

Development of reporter RVs has been hampered by the lack of comprehensive reverse genetics systems. Recently, we developed a plasmid-based reverse genetics system that enables generation of reporter RVs expressing NLuc luciferase. The prototype reporter RV had some disadvantages (i.e., the transgene was unstable and was expressed as a fusion protein with a partial NSP1 peptide); however, the improved reporter RV overcomes these problems through modification of the untranslated region of the reporter-NSP1 chimeric gene. This strategy for generating stable reporter RVs could be expanded to diverse transgenes and be used to develop RV transduction vectors. Also, the data improve our understanding of the importance of 5′- and 3′-terminal sequences in terms of genome replication, assembly, and packaging.
KEYWORDS: reporter virus, virus vector, rotavirus
ABSTRACT
Engineered recombinant viruses expressing reporter genes have been developed for real-time monitoring of replication and for mass screening of antiviral inhibitors. Recently, we reported using a reverse genetics system to develop the first recombinant reporter rotaviruses (RVs) that expressed NanoLuc (NLuc) luciferase. Here, we describe a strategy for developing stable reporter RVs expressing luciferase and green or red fluorescent proteins. The reporter genes were inserted into the open reading frame of NSP1 and expressed as a fusion with an NSP1 peptide consisting of amino acids 1 to 27. The stability of foreign genes within the reporter RV strains harboring a shorter chimeric NSP1-reporter gene was greater than that of those in the original reporter RV strain, independent of the transgene inserted. The improved reporter RV was used to screen for neutralizing monoclonal antibodies (MAbs). Sequence analysis of escape mutants from one MAb clone (clone 29) identified an amino acid substitution (arginine to glycine) at position 441 in the VP4 protein, which resides within neutralizing epitope 5-1 in the VP5* fragment. Furthermore, to express a native reporter protein lacking NSP1 amino acids 1 to 27, the 5′- and 3′-terminal region sequences were modified to restore the predicted secondary RNA structure of the NSP1-reporter chimeric gene. These data demonstrate the utility of reporter RVs for live monitoring of RV infections and also suggest further applications (e.g., RV vaccine vectors, which can induce mucosal immunity against intestinal pathogens).
IMPORTANCE Development of reporter RVs has been hampered by the lack of comprehensive reverse genetics systems. Recently, we developed a plasmid-based reverse genetics system that enables generation of reporter RVs expressing NLuc luciferase. The prototype reporter RV had some disadvantages (i.e., the transgene was unstable and was expressed as a fusion protein with a partial NSP1 peptide); however, the improved reporter RV overcomes these problems through modification of the untranslated region of the reporter-NSP1 chimeric gene. This strategy for generating stable reporter RVs could be expanded to diverse transgenes and be used to develop RV transduction vectors. Also, the data improve our understanding of the importance of 5′- and 3′-terminal sequences in terms of genome replication, assembly, and packaging.
INTRODUCTION
Reporter viruses that express reporter genes (i.e., chemiluminescent or fluorescent proteins) are useful tools for studying replication and spread of diverse RNA viruses in living cells or animals. In general, transgenes harbored by RNA viruses are unstable; therefore, they are lost gradually over several replication cycles (1–6). Moreover, insertion of transgenes into viral genes often attenuates the reporter virus. Thus, a revertant virus that recovers its ability to replicate by deleting or mutating the transgene(s) will replace the entire transgenic population within a few replication cycles.
Rotavirus (RV) is an intestinal pathogen that causes severe gastroenteritis in infants and young children under 5 years of age (7, 8). RV belongs to the family Reoviridae, which includes mammalian orthoreovirus (MRV), a highly tractable experimental model for Reoviridae viruses, and bluetongue virus (BTV), which is an arthropod-transmitted virus fatal to ruminants (9, 10). Viruses belonging to the family Reoviridae harbor a 9- to 12-segment double-stranded RNA (dsRNA) genome within a nonenveloped capsid (11, 12). The dsRNA genome of Reoviridae viruses comprises a minimum of two components: a 5′ and 3′ untranslated region (UTR) and an open reading frame (ORF). Therefore, insertion of foreign genes may compromise viral protein expression and genome packaging signals by disrupting the ordered dsRNA structure; therefore, sites for transgene insertion are limited. One prototype reporter MRV was generated by replacing the σ3 ORF in the S4 gene segment with the green fluorescent protein ORF; infectious viruses were rescued and amplified only in cells stably expressing the σ3 protein (13). Subsequently, autonomously replicating reporter viruses were generated, including MRV expressing iLOV fluorescent protein and Nelson Bay orthoreovirus (NBV) expressing yellow fluorescent protein (14, 15). The strategy for generating reporter viruses has been used to develop a transduction vector expressing large foreign genes; an example is an MRV expressing the simian immunodeficiency virus (SIV) Gag protein (16).
RV has an 11-segment dsRNA genome encoding six structural and six nonstructural proteins. Despite the importance of RV to public health, lack of a comprehensive reverse genetics system has prevented development of reporter RVs or RV transduction vectors harboring a foreign transgene. Recently, we developed a plasmid-based reverse genetics system to generate a recombinant RV strain from 14 plasmids encoding 11 dsRNA RV genomes, a cell-cell fusion-inducing FAST (fusion-associated small transmembrane) protein, and vaccinia virus capping enzyme proteins D1R and D12L (17). The first recombinant reporter RVs generated by a reverse genetics system harbored the NanoLuc (NLuc) gene ORF within the NSP1 gene; in this system, NLuc was expressed as a fusion protein with a 27-amino-acid segment of the NSP1 N-terminal region (17). RV has significant advantages in terms of developing a transduction vector because the RV NSP1 gene is not essential for viral replication, meaning that a whole gene segment is available for insertion of a foreign gene. Thus, development of potentially useful RV vectors, such as vaccine vectors, can be explored.
Further analysis of the prototype reporter RV-NLuc strain revealed that the NLuc transgene was lost after long-term passage in cell culture. Here, we demonstrate that deleting the UTR of the NSP1 gene from NSP1-NLuc chimeric viruses increases the stability of reporter genes by preventing reconstitution of an intact NSP1 ORF. Using the same strategy, we generated other reporter RV strains expressing green or red fluorescent proteins. The stable RV-NLuc virus was used to screen for neutralizing monoclonal antibodies (MAbs). Analysis of escape mutants from the neutralizing MAb screen revealed that the neutralizing epitope resides within the VP5* fragment of the VP4 protein. Furthermore, we modified the 5′- and 3′-terminal regions of the reporter-NSP1 chimeric gene to enable expression of a native reporter protein by abolishing unnecessary ATG codons and restoring the appropriate RNA secondary structure generated by interaction between the 5′ and 3′ termini.
RESULTS
Stability of reporter genes in different RV expression systems.
In a previous study, we generated a recombinant strain SA11 (rsSA11)-NLuc-Full virus encoding the NLuc protein fused a peptide consisting of amino acids 1 to 27 of the NSP1 N-terminal region (NSP11–27) (Fig. 1A) (17). To increase the versatility of the RV reporter systems, we attempted to generate an rsSA11-ZsG-Full virus expressing green fluorescent protein (ZsGreen) using the same method (Fig. 1A). Expression of ZsGreen fused to the NSP11–27 peptide was detected in MA104 cells infected with rsSA11-ZsG-Full (Fig. 1B). To examine the stability of RV-expressing reporter genes, we serially passaged (10 times) rsSA11-NLuc-Full in MA104 cells. Examination of the electropherotypes of rsSA11-NLuc-Full viruses after five passages (P5) revealed emergence of a truncated NSP1-NLuc gene in two of five clones (Fig. 1C, arrows). All five clones among P10 rsSA11-NLuc-Full viruses harbored the truncated NSP1-NLuc gene (Fig. 1C, arrows). In addition, we detected the truncated NSP1-ZsGreen protein after 10 passages in MA104 cells (Fig. 1D, arrows). These results indicate that the NSP1 reporter gene is unstable after long-term serial passage.
FIG 1.
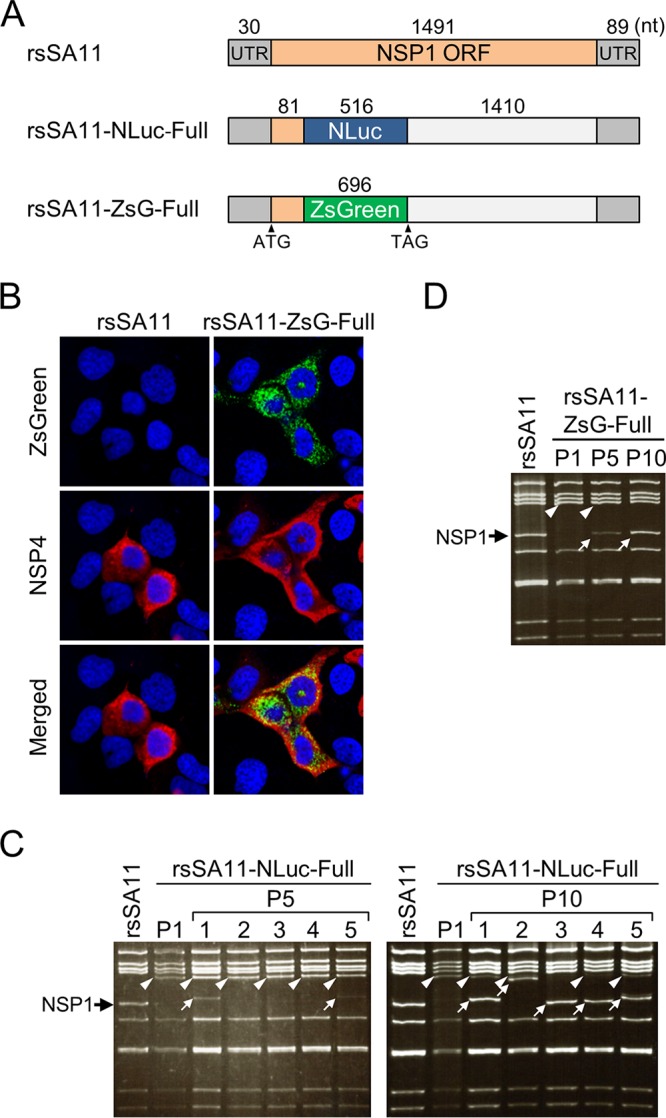
Transgene stability of reporter RV SA11 expressing NanoLuc luciferase (NLuc) or ZsGreen (ZsG). (A) Construction of the NSP1-NLuc-Full and NSP1-ZsG-Full genes. The NLuc or ZsG gene was inserted into the SA11 NSP1 gene between nucleotides 111 and 112. (B) Fluorescence imaging of rsSA11-ZsG-infected cells. MA104 cells were infected with rsSA11 or rsSA11-ZsG-Full, and ZsG expression was observed under a fluorescence microscope. As a control, NSP4 was detected in an indirect immunofluorescence assay using rabbit anti-NSP4 serum and a CF594-conjugated anti-rabbit IgG antibody. (C and D) Stability of the NLuc and ZsG genes after serial passage of reporter RVs. Electrophoresis of the dsRNA genome purified from rsSA11-NLuc-Full and rsSA11-ZsG-Full viruses, as indicated, after passages 5 and 10. Black arrow, NSP1; white arrowheads, the NSP1-NLuc gene (C) or the NSP1-ZsG gene (D); white arrows, truncated NSP1-NLuc (C) or NSP1-ZsG (D) gene.
To examine the mechanisms underlying instability of reporter transgenes introduced into the NSP1 gene segment, we analyzed the NSP1-NLuc and NSP1-ZsGreen dsRNA genomes derived from P1 and P10 rsSA11-NLuc-Full and rsSA11-ZsG-Full viruses, respectively. Sequence analysis of P10 viruses revealed a deletion in both the NLuc and ZsGreen genes, resulting in reconstitution of the NSP1 ORF (Fig. 2A and B). Deletion of most nucleotides spanning bp 31 to 516 of the NLuc ORF and of those spanning bp 112 to 114 of the NSP1 ORF was noted in the NSP1 gene of the rsSA11-NLuc-Full P10 virus (Fig. 2A). The entire NSP1-ZsGreen transgene and the nucleotide sequences spanning bp 112 to 114 of the NSP1 ORF were lost from the rsSA11-ZsG-Full P10 virus, leading to reconstitution of the NSP1 ORF (Fig. 2B). To confirm reconstitution of the NSP1 ORF in these P10 viruses, we used an anti-NSP1 antibody to examine expression of NSP1 in MA104 cells infected with the rsSA11 (wild-type) virus or the P1 and P10 rsSA11-NLuc-Full and rsSA11-ZsG-Full viruses. Western blot analysis showed that the rsSA11-NLuc-Full and rsSA11-ZsG-Full P10 viruses expressed the NSP1 protein, whereas the P1 viruses did not (Fig. 2C). Expression of reconstituted NSP1 by both P10 viruses was significantly higher than that by the wild-type SA11 virus although expression of the VP7 protein was comparable (Fig. 2C).
FIG 2.
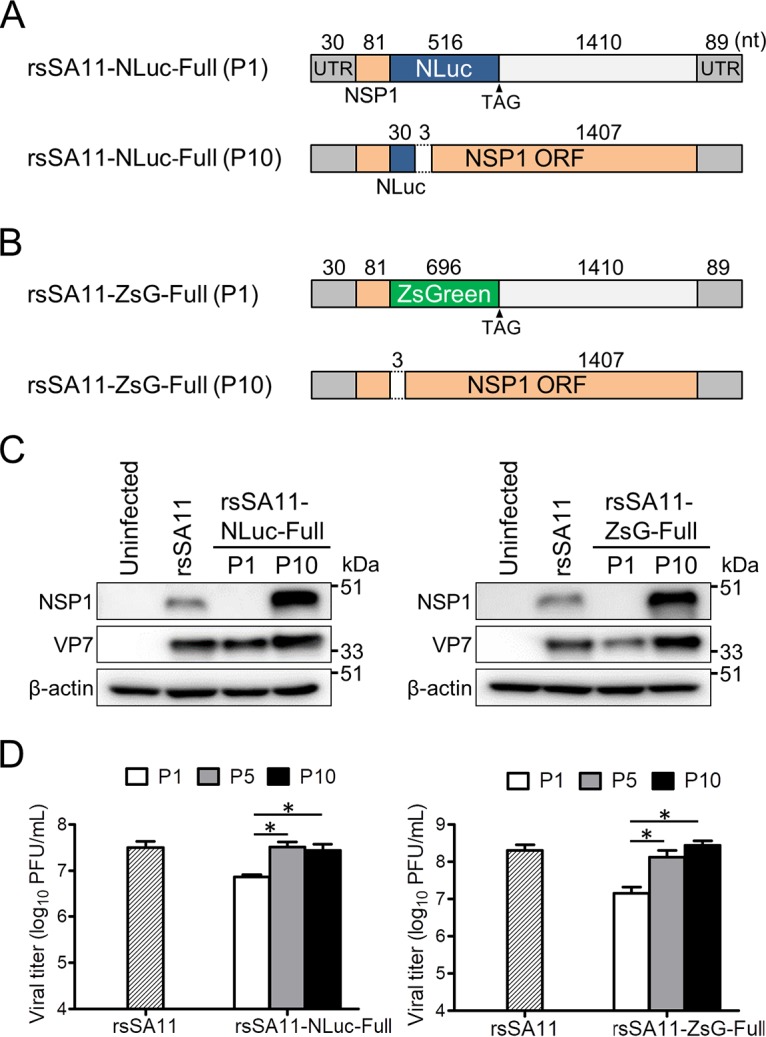
Examination of revertant reporter viruses after 10 passages. (A and B) Schematic images of reporter genes before and after serial passage. Nucleotide sequences of dsRNA genomes purified from rsSA11-NLuc-Full (P10) and rsSA11-ZsG-Full (P10), as indicated, were analyzed. (C) Expression of NSP1 from reporter SA11 virus-infected cells (P1 or P10). MA104 cells were infected with rsSA11 or rsSA11-NLuc-Full (P1 and P10) or rsSA11-ZsG-Full (P1 and P10) at an MOI of 1 PFU/cell. NSP1 and VP7 in cell lysates were detected using a rabbit anti-NSP1 antibody and a monoclonal antibody specific for VP7, followed by appropriate HRP-conjugated secondary antibodies. An anti-β-actin antibody was used as a loading control. (D) Replication of rsSA11-NLuc and rsSA11-ZsG P1 (P5 and P10) viruses. MA104 cells were infected with each reporter virus at an MOI of 0.01 PFU per cell and incubated for 48 h. Infectious virus titers were measured in a plaque assay. Data are expressed as the means ± standard deviations (n = 3). *, P < 0.05 (Tukey’s multiple-comparison test).
RV NSP1 functions as an antagonist of interferon-mediated responses (18). Recently, we demonstrated that replication of an NSP1 deletion mutant virus (lacking the C-terminal region of NSP1, which is associated with interferon regulatory factor 3 [IRF3] degradation) was attenuated in cultured cells (17). Thus, we expected that both P10 viruses would replicate more efficiently than P1 viruses due to restoration of NSP1 expression. As expected, replication rates of rsSA11-NLuc-Full P10 and rsSA11-ZsG-Full P10 viruses were higher than those of the respective P1 viruses and wild-type rsSA11 (Fig. 2D). These results suggest that the rsSA11-NLuc-Full and rsSA11-ZsG-Full viruses that harbor the full-length NSP1 gene segment reconstituted NSP1 function by removing foreign genes and that selection of viral genomes expressing a functional NSP1 protein in the reporter gene cassette may be due to efficient viral replication during long-term passage.
Generation of stable recombinant RVs expressing reporter genes.
To improve the stability of transgenes by avoiding reconstitution of a functional NSP1 gene, we generated truncated forms of the NSP1-NLuc chimeric gene by deleting several fragments of NSP1 (nucleotides [nt] 134 to 465 [332 nt], 134 to 855 [722 nt], and 134 to 1243 [1,110 nt]) downstream of the NLuc gene (Fig. 3A). The three truncated NSP1-NLuc cDNAs were used to generate rsSA11-NLuc-Δ332, rsSA11-NLuc-Δ722, and rsSA11-NLuc-Δ1110, respectively, using an RV reverse genetics system. The three reporter SA11 viruses were passaged up to 10 times in MA104 cells. The electropherotypes of dsRNA genomes purified from P5 and P10 viruses demonstrated that the NSP1-NLuc chimeric genes were maintained and intact after serial passage (Fig. 3B). Consistent with this, we found that the NLuc activities of rsSA11-NLuc-Δ332, -Δ722, and -Δ1110 were comparable between P1, P5, and P10, indicating that these truncated reporter viruses did not undergo genetic or phenotypic changes (Fig. 3C). Single-step and multistep replication kinetics of the deletion mutant viruses were lower than those of the rsSA11 virus in MA104 cells because of lack of functional NSP1 expression (Fig. 3D and E). There were no significant differences in the replication kinetics of the rsSA11-NLuc viruses, indicating that truncation of NSP1 genes did not affect viral replication. To confirm the effect of deleting NSP1 gene segments on foreign gene stability, we prepared three NSP1-ZsGreen chimeric gene cassette cDNAs, each harboring an NSP1 gene of a different length. These were used to rescue rsSA11-ZsG-Δ332, rsSA11-ZsG-Δ722, and rsSA11-ZsG-Δ1110 (Fig. 4A). To investigate the stability of the ZsGreen gene within the NSP1 gene segment, we passaged rsSA11-ZsG-Δ332, rsSA11-ZsG-Δ722, and rsSA11-ZsG-Δ1110 10 times in MA104 cells. The electropherotypes of the dsRNA genomes from rsSA11-ZsG-Δ332, rsSA11-ZsG-Δ722, and rsSA11-ZsG-Δ1110 at P1, P5, and P10 revealed that an NSP1-ZsGreen chimeric gene was maintained (Fig. 4B). To verify ZsGreen activity in P10 viruses of each recombinant strain, MA104 cells were infected with P1 and P10 rsSA11-ZsG viruses. The number of cells expressing ZsGreen per total number of virus-infected cells was measured in an indirect immunofluorescence assay using an NSP4-specific antibody. After infection by prototype rsSA11-ZsG-Full virus, 100% of P1 virus-infected cells expressed ZsGreen, whereas only 8.4% of P10 virus-infected cells expressed ZsGreen (Fig. 4C). In contrast, almost 100% of MA104 cells infected with P1 and P10 rsSA11-ZsG-Δ332, rsSA11-ZsG-Δ722, or rsSA11-ZsG-Δ1110 viruses displayed a ZsGreen fluorescent signal (Fig. 4C). Using the same strategy, we generated another reporter RV, rsSA11-AsR-Δ332, in which the NLuc gene in rsSA11-NLuc-Δ332 was replaced with AsRed2 (Fig. 5A). Electropherotypic analysis of the dsRNA genome of rsSA11-AsR-Δ332 confirmed the presence of the NSP1-AsRed2 chimeric gene segment (Fig. 5B). As expected, MA104 cells infected with rsSA11-AsR-Δ332 exhibited a red fluorescence signal (Fig. 5C). The multistep and single-step replication kinetics of the three reporter viruses, rsSA11-NLuc-Δ332, rsSA11-ZsG-Δ332, and rsSA11-AsR-Δ332, were comparable to each other, indicating that insertion of different reporter genes did not affect the replication of the SA11 virus (Fig. 5D and E). These results suggest that the strategy used to generate a stable recombinant RV vector using truncated forms of the NSP1 gene may be extremely useful, regardless of the transgene inserted.
FIG 3.
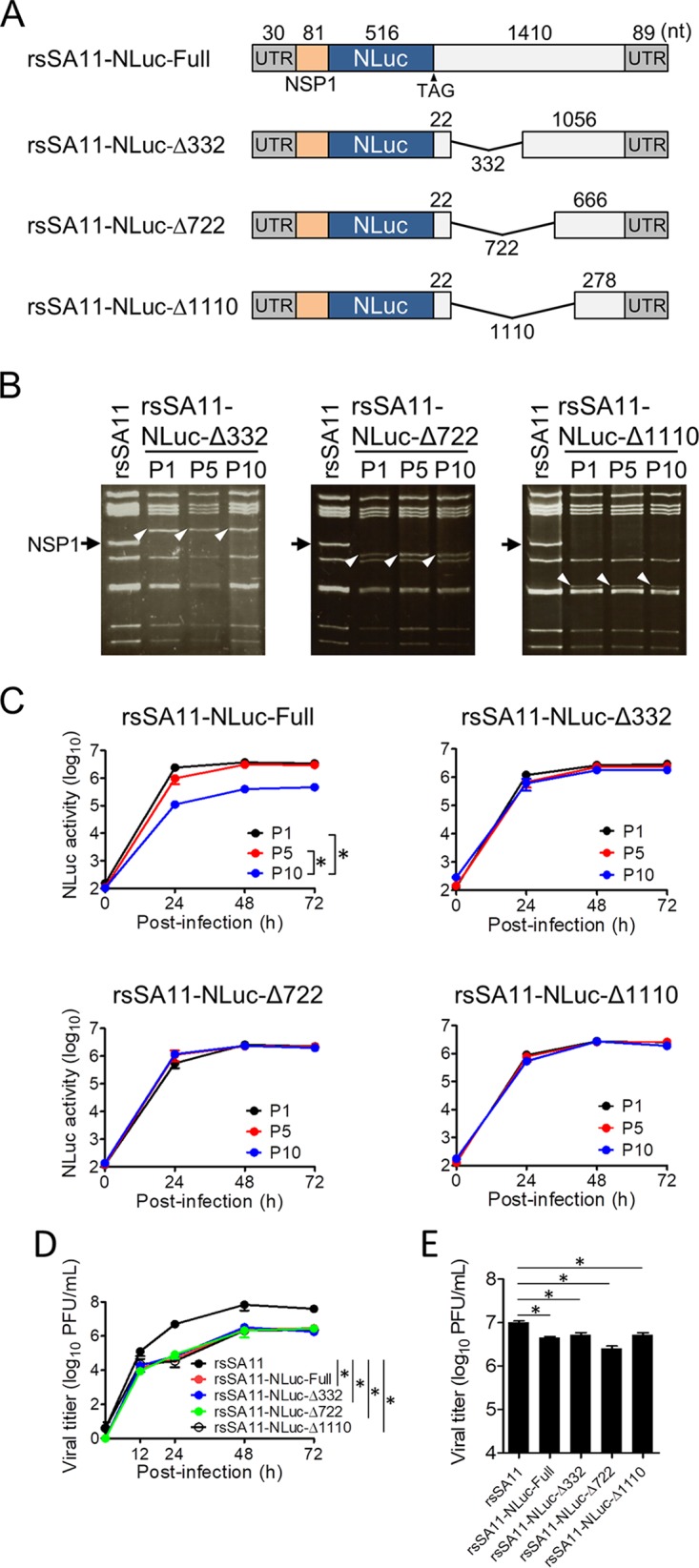
Generation of reporter viruses with improved transgene stability. (A) NSP1-NLuc-Δ332, -Δ722, and -Δ1110 were generated by deleting the NSP1 gene region downstream of the NLuc ORF. (B and C) Stability of NSP1-NLuc reporter genes after serial passage. Panel B shows electropherotypes of dsRNA purified from P1, P5, and P10 reporter viruses. Black arrow, wild-type NSP1; white arrowheads, NSP1-NLuc gene. The kinetics of NLuc activity in rsSA11-NLuc viruses is shown in panel C. MA104 cells were infected with each rsSA11-NLuc virus (P1, P5, and P10) at an MOI of 0.01 PFU per cell and incubated for various times. NLuc activity in the cell lysate was measured by luminometry. Data are expressed as the means ± standard deviations (n = 3). NLuc activity values at 72 h postinfection were compared statistically. *, P < 0.05 (t test). (D and E) Replication kinetics of rsSA11-NLuc viruses. MA104 cells were infected with rsSA11 and rsSA11-NLuc-Full -Δ332, -Δ722, or -Δ1110 viruses at an MOI of 0.001 PFU per cell (D) and incubated for various times or at an MOI of 5 PFU per cell (E) and incubated for 16 h. Infectious virus titers in cell lysates were determined by plaque assay. Data are expressed as the means ± standard deviations (n = 3). Statistical significance was determined by one-way analysis of variance and Tukey’s multiple-comparison posttest. *, P < 0.05.
FIG 4.
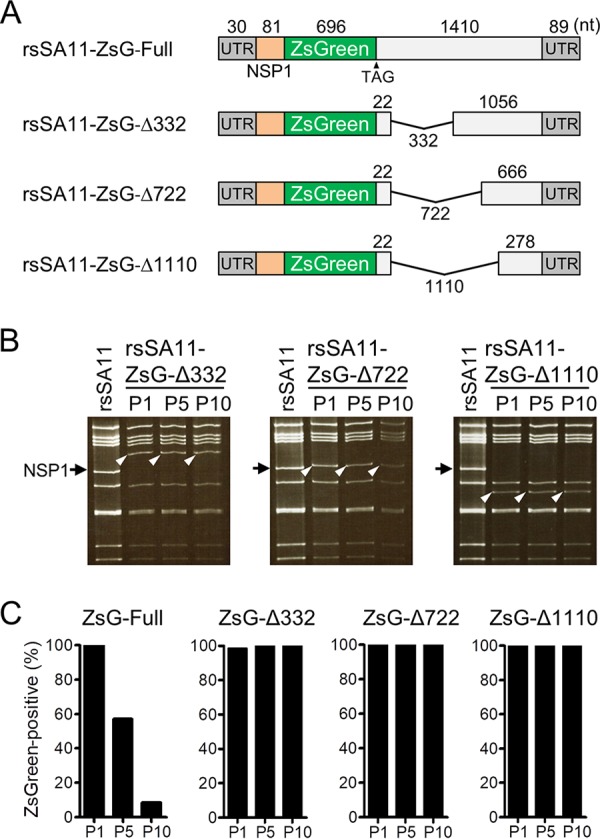
Generation of ZsG-expressing reporter viruses with improved transgene stability. (A) NSP1-ZsG-Δ332, -Δ722, and -Δ1110 were generated by deleting the NSP1 gene region downstream of the ZsG ORF. (B and C) Stability of NSP1-ZsG reporter genes after serial passage. (B) Electropherotypes of dsRNA purified from P1, P5, and P10 reporter viruses. Black arrow, wild-type NSP1; white arrowhead, NSP1-ZsG gene. (C) Quantitative examination of transgene stability after serial passage. MA104 cells were infected with each rsSA11-ZsG virus (P1, P5, and P10). At 24 h postinfection, the viral NSP4 antigen was detected in an indirect immunofluorescence assay using rabbit anti-NSP4 serum and a CF594-conjugated anti-rabbit IgG antibody. The number of ZsG-positive cells within the total NSP4-positive cell population was examined.
FIG 5.
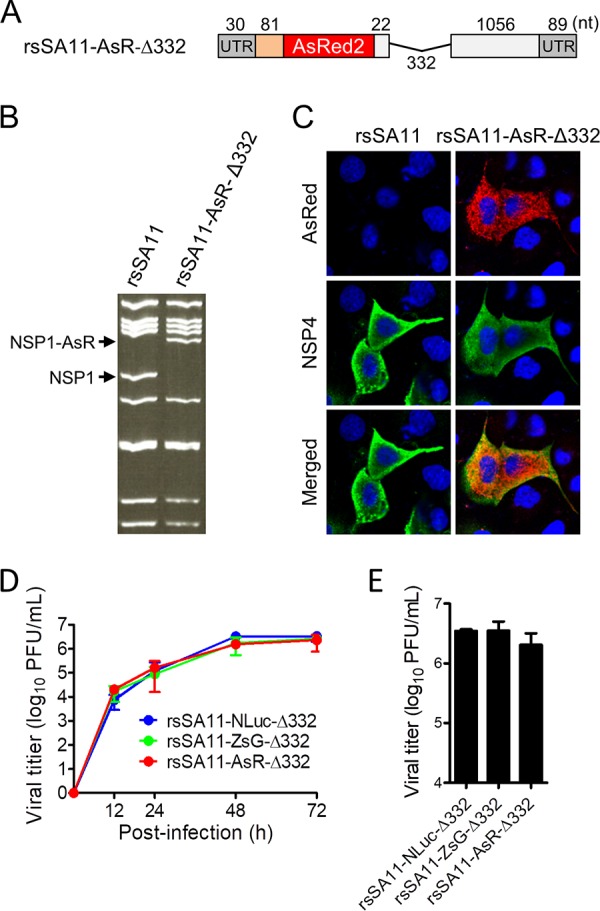
Generation of reporter RVs expressing AsRed2 (AsR). (A) Construction of the NSP1 gene containing the AsRed2 gene. The AsRed2 gene was inserted into the SA11 NSP1 gene between nucleotides 111 and 112. NSP1-AsR-Δ332 was generated by deleting the NSP1 gene fragment after the AsRed2 ORF. (B) Electropherotype of the dsRNA genome purified from rsSA11-AsR-Δ332. Viral dsRNAs were separated in 8% polyacrylamide gels and stained with ethidium bromide. (C) MA104 cells were infected with rsSA11 or rsSA11-AsR-Δ332. Expression of AsRed2 was observed under a fluorescence microscope. As a control, NSP4 was detected in an indirect immunofluorescence assay using rabbit anti-NSP4 serum and an anti-rabbit IgG antibody-CF488 conjugate. (D and E) Replication kinetics of reporter viruses. MA104 cells were infected with rsSA11-NLuc-Δ332, rsSA11-ZsG-Δ332, or rsSA11-AsR-Δ332 virus at an MOI of 0.001 PFU per cell and incubated for various times (D) or at an MOI of 5 PFU per cell and incubated for 16 h (E). Infectious virus titers in cell lysates were determined by plaque assay. Data are expressed as the means ± standard deviations (n = 3). Data were analyzed by one-way ANOVA and Tukey’s multiple-comparison posttest. A P value of <0.05 was considered statistically significant.
Use of the stable reporter RV to screen for NT MAbs specific for strain SA11 virions.
To demonstrate the utility of this stable RV reporter expression system, we used rsSA11-NLuc-Δ332 to screen for neutralizing (NT) antibodies. We generated 91 hybridoma clones from the lymphocytes of mice immunized with wild-type SA11 virions. Next, rsSA11-NLuc-Δ332 was incubated with hybridoma culture supernatant and used to infect MA104 cells. At 24 h postinfection, the NLuc titer in cell lysates from infected cells was measured. Seven MAbs (clones 5, 26, 29, 47, 67, 68, and 91) inhibited infection by rsSA11-NLuc-Δ332 (Fig. 6A). Among the seven NT clones, three (clones 26, 29, and 47) were purified and subjected to further analysis. One NT-negative clone (clone 11) was also analyzed as a control. An NT assay performed with diluted purified MAbs revealed that each MAb had a different NT titer (Fig. 6B). Clone 29, which showed the strongest NT activity, was selected for further analysis. An indirect immunofluorescence assay revealed that clone 29 was specific for a recombinant VP4 protein expressed in cultured cells (Fig. 6C). To identify the neutralizing epitope recognized by clone 29, we passaged the stable rsSA11-NLuc-Δ332 virus in the presence of subneutralizing concentrations (0.1 μg/ml) of clone 29. After serial passage, escape mutants were purified using a plaque cloning method, and a single clone was obtained. The escape mutant virus, named rsSA11-NLuc-esc29, was resistant to neutralization by clone 29 (Fig. 6D). Sequence analysis of the VP4 gene segment of rsSA11-NLuc-esc29 revealed an amino acid substitution (arginine [R] to glycine [G]) at position 441 of the VP4 protein. To confirm the effect of this R441G substitution, we generated a recombinant rsSA11 virus harboring the R441G amino acid mutation in the VP4 gene segment (named rsSA11-R441G-NLuc). The rsSA11-R441G-NLuc virus was not neutralized by clone 29 (Fig. 6D). Furthermore, clone 29 MAb showed markedly reduced activity against VP4 proteins expressed in rsSA11-R441G-NLuc-infected cells (Fig. 6E). This indicates that amino acid R441 in the VP4 protein of strain SA11 plays a critical role in binding to clone 29. VP4 proteins on the mature virion surface are cleaved by enzymatic digestion to yield VP8* and VP5* fragments. R441 is located in the loop structure of the VP5* fragment within neutralizing epitope 5-1; this is one of five neutralizing domains (5-1, 5-2, 5-3, 5-4, and 5-5) identified in the VP5* fragment (Fig. 6F) (19–25). Because R441 is located within the variable region of the VP4 gene (Fig. 6G), this result implies that clone 29 may show specific neutralizing activity against particular RV strains.
FIG 6.

Screening for neutralizing monoclonal antibodies specific for SA11. (A) rsSA11-NLuc-Δ332 (1.0 × 102 PFU) was incubated at 37°C for 1 h with hybridoma culture supernatant (1:5 dilution). The virus-antibody mixture was inoculated onto MA104 cells, and NLuc titers in cell lysates were measured at 24 h postinfection. PBS was used as a negative control. Candidate NT-positive hybridoma clones are indicated by asterisks. Murine anti-SA11 serum raised after oral infection with SA11 virus was used as a positive control. (B) rsSA11-NLuc-Δ332 (1.0 × 102 PFU) was incubated with different concentrations of purified MAb clones 26, 29, 47, and 11. The virus-MAb mixture was inoculated onto MA104 cells, and NLuc titers in the cell lysates were measured at 24 h postinfection. MAb clone 11 was used as a negative control. (C) Hemagglutinin (HA)-tagged recombinant SA11 VP4 protein expressed in 293T cells was detected in an indirect immunofluorescence assay using MAb clone 29 and a rabbit anti-hemagglutinin peptide antibody, followed by anti-mouse IgG-CF594 and anti-rabbit IgG-CF488 secondary antibodies, respectively. (D) rsSA11-NLuc-Δ332, rsSA11-NLuc-esc29, and rsSA11-R441G-NLuc were incubated with various concentrations of MAb clone 29 and then inoculated onto MA104 cells. NLuc titers in the cell lysates were measured at 24 h postinfection. (E) MA104 cells were infected with rsSA11-NLuc-Δ332 or rsSA11-R441G-NLuc, and viral antigens were detected in an indirect immunofluorescence assay using a rabbit anti-NSP4 antibody and MAb clone 29, followed by an anti-rabbit IgG-CF488 conjugate or an anti-mouse IgG-CF594 conjugate, respectively. (F) Crystal structure of the trimeric VP5*CT (VP4 residues N252-L523) from a rhesus rotavirus (RRV) strain (PDB accession number 1SLQ). R441 is indicated in red. (G) Alignment of amino acid residues 428 to 455 of the VP4 proteins of RV reference strains. The P genotypes of the VP4 gene of each strain are shown. Amino acids identical to the consensus sequence (SA11) are indicated by dots. R441 is underlined.
Improving reporter gene expression systems by modifying the 5′- and 3′-terminal sequences of the NSP1 gene segment.
Although expression of the native ZsGreen protein exhibited a uniform distribution throughout the cytosol and nucleus of transfected cells (Fig. 7A), the NSP11–27-ZsGreen fusion protein was detected as scattered and aggregated structures in the cytoplasm of cells after transfection with an expression plasmid (Fig. 7A) and infection with rsSA11-ZsG-Full (Fig. 1B). These results raise concerns that the N-terminal NSP11–27 peptide can affect the subcellular localization and/or structural function of foreign reporter proteins. To improve reporter gene expression systems without affecting the subcellular localization and/or function of the targeted proteins, we attempted to generate recombinant reporter RVs lacking expression of the fused NSP11–27 peptide. The sequence encoding the NSP11-27 peptide includes four ATG codons (ATG1, ATG2, ATG3, and ATG4), each of which can act as a translation initiation codon (Fig. 7B). First, we tried to rescue two reporter viruses encoding the NSP11–27 peptide sequence containing mutated versions of the ATG codons (31ATG33 → ATC, 47ATG49 → ATC, 51ATG53 → ATC, and 93ATG95 → ATC) or a virus harboring an NSP11–27 peptide in which only the first ATG start codon was mutated (31ATG33 → ATC); however, we failed to rescue any of them after five attempts (Fig. 7B). In contrast, an NSP1-ZsGreen reporter virus harboring mutated 2nd and 3rd ATG codons could be rescued, suggesting that mutations in the 2nd and 3rd ATG codons do not affect RV propagation (Fig. 7B). Although the functions of the 5′- and 3′-terminal sequences, which include the UTRs and parts of the ORFs, are not entirely clear, it is thought that they include cis-acting signals associated with genome replication and packaging and that complementary 5′- and 3′-terminal sequences of RV mRNA form secondary structures, including a panhandle structure (26–29). The predicted RNA secondary structure of the NSP1 mRNA revealed the potential for high-average base pairing between the 5′- and 3′-terminal sequences (positions 2 to 37 and 1552 to 1581; based on the numbering of wild-type NSP1), which include the NSP1 start codon (Fig. 7C). This suggests that a mutation in the NSP1 initiation codon may disrupt any putative and critical secondary structure formed between the 5′ and 3′ termini of NSP1 mRNA during replication, transcription, and packaging. Therefore, to generate a recombinant RV harboring a mutated NSP1 initiation codon that maintains the stem structure formed by interaction between the 5′ and 3′ termini, we attempted to rescue the rsSA11-ZsG-1556G virus harboring a C-to-G mutation in all four ATG codons within the NSP11–27 peptide sequence (nt 1556 in the 3′ terminus) (Fig. 7D). As expected, we rescued NSP1-ZsGreen start codon-mutant viruses harboring compensatory mutations that do not disrupt the base paring at positions 33 and 1556 in the stem structure (Fig. 7D). To better understand the importance of NSP1 5′-terminal gene sequences for viral replication and genome assembly, we attempted to generate an NSP1-ZsGreen mutant virus (rsSA11-ZsG-ΔN1556G) possessing the truncated NSP1 gene segment with further deletions in the 5′-terminal sequences (Fig. 7D). The N-terminal deletion mutants were rescued, demonstrating that the N-terminal nucleotide sequences (residues 1 to 40) of NSP1 play an important role in viral replication and packaging. Finally, cells infected with the rsSA11-ZsG-ΔN1556G virus showed normal subcellular localization of the ZsGreen protein, similar to that in cells infected with wild-type ZsGreen (Fig. 7E). Multistep and single-step growth of rsSA11-ZsG-ΔN1556G was comparable to that of rsSA11-ZsG-Δ722, indicating that viral replication was not affected by G33C and C1556G mutations (Fig. 7F and G). Therefore, the data suggest that the predicted secondary structures formed by the 5′ and 3′ termini play critical roles in viral replication, transcription, and packaging and demonstrate that genetic modification of the 5′ and 3′ termini of NSP1 mRNA is a very powerful approach to stabilizing expression of foreign genes.
FIG 7.

Modification of the reporter gene. (A) Cellular localization of a recombinant ZsG protein expressed with/without the NSP11–27 peptide plasmid vector. (B) Partial nucleotide sequence of the NSP11–27-ZsG gene used for reverse genetics. Four ATG triplets (bold) and ATG-to-ATC substitutions (red, underlined) in the NSP11–27 coding sequence are shown. The results of virus rescue and the rescue efficiencies of recombinant viruses harboring each reporter gene are shown. Data represent the mean values of five independent rescue experiments. (C) Predicted secondary structure of wild-type NSP1 mRNA. Double-strands formed by the 5′- and 3′-terminal regions are shown in the insets. Base paring at positions 33 and 1556 within the stem structure are shown in the square. Nucleotide positions are numbered according to the wild-type NSP1 gene sequence. (D) Schematic showing the NSP1-ZsG-1556G and NSP1-ZsG-ΔN1556G constructs. Nucleotide positions are numbered according to the wild-type NSP1 sequence. (E) MA104 cells were infected with rsSA11-ZsG-Δ722 or rsSA11-ZsG-ΔN1556G. Viral NSP4 was detected using rabbit anti-NSP4 and an anti-rabbit-IgG-CF594 conjugate. Nuclei were stained with 4′,6-diamidino-2-phenylindole. (F and G) Replication kinetics of rsSA11-ZsG-ΔN1556G. MA104 cells were infected with rsSA11, rsSA11-NLuc-Δ722, or rsSA11-ZsG-ΔN1556G virus at MOI of 0.001 PFU per cell and incubated for various times (F) or at an MOI of 5 PFU per cell and incubated for 16 h (G). Infectious virus titers in cell lysates were determined by the plaque assay. Data are expressed as the means ± standard deviations (n = 3). Statistical significance was determined by one-way ANOVA and Tukey’s multiple-comparison posttest. *, P < 0.05.
DISCUSSION
Luciferase family proteins have been used to develop reporter viruses belonging to various virus families; this is due to the high sensitivity of luciferase, which is especially useful for reporter viruses developed for in vivo imaging (30–34). However, luciferase requires an additional substrate for activation, so it is not suitable for tracking cell-cell virus transmission under a microscope. To overcome these disadvantages, we constructed rsSA11-ZsG by expressing the ZsGreen protein as a fusion with the NSP11–27 peptide. Here, we improved the stability of the foreign gene inserted into the RV NSP1 gene by truncating the UTR of the NSP1 gene. Sequence analysis of prototype reporter viruses (rsSA11-NLuc-Full and rsSA11-ZsG-Full) after the 10th passage revealed that almost the entire foreign gene ORF was removed, leading to reconstitution of the NSP1 ORF. Because NSP1 functions to impede interferon (IFN) signaling (35), a revertant virus harboring the intact NSP1 ORF would propagate more rapidly than the original reporter viruses; therefore, the entire virus population would be replaced by revertant viruses during long-term passage. In contrast, a modified reporter virus lacking almost all of the NSP1 nucleotide sequence harbored the foreign gene in a stable manner. Even if a mutant reporter virus that lacked a foreign gene was generated, it would not replace the whole population unless it had a marked replication advantage over the original virus. Furthermore, rsSA11-AsR-Δ332 (which expresses the AsRed2 fluorescent protein) was based on the stable rsSA11-ZsG-Δ332 virus. The green and red fluorescent protein-expressing viruses enable live imaging of coinfection by different RV strains, which is an important process during evolution of segmented RNA viruses through gene reassortment (36). Increasing the total genome size is a concern when a stable reporter virus is generated. The 9- to 12-segment dsRNA genomes of the viruses belonging to the Reoviridae family are packed tightly within single-, double-, or triple-layered viral capsids, suggesting that there is a maximum limit to the size of genome that can be contained within the virion (37, 38); increasing the total genome size may affect genome packaging efficiency. This is one of the reasons why we used a small reporter gene (NLuc luciferase, ZsGreen, or AsRed2; all, 516 to 699 bp in length) and reduced the length of the NSP1 reporter genes by truncating the untranslated region (39). A previous study obtained a natural NSP1 mutant gene segment from an immunocompromised patient; this segment possessed an additional 1,800 bp due to gene rearrangement, indicating that a transgene of up to 1,800 bp could be inserted into the genome (40, 41).
Here, we showed that a reporter RV could be a useful tool for screening neutralizing antibodies. The escape mutant virus from MAb clone 29 harbored an amino acid substitution in the VP5 fragment. The VP4 spike protein on the surface of the synthesized virion is cleaved by trypsin to yield VP8* and VP5* fragments. VP8* forms the globular domain of the spike protein and mediates virus-cell attachment via cell surface sialic acids; therefore, VP8* harbors a number of serotype-specific domains. VP5* plays a role in integrin binding and membrane penetration. The function of VP5* is both essential and common among RV strains; therefore, the amino acid sequences of VP5* are conserved and rich in cross-neutralizing domains (25). However, the VP5* loop region containing R441G is relatively variable among RV group A strains. Further investigation of the cross-reactivity of MAb clone 29 with other RV strains would help to develop a diagnostic tool for particular RV genotypes.
The effect of transgene insertion on packaging of the native genome must also be considered in engineering the genome of segmented RNA viruses. cis-acting elements, including the packaging signal, are thought to contain sequences necessary for intergene segment interaction, which enables packaging of complete sets of gene segments into progeny virions. Although the mechanisms underlying packaging of RV gene segments are poorly understood due to lack of an in vitro packaging assay, a prediction based on sequence and structural analysis and on interactions between RV and BTV gene segments (36, 42) suggests that segment-specific packaging signals are located within several hundred nucleotides, including the 5′ and 3′ untranslated terminal regions and part of the ORF of each gene segment. Therefore, we left 150 bp of the 5′-terminal region, including the NSP11–27 peptide, to maintain the packaging signal of the NSP1 gene segment even though addition of the NSP11–27 peptide clearly affected localization of the reporter proteins. To generate the reporter SA11 expressing a native reporter protein without the NSP1 peptide at the N terminus, we developed improved reporter viruses harboring not only the ATG-to-ATC substitution but also a complemental substitution in the 5′- and 3′-terminal regions (in accordance with the predicted secondary structure of the wild-type NSP1 gene). This demonstrated the importance of the predicted RNA secondary structure formed between the 5′ and 3′ termini and the importance of the 5′-terminal region of NSP1 consisting of residues 1 to 40 for rescuing recombinant viruses. Further mutational studies of the 5′- and 3′-terminal regions will help to define the molecular mechanisms underlying RV genome replication and packaging.
The current strategy for generating stable reporter RVs could be used to develop RV transduction vectors. Since RV infection induces strong mucosal immunity in the intestine (43, 44), reporter RVs could be used to generate vaccine vectors for intestinal pathogens, including norovirus, Vibrio cholerae, and human immunodeficiency virus (which can infect subjects via the intestinal mucosa). NSP1 antagonizes IFN and IFN-stimulated gene expression by interacting with IRF3 and/or β-TrCP at the C-terminal region of NSP1, leading to degradation of these molecules. Thus, improved strategies to express foreign genes using attenuated NSP1 mutant viruses will provide a platform for developing useful vaccine vectors. However, to better understand the functions of NSP1 during viral replication and pathogenesis, other strategies to develop different types of RV vector systems that express both a reporter gene and an intact NSP1 protein should be considered in the future.
Different methods have been used to generate recombinant Reoviridae viruses that express transgenes (13–15, 45–47). A more robust method is the bi-cistronic expression system based on the self-cleaving Thosea asigna virus (TaV) 2A-like peptide. In this system, a recombinant MRV transduction vector harboring the SIV Gag protein was inserted into the MRV L1 gene encoding the λ3 protein. Insertion of the TaV 2A-like peptide between λ3 and the SIV Gag ORF enabled expression of both intact λ3 and the SIV Gag protein (16). Theoretically, these methods could be used to insert any transgene into RV gene segments. The recent development of comprehensive reverse genetics systems for RV has enabled arbitrary modification of viral genes. The reporter RVs expressing NanoLuc, ZsGreen, and AsRed2 allow us to monitor viral replication and transmission both in vitro and in vivo. The strategy of inserting a foreign gene into the NSP1 gene segment provides a general platform for developing not only reporter RVs expressing different reporter genes but also RV transduction vectors.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
Epithelial monkey kidney MA104 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Nacalai Tesque) supplemented with 5% fetal bovine serum (FBS) (Gibco). Murine myeloma PAI cells were cultured in RPMI 1640 medium (Nacalai Tesque) supplemented with 10% FBS. Baby hamster kidney BHK-T7 cells (17, 48) stably expressing T7 RNA polymerase were cultured in DMEM supplemented with 5% FBS. Recombinant strains of simian RV strain SA11 (rsSA11), rescued by a reverse genetics system using cDNA from a wild-type SA11 strain (G3P[2]), were propagated in MA104 cells cultured in DMEM supplemented with 0.5 μg/ml trypsin (Sigma-Aldrich). Infectious titers of RVs were determined in a plaque assay using MA104 cells, as previously described (49). Rabbit anti-NSP1 antiserum was prepared as described previously (17). Rabbit anti-NSP4 antiserum was raised against a synthetic SA11 NSP4 peptide spanning amino acid residues 158 to 171 (Eurofins Genomics, Tokyo, Japan).
Plasmid construction.
RV rescue plasmids pT7-VP1SA11, pT7-VP2SA11, pT7-VP3SA11, pT7-VP4SA11, pT7-VP6SA11, pT7-VP7SA11, pT7-NSP1SA11, pT7-NSP2SA11, pT7-NSP3SA11, pT7-NSP4SA11, and pT7-NSP5SA11 were prepared as described previously (17). The pT7-NSP1-NLuc-Full plasmid, which expresses a fusion protein consisting of NSP1 amino acids 1 to 27 (NSP11–27) and NLuc, was prepared as described previously (17). The pT7-NSP1-ZsG-Full plasmid was generated by replacing the NLuc ORF region of pT7-NSP1-NLuc-Full with the ZsGreen gene (GenBank accession number JQ071441). Truncated forms of the pT7-NSP1-NLuc and pT7-NSP1-ZsG plasmids were generated by deleting 332 nt (positions 134 to 465), 722 nt (positions 134 to 855), and 1,110 nt (positions 134 to 1243) from the NSP1 regions in pT7-NSP1-NLuc (ZsG)-Full to yield pT7-NSP1-NLuc (ZsG)-Δ332, pT7-NSP1-NLuc (ZsG)-Δ722, and pT7-NSP1-NLuc (ZsG)-Δ1110, respectively. pT7-NSP1-AsR-Δ332 was generated by replacing the NLuc gene of pT7-NSP1-NLuc-Δ332 with the AsRed2 gene (Clontech). The gene encoding the NSP11–27-ZsG fusion protein was amplified by PCR and inserted into the EcoRI site of the pCAGGS vector (50) using an In-Fusion HD cloning kit to create pCAG-NSP1-ZsG. The NSP2 and NSP5 genes from SA11 were amplified by reverse transcription-PCR (RT-PCR) and inserted into the EcoRI site of the pCAGGS vector to yield pCAG-NSP2SA11 and pCAG-NSP5SA11, respectively. Primers and plasmid sequences are available upon request.
Recovery of reporter RVs from cloned cDNAs.
To rescue recombinant strain SA11 (rsSA11), monolayers of BHK-T7 cells (4.0 × 105) cultured in 12-well plates were cotransfected with plasmids using 2 μl of TransIT-LT1 transfection reagent per microgram of plasmid DNA. Each mixture consisted of 0.25 μg of each SA11 rescue plasmid (pT7-VP1SA11, pT7-VP2SA11, pT7-VP3SA11, pT7-VP4SA11, pT7-VP6SA11, pT7-VP7SA11, pT7-NSP1SA11, pT7-NSP2SA11, pT7-NSP3SA11, pT7-NSP4SA11, and pT7-NSP5SA11), 0.001 μg of pCAG-FAST, and 0.25 μg of pCAG-D1R, pCAG-D12L, pCAG-NSP2SA11, and pCAG-NSP5SA11 expression plasmids. pCAG-NSP2SA11 and pCAG-NSP5SA11 encoding SA11 NSP2 and NSP5 proteins, both of which are required for viral inclusion formation, were cotransfected to increase rescue efficiency (47, 51). To rescue reporter RVs, reporter plasmids encoding chimeric NSP1-reporter genes were used instead of pT7-NSP1. At 24 h posttransfection, cell culture medium was replaced with FBS-free medium. At 48 h posttransfection, MA104 cells (5.0 × 104 cells) were added to transfected cells and cocultured for 5 days in FBS-free medium supplemented with trypsin (0.5 μg/ml). After incubation, transfected cells were lysed by freeze-thawing, and the lysates were transferred to fresh MA104 cells. After adsorption at 37°C for 1 h, cells were washed and cultured at 37°C for 3 days in FBS-free DMEM supplemented with 0.5 μg/ml trypsin. Recombinant viruses were isolated and cloned by plaque purification using MA104 cells.
Serial passage of reporter viruses.
To examine the stability of the transgene, a monolayer of MA104 cells was infected with reporter SA11 viruses at a multiplicity of infection (MOI) of 0.001 PFU/cell and cultured for 48 h (passage 1 [P1]). Virus-containing culture supernatants (diluted 1:1,000 in culture medium) were transferred to fresh MA104 cells and cultured for another 48 h (P2). Serial passage was continued up to P10. The dsRNA genomes of each reporter virus at P1 and P10 were purified and subjected to electropherotyping and sequence analysis as described previously (17).
Replication kinetics of reporter viruses.
A monolayer of MA104 cells was infected with rsSA11 or reporter SA11 viruses at an MOI of 0.001 PFU/cell for multistep growth curves or at an MOI of 5 PFU/cell for single-step growth curves. After adsorption for 1 h at 37°C, cells were washed twice with phosphate-buffered saline (PBS), and the medium was replaced with DMEM supplemented with 0.5 μg/ml trypsin. After incubation at 37°C for various times, cells were disrupted by freeze-thawing. The virus titer in the cell lysates was measured in a plaque assay. NLuc activity in the cell lysates after freeze-thawing was measured using a Nano-Glo luciferase assay system (Promega).
Immunofluorescence assay.
To analyze transgene stability, a monolayer of MA104 cells was infected with rsSA11, rsSA11-ZsG-Full, -Δ332, -Δ722, or -Δ1110 at an MOI of 0.1 PFU/cell, overlaid with 0.5% agarose gel, and incubated for 48 h. Cells were fixed with 10% formalin, and virus-infected foci were visualized in an indirect immunofluorescence assay using a rabbit anti-NSP4 antibody (1:3,000) and an anti-rabbit IgG antibody-CF594 conjugate (Nacalai Tesque). The number of cells emitting green, red, and green-red fluorescent signals was counted. To observe expression of AsRed2, MA104 cells were infected with rsSA11-AsR-Δ332 at an MOI of 0.1 PFU/cell. At 24 h postinfection, cells were fixed with 10% formalin, and virus-infected foci were visualized in an indirect immunofluorescence assay using a rabbit anti-NSP4 antibody (1:3000) and an anti-rabbit IgG antibody-CF488 conjugate (Nacalai Tesque).
Western blotting.
MA104 cells were infected with rsSA11-NLuc or rsSA11-ZsG (P1 or P10) at an MOI of 1 PFU/cell. Twenty-four hours later, cells were lysed, and proteins were size fractionated by SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes. Viral proteins were detected using anti-NSP1 antiserum (1:2,000), followed by horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:2,000; Sigma). Proteins were detected using Super Signal West Femto Maximum Sensitivity Substrate (Pierce) and an Amersham imager 600 (Amersham/GE Healthcare).
Generation of monoclonal antibodies.
ICR mice (Japan CLEA) were immunized in the footpads with purified rsSA11 virions conjugated to an alhydrogel adjuvant (Invivogen). Immunization was repeated six times at 5- to 10-day intervals. At 2 days after the final immunization, popliteal lymph nodes were removed. Lymphocytes were fused with PAI myeloma cells using 50% polyethylene glycol (Hybri-Max; Sigma-Aldrich). Hybridoma cells were cultured in RPMI 1640 medium supplemented with 10% FBS and 1× hypoxanthine-aminopterin-thymidine (HAT) medium (Hybri-Max; Sigma-Aldrich). Ninety-one hybridoma clones derived from a single cell colony were used for the experiment. MAbs from hybridoma clones 11, 26, 29, and 47 were purified from hybridoma culture supernatant using HiTrap protein G columns (GE Health Care) and used for further analysis. To examine viral proteins targeted by MAb clone 29, MA104 cells were transfected with an SA11 VP4, VP6, or VP7 expression plasmid vector. At 24 h posttransfection, cells were fixed in 10% formalin and incubated with 1 μg/ml of MAb clone 11, 26, 29, or 47, followed by an anti-mouse IgG-CF488 conjugate (Nacalai Tesque). Green fluorescence signals were observed under a fluorescence microscope. MAb clone 11 which recognized the VP7 protein, was used for Western blot analysis.
Neutralization assay.
Equal volumes of hybridoma culture supernatant (1:5 dilution) and 1.0 × 102 PFU of rsSA11-ZsG-Δ332 were mixed and incubated at 37°C for 1 h. The MAb-virus mixture was inoculated onto MA104 cells. After absorption for 30 min, cells were washed with PBS and cultured in DMEM–5% FBS. At 24 h postinfection, cells were lysed, and NLuc activity measured using a Nano-Glo luciferase assay system (Promega) and a microplate reader (PowerScan HT; DS Pharma Biomedical, Osaka, Japan).
Generation of escape mutants.
MA104 cells were infected with rsSA11-NLuc-Δ332 at an MOI of 0.001 PFU/cell and cultured in the presence of 1 ng/ml neutralizing MAb clone 29. After 3 to 5 days, the culture supernatant (1:1,000 dilution) was transferred to fresh MA104 cells and cultured in the presence of the MAb. The MAb concentration was increased gradually up to 100 ng/ml by the 5th passage. The escape mutant virus from SA11-MAb clone 29 was purified by plaque purification, and cDNA was generated from genomic dsRNA using primers specific for the determined genome sequences to identify the responsible gene segment. The mutant VP4 gene (which harbored an R441G substitution) from the escape mutant virus was amplified, cloned into an RV rescue plasmid, and subjected to RV reverse genetics to generate rsSA11-R441G-NLuc.
Prediction of the secondary structure of the NSP1 gene segment.
The secondary structure of the NSP1 gene segment was predicted by the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) (52). The nucleotide positions of the NSP1-ZsGreen genes (Fig. 7) were based on the numbering of the wild-type NSP1 gene.
Ethics statement.
The study was approved by the Animal Research Committee of the Research Institute for Microbial Diseases, Osaka University (approval number Bi-Dou-28-05-0). The experiments were conducted according to the guidelines for the Care and Use of Laboratory Animals of the Ministry of Education, Culture, Sports, Science and Technology, Japan.
VP4 sequences of the RV reference strains.
The amino acid sequences of the VP4 protein deduced from representative RV reference strains were retrieved from GenBank, as follows: rhesus rotavirus (RRV) (AY033150), neonatal calf diarrhea virus (NCDV) (JF693029), L26 (EF672591), DS-1 (HQ650119), WC3 (AY050271), ST3 (L33895), US1205 (AF079356), OSU (KR052770), Hochi (AB008295), Wa (KT694942), AU-1 (D10970), 69 M (M60600), I321 (L07657), 116E (FJ361204), EDIM (AF039219), and L338 (KR086410).
Availability of data.
Nucleotide sequences of plasmid constructs and primer sequences are available upon request.
ACKNOWLEDGMENTS
We thank Y. Saioka and N. Negishi for technical assistance, N. Ito for kindly providing BHK-T7 cells, and K. Yukawa and M. Yoshida for secretarial work.
This work was supported in part by AMED grant numbers JP18im0210610, JP18fk0108018, and JP18fk0108001 and by JSPS KAKENHI grant numbers JP18H02663, JP18K07145, JP18K15167, JP17H05814, and JP18K19444.
REFERENCES
- 1.Agapov EV, Frolov I, Lindenbach BD, Pragai BM, Schlesinger S, Rice CM. 1998. Noncytopathic Sindbis virus RNA vectors for heterologous gene expression. Proc Natl Acad Sci U S A 95:12989–12994. doi: 10.1073/pnas.95.22.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsetsarkin K, Higgs S, McGee CE, De Lamballerie X, Charrel RN, Vanlandingham DL. 2006. Infectious clones of Chikungunya virus (La Reunion isolate) for vector competence studies. Vector Borne Zoonotic Dis 6:325–337. doi: 10.1089/vbz.2006.6.325. [DOI] [PubMed] [Google Scholar]
- 3.McGee CE, Shustov AV, Tsetsarkin K, Frolov IV, Mason PW, Vanlandingham DL, Higgs S. 2010. Infection, dissemination, and transmission of a West Nile virus green fluorescent protein infectious clone by Culex pipiens quinquefasciatus mosquitoes. Vector Borne Zoonotic Dis 10:267–274. doi: 10.1089/vbz.2009.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, García-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A 107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou G, Xu HY, Qing M, Wang QY, Shi PY. 2011. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antiviral Res 91:11–19. doi: 10.1016/j.antiviral.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Ozawa M, Victor ST, Taft AS, Yamada S, Li C, Hatta M, Das SC, Takashita E, Kakugawa S, Maher EA, Neumann G, Kawaoka Y. 2011. Replication-incompetent influenza A viruses that stably express a foreign gene. J Gen Virol 92:2879–2888. doi: 10.1099/vir.0.037648-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tate JE, Burton AH, Boschi-Pinto C, Steele AD, Duque J, Parashar UD. 2012. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect Dis 12:136–141. doi: 10.1016/S1473-3099(11)70253-5. [DOI] [PubMed] [Google Scholar]
- 8.Desselberger U. 2014. Rotaviruses. Virus Res 190:75–96. doi: 10.1016/j.virusres.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 9.Santman-Berends IM, van Schaik G, Bartels CJ, Stegeman JA, Vellema P. 2011. Mortality attributable to bluetongue virus serotype 8 infection in Dutch dairy cows. Vet Microbiol 148:183–188. doi: 10.1016/j.vetmic.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Dermody TS, Parker JS, Barbara S. 2013. Orthoreoviruses, p 1304–1346. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 11.Attoui H, Mohd Jaafar F, Belhouchet M, Biagini P, Cantaloube JF, de Micco P, de Lamballerie X. 2005. Expansion of family Reoviridae to include nine-segmented dsRNA viruses: isolation and characterization of a new virus designated Aedes pseudoscutellaris reovirus assigned to a proposed genus (Dinovernavirus). Virology 343:212–223. doi: 10.1016/j.virol.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 12.Mertens PPC, Attoui H, Duncan R, Dermody TS. 2005. Reoviridae, p 447–454. In Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA(ed), Virus taxonomy. Eighth report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press, Amsterdam, Netherlands. [Google Scholar]
- 13.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawagishi T, Kanai Y, Tani H, Shimojima M, Saijo M, Matsuura Y, Kobayashi T. 2016. Reverse genetics for fusogenic bat-borne orthoreovirus associated with acute respiratory tract infections in humans: role of outer capsid protein sigmaC in viral replication and pathogenesis. PLoS Pathog 12:e1005455. doi: 10.1371/journal.ppat.1005455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Wollenberg DJM, Dautzenberg IJC, Ros W, Lipińska AD, van den Hengel SK, Hoeben RC. 2015. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene Ther 22:267–279. doi: 10.1038/gt.2014.126. [DOI] [PubMed] [Google Scholar]
- 16.Demidenko AA, Blattman JN, Blattman NN, Greenberg PD, Nibert ML. 2013. Engineering recombinant reoviruses with tandem repeats and a tetravirus 2A-like element for exogenous polypeptide expression. Proc Natl Acad Sci U S A 110:E1867. doi: 10.1073/pnas.1220107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanai Y, Komoto S, Kawagishi T, Nouda R, Nagasawa N, Onishi M, Matsuura Y, Taniguchi K, Kobayashi T. 2017. Entirely plasmid-based reverse genetics system for rotaviruses. Proc Natl Acad Sci U S A 114:2349–2354. doi: 10.1073/pnas.1618424114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morelli M, Ogden KM, Patton JT. 2015. Silencing the alarms: innate immune antagonism by rotavirus NSP1 and VP3. Virology 479–480:75–84. doi: 10.1016/j.virol.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi K, Maloy WL, Nishikawa K, Green KY, Hoshino Y, Urasawa S, Kapikian AZ, Chanock RM, Gorziglia M. 1988. Identification of cross-reactive and serotype 2-specific neutralization epitopes on VP3 of human rotavirus. J Virol 62:2421–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirkwood CD, Bishop RF, Coulson BS. 1996. Human rotavirus VP4 contains strain-specific, serotype-specific and cross-reactive neutralization sites. Arch Virol 141:587–600. doi: 10.1007/BF01718319. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi N, Taniguchi K, Urasawa S. 1990. Identification of operationally overlapping and independent cross-reactive neutralization regions on human rotavirus VP4. J Gen Virol 71:2615–2623. doi: 10.1099/0022-1317-71-11-2615. [DOI] [PubMed] [Google Scholar]
- 22.Mackow ER, Shaw RD, Matsui SM, Vo PT, Dang MN, Greenberg HB. 1988. The rhesus rotavirus gene encoding protein VP3: location of amino acids involved in homologous and heterologous rotavirus neutralization and identification of a putative fusion region. Proc Natl Acad Sci U S A 85:645–649. doi: 10.1073/pnas.85.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorziglia M, Larralde G, Ward RL. 1990. Neutralization epitopes on rotavirus SA11 4fM outer capsid proteins. J Virol 64:4534–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Padilla-Noriega L, Dunn SJ, Lopez S, Greenberg HB, Arias CF. 1995. Identification of two independent neutralization domains on the VP4 trypsin cleavage products VP5* and VP8* of human rotavirus ST3. Virology 206:148–154. doi: 10.1016/S0042-6822(95)80029-8. [DOI] [PubMed] [Google Scholar]
- 25.Nair N, Feng N, Blum LK, Sanyal M, Ding S, Jiang B, Sen A, Morton JM, He XS, Robinson WH, Greenberg HB. 2017. VP4- and VP7-specific antibodies mediate heterotypic immunity to rotavirus in humans. Sci Transl Med 9:eaam5434. doi: 10.1126/scitranslmed.aam5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen D, Patton JT. 1998. Rotavirus RNA replication requires a single-stranded 3′ end for efficient minus-strand synthesis. J Virol 72:7387–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W, Manktelow E, von Kirchbach JC, Gog JR, Desselberger U, Lever AM. 2010. Genomic analysis of codon, sequence and structural conservation with selective biochemical-structure mapping reveals highly conserved and dynamic structures in rotavirus RNAs with potential cis-acting functions. Nucleic Acids Res 38:7718–7735. doi: 10.1093/nar/gkq663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki Y. 2014. A possible packaging signal in the rotavirus genome. Genes Genet Syst 89:81–86. doi: 10.1266/ggs.89.81. [DOI] [PubMed] [Google Scholar]
- 29.McDonald SM, Patton JT. 2011. Assortment and packaging of the segmented rotavirus genome. Trends Microbiol 19:136–144. doi: 10.1016/j.tim.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burke CW, Mason JN, Surman SL, Jones BG, Dalloneau E, Hurwitz JL, Russell CJ. 2011. Illumination of parainfluenza virus infection and transmission in living animals reveals a tissue-specific dichotomy. PLoS Pathog 7:e1002134. doi: 10.1371/journal.ppat.1002134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez JF, Rodriguez D, Rodriguez JR, McGowan EB, Esteban M. 1988. Expression of the firefly luciferase gene in vaccinia virus: a highly sensitive gene marker to follow virus dissemination in tissues of infected animals. Proc Natl Acad Sci U S A 85:1667–1671. doi: 10.1073/pnas.85.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoggins JW, Dorner M, Feulner M, Imanaka N, Murphy MY, Ploss A, Rice CM. 2012. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc Natl Acad Sci U S A 109:14610–14615. doi: 10.1073/pnas.1212379109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook SH, Griffin DE. 2003. Luciferase imaging of a neurotropic viral infection in intact animals. J Virol 77:5333–5338. doi: 10.1128/JVI.77.9.5333-5338.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luker GD, Bardill JP, Prior JL, Pica CM, Piwnica-Worms D, Leib DA. 2002. Noninvasive bioluminescence imaging of herpes simplex virus type 1 infection and therapy in living mice. J Virol 76:12149–12161. doi: 10.1128/JVI.76.23.12149-12161.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis KA, Morelli M, Patton JT. 2017. Rotavirus NSP1 requires casein kinase II-mediated phosphorylation for hijacking of Cullin-RING ligases. mBio 8:e01213-17. doi: 10.1128/mBio.01213-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald SM, Nelson MI, Turner PE, Patton JT. 2016. Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat Rev Microbiol 14:448–460. doi: 10.1038/nrmicro.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Ding K, Yu X, Chang W, Sun J, Zhou ZH. 2015. In situ structures of the segmented genome and RNA polymerase complex inside a dsRNA virus. Nature 527:531–534. doi: 10.1038/nature15767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nibert ML. 1998. Structure of mammalian orthoreovirus particles. Curr Top Microbiol Immunol 233:1–30. [DOI] [PubMed] [Google Scholar]
- 39.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. 2012. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol 7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hundley F, McIntyre M, Clark B, Beards G, Wood D, Chrystie I, Desselberger U. 1987. Heterogeneity of genome rearrangements in rotaviruses isolated from a chronically infected immunodeficient child. J Virol 61:3365–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McIntyre M, Rosenbaum V, Rappold W, Desselberger M, Wood D, Desselberger U. 1987. Biophysical characterization of rotavirus particles containing rearranged genomes. J Gen Virol 68:2961–2966. doi: 10.1099/0022-1317-68-11-2961. [DOI] [PubMed] [Google Scholar]
- 42.Boyce M, McCrae MA, Boyce P, Kim JT. 2016. Inter-segment complementarity in orbiviruses: a driver for co-ordinated genome packaging in the Reoviridae? J Gen Virol 97:1145–1157. doi: 10.1099/jgv.0.000400. [DOI] [PubMed] [Google Scholar]
- 43.Yuan L, Saif LJ. 2002. Induction of mucosal immune responses and protection against enteric viruses: rotavirus infection of gnotobiotic pigs as a model. Vet Immunol Immunopathol 87:147–160. doi: 10.1016/S0165-2427(02)00046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conner ME, Gilger MA, Estes MK, Graham DY. 1991. Serologic and mucosal immune response to rotavirus infection in the rabbit model. J Virol 65:2562–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw AE, Veronesi E, Maurin G, Ftaich N, Guiguen F, Rixon F, Ratinier M, Mertens P, Carpenter S, Palmarini M, Terzian C, Arnaud F. 2012. Drosophila melanogaster as a model organism for bluetongue virus replication and tropism. J Virol 86:9015–9024. doi: 10.1128/JVI.00131-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Navarro A, Trask SD, Patton JT. 2013. Generation of genetically stable recombinant rotaviruses containing novel genome rearrangements and heterologous sequences by reverse genetics. J Virol 87:6211–6220. doi: 10.1128/JVI.00413-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komoto S, Fukuda S, Ide T, Ito N, Sugiyama M, Yoshikawa T, Murata T, Taniguchi K. 2018. Generation of recombinant rotaviruses expressing fluorescent proteins by using an optimized reverse genetics system. J Virol 92:e00588-18. doi: 10.1128/JVI.00588-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito N, Takayama-Ito M, Yamada K, Hosokawa J, Sugiyama M, Minamoto N. 2003. Improved recovery of rabies virus from cloned cDNA using a vaccinia virus-free reverse genetics system. Microbiol Immunol 47:613–617. doi: 10.1111/j.1348-0421.2003.tb03424.x. [DOI] [PubMed] [Google Scholar]
- 49.Arnold M, Patton JT, McDonald SM. 2009. Culturing, storage, and quantification of rotaviruses. Curr Protoc Microbiol Chapter 15: Unit 15C.3. doi: 10.1002/9780471729259.mc15c03s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. [DOI] [PubMed] [Google Scholar]
- 51.van Rijn PA, van de Water SG, Feenstra F, van Gennip RG. 2016. Requirements and comparative analysis of reverse genetics for bluetongue virus (BTV) and African horse sickness virus (AHSV). Virol J 13:119. doi: 10.1186/s12985-016-0574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hofacker IL. 2003. Vienna RNA secondary structure server. Nucleic Acids Res 31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]