

As the worldwide HIV pandemic continues, there is a continued need for novel drugs and therapies. A new class of drug, the attachment inhibitors, will soon be approved for the treatment of HIV. Broadly neutralizing antibodies are also promising candidates for HIV prevention and therapy. We investigated how this drug might work with these exciting antibodies that are very potent in blocking HIV infection of cells. These antibodies worked against virus known to be resistant to the new drug. In addition, a specific type of antibody worked really well with the new drug in blocking virus infection of cells. This work has implications for both the new drug and the antibodies that are poised to be used against HIV.
KEYWORDS: HIV-1, attachment inhibitor, bNAbs, escape mutants, synergy
ABSTRACT
Attachment inhibitor (AI) BMS-626529 (fostemsavir) represents a novel class of antiretrovirals which target human immunodeficiency virus type 1 (HIV-1) gp120 and block CD4-induced conformational changes required for viral entry. It is now in phase III clinical trials and is expected to be approved by the U.S. Food and Drug Administration (FDA) in the near future. Although fostemsavir is very potent against HIV in vitro and in vivo, a number of resistant mutants have already been identified. Broadly neutralizing HIV antibodies (bNAbs) can potently inhibit a wide range of HIV-1 strains by binding to viral Env and are very promising candidates for HIV-1 prevention and therapy. Since both target viral Env to block viral entry, we decided to investigate the relationship between these two inhibitors. Our data show that Env mutants resistant to BMS-626529 retained susceptibility to bNAbs. A single treatment of bNAb NIH45-46G54W completely inhibited the replication of these escape mutants. Remarkable synergy was observed between BMS-626529 and CD4 binding site (CD4bs)-targeting bNAbs in neutralizing HIV-1 strains at low concentrations. This synergistic effect was enhanced against virus harboring mutations conferring resistance to BMS-626529. The mechanistic basis of the observed synergy is likely enhanced inhibition of CD4 binding to the HIV-1 Env trimer by the combination of BMS-626529 and CD4bs-targeting bNAbs. This work highlights the potential for positive interplay between small- and large-molecule therapeutics against HIV entry, which may prove useful as these agents enter clinical use.
IMPORTANCE As the worldwide HIV pandemic continues, there is a continued need for novel drugs and therapies. A new class of drug, the attachment inhibitors, will soon be approved for the treatment of HIV. Broadly neutralizing antibodies are also promising candidates for HIV prevention and therapy. We investigated how this drug might work with these exciting antibodies that are very potent in blocking HIV infection of cells. These antibodies worked against virus known to be resistant to the new drug. In addition, a specific type of antibody worked really well with the new drug in blocking virus infection of cells. This work has implications for both the new drug and the antibodies that are poised to be used against HIV.
INTRODUCTION
Although there are already many antiretroviral drugs that are approved by the U.S. Food and Drug Administration (FDA) for HIV-1 therapy, the development of novel medications with different mechanisms of action is needed, mainly due to the specter of virus resistance to current drugs and also to the demand for better-tolerated, longer-lasting medications with a higher genetic barrier to resistance (1). Infection of host cells by HIV-1 requires multiple steps, the first being Env attachment to the cell surface CD4 receptor. This causes conformational changes in Env, allowing coreceptor binding, which initiates virus-host membrane fusion (2). As viral entry is the first step of viral replication in cells, it is an important target for the development of antivirals, two of which are on the market today. Enfuvirtide, or T-20, is a peptide fusion inhibitor that competitively prevents the formation of the GP41 6-helix bundle, necessary for virus-cell membrane apposition. Maraviroc is a small-molecule allosteric inhibitor of CCR5, the major coreceptor for R5-tropic HIV strains.
Attachment inhibitors (AI) represent a novel class of small-molecule antiretrovirals that bind to HIV-1 gp120 and selectively block the CD4-induced conformational changes needed for coreceptor binding, thereby preventing viral entry into host cells (3–5). The latest AI, fostemsavir (or BMS-663068 [prodrug of BMS-626529]), is now in phase III clinical trials (https://aidsinfo.nih.gov/drugs/508/fostemsavir/0/patient) (6, 7) and is expected to be approved by the FDA in the near future. Although both in vitro and in vivo studies have shown that BMS-626529 and BMS-663068 are very potent against HIV-1 (6, 8), there are already a number of escape mutants (MT) corresponding to BMS-626529 identified through in vitro replication assays or from treated human subjects (9–11). Because BMS-626529 targets different sites of Env to block viral entry, there is low-level cross-resistance between BMS-626529 and other entry inhibitors such as enfuvirtide and ibalizumab (iMab—a monoclonal antibody [MAb] that targets D2 of CD4). Some resistant mutants of maraviroc have shown reduced susceptibility to BMS-626529 (12).
Broadly neutralizing HIV-1 antibodies (bNAbs) are relatively potent monoclonal antibodies that neutralize multiple HIV-1 strains. HIV-1 bNAbs target different epitopes on the HIV-1 Env trimer to block viral entry, some by steric effect and others by presumably preventing necessary conformational changes in Env trimer (13). Some of the latest next-generation bNAbs, e.g., N6 and VRC07-523, are capable of neutralizing >90% viral strains with 50% inhibitory concentrations (IC50) at low values of micrograms per milliliter in vitro (14, 15). Given their excellent properties, the use of bNAbs for the prevention and treatment of HIV-1 infection is being considered seriously (16). It is noteworthy that in addition to neutralizing HIV-1 infection, bNAbs can also induce long-lasting host immunity capable of suppressing viral replication in macaques and man (17, 18). Another extraordinary effect of bNAbs is that they can help clear virally infected cells, with the effect likely mediated by antibody-dependent cellular cytotoxicity (ADCC) (19, 20). In combination with latency-reversing agents (LRAs), bNAbs decreased rebound from latent reservoirs of HIV-1 in humanized mice (21), representing a promising strategy to target and eradicate this pool of virus.
As both BMS-626529 and the bNAbs target Env and viral entry, we initially asked whether the highly potent bNAbs are capable of neutralizing Env escape mutants resistant to BMS-626529. Furthermore, we wished to determine whether there is any synergy between BMS-626529 and bNAbs in terms of neutralizing HIV-1, which could then open up other possible treatment options.
RESULTS
Escape mutants of attachment inhibitor BMS-626529.
Following reports in the literature, 11 escape mutants resistant to BMS-626529 were constructed based on HIV-1 strain ADA Env (10). Eight of them were single mutations, and three were double-amino-acid mutations (Table 1) (Fig. 1). An HIV envelope structure indicating the important escape mutations corresponding to BMS-626529 is shown in Fig. 1a (5, 9). Pseudotyped viruses using Env containing these mutations were produced and tested for infectivity using GHOST.Hi5 cells as targets. Two of the mutants, L116P and A204D, showed markedly reduced infectivity of GHOST.Hi5 cells (Fig. 1b). Neutralization assays demonstrated that these escape mutants were less susceptible to BMS-626529 to various extents (Fig. 1c). Among them, M426L, S375M, M426L/M434I, and S375M/M434I were the most resistant escape mutants. The susceptibility of S375M to BMS-626529 and S375M/M434I was decreased over 200-fold compared to wild-type ADA Env. Surprisingly, mutant V506M on the ADA pseudotype background was not resistant to BMS-626529 (Table 1).
TABLE 1.
Susceptibility of wild-type and mutant ADA Envs to BMS-626529 and bNAbsa
| Antiviral | IC50 (nM) (fold change) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WT | M426L | M434I | S375M | S375I | M475I | V506M | M426L/ M434I |
M426L/ M475I |
S375M/ M434I |
|
| BMS-626529 | 0.14 | 6.67 (49) | 0.26 (1.9) | 30.0 (221) | 1.78 (13.1) | 0.60 (4.4) | 0.11 (0.8) | 15.04 (111) | 5.81 (43) | 29.58 (218) |
| VRC03 | 0.18 | 0.16 (0.9) | 0.10 (0.6) | 0.08 (0.4) | 0.21 (1.2) | 0.09 (0.5) | 0.09 (0.5) | 0.107 (0.58) | 0.16 (0.9) | 0.11 (0.6) |
| VRC07-523 | 0.29 | 0.25 (0.8) | — | 0.16 (0.6) | — | — | — | — | — | 0.23 (0.8) |
| 4Dm2m | 0.06 | 0.08 (1.3) | 0.52 (8.1) | 0.03 (0.5) | 0.07 (1.1) | 0.73 (11) | 0.22 (3.5) | 0.373 (5.83) | 0.23 (3.8) | 0.05 (0.7) |
| 6Dm2m | 0.09 | 0.30 (3.2) | — | 0.06 (0.6) | — | — | — | — | — | 0.08 (0.8) |
| 35O22 | 0.06 | 0.15 (2.7) | 0.15 (2.5) | 0.05 (0.8) | 0.12 (2.0) | 0.13 (2.3) | 0.06 (1.1) | 0.06 (1.03) | 0.21 (3.5) | 0.12 (2.1) |
| PG16 | 0.08 | 0.08 (1.0) | — | 0.05 (0.6) | — | — | — | — | — | 0.06 (0.8) |
| PG16-iMab | 0.04 | 0.01 (0.2) | 0.01 (0.3) | 0.01 (0.4) | 0.01 (0.3) | 0.02 (0.6) | 0.02 (0.4) | 0.004 (0.10) | 0.03 (0.8) | 0.001 (0.0) |
| PGT128 | 0.09 | 0.03 (0.4) | 0.07 (0.7) | 0.02 (0.3) | 0.06 (0.6) | 0.11 (1.1) | 0.13 (1.4) | 0.076 (0.82) | 0.10 (1.2) | 0.04 (0.4) |
| PGT128-iMab | 0.02 | 0.01 (0.3) | 0.01 (0.4) | 0.01 (0.5) | 0.02 (0.8) | 0.02 (1.1) | 0.01 (0.7) | 0.001 (0.05) | 0.02 (0.9) | 0.004 (0.2) |
| NIH45-46G54W | 0.08 | 0.06 (0.7) | 0.09 (1.1) | 0.04 (0.5) | 0.06 (0.8) | 0.07 (0.8) | 0.07 (0.8) | 0.047 (0.56) | 0.13 (1.6) | 0.09 (1.1) |
| PG16-NIH45-46G54W | 0.03 | 0.03 (0.8) | 0.03 (0.9) | 0.03 (0.9) | 0.07 (2.1) | 0.04 (1.3) | 0.04 (1.2) | 0.03 (0.8) | 0.06 (1.6) | 0.12 (3.4) |
Data represent results of tests of GHOST-Hi5 cells with HIV-LIB pseudotyped viruses. Each entry shows the IC50 in nanomoles for the indicated strain; fold change in susceptibility compared to the WT is shown in parentheses. —, not determined.
FIG 1.
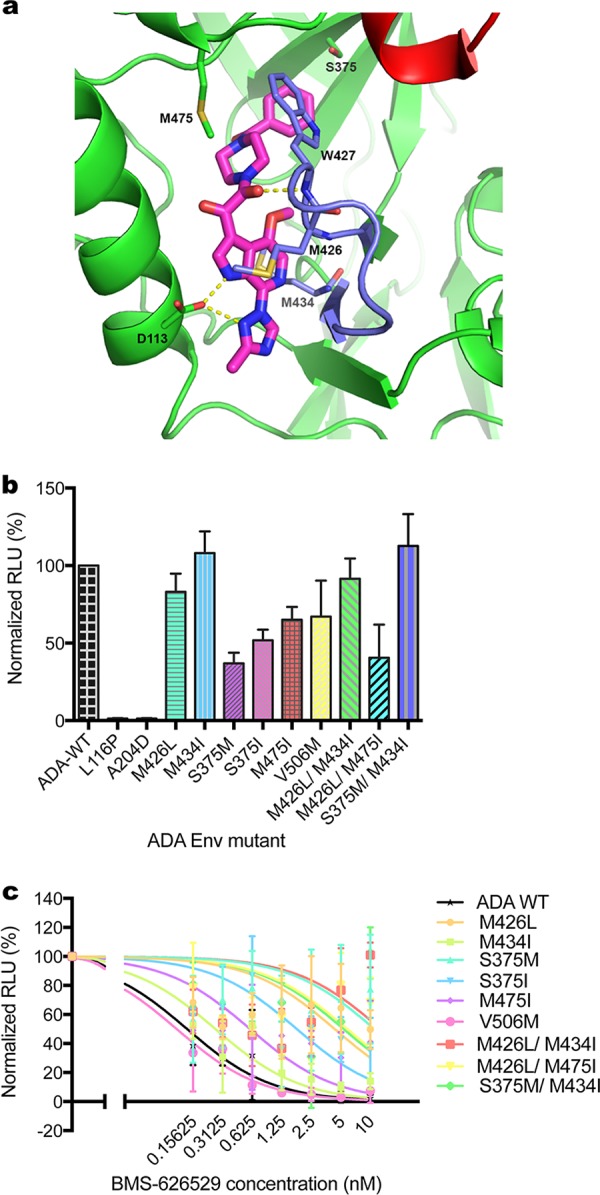
Reduced susceptibility of escape mutants to BMS-626529. (a) Crystal structure of HIV-1 BG505 SOSIP.664 prefusion Env trimer bound to BMS-626529, in complex with bNAb PGT-122 and 35O22 at 3.8-Angstrom resolution (PDB code: 5U7O) (5). BMS-626529 predominantly binds to gp120 via hydrophobic interactions and forms hydrogen bonds with D113 from the α1-helix of the gp120 inner domain and W427 of the outer domain. The benzamide group of BMS-626529 occupies the site of gp120 that is occupied by W427 in the open state such that W427 and the β20-β21 loop are extended toward the CD4 binding loop (red), thereby blocking CD4 binding. Major escape mutation sites of BMS-626529 including M426, S375, M434 and M475, are shown. (b) Pseudotype titer of reconstructed BMS-626529-resistant mutations introduced in ADA Env. GHOST.Hi5 cells were transduced with identical volumes of supernatant of HIV-LIB pseudotyped with each mutant Env. RLU levels in transduced cells were measured and normalized to those of wild-type Env. The data represent means ± standard deviations (SD) of results from three experiments. (c) Neutralization of wild-type and mutant ADA Env pseudotyped virus by BMS-626529. The means ± SD of results from three independent experiments are shown.
BMS-626529 escape mutants were still sensitive to bNAbs.
We then tested whether the latest bNAbs, including bispecific bNAbs (bibNAbs), could effectively inhibit these BMS-626529-resistant mutants. For the wild-type ADA Env, both BMS-626529 and the bNAbs efficiently neutralized pseudotyped particles (Fig. 2a). For the escape mutants, with the exception of V506M, BMS-626529 failed to effectively inhibit viral entry at a concentration of 2 nM, whereas most bNAbs, except for 35O22, still completely neutralized the mutants at that concentration. Data corresponding to the changes in the susceptibility of these escape mutants resistant to BMS-626529 and bNAbs, compared to the wild-type envelope, are shown in Fig. 2b. Different mutants showed increased resistance to BMS-626529 at levels ranging from 1-fold to 200-fold. As mentioned previously, S375M and S375M/M434I are the escape mutations most resistant to BMS-626529, whereas the change in the susceptibility of the mutants to most bNAbs was less than 2-fold. Some mutants became more resistant to 4Dm2m, a bispecific multivalent fusion protein of a single-domain CD4 (sCD4) (mD1.22) fused with an antibody domain (m36.4) that targets the coreceptor binding site on gp120 (22). Other CD4 binding site (CD4bs)-targeting bNAbs (e.g., VRC03, VRC07-523, and NIH45-46G54W), however, were still quite sensitive to these BMS-626529-resistant mutants, suggesting that their targeting sites differ from that of BMS-626529.
FIG 2.

Neutralization of BMS-626529 escape mutants with bNAbs. (a) Neutralization of wild-type and mutant ADA pseudotypes by BMS-626529 and bNAbs. The starting concentration of BMS-626529 and each bNAb was 2 nM, followed by serial 2-fold dilutions. The data show the means ± SD of results from three experiments. (b) Calculated IC50 values of ADA Env-pseudotype with escape mutations, normalized to wild-type values, for BMS-626529 and each bNAb. The solid horizontal bars indicate mean fold change in IC50. The data were derived from three independent experiments.
bNAbs inhibit replication-competent virus that contain BMS-626529 resistant mutations.
For pseudotyping, cells are infected with cell-free, replication-defective virus, which is easily neutralized by bNAbs. In the case of replication-competent virus, however, cells are also infected via cell-to-cell transmission, which is both highly efficient and more difficult for bNAbs to neutralize (23). The mutations most resistant to BMS-626529, i.e., M426L, S375M, and S375M/M434I, were introduced into Env sequence of HIV-1NL43-ΔR1, which has a frameshift in vpr, rendering that gene nonfunctional. C8166 T cells were infected with each of these mutant viruses and treated with BMS-626529 or bNAb NIH45-46G54W at 1 nM every 6 days (Fig. 3a). In the absence of treatment, replication of wild-type virus peaked at day 6 postinfection, with a 2-log increase of viral production (Fig. 3b). Treatment with BMS-626529 reduced viral replication by ∼1 log and delayed the time to peak replication. bNAb NIH45-46G54W completely inhibited viral replication. With the escape mutants, BMS-626529 failed to inhibit viral replication; results of treatment with bNAb NIH45-46G54W were similar to those seen with wild-type virus (Fig. 3b). These data are thus consistent with bNAb NIH45-46G54W retaining activity against Env BMS-626529 escape mutants and being very potent in inhibiting replication-competent virus. The level of NIH45-46G54W antibody present in supernatant of infected C8166 cells was very stable during the course of infection (data not shown), considering that half of the cell supernatant was harvested every 3 days, thus resulting in a 2-fold dilution of bNAb every 72 h. A single addition of NIH45-46G54W at 1 nM, nonetheless, completely suppressed wild-type and mutant virus replication (Fig. 3c and d). One dose of BMS-626529 also inhibited the replication of wild-type but not mutant virus for a week. The superior in vitro stability and activity against spreading infection of bNAb NIH45-46G54W may bode well for treatment and prophylaxis.
FIG 3.

bNAb NIH45-46G54W potently inhibits replication-competent HIV-1NL43-ΔR1 strains harboring escape mutations to BMS-626529. (a) Schematic of the infection of C8166 T cells with wild-type or mutant HIV-1NL43-ΔR1 and treatment with BMS-626529 or bNAb, which was added to cell supernatant at 1 nM every 6 days. D, day. (b) Replication curves of WT/mutant HIV-1NL43-ΔR1 with the indicated treatment. RLU levels were determined by infecting TZM-bl cells with cell supernatant collected at the indicated time points after infection. Data represent results of two independent experiments performed in duplicate. (c) Schematic of the D0 infection of C8166 T cells with wild-type or mutant HIV-1NL43-ΔR1 and D0 treatment with 1 nM BMS-626529 or bNAb. (d) Replication curves of mutant NL43-ΔR1 in the presence of BMS-626529 or bNAb, as indicated. RLU levels were determined by infecting TZM-bl cells with cell supernatant collected at the indicated time points postinfection. Data represent results of two independent experiments performed in duplicate.
Synergy of BMS-626529 and CD4 binding site-targeting bNAbs at low concentrations in neutralizing HIV-1.
Since both BMS-626529 and the bNAbs target cellular binding/viral entry, we decided to test the bNAbs together with BMS-626529. We first combined BMS-626529 and each bNAb at a 1:1 molar ratio and assessed their ability to neutralize R5 HIV-1ADA pseudotyped particles. Starting at 5 nM, both BMS-626529 and each bNAb showed typical neutralization curves (Fig. 4). Most bNAbs, especially bibNAbs PG16-iMab and PGT128-iMab, were more potent than BMS-626529, indicated by the leftwise shift of the neutralization curves. When two agents were combined at equimolar amounts and the total concentration remained unchanged, a leftwise shift of the neutralization curve revealed a combinatorial synergistic effect (24, 25). Used in this manner, the combination of BMS-626629 plus bNAb at low concentrations showed left-shifted neutralization curves with most CD4 binding site-targeting bNAbs, including NIH45-46G54W and 4Dm2m and, to a lesser degree, VRC07-523 (Fig. 4). On the other hand, bNAbs targeting other epitopes did not demonstrate this pattern. In addition, we observed similar but not identical results using the X4-tropic HIV-1 pseudotype strain NL4-3 (data not shown). Here, CD4bs-targeting bNAbs, such as NIH45-46G54W and 4Dm2m and, to a lesser degree, VRC03, showed left-shifted neutralization curves in combination with BMS-626529 against NL4-3; such effects were not observed for bNAbs targeting non-CD4bs, including PG16, PGT128, 35O22, and 10E8.
FIG 4.

Combinatorial effects of BMS-626529 and CD4bs-targeting bNAb in neutralizing ADA Env pseudotypes. Data are presented as neutralization curves of the combination of BMS-626529 and the indicated bNAb in equimolar amounts against ADA pseudotyped particles. n = 3.
The combination index (CI) provides a quantitative method to define the combinatorial effect of two agents. BMS-626529 and bNAbs were tested individually and in combination at equimolar ratios over a range of concentrations. Synergy, additivity, and antagonism are indicated by CI values of <1, 1, and >1, respectively (24). The CI values at 50% inhibition (CI50) and 90% inhibition (CI90) of each combination of BMS-626529 and bNAb were calculated (Table 2). CD4bs-targeting bNAbs NIH45-46G54W, VRC07-523, 3BNC117, N6, CH106, and 4Dm2m demonstrated significant synergy with BMS-626529, with mean values for both CI50 and CI90 of less than 1, whereas the mean CI values of the V1/V2 domain-targeting bNAb PG16, V3 domain-targeting bNAb PGT128, and the gp41-gp120 interface-targeting bNAb 35O22 were not consistently less than 1. One exception was the CD4bs bNAb VRC03, with a mean CI90 value of slightly more than 1. The CI50/CI90 values for the BMS-626529/PG16-iMab combination were also below 1, suggesting that it too had a synergistic effect with BMS-626529, representing a result not seen with another bibNAb, PGT128-iMab. Similar results were observed with HIV-1NL4-3 pseudotyped particles (data not shown). The differences in the CI50/CI90 values between CD4bs-targeting and non-CD4bs-targeting bNAbs were statistically significant (Fig. 5). Collectively, these data suggest that the synergistic effect of the combination of BMS-626529 and bNAbs in neutralizing HIV-1 is preferentially observed for the CD4bs-targeting ones.
TABLE 2.
Combination indices of BMS-626529 and bNAbs against ADA pseudotypea
| First inhibitor | IC50
(nM) |
Second inhibitor |
Target(s) | IC50
(nM) |
Combination ratio |
Combination IC50 (nM) |
CI50b | CI90c |
|---|---|---|---|---|---|---|---|---|
| BMS-626529 | 0.35 | NIH 45-46 G54W | CD4bs | 0.09 | 1:1 | 0.06 | 0.30 (0.09) | 0.21 (0.10) |
| VRC03 | CD4bs | 0.24 | 1:1 | 0.45 | 0.47 (0.20) | 1.14 (0.55) | ||
| VRC07-523 | CD4bs | 0.64 | 1:1 | 0.21 | 0.49 (0.22) | 0.35 (0.31) | ||
| 3BNC117 | CD4bs | 0.21 | 1:1 | 0.13 | 0.44 (0.12) | 0.52 (0.19) | ||
| N6 | CD4bs | 0.98 | 1:1 | 0.11 | 0.26 (0.08) | 0.47 (0.26) | ||
| CH106 | CD4bs | 1.13 | 1:1 | 0.18 | 0.64 (0.70) | 0.75 (0.53) | ||
| 4Dm2m | CD4bs | 3.88 | 1:1 | 0.32 | 0.49 (0.40) | 0.32 (0.37) | ||
| PG16 | V1V2 | 0.006 | 1:1 | 0.02 | 1.88 (0.41) | 13.73 (5.70) | ||
| PGT128 | V3 | 0.07 | 1:1 | 0.22 | 1.39 (0.57) | 1.41 (0.49) | ||
| 35O22 | gp41-gp120 interface | 0.20 | 1:1 | 0.13 | 0.85 (0.74) | 1.65 (1.17) | ||
| PG16-iMab | V1V2, CD4 | 0.01 | 1:1 | 0.01 | 0.81 (0.09) | 0.7 (0.10) | ||
| PGT128-iMab | V3, CD4 | 0.003 | 1:1 | 0.03 | 1.30 (0.47) | 3.01 (2.29) |
Compounds were tested individually and in combination at a fixed molar ratio (1:1) over a range of serial dilutions. Synergy, additivity, and antagonism are indicated by combination index (CI) values of <1, 1, and >1, respectively.
Data represent means (SD) of results from three independent experiments. Bold numbers indicate CI values of <1 at 50% inhibition (CI50).
Data represent means (SD) of results from three independent experiments. Bold numbers indicate CI values of <1 at 90% inhibition (CI90).
FIG 5.

Combination index (CI) of BMS-626529 and bNAb in neutralizing HIV-1ADA pseudotyped particles. (a) CI at 50% neutralization (CI50) plot of combination of BMS-626529 and CD4bs-targeting bNAb versus non-CD4bs-targeting bNAb. Unpaired t test with two-tailed P value was applied. ***, P < 0.0001. (b) CI at 90% neutralization (CI90) plot of combination of BMS-626529 and CD4bs-targeting bNAb versus non-CD4bs-targeting bNAb. Unpaired t test with two-tailed P value was applied. **, P < 0.01. The data represent results from three independent experiments.
To further confirm the synergistic effects of combinations of BMS-626529 and CD4bs-targeting bNAbs, we conducted a combination matrix neutralization assay. The synergistic or antagonistic effect of the combination of BMS-626529 and NIH45-46G54W or PG16 at different concentrations was determined using classical synergy models (Bliss and Loewe) implemented in the program Combenefit (26–28). Both the Bliss and Loewe models suggested synergistic effects between bNAb NIH45-46G54W and BMS-626529 in neutralizing HIV-1ADA pseudotype when used at low concentrations (Fig. 6a and b). In contrast, a moderately antagonistic effect was suggested between bNAb PG16 and BMS-626529 at low concentrations (Fig. 6c and d), which is consistent with the combination index analysis. Since both BMS-626529 and the bNAbs are extremely potent in neutralizing HIV-1, combinatorial effects were not significant at high concentrations.
FIG 6.

Synergistic effects in neutralizing ADA Env pseudotypes with BMS-626529 and CD4bs-targeting bNAb NIH45-46G54W. (a) Bliss synergy and antagonism matrix of BMS-626529 and bNAb NIH45-46G54W at indicated concentrations against ADA pseudotyped particles. (b) Loewe synergy and antagonism surface of BMS-626529 and NIH45-46G54W at indicated concentrations in neutralizing ADA Env pseudotypes. (c) Bliss synergy and antagonism matrix of BMS-626529 and bNAb PG16 at indicated concentrations against ADA pseudotyped particles. (b) Loewe synergy and antagonism surface of BMS-626529 and PG16 at indicated concentrations in neutralizing ADA Env pseudotypes. Data represent three independent experiments.
Synergy of BMS-626529 and CD4 binding site-targeting bNAb NIH45-46G54W in neutralizing BMS-626529 escape mutant and tier 2 HIV-1 isolate.
Using the escape mutant of BMS-626529 M426L, enhanced synergy between BMS-626529 and NIH45-46G54W was observed compared to the wild-type ADA enveloped pseudovirus (Fig. 7a and b). We also examined their combinatorial effects against a representative tier 2 HIV-1 isolate, REJO4541.67, which exhibits moderate sensitivity to antibody-mediated neutralization (29). The dose-response curve indicated that this isolate was resistant to BMS-626529 but was sensitive to the neutralization of NIH45-46G54W (Fig. 7c and d). However, the combination of BMS-626259 and NIH45-46G54W showed a strong synergistic effect in neutralizing this tier 2 isolate (Fig. 7e and f). Sequence analysis indicated that tier 2 isolate REJO4541.67 harbors two escape mutations, S375T and M434I, with respect to BMS-626529 (Fig. 7g), which likely explains why this isolate is resistant to BMS-626529. Thus, these data suggest that the combination of BMS-626529 and NIH45-46G54W is capable of synergistically neutralizing a tier 2 HIV-1 isolate carrying escape mutations of BMS-626529.
FIG 7.

Synergy between BMS-626529 and NIH45-46G54W in neutralizing escape mutant of BMS-626529 and tier 2 HIV-1 isolate. (a) Bliss synergy and antagonism matrix of BMS-626529 and bNAb NIH45-46G54W at indicated concentrations against ADA pseudotyped particles with M426L mutation. (b) Loewe synergy and antagonism surface of BMS-626529 and NIH45-46G54W at indicated concentrations in neutralizing ADA Env with M426L mutation. (c and d) Dose-response curves of neutralization of tier 2 HIV-1 isolate REJO4541.67 by BMS-626529 (c) and NIH45-46G54W (d). (e) Bliss synergy and antagonism matrix of BMS-626529 and bNAb NIH45-46G54W at indicated concentrations against REJO4541.67 Env pseudotypes. (f) Loewe synergy and antagonism surface of BMS-626529 and NIH45-46G54W at indicated concentrations in neutralizing REJO4541.67 Env pseudotypes. (g) Sequence alignment between REJO4541.67 and HIV-1 AD8 Env. Resistance mutations to BMS-626529 are indicated in red.
Treatment of replication-competent HIV-1 with both BMS-626529 and bNAb.
It was of interest to examine the effect of the combination of BMS-626529 and NIH45-46G54W on replication-competent virus, which is more physiological. X4 virus HIV-1NL43-ΔR1 was used to infect C8166 cells; at the time of infection, cells were treated with 0.3 nM BMS-626529 or with 0.1 nM NIH45-46G54W or with the two in combination. In the absence of either agent, viral replication peaked at day 6 and then decreased due to massive cell death (Fig. 8a). BMS-626529 or NIH45-46G54W alone efficiently reduced viral replication at day 3 and day 6. At day 9, however, neither agent alone could completely suppress viral replication; however, the combination of BMS-626529 and NIH45-46G54W was more inhibitory (Fig. 8a). We calculated the inhibitory efficiency of each single agent at days 3, 6, and 9 and the expected additive effect in comparison to the actual effects of the combination (Fig. 8b). The data suggest that early on, i.e., at days 3 and 6, both single agents were very effective and that there were no significant differences in inhibition with respect to the expected additivity for the combination. At day 9, the combination had a greater than additive effect. The results of an area under the curve (AUC) analysis also suggested that the antiviral effect of BMS-626529 plus NIH45-46G54W in combination was greater than that of each agent alone (Fig. 8c). These data lend further support to the concept of synergy between BMS-626529 and NIH45-46G54W and other CD4bs-targeting bNAbs.
FIG 8.
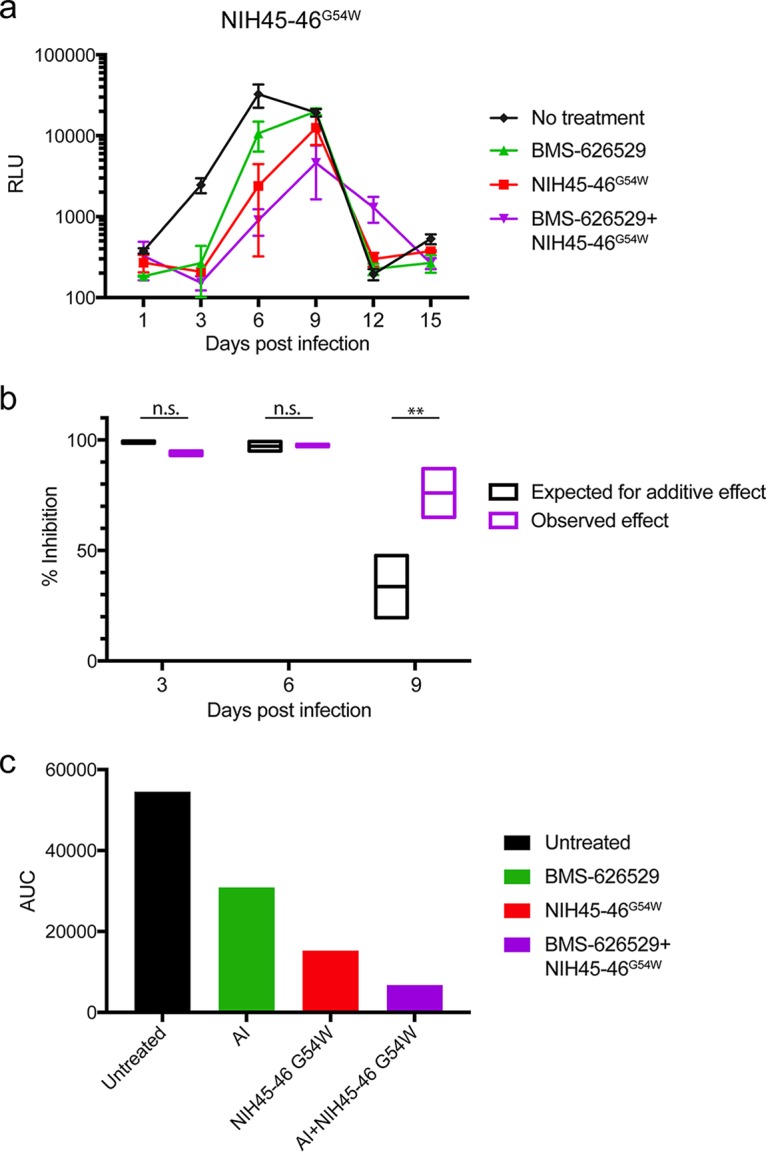
Synergistic effect between BMS-626529 and NIH45-46G54W against replication-competent HIV-1NL43-ΔR1. (a) Replication of NL43-ΔR1 in the presence of each single agent or the combination of BMS-626529 (0.3 nM) and NIH45-46G54W (0.1 nM). BMS-626529 and bNAb were added every 6 days. These data represent results of two independent experiments performed in duplicate. (b) Comparison of inhibition data corresponding to the expected additive effect and the observed effect during the robust replication phase. The expected additive inhibition effect was calculated for days 3, 6, and 9 based on the effect of each single agent and was compared to that of the combination of both agents. These data represent results of two independent experiments performed in duplicate. Data were analyzed with t tests. **, P < 0.01. (c) Area under the curve (AUC) analysis of data in panel a.
Mechanistic investigation of the synergistic effect between BMS-626529 and CD4 binding site-targeting bNAbs.
To investigate the mechanism of synergy between BMS-626529 and CD4bs-targeting bNAbs, we first examined whether BMS-626529 can affect bNAb binding to HIV-1 Env-expressing cells by flow cytometry. bNAbs varied in their levels of binding to HIV-1 YU2 Env (data not shown). In the presence of 100 nM BMS-626529, all bNAbs showed an insignificant change in the level of binding to HIV-1 YU2 Env, although CD4 binding site-targeting bNAbs showed a subtle increase in binding (data not shown). At the concentration of 1 μM, BMS-626529 minimally decreased soluble CD4-Ig binding to YU2 Env (data not shown), consistent with prior reports (5, 30, 31). Notably, the binding to HIV-1 Env by bNAbs at low concentrations, e.g., lower than 100 ng/ml, is subtle. Thus, it was difficult to assess any interaction between BMS-626529 and bNAb in terms of viral Env binding at low concentrations by flow cytometry.
We next used HIV-1 Env trimer binding enzyme-linked immunosorbent assays (ELISA) to examine the binding of CD4 to HIV-1 Env. As shown in Fig. 9a, the binding of CD4-IgG2 to HIV-1 JRFL gp140 was sensitive and specific. Without CD4-IgG2 coating or JRFL gp140, very low background was observed (Fig. 9a). Addition of CD4-IgG2 or soluble CD4 (sCD4) markedly decreased the binding of CD4-IgG2 to the viral Env (Fig. 9a). Treatment with BMS-626529 or CD4bs-targeting bNAbs NIH45-46G54W and N6 strikingly blocked CD4 binding to viral Env, while the inhibitory effect of other bNAbs were less substantial (Fig. 9a). Added in combination at different concentrations, BMS-626529 and NIH45-46G54W showed an enhanced inhibitory effect on CD4 binding to HIV-1 JRFL gp140 compared to the results seen with each single agent (Fig. 9b). In addition, a combination matrix experiment using BMS-626529 and NIH45-46G54W demonstrated a synergistic effect in blocking CD4 binding to HIV-1 Env (Fig. 9c). The mean effect value for all combinations was 3.14, with a 95% confidence interval of 1.63 to 4.65, which indicates a result significantly greater than an additive effect (0). These data suggest that BMS-626529 and CD4bs-targeting bNAb NIH45-46G54W are able to cooperatively inhibit the binding of CD4 to HIV-1 Env.
FIG 9.

CD4 binding to HIV-1 Env and the inhibitory effect of BMS-626529 and bNAbs. (a) CD4 binding to HIV-1 JRFL Env trimer was measured by Env binding ELISA. Competitors, including CD4-IgG2, sCD4, BMS-626529, and bNAbs, were added at 25 nM final concentration. Samples without CD4-IgG2 coating or JRFL Env trimer were controls. Binding data of sample with competitor were normalized to the no-competitor control. (b) Binding of CD4 to HIV-1 JRFL Env trimer with treatment of BMS-626529 or NIH45-46G54W or the two in combination at the indicated concentrations. The binding data of sample with competitor(s) were normalized to the no-competitor control. The data represent results from three independent assays performed with duplicates. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (multiple t tests). (c) Highest single agent (HSA) synergy and antagonism matrix of BMS-626529 and bNAb NIH45-46G54W at indicated concentrations in blocking CD4 binding to HIV-1 JRFL gp140. The data represent results of two independent assays.
DISCUSSION
Given that new drug development is an extremely time-consuming and expensive process, study of the interaction between small molecules and biologics already known to be efficacious may be a more time- and money-saving route to the development of novel antiviral therapeutics. In the past decade, over 60 anti-HIV bNAbs have been identified which target different epitopes on the viral Env trimer (32). These bNAbs represent an invaluable resource for the study of the structure of Env and of the interaction between different classes of antivirals. Here, we investigated the interaction between a member of a novel class of anti-HIV small-molecule drugs—attachment inhibitor BMS-626529—and several of the latest bNAbs. We found that the escape mutants resistant to BMS-626529 were still very sensitive to most bNAbs tested here, even the CD4bs-targeting bNAbs, suggesting that their targeting sites do not overlap. This finding also implies that discovery of small molecules that are more specifically targeted against the CD4 binding site of HIV Env is feasible, as demonstrated by the work on CD4 mimetics (33–35).
There is still debate as to how to best measure synergy/antagonism of drug combinations (36). Such efforts were even more challenging in this study, considering that BMS-626529 and bNAbs are very potent against HIV-1 with low-nanomole IC50 values. Here, we employed multiple tests or models to confirm the combinatorial effects of BMS-626529 and bNAbs. The combination index method is based on the median-effect principle (MEP) that developed from the physicochemical principle of the mass-action law, which is independent of the mechanism of each agent (24). The two agents in combination can be mixed at fixed or variable ratios, and limited data points are needed for analysis. These features make it possible to perform a wide screen for synergy with many drug combinations. By this method, we examined the combinatorial effect of BMS-626529 with more than 10 bNAbs at a 1:1 molar ratio in neutralizing two HIV-1 strains with R5 and X4 tropisms, i.e., ADA and NL4-3. The results were quite similar in terms of synergistic effects of BMS-626529 and CD4bs-targeting bNAbs (Table 2; see also Fig. 4). Notable discrepancies with these viruses included the combinatorial effects of BMS-626529 and VRC07-523 or VRC03; the former combination exhibited synergy against ADA but not NL4-3 pseudotyped particles, while the latter combination showed the opposite effect. This suggests, unsurprisingly, that the synergistic effect of combinations of BMS-626529 and CD4bs-targeting bNAbs depends on the viral Env. We also examined the combinatorial effect between BMS-626529 and bNAb NIH45-45G54W by combination matrix of different concentrations and analyzed the data with another two established synergy models, namely, Bliss and Loewe models (26). These methods are more extensively geared toward investigating specific drug combinations in greater detail, and the results from these two models were quite consistent (Fig. 6 and 7). That we used different experimental settings and analytical models to examine the combinatorial effect between BMS-626529 and bNAbs and observed similar results suggests that there is true synergy between the small molecule and certain CD4bs bNAbs that was not consistently seen with other bNAbs tested here. Another interesting finding reported here is the enhanced synergistic effect between BMS-626529 and bNAb on escape mutants resistant to BMS-626529 compared to the wild-type results (Fig. 7). This reinforces the notion that escape mutants resistant to BMS-626529 can be natural variants (11). It also suggests that patients who fail BMS-626529 therapy due to viral resistance may do well on the combination of BMS-626529 and bNAbs, although this will require further study.
Multiple mechanisms of action have been suggested for BMS-626529 (37, 38). The crystal structure of trimeric HIV envelope with BMS-626529 attachment inhibitors was recently reported (5). Based on this binding mode, the following concentration-dependent mechanism of HIV-1 inhibition by BMS-626529 was proposed: at low concentrations, BMS-626529 efficiently inhibits CD4-induced conformational changes needed for coreceptor binding and subsequent steps of viral entry but not sCD4 binding; at high concentrations, BMS compounds are able to inhibit the sCD4 binding by competing for certain residues that contact both BMS compounds and CD4, e.g., Trp427 (5). We showed for the first time that when BMS-626529 was combined with CD4bs-targeting bNAb NIH45-46G54W, there was a significant enhancement of inhibition of CD4 binding to viral Env (Fig. 9). This observation may well explain the synergy of BMS-626529 and CD4bs-targeting bNAbs observed by us in neutralizing HIV-1 at low concentrations. The precise molecular mechanism of this effect is not known; structural studies may be useful in this regard. Irrespective of the precise mechanism of action, the observed synergy between BMS-626529 and CD4-bs bNAbs opens up additional therapeutic avenues and highlights the potential of complex interplay between small molecules and biologics in the treatment of HIV disease.
MATERIALS AND METHODS
Plasmids and cells.
The HIV-1 ADA Env clone was from the laboratory of Dan Littman at the New York University (NYU) School of Medicine. HIV-LIB and HIV-CIY backbone plasmids have been described previously (39, 40). A full-length replication-competent HIV-1NL43-ΔR1 clone was derived from NL4-3 by frameshifting vpr at the unique EcoR1 site. PCR-based site-directed mutagenesis was used to generate BMS-626529 escape mutants in ADA Env and separately in replication-competent HIV-1NL43-ΔR1, and the results were confirmed by Sanger sequencing. Tier 2 HIV-1 subtype B Env clone REJO4541.67 was from the NIH AIDS Reagent Program.
HEK293T (ATCC CRL-11268™) cells were from the American Type Culture Collection (ATCC); TZM-bl, C8166, GHOST-Hi5, and GHOST-X4 cells were from the AIDS Reagent Repository. HEK293T, TZM-bl, GHOST-Hi5, and GHOST-X4 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with penicillin/streptomycin and 10% fetal bovine serum (FBS; Invitrogen). C8166 cells were cultured in similarly supplemented RPMI 1640.
Viruses and reagents.
HIV-1 Env pseudotyped viruses were produced as described previously (41). Replication-competent HIV-1NL43-ΔR1 wild-type and mutant viruses were produced by transfecting HEK293T cells with full-length HIV-1NL43-ΔR1 plasmid or Env mutants. HIV-1 JRFL gp140 (B.JRFL gp140CF), MAb 2G12, CD4-IgG2, sCD4, and purified bNAbs were from the NIH AIDS Reagent Program. Attachment inhibitor BMS-626529 was provided by Mark Krystal (formerly of Bristol Myers-Squibb, now at ViiV HealthCare).
Neutralization assay with wild-type or escape mutant Env pseudotype HIV.
BMS-626529 or bNAb was serially diluted 2-fold as indicated and then added to HIV-1 ADA wild-type/mutant (WT/MT) pseudotypes. After a 1-h incubation at 37°C, the mixture was added to GHOST-Hi5 target cells. Luciferase activity was measured 48 to 72 h postinfection by luminometry using luciferin (Sigma) as the substrate.
Assay of combinatorial effect between BMS-626529 and bNAb.
For fixed-ratio combination tests, BMS-626529 was mixed with each bNAb at a 1:1 molecular ratio at a total concentration of 5 nM, and the mixture was serially diluted 5-fold. Each single agent (BMS-626529 or bNAb) was included in the same assay at 5 nM. The serially diluted agents were mixed with HIV-1 pseudotypes and added to GHOST-CCR5+/CXCR4+ or TZM-bl cells for luciferase activity assay. For combination matrix assay, BMS-626529 and each bNAb were mixed at different concentrations as indicated. These were then added to HIV-1 pseudotypes and then to target cells for luciferase activity assay as described above. For each test, the luciferase activity (expressed in relative light units [RLU]) of pseudotype-infected cells in the absence of any inhibitor was normalized to that of uninfected cells (considered the background); data were used for further analysis only when the signal/noise ratio was >10.
HIV-1 Env binding assay by Env trimer binding ELISA.
HIV-1 Env trimer binding ELISA was performed according to the method described in a previous report (5), with some modifications. Briefly, 96-well polystyrene plates were coated with phosphate-buffered saline (PBS)-diluted CD4-IgG2 (1 μg/ml) at 4°C overnight. The next day, the plates were washed three times with wash buffer (0.1% phosphate-buffered saline with Tween 20 [PBS-T]) and blocked with blocking buffer (5% milk–wash buffer) for 1 h. HIV-1 JRFL gp140 (3 nM) was pretreated with single or combined competitors at the indicated concentration for 1 h at room temperature; the mixture was then transferred to a CD4-IgG2-coated plate and incubated for 2 h at room temperature. After three washes with blocking buffer and another three with wash buffer, horseradish peroxidase (HRP)-conjugated 2G12 MAb was added to the plate at a 1:9,000 dilution with blocking buffer and incubated for 1 h. After another washing, 3,3',5,5'-tetramethylbenzidine (TMB) substrate was added to the plate and the reaction mixture was incubated for 10 min; the reaction was then stopped by the use of 1 M H3PO4, and the readout was measured (optical density at 450 nm [OD450]).
Replication-competent HIV.
C8166 T cells (1.0 × 105) were infected with 10 μl of HIV-1NL43-ΔR1 wild-type or mutant virus in a 96-well format. At the time of infection, 1 nM BMS-626529 or bNAb was added to cell culture medium for single-agent treatment. BMS-626529 (0.3 nM) or bNAb (0.1 nM) or a combination of the two was added to cell culture for combinatorial-agent assay. Cells were treated with the same concentration of BMS-626529 or bNAb every 6 days. Half of the culture supernatant (100 μl) was collected, and cells were refed with same volume of fresh, complete RPMI 1640 medium at the indicated time points. TZM-bl cells were infected with culture supernatant, and firefly luciferase activity was measured 48 h later, subtracting uninfected-cell background RLU.
Western blotting.
Western blotting of bNAbs in supernatants of transfected cells was performed as described previously (41). Culture supernatants were mixed 1:1 with Laemmli sample buffer (Bio-Rad, Hercules, CA), supplemented with 2-mercaptoethanol, and heated at 100°C for 10 min. Samples were loaded onto 4% to 20% Mini-Protean precast gels (Bio-Rad) and run for 1 h at 120 V. Gels were transferred onto polyvinylidene difluoride (PVDF) membranes for 1 h at 200 mA. Membranes were blocked in PBS-T–5% milk, washed three times with PBS-T, probed with secondary goat anti-human IgG H+L HRP-conjugated antibody (Bethyl Laboratories) (1:5,000 dilution), washed three times, allowed to react with Hy-GLO quick-spray chemiluminescent substrate (Denville Scientific, Metuchen, NJ), and developed with autoradiographic film.
HIV-1 Env binding assay by flow cytometry.
HEK293T cells were transfected with HIV-1YU2 Env-expressing plasmid or mock transfected using Lipofectamine 2000 (Invitrogen) in a six-well format. At 48 h posttransfection, cells were harvested and washed with cold PBS. A total of 2 × 105 cells were blocked in 100 μl PBS–5% milk on ice for 20 min. bNAbs were then added to the cells at a final concentration of 10 μg/ml (or as indicated) and incubated for 1 h on ice. Mock-transfected cells were also incubated with each bNAb as a control. For BMS-626529 treatment, BMS-626529 was added to the cells at a final concentration of 100 nM, along with bNAbs. Cells were then washed twice with cold PBS and incubated 30 min on ice with anti-human IgG1 (H+L) Alexa Fluor 647 (Life Technologies) (1:400 dilution). Cells were then treated with fixation buffer (eBioscience), and samples were run on an LSR II cytometer (BD). Fluorescence-activated cell sorter (FACS) data were analyzed using FlowJo software. Gating for bNAb binding to HIV-1 Env was set based on the mock-transfected cells serving as a negative control.
Data analysis.
The IC50 of BMS-626529 or bNAb was calculated according to normalized RLU data from the neutralization assays using GraphPad Prism software. Nonlinear regression was applied with a normalized response model. The combination index (CI) of each BMS-626529 and bNAb mixture was calculated using the CompuSyn program (25). The Bliss and Loewe synergy models for drug combinations were analyzed with the program Combenefit according to the corresponding manual (26).
ACKNOWLEDGMENTS
We thank Mark Krystal, formerly of Bristol Myers-Squibb and now at ViiV HealthCare, for the generous gift of BMS-626529. We thank Eugene Stewart (GlaxoSmithKline) for providing the source picture for Fig. 1a. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: human CD4-Ig expression vector from Xueling Wu; TZM-bl cells from John C. Kappes, Xiaoyun Wu, and Tranzyme Inc., and GHOST.X4 and GHOST.Hi5 cells from Vineet N. KewalRamani and Dan R. Littman. We thank David Ho and Craig Pace (Rockefeller University) for the generous gift of PG16-iMab; Pamela Bjorkman (California Institute of Technology) for NIH45-46G54W; Heinrich Gottlinger (University of Massachusetts [UMass]–Worcester) for PG16; Dimiter S. Dimitrov (formerly of NIH-NCI, now at the University of Pittsburgh Medical Center [UPMC]) for 4Dm2m and 6Dm2m; and the Vaccine Research Center (NIH) for VRC03, VRC07-523, 35O22, and 10E8 plasmid clones.
This study was funded in part by DP1DA036463, R21AI122384, and R33AI122384. R.E.S. is a NIDA Avant Garde awardee and a paid consultant of ViiV Healthcare.
REFERENCES
- 1.De Clercq E. 2009. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents 33:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Wilen CB, Tilton JC, Doms RW. 10 April 2012. HIV: cell binding and entry. Cold Spring Harb Perspect Med doi: 10.1101/cshperspect.a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo Q, Ho HT, Dicker I, Fan L, Zhou N, Friborg J, Wang T, McAuliffe BV, Wang HG, Rose RE, Fang H, Scarnati HT, Langley DR, Meanwell NA, Abraham R, Colonno RJ, Lin PF. 2003. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J Virol 77:10528–10536. doi: 10.1128/JVI.77.19.10528-10536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A 100:11013–11018. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pancera M, Lai YT, Bylund T, Druz A, Narpala S, O'Dell S, Schon A, Bailer RT, Chuang GY, Geng H, Louder MK, Rawi R, Soumana DI, Finzi A, Herschhorn A, Madani N, Sodroski J, Freire E, Langley DR, Mascola JR, McDermott AB, Kwong PD. 21 August 2017. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat Chem Biol doi: 10.1038/nchembio.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lalezari JP, Latiff GH, Brinson C, Echevarría J, Treviño-Pérez S, Bogner JR, Thompson M, Fourie J, Sussmann Pena OA, Mendo Urbina FC, Martins M, Diaconescu IG, Stock DA, Joshi SR, Hanna GJ, Lataillade M. 2015. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial. Lancet HIV 2:e427–e437. doi: 10.1016/S2352-3018(15)00177-0. [DOI] [PubMed] [Google Scholar]
- 7.Landry I, Zhu L, Abu Tarif M, Hruska M, Sadler BM, Pitsiu M, Joshi S, Hanna GJ, Lataillade M, Boulton DW, Bertz RJ. 2016. Model-based phase 3 dose selection for HIV-1 attachment inhibitor prodrug BMS-663068 in HIV-1-infected patients: population pharmacokinetics/pharmacodynamics of the active moiety, BMS-626529. Antimicrob Agents Chemother 60:2782–2789. doi: 10.1128/AAC.02503-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nowicka-Sans B, Gong YF, McAuliffe B, Dicker I, Ho HT, Zhou N, Eggers B, Lin PF, Ray N, Wind-Rotolo M, Zhu L, Majumdar A, Stock D, Lataillade M, Hanna GJ, Matiskella JD, Ueda Y, Wang T, Kadow JF, Meanwell NA, Krystal M. 2012. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob Agents Chemother 56:3498–3507. doi: 10.1128/AAC.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou N, Nowicka-Sans B, Zhang S, Fan L, Fang J, Fang H, Gong YF, Eggers B, Langley DR, Wang T, Kadow J, Grasela D, Hanna GJ, Alexander L, Colonno R, Krystal M, Lin PF. 2011. In vivo patterns of resistance to the HIV attachment inhibitor BMS-488043. Antimicrob Agents Chemother 55:729–737. doi: 10.1128/AAC.01173-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou N, Nowicka-Sans B, McAuliffe B, Ray N, Eggers B, Fang H, Fan L, Healy M, Langley DR, Hwang C, Lataillade M, Hanna GJ, Krystal M. 2014. Genotypic correlates of susceptibility to HIV-1 attachment inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J Antimicrob Chemother 69:573–581. doi: 10.1093/jac/dkt412. [DOI] [PubMed] [Google Scholar]
- 11.Ray N, Hwang C, Healy MD, Whitcomb J, Lataillade M, Wind-Rotolo M, Krystal M, Hanna GJ. 2013. Prediction of virological response and assessment of resistance emergence to the HIV-1 attachment inhibitor BMS-626529 during 8-day monotherapy with its prodrug BMS-663068. J Acquir Immune Defic Syndr 64:7–15. doi: 10.1097/QAI.0b013e31829726f3. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Zhou N, Sun Y, Ray N, Lataillade M, Hanna GJ, Krystal M. 2013. Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors. Antimicrob Agents Chemother 57:4172–4180. doi: 10.1128/AAC.00513-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wibmer CK, Moore PL, Morris L. 1 May 2015. HIV broadly neutralizing antibody targets. Curr Opin HIV AIDS doi: 10.1097/COH.0000000000000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J, Kang BH, Ishida E, Zhou T, Griesman T, Sheng Z, Wu F, Doria-Rose NA, Zhang B, McKee K, O’Dell S, Chuang G-Y, Druz A, Georgiev IS, Schramm CA, Zheng A, Joyce MG, Asokan M, Ransier A, Darko S, Migueles SA, Bailer RT, Louder MK, Alam SM, Parks R, Kelsoe G, Von Holle T, Haynes BF, Douek DC, Hirsch V, Seaman MS, Shapiro L, Mascola JR, Kwong PD, Connors M. 2016. Identification of a CD4-binding-site antibody to HIV that evolved near-pan neutralization breadth. Immunity 45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudicell RS, Kwon YD, Ko SY, Pegu A, Louder MK, Georgiev IS, Wu X, Zhu J, Boyington JC, Chen X, Shi W, Yang ZY, Doria-Rose NA, McKee K, O'Dell S, Schmidt SD, Chuang GY, Druz A, Soto C, Yang Y, Zhang B, Zhou T, Todd JP, Lloyd KE, Eudailey J, Roberts KE, Donald BR, Bailer RT, Ledgerwood J, NISC Comparative Sequencing Program, Mullikin JC, Shapiro L, Koup RA, Graham BS, Nason MC, Connors M, Haynes BF, Rao SS, Roederer M, Kwong PD, Mascola JR, Nabel GJ. 2014. Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J Virol 88:12669–12682. doi: 10.1128/JVI.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Escolano A, Dosenovic P, Nussenzweig MC. 21 December 2016. Progress toward active or passive HIV-1 vaccination. J Exp Med doi: 10.1084/jem.20161765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimura Y, Gautam R, Chun TW, Sadjadpour R, Foulds KE, Shingai M, Klein F, Gazumyan A, Golijanin J, Donaldson M, Donau OK, Plishka RJ, Buckler-White A, Seaman MS, Lifson JD, Koup RA, Fauci AS, Nussenzweig MC, Martin MA. 13 March 2017. Early antibody therapy can induce long-lasting immunity to SHIV. Nature doi: 10.1038/nature21435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoofs T, Klein F, Braunschweig M, Kreider EF, Feldmann A, Nogueira L, Oliveira T, Lorenzi JC, Parrish EH, Learn GH, West AP Jr, Bjorkman PJ, Schlesinger SJ, Seaman MS, Czartoski J, McElrath MJ, Pfeifer N, Hahn BH, Caskey M, Nussenzweig MC. 2016. HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science 352:997–1001. doi: 10.1126/science.aaf0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruel T, Guivel-Benhassine F, Amraoui S, Malbec M, Richard L, Bourdic K, Donahue DA, Lorin V, Casartelli N, Noel N, Lambotte O, Mouquet H, Schwartz O. 2016. Elimination of HIV-1-infected cells by broadly neutralizing antibodies. Nat Commun 7:10844. doi: 10.1038/ncomms10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu CL, Murakowski DK, Bournazos S, Schoofs T, Sarkar D, Halper-Stromberg A, Horwitz JA, Nogueira L, Golijanin J, Gazumyan A, Ravetch JV, Caskey M, Chakraborty AK, Nussenzweig MC. 2016. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 352:1001–1004. doi: 10.1126/science.aaf1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halper-Stromberg A, Lu CL, Klein F, Horwitz JA, Bournazos S, Nogueira L, Eisenreich TR, Liu C, Gazumyan A, Schaefer U, Furze RC, Seaman MS, Prinjha R, Tarakhovsky A, Ravetch JV, Nussenzweig MC. 2014. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell 158:989–999. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W, Feng Y, Prabakaran P, Ying T, Wang Y, Sun J, Macedo CD, Zhu Z, He Y, Polonis VR, Dimitrov DS. 2014. Exceptionally potent and broadly cross-reactive, bispecific multivalent HIV-1 inhibitors based on single human CD4 and antibody domains. J Virol 88:1125–1139. doi: 10.1128/JVI.02566-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Zony C, Chen P, Chen BK. 13 April 2017. Reduced potency and incomplete neutralization of broadly neutralizing antibodies against cell-to-cell transmission of HIV-1 with transmitted founder Envs. J Virol doi: 10.1128/JVI.02425-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou TC. 2010. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 25.Chou TC. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 26.Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, Jodrell DI. 2016. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 32:2866–2868. doi: 10.1093/bioinformatics/btw230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loewe S. 1953. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 3:285–290. [PubMed] [Google Scholar]
- 28.Bliss C. 1939. The toxicity of poisons applied jointly 1. Ann Appl Biol 26:585–615. doi: 10.1111/j.1744-7348.1939.tb06990.x. [DOI] [Google Scholar]
- 29.Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, Bhattacharya T, Lapedes A, Polonis VR, McCutchan FE, Gilbert PB, Self SG, Korber BT, Montefiori DC, Mascola JR. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol 84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herschhorn A, Ma X, Gu C, Ventura JD, Castillo-Menendez L, Melillo B, Terry DS, Smith AB, Blanchard SC, Munro JB, Mothes W, Finzi A, Sodroski J. 2016. Release of gp120 restraints leads to an entry-competent intermediate state of the HIV-1 envelope glycoproteins. mBio 7:e01598-16. doi: 10.1128/mBio.01598-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herschhorn A, Gu C, Espy N, Richard J, Finzi A, Sodroski JG. 2014. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat Chem Biol 10:845–852. doi: 10.1038/nchembio.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eroshkin AM, LeBlanc A, Weekes D, Post K, Li Z, Rajput A, Butera ST, Burton DR, Godzik A. 2014. bNAber: database of broadly neutralizing HIV antibodies. Nucleic Acids Res 42:D1133–D1139. doi: 10.1093/nar/gkt1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madani N, Princiotto AM, Mach L, Ding S, Prevost J, Richard J, Hora B, Sutherland L, Zhao CA, Conn BP, Bradley T, Moody MA, Melillo B, Finzi A, Haynes BF, Smith AB III, Santra S, Sodroski J. 2018. A CD4-mimetic compound enhances vaccine efficacy against stringent immunodeficiency virus challenge. Nat Commun 9:2363. doi: 10.1038/s41467-018-04758-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Princiotto AM, Vrbanac VD, Melillo B, Park J, Tager AM, Smith AB III, Sodroski J, Madani N. 2018. A small-molecule CD4-mimetic compound protects bone marrow-liver-thymus humanized mice from HIV-1 infection. J Infect Dis 218:471–475. doi: 10.1093/infdis/jiy174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quinlan BD, Joshi VR, Gardner MR, Ebrahimi KH, Farzan M. 2014. A double-mimetic peptide efficiently neutralizes HIV-1 by bridging the CD4- and coreceptor-binding sites of gp120. J Virol 88:3353–3358. doi: 10.1128/JVI.03800-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foucquier J, Guedj M. 2015. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect 3:e00149. doi: 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho HT, Fan L, Nowicka-Sans B, McAuliffe B, Li CB, Yamanaka G, Zhou N, Fang H, Dicker I, Dalterio R, Gong YF, Wang T, Yin Z, Ueda Y, Matiskella J, Kadow J, Clapham P, Robinson J, Colonno R, Lin PF. 2006. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J Virol 80:4017–4025. doi: 10.1128/JVI.80.8.4017-4025.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schön A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB III, Sodroski JG. 6 April 2006. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc Nat Acad Sci U S A doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sutton RE, Wu HT, Rigg R, Bohnlein E, Brown PO. 1998. Human immunodeficiency virus type 1 vectors efficiently transduce human hematopoietic stem cells. J Virol 72:5781–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elinav H, Wu Y, Coskun A, Hryckiewicz K, Kemler I, Hu Y, Rogers H, Hao B, Ben Mamoun C, Poeschla E, Sutton R. 2012. Human CRM1 augments production of infectious human and feline immunodeficiency viruses from murine cells. J Virol 86:12053–12068. doi: 10.1128/JVI.01970-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu S, Jackson A, Beloor J, Kumar P, Sutton RE. 2015. Adenovirus-vectored broadly neutralizing antibodies directed against gp120 prevent human immunodeficiency virus type 1 acquisition in humanized mice. Hum Gene Ther 26:622–634. doi: 10.1089/hum.2014.146. [DOI] [PMC free article] [PubMed] [Google Scholar]