cell carcinoma. Epigenetics. 2011;6:703–709. doi: 10.4161/epi.6.6.16158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavey M., Lecuit T. Molecular bases of cell-cell junctions stability and dynamics. Cold Spring Harb. Perspect. Biol. 2009;1:a002998. doi: 10.1101/cshperspect.a002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A., Silva T.C., Olsen C., Garofano L., Cava C., Garolini D., Sabedot T.S., Malta T.M., Pagnotta S.M., Castiglioni I. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaurex N., Poburko D., Frieden M. Regulation of plasma membrane calcium fluxes by mitochondria. Biochim. Biophys. Acta. 2009;1787:1383–1394. doi: 10.1016/j.bbabio.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413. doi: 10.1038/sj.onc.1205651. [DOI] [PubMed] [Google Scholar]
- Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–259. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M., Sheffield N.C., Nuzzo A., Datlinger P., Schönegger A., Klughammer J., Bock C. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015;10:1386–1397. doi: 10.1016/j.celrep.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M., Halbritter F., Müller F., Choudry F.A., Ebert P., Klughammer J., Farrow S., Santoro A., Ciaurro V., Mathur A. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell. 2016;19:808–822. doi: 10.1016/j.stem.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A.P., Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- Feinberg A.P., Ohlsson R., Henikoff S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- Fidler I.J. The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur. J. Cancer. 1973;9:223–227. doi: 10.1016/s0014-2964(73)80022-2. [DOI] [PubMed] [Google Scholar]
- Herrmann C., Van de Sande B., Potier D., Aerts S. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012;40:e114. doi: 10.1093/nar/gks543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S., Noh H., Teng Y., Shao J., Rehmani H., Ding H.F., Dong Z., Su S.B., Shi H., Kim J. SHOX2 is a direct miR-375 target and a novel epithelial-to-mesenchymal transition inducer in breast cancer cells. Neoplasia. 2014;16:279–290.e1-5. doi: 10.1016/j.neo.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janky R., Verfaillie A., Imrichová H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Herten K., Naval Sanchez M., Potier D. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014;10:e1003731. doi: 10.1371/journal.pcbi.1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X., Katiyar S., Willmarth N.E., Liu M., Ma X., Flomenberg N., Lisanti M.P., Pestell R.G. c-Jun induces mammary epithelial cellular invasion and breast cancer stem cell expansion. J. Biol. Chem. 2010;285:8218–8226. doi: 10.1074/jbc.M110.100792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Chu J., Shen X., Wang J., Orkin S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Woo A.J., Chu J., Snow J.W., Fujiwara Y., Kim C.G., Cantor A.B., Orkin S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.A., Tai C.Y., Mok L.P., Mosser E.A., Schuman E.M. Calcium-dependent dynamics of cadherin interactions at cell-cell junctions. Proc. Natl. Acad. Sci. USA. 2011;108:9857–9862. doi: 10.1073/pnas.1019003108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klutstein M., Nejman D., Greenfield R., Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016;76:3446–3450. doi: 10.1158/0008-5472.CAN-15-3278. [DOI] [PubMed] [Google Scholar]
- Kron K., Trudel D., Pethe V., Briollais L., Fleshner N., van der Kwast T., Bapat B. Altered DNA methylation landscapes of polycomb-repressed loci are associated with prostate cancer progression and ERG oncogene expression in prostate cancer. Clin. Cancer Res. 2013;19:3450–3461. doi: 10.1158/1078-0432.CCR-12-3139. [DOI] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo G.S., Alonzo T.A., Gudgeon C.J., Harrington K.H., Kentsis A., Gerbing R.B., Wang Y.C., Ries R.E., Raimondi S.C., Hirsch B.A. High expression of myocyte enhancer factor 2C (MEF2C) is associated with adverse-risk features and poor outcome in pediatric acute myeloid leukemia: a report from the Children’s Oncology Group. J. Hematol. Oncol. 2015;8:115. doi: 10.1186/s13045-015-0215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauss M., Aine M., Sjödahl G., Veerla S., Patschan O., Gudjonsson S., Chebil G., Lövgren K., Fernö M., Månsson W. DNA methylation analyses of urothelial carcinoma reveal distinct epigenetic subtypes and an association between gene copy number and methylation status. Epigenetics. 2012;7:858–867. doi: 10.4161/epi.20837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.S., Shin J.Y., Tonge P.D., Puri M.C., Lee S., Park H., Lee W.C., Hussein S.M., Bleazard T., Yun J.Y. An epigenomic roadmap to induced pluripotency reveals DNA methylation as a reprogramming modulator. Nat. Commun. 2014;5:5619. doi: 10.1038/ncomms6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Zhou J., Wang L., Shin M.E., Su P., Lei X., Kuang H., Guo W., Yang H., Cheng L. Integrated biochemical and mechanical signals regulate multifaceted human embryonic stem cell functions. J. Cell Biol. 2010;191:631–644. doi: 10.1083/jcb.201006094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Wang S., Jezierski A., Moalim-Nour L., Mohib K., Parks R.J., Retta S.F., Wang L. A unique interplay between Rap1 and E-cadherin in the endocytic pathway regulates self-renewal of human embryonic stem cells. Stem Cells. 2010;28:247–257. doi: 10.1002/stem.289. [DOI] [PubMed] [Google Scholar]
- Li L., Bennett S.A., Wang L. Role of E-cadherin and other cell adhesion molecules in survival and differentiation of human pluripotent stem cells. Cell Adhes. Migr. 2012;6:59–70. doi: 10.4161/cam.19583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liotta L.A., Saidel M.G., Kleinerman J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 1976;36:889–894. [PubMed] [Google Scholar]
- Liu J., Lichtenberg T., Hoadley K.A., Poisson L.M., Lazar A.J., Cherniack A.D., Kovatich A.J., Benz C.C., Levine D.A., Lee A.V. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T., Bach K., Marioni J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016;17:75. doi: 10.1186/s13059-016-0947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay I.C., Teng M.J., Haerty W., Kumar P., Ponting C.P., Voet T. Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq. Nat. Protoc. 2016;11:2081–2103. doi: 10.1038/nprot.2016.138. [DOI] [PubMed] [Google Scholar]
- Martin H.L., Adams M., Higgins J., Bond J., Morrison E.E., Bell S.M., Warriner S., Nelson A., Tomlinson D.C. High-content, high-throughput screening for the identification of cytotoxic compounds based on cell morphology and cell proliferation markers. PLoS ONE. 2014;9:e88338. doi: 10.1371/journal.pone.0088338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonel P., Demmers J., Tan D.W., Watt F., Hendrich B.D. Sin3a is essential for the genome integrity and viability of pluripotent cells. Dev. Biol. 2012;363:62–73. doi: 10.1016/j.ydbio.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson A., Roth A., Laks E., Masud T., Bashashati A., Zhang A.W., Ha G., Biele J., Yap D., Wan A. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016;48:758–767. doi: 10.1038/ng.3573. [DOI] [PubMed] [Google Scholar]
- Minn A.J., Gupta G.P., Siegel P.M., Bos P.D., Shu W., Giri D.D., Viale A., Olshen A.B., Gerald W.L., Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam S.K., Rodriguez-Boulan E., Silver R.B. Changes in intracellular calcium during the development of epithelial polarity and junctions. Proc. Natl. Acad. Sci. USA. 1992;89:6162–6166. doi: 10.1073/pnas.89.13.6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H. How is pluripotency determined and maintained? Development. 2007;134:635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Pieters T., van Roy F. Role of cell-cell adhesion complexes in embryonic stem cell biology. J. Cell Sci. 2014;127:2603–2613. doi: 10.1242/jcs.146720. [DOI] [PubMed] [Google Scholar]
- Rajasekaran S.A., Palmer L.G., Moon S.Y., Peralta Soler A., Apodaca G.L., Harper J.F., Zheng Y., Rajasekaran A.K. Na,K-ATPase activity is required for formation of tight junctions, desmosomes, and induction of polarity in epithelial cells. Mol. Biol. Cell. 2001;12:3717–3732. doi: 10.1091/mbc.12.12.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond A., Liu B., Liang H., Wei C., Guindani M., Lu Y., Liang S., St John L.S., Molldrem J., Nagarajan L. A role for BMP-induced homeobox gene MIXL1 in acute myelogenous leukemia and identification of type I BMP receptor as a potential target for therapy. Oncotarget. 2014;5:12675–12693. doi: 10.18632/oncotarget.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddington J.P., Sproul D., Meehan R.R. DNA methylation reprogramming in cancer: does it act by re-configuring the binding landscape of Polycomb repressive complexes? BioEssays. 2014;36:134–140. doi: 10.1002/bies.201300130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J.G., Makohon-Moore A.P., Gerold J.M., Bozic I., Chatterjee K., Iacobuzio-Donahue C.A., Vogelstein B., Nowak M.A. Reconstructing metastatic seeding patterns of human cancers. Nat. Commun. 2017;8:14114. doi: 10.1038/ncomms14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A., Huang X., Fidalgo M., Reimer M.H., Jr., Faiola F., Ding J., Sánchez-Priego C., Guallar D., Sáenz C., Li D., Wang J. The SIN3A/HDAC corepressor complex functionally cooperates with NANOG to promote pluripotency. Cell Rep. 2017;18:1713–1726. doi: 10.1016/j.celrep.2017.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz A. Is the cell membrane Na+, K+ -ATPase enzyme system the pharmacological receptor for digitalis? Circ. Res. 1976;39:1–7. doi: 10.1161/01.res.39.1.1. [DOI] [PubMed] [Google Scholar]
- Sheffield N.C., Bock C. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2016;32:587–589. doi: 10.1093/bioinformatics/btv612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart R.O., Sun A., Panichas M., Hebert S.C., Brenner B.M., Nigam S.K. Critical role for intracellular calcium in tight junction biogenesis. J. Cell. Physiol. 1994;159:423–433. doi: 10.1002/jcp.1041590306. [DOI] [PubMed] [Google Scholar]
- van den Berg D.L., Snoek T., Mullin N.P., Yates A., Bezstarosti K., Demmers J., Chambers I., Poot R.A. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell. 2010;6:369–381. doi: 10.1016/j.stem.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vocci F.J., London E.D. Assessment of neurotoxicity from potential medications for drug abuse: ibogaine testing and brain imaging. Ann. N Y Acad. Sci. 1997;820:29–39, discussion 39–40. doi: 10.1111/j.1749-6632.1997.tb46187.x. [DOI] [PubMed] [Google Scholar]
- Wang H.Q., Tuominen L.K., Tsai C.J. SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics. 2011;27:225–231. doi: 10.1093/bioinformatics/btq650. [DOI] [PubMed] [Google Scholar]
- Wang L., Wang S., Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Wolff E.M., Chihara Y., Pan F., Weisenberger D.J., Siegmund K.D., Sugano K., Kawashima K., Laird P.W., Jones P.A., Liang G. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010;70:8169–8178. doi: 10.1158/0008-5472.CAN-10-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D.J., Liu H., Ridky T.W., Cassarino D., Segal E., Chang H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Mao X., Imrali A., Syed F., Mutsvangwa K., Berney D., Cathcart P., Hines J., Shamash J., Lu Y.J. Optimization and evaluation of a novel size based circulating tumor cell isolation system. PLoS ONE. 2015;10:e0138032. doi: 10.1371/journal.pone.0138032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Bardia A., Aceto N., Bersani F., Madden M.W., Donaldson M.C., Desai R., Zhu H., Comaills V., Zheng Z. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216–220. doi: 10.1126/science.1253533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Cunningham J.J., Brown J.S., Gatenby R.A. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat. Commun. 2017;8:1816. doi: 10.1038/s41467-017-01968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data Analysis and Data Availability
Data analysis, statistical test and visualization were conducted in R (version 3.4.0; R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism (v7.0). RNA sequencing data have been deposited to Gene Expression Omnibus (Edgar et al., 2002). The accession number for the RNA sequencing data reported in this paper is GEO: GSE111065. Bisulfite sequencing data have been deposited to the European Nucleotide Archive. The accession number for the bisulfite sequencing data reported in this paper is ENA: PRJEB25101.