

A few individuals can control HIV infection without the need for antiretroviral treatment and are referred to as HIV controllers. We have studied HIV controllers who suddenly lose this ability and present with high in vivo viral replication and decays in their CD4+ T-cell counts to identify potential immune and virological factors that were responsible for initial virus control. We identify in vitro-determined reductions in the ability of CD8 T cells to suppress viral control and the presence of PD-1-expressing CD8+ T cells with a naive immune phenotype as potential predictors of in vivo loss of virus control. The findings could be important for the clinical management of HIV controller individuals, and it may offer an important tool to anticipate viral rebound in individuals in clinical studies that include combination antiretroviral therapy (cART) treatment interruptions and which, if not treated quickly, could pose a significant risk to the trial participants.
KEYWORDS: HIV-1 control, HIV-1 progression, cell tropism, host genetics, in vitro virus inhibition, loss of control
ABSTRACT
Elite and viremic HIV controllers are able to control their HIV infection and maintain undetectable or low-level viremia in the absence of antiretroviral treatment. Despite extensive studies, the immune factors responsible for such exclusive control remain poorly defined. We identified a cohort of 14 HIV controllers that suffered an abrupt loss of HIV control (LoC) to investigate possible mechanisms and virological and immunological events related to the sudden loss of control. The in-depth analysis of these subjects involved the study of cell tropism of circulating virus, evidence for HIV superinfection, cellular immune responses to HIV, as well as an examination of viral adaptation to host immunity by Gag sequencing. Our data demonstrate that a poor capacity of T cells to mediate in vitro viral suppression, even in the context of protective HLA alleles, predicts a loss of viral control. In addition, the data suggest that inefficient viral control may be explained by an increase of CD8 T-cell activation and exhaustion before LoC. Furthermore, we detected a switch from C5- to X4-tropic viruses in 4 individuals after loss of control, suggesting that tropism shift might also contribute to disease progression in HIV controllers. The significantly reduced inhibition of in vitro viral replication and increased expression of activation and exhaustion markers preceding the abrupt loss of viral control may help identify untreated HIV controllers that are at risk of losing control and may offer a useful tool for monitoring individuals during treatment interruption phases in therapeutic vaccine trials.
IMPORTANCE A few individuals can control HIV infection without the need for antiretroviral treatment and are referred to as HIV controllers. We have studied HIV controllers who suddenly lose this ability and present with high in vivo viral replication and decays in their CD4+ T-cell counts to identify potential immune and virological factors that were responsible for initial virus control. We identify in vitro-determined reductions in the ability of CD8 T cells to suppress viral control and the presence of PD-1-expressing CD8+ T cells with a naive immune phenotype as potential predictors of in vivo loss of virus control. The findings could be important for the clinical management of HIV controller individuals, and it may offer an important tool to anticipate viral rebound in individuals in clinical studies that include combination antiretroviral therapy (cART) treatment interruptions and which, if not treated quickly, could pose a significant risk to the trial participants.
INTRODUCTION
There is a small proportion of HIV-1-infected individuals that spontaneously control HIV infection (1, 2). Due to the heterogeneity among individuals with this clinical course (3, 4) they are referred to as long-term nonprogressors (LTNP), HIV controllers, or, in the case of undetectable viremia, elite controllers. Several factors have been postulated to play a role in this viral control, including host genetic, immunological, and viral factors. In particular, host genetic markers have been associated with disease progression, yet their mechanistic action remains uncertain (5). Possibly, the strongest predictors of HIV control include polymorphisms in HLA class I alleles, which alone or in combination with killer cell immunoglobulin-like receptors (KIR) have been linked to sustained low-level viremia in the absence of combination antiretroviral therapy (cART) (6–8). Since the HLA class I-encoded gene products present virus-derived T-cell epitopes to CD8+ T cells, an extensive number of studies exist that have also linked T-cell responses and their specificities to HIV control (2, 9, 10). Aside from host genetics and immune factors, viral factors, such as viral replication capacity and cell tropism, have been associated with HIV control, although cell tropism has not been consistently documented (11, 12).
During the course of HIV infection, a proportion of nonprogressor individuals may suffer a disruption of their capacity to control infection, which can manifest itself in different ways, as follows: clinical progression defined as a new AIDS-defining event, immunological progression defined as an abrupt decrease of CD4+ T-cell counts, and/or virological progression as a significant increase in viral loads (13–16). In addition, HIV superinfection has been identified as a possible explanation for sudden signs of uncontrolled HIV infection (17). Specific plasma cytokine profiles and Gag-specific T-cell responses have been linked as well to eventual loss of control in an elite controller cohort (18). However, the contribution of these factors to a sudden loss of control is poorly defined, in part due to the scarce availability of longitudinal samples from such individuals, especially of samples close to the time point before loss of viral control (LoC). Here, we have identified and longitudinally followed 14 individuals who experienced an abrupt transition to progressive HIV infection with pre- and post-LoC samples available within 1 year, and we have integrated host, virological, and immune parameters in order to better define the mechanisms underlying the progression of HIV-1 infection.
RESULTS
Clinical characteristics of HIV controllers experiencing abrupt LoC.
For the present analyses, 14 individuals with LoC were identified after reviewing clinical criteria and sample availability for HIV-infected individuals with elite controller (EC) or viremic controller (VC) status from the IrsiCaixa and the BioBank Controllers cohorts (Table 1). The evolution of viral load and CD4 counts is shown in Fig. S1 in the supplemental material. Samples prior to LoC were obtained with a median of 20 months before the first signs of LoC defined either as detection of uncontrolled viral load (n = 4) or a concomitant raise in viral load and reduction in CD4 counts (n = 10, Fig. S1). Follow-up samples included samples taken at first diagnosis of LoC (n = 11) or within the following 6 months (n = 3). As HIV control has been linked to HLA class I genotypes, high-resolution HLA class I typing was performed on all 14 individuals included in the study. In line with data from other cohorts of elite and viremic controllers (19, 20), protective HLA class I alleles B57 and B58 were more common in this group of individuals (Fig. 1) than in the general population in Spain and similar to the frequencies seen in the EC and VC comparison group established at our center. Thus, in this relatively small cohort, no specific class I HLA was associated with LoC in individuals with previous HIV control.
TABLE 1.
Clinical characteristics of the 14 controllers with loss of HIV control
| IDa | Age (yr)b | Sex | Time since known HIV-1 infection (yr)b | Log10 of viral load at control timepoint studiedc | Log10 of viral load at peak of loss of control | No. (%) of CD4 cells/mm3 at control time point studied | No. (%) of CD4 cells/mm3 at loss of control | HLA-Ae | HLA-Be | HLA-Ce | HLA-DRBe | HLA-DQBd,e |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LP1 | 44 | Male | 16 | UD | 3.66 | 1,007 (37) | 426 (18) | 0201/3002 | 1801/5701 | 0501/0602 | 0301/0701 | 0201/0303 |
| LP2 | 54 | Female | 25 | 3.13 | 5.18 | 721 (44) | 171 (19) | 0201/3402 | 1401/4402 | 0501/0802 | 1301/1454 | NA |
| LP3 | 42 | Male | 3 | 2.74 | 4.36 | 507 (26) | 265 (17) | 0101/1101 | 3508/5201 | 0401/1202 | 0801/1502 | 0402/0601 |
| LP4 | 23 | Male | 3 | UD | 4.90 | 1148 (32) | 858 (29) | 0201/2402 | 1501/4403 | 0303/1502 | 0103/1103 | 0301/0501 |
| LP5 | 37 | Male | 13 | 2.84 | 5.63 | 495 (25) | 266 (15) | 0201/0301 | 5101/5101 | 1502/1502 | 1301/1301 | 1603/1603 |
| LP6 | 47 | Female | 25 | UD | 3.70 | 438 (25) | 282 (18) | 0201/2902 | 4001/5701 | 0304/0602 | 0401/0701 | 0302/0303 |
| LP7 | 36 | Male | 4 | 2.40 | 4.72 | 1,128 (47) | 555 (31) | 0101/3101 | 4001/5701 | 0304/0602 | 0404/0701 | 0302/0303 |
| LP8 | 43 | Female | 18 | 3.49 | 5.20 | 534 (33) | 166 (11) | 0301/2501 | 0702/5101 | 0702/0303 | 0901/1101 | 0301/0303 |
| LP9 | 44 | Male | 14 | 3.66 | 5.02 | 748 (21) | 402 (22) | 0301/6802 | 0702/5301 | 0702/0401 | 1301/1302 | 0603/0604 |
| LP10 | 38 | Male | 13 | UD | 5.90 | 913 (32) | 400 (21) | 1101/2402 | 0702/5101 | 0702/1502 | 0803/1501 | 0301/0602 |
| LP11 | 44 | Male | 14 | 2.96 | 5.36 | 920 | 464 | 0201/3001 | 4501/5701 | 0602/0701 | 0405/1502 | 0302/0601 |
| LP12 | 45 | Male | 23 | 2.93 | 4.00 | 810 | 493 | 0201/3201 | 1302/1501 | 0303/0602 | 0701/1401 | 0202/0301 |
| LP13 | 45 | Male | 16 | 2.52 | 3.64 | 434 (32) | 266 (22) | 0301/1101 | 0702/5201 | 0702/1202 | 1301/1502 | 0601/0603 |
| LP14 | 35 | Male | 1 | 2.63 | 5.20 | 430 | 465 | 0205/2402 | 0801/5801 | 0701/0702 | 0301/1102 | 0201/0301 |
ID, identifier; LP, late progressor.
Age and HIV duration are shown for the latest time point analyzed.
UD, undetermined.
NA, not available.
Individual HLA class I typing is shown (A, B, C, DRB, and DQB).
FIG 1.

HLA class I allele frequencies. HLA-B allele frequencies are shown for the general Spanish population (64), a cohort of elite controllers (EC, n = 38), viremic controllers (VC, n = 27), and individuals that have abruptly lost their capacity to control the infection (LoC, n = 14).
Evidence for virus tropism change and superinfection between pre- and post-LoC time points.
In order to determine whether changes in the cellular tropism of plasma virus preceded the abrupt loss of virus control, we determined viral tropism in plasma samples drawn before (n = 9; median, 21 months; range, 5 to 74 months) and after (n = 11), at diagnosis (n = 8), or during the peak of viremia (n = 3) clinical progression. Of the 14 samples obtained while the individuals had controlled infection, viral sequences covering the V3-loop (gp160) in all cases were indicative of a dominant CCR5-tropic viral population. In contrast, CXCR4-tropic plasma viruses were detected in four individuals post-LoC (although one of the individual was approaching the false-positive threshold of 10%), of which three had confirmed R5 virus pre-LoC (one did not amplify for env; Table 2). Based on phylogenetic analyses of Gag sequences, all pre- and post-LoC sequences showed close clustering for each individual except for one (subject LP4, Fig. 2), suggesting a superinfection event. Overall, clear evidence for changes in cell tropism were observed in one-quarter of the studied cohort, while superinfection was not a generalized mechanism for LoC in this group of patients.
TABLE 2.
HIV tropism before and after loss of control HIV tropism was estimated by Gp120 V3-loop region sequencing before and after late progressiona
| ID | Control-Pre (%FPR) | LoC-Post (%FPR) |
|---|---|---|
| LP1 | NA | CCR5 (12.5) |
| LP2 | CCR5 (36.2) | CXCR4 (0.1) |
| LP3 | CCR5 (35.5) | CCR5 (35.3) |
| LP4 | NA | CXCR4 (3.2) |
| LP5 | CCR5 (13.2) | CXCR4 (8.6) |
| LP6 | NA | NA |
| LP7 | CCR5 (36.2) | CXCR4 (4.8) |
| LP8 | CCR5 (10) | CCR5 (41.4) |
| LP9 | CCR5 (55) | CCR5 (28.8) |
| LP10 | CCR5 (42.3) | CCR5 (28.8) |
| LP11 | CCR5 (38) | CCR5 (37.4) |
| LP12 | NA | CCR5 (86.5) |
| LP13 | NA | CCR5 (42.2) |
| LP14 | CCR5 (55.1) | CCR5 (55.1) |
The false-positive rate (FPR) was set to 10% to identify X4 tropism. NA, sample not amplified.
FIG 2.

Evolutionary relationships of taxa. The evolutionary history was inferred using the neighbor-joining method (65). The optimal tree with the sum of branch length of 1.91362276 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches (66). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Tamura-Nei method (67) and are in the units of the number of base substitutions per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). The analysis was based on a 66-nucleotide amplicon in the V3-loop of HIV gp120. All ambiguous positions were removed for each sequence pair. Evolutionary analyses were conducted in MEGA5 (68).
Virus maintained HLA-associated escape mutations.
Viral breakthrough from immune surveillance has been described for both T- and B-cell-mediated immune control and could contribute to the sudden raise in viral loads and clinical HIV disease progression (21, 22). In order to evaluate whether the presence and accumulation of T-cell escape mutations contribute to LoC, HIV Gag sequences were obtained from samples drawn before and after LoC and analyzed for the presence of HLA class I footprints (23) (Fig. 2). All individuals yielded sequences in post-LoC samples, while HIV Gag sequences were successfully amplified in 9 out of 14 pre-LoC samples when subjects presented with effective in vivo virus control and low or undetectable viral loads. Gag sequences were compared with described HLA-associated escape mutations specific to the individuals’ HLA class I genotype (23). In 67% of patients with pre- and post-LoC sequences available, the number of patient-HLA specific footprints increased from pre- to post-LoC sequences. Despite this trend, the median number of footprints post-LoC (median, 3 footprints; range 1 to 8 footprints) did not differ significantly from the frequency of HLA footprints pre-LoC (median, 2 footprints; range, 1 to 5 footprints; Fig. 3, P = 0.2682). Although limited to Gag, the target of some of the most effective antiviral T-cell responses (10), and although single epitope mutations can lead to a complete loss of control, these data do not suggest that broad cytotoxic T lymphocyte (CTL) escape on multiple epitopes is the major driving force for LoC in the present cohort.
FIG 3.
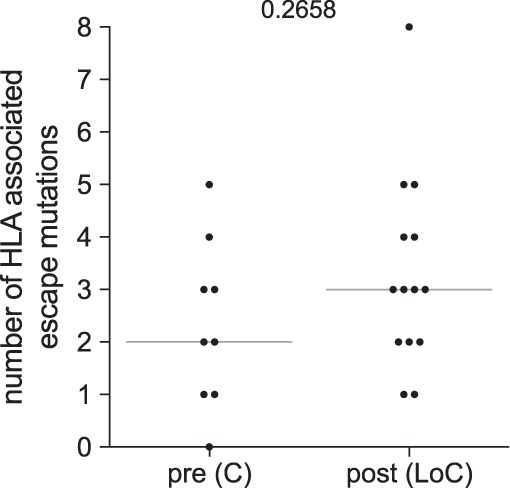
Number of HLA escape mutations in Gag (23) detected in viral population from samples before and after loss of control.
Increased number of HIV-specific T-cell responses post-LoC.
Individuals that control HIV infection have been shown to have a robust HIV-1-specific CD8+ T-cell response, especially against the Gag protein, compared to responses observed in noncontrollers (10, 24). The HIV-specific CD8+ T cells responses before LoC were thus measured and compared to those of post-LoC samples and to responses seen in 65 individuals from the IrsiCaixa Controllers cohort. Responses to the entire HIV proteome were measured by gamma interferon (IFN-γ) enzyme-linked immunosorbent spot assay (ELISPOT) using a 410-overlapping-peptide set, covering the complete HIV genome, as described previously (25). Twelve out of the 14 individuals had sufficient cell viability to conduct longitudinal full-proteome HIV ELISPOT screens (Fig. 3). In nine of them, the number of individual responses remained stable (n = 1 individual) or increased (n = 8 individuals), with a significant increase in the median breadth of response from 6 responses (range, 1 to 14 responses) pre-LoC to a median of 11 responses (range, 2 to 20 responses; Fig. 4A; P = 0.0488). Even though the appearance of more responses post-LoC may be due to higher viral replication and antigen availability post-LoC, the increase in the breadth of responses did not result in a significant increase in the total magnitude (median magnitude from pre-LoC, 3,145 spot-forming cells [SFC]/million peripheral blood mononuclear cells [PBMC; range, 100 to 7,950 SFC/million PBMC] to median post-LoC of 4,745 SFC/million PBMC [range, 690 to 27,420 SFC/million PBMC; Fig. 4B; P = 0.6221]. Of note, the dominance of Gag-specific responses which characterizes naturally controlling individuals was not lost after LoC and was similar to levels tested in the comparison cohort of elite and viremic controllers (Fig. 4C andS3). Furthermore, there was no consistent pattern in the emergence of new and expansion/reduction of preexisting responses from pre- to post-LoC; however, while some individuals showed a clear loss of immunodominant responses to certain HLA-restricted epitopes in the absence of detectable mutations, other patients maintained a stable response pattern (Fig. S3). Furthermore, there were no differences in the changes in responses between individuals in whom the virus changed tropism and individuals that maintained R5-tropic virus. Interestingly, the one individual (LP4) that showed evidence of superinfection had a limited breadth of responses pre-LoC (4 responses), of which only 2 were still detectable post-LoC, while there were an additional 15 responses detected post-LoC, possibly reflecting new targets in the superinfecting virus. In this individual, viral sequence data for Gag indicated that the pre-LoC virus contained all described optimal epitopes for his HLA class I alleles in wild-type immunogenic sequence, yet the subjects only mounted these Gag responses upon uncontrolled superinfection (17).
FIG 4.

HIV-specific T-cell responses. (A) Breadth of HIV-specific T-cell responses before and after loss of control (n = 12). (B) Magnitude of responses, expressed as SFC/million PBMC, is shown longitudinally (C) Gag dominance (magnitude of Gag responses/total magnitude) is shown before and after evolution in the 12 LoC subjects and compared to a cohort of elite (EC) and viremic (VC) controllers (n = 65). Individuals that maintained an CCR5 tropism are shown in black dots, individuals that changed to a CXCR4 are shown in gray dots, and the individual from which tropism was not available is shown in white dots. P values are shown for Wilcoxon signed rank test in a comparison of paired data and for Mann-Whitney test to compare between groups.
Reduced antiviral capacity of CD8+ T cells after loss of virological control.
In vitro inhibition of viral replication by autologous CD8+ T cells has become a standard measure to determine the ex vivo antiviral activity of cytotoxic T lymphocyte responses. We asked whether the viral inhibition assay (VIA) activity in individuals suffering an abrupt loss of control would be reduced at pre-LoC in comparison to persistent elite and viremic controllers and how VIA activity would change after LoC. In the absence of autologous virus isolates, the HIV IIIB laboratory strain was used to test in vitro inhibitory capacity of PBMC-derived CD8+ T cells. As a comparison, isolated CD8+ T cells from longitudinal PBMC samples of eight long-term HIV controllers with persistent in vivo HIV control (n = 6 EC and n = 2 VC, >15 years of diagnosed HIV-2 infection; longitudinal samples were 102 [range, 45 to 156] months apart between both follow-up time points) were tested (Table S1). Viral replication was measured by flow cytometry as a percentage of HIV Gag p24-positive cells in culture. The results showed a strong capacity of CD8+ T cells from pre-LoC samples to inhibit HIV replication at a 1:1 E/T (effector-to-target cells) ratio. However, this activity at the 1:1 ratio among the LoC patients studied here was somewhat lower than that of HIV controllers (median, 65% of LoC individuals compared to 84% of controllers, P = 0.0650; Fig. 5A) and significantly weaker at the 1:10 E/T ratio (median, 23% of LoC compared to 50% of controllers, P = 0.0104; Fig. 5B). While virus-suppressive activity in long-term controllers was maintained over time (P = 0.8438; median, 102 months between both time points tested), the suppressive capacity in LoC further decreased, especially detectable at the 1:1 ratio (P = 0.0156, Fig. 5A) upon loss of control. Thus, the data suggest that declining VIA activity over pre-LoC time points may be a prognostic marker for an increased risk of LoC.
FIG 5.

Viral inhibition capacity. Levels of CD8+ viral inhibitory capacity are shown for IIIB E/T 1:1 (A) and E/T 1:10 (B) in individuals who presented a loss of control (n = 8) and long-term controllers (n = 8). Comparison of antiviral capacity is expressed as % inhibition = [(fraction of p24+cells in CD4+ T cells cultured alone) − (fraction of p24+cells in CD4+ T cells cultured with CD8+ T cells)]/(fraction of p24+ in CD4+ T cells cultured alone) × 100.
Elevated frequencies of CD38 and PD-1 expression in CD8+ T cells after LoC.
The expression of CD38 and HLA-DR on T cells has been shown to be lower in treated HIV-infected individuals under cART and in HIV controllers than in individuals with progressive HIV disease. We therefore asked whether individuals experiencing LoC had elevated levels of activated cells right before the loss of control and how the increased virus replication post-LoC may impact CD38 and HLA-DR expression. The expression of surface activation markers on CD4+ and CD8+ T cells was assessed by flow cytometry before and after a loss of HIV control, along with additional activation markers (HLA-DR, CD38, CD25, and CD69) and an exhaustion marker (PD-1). Longitudinal samples from persistent HIV controllers were tested for comparison (102 months apart; range, 45 to 156 months). The expression of activation markers pre-LoC was similar to that in long-term controllers (data not shown); therefore, there was no evidence for elevated residual viral replication in LoC that could drive higher levels of activated T cells. In contrast, an almost 2-fold increase in the percentage of CD38+ and a trend toward higher levels of HLA-DR+ CD8 cells were observed after a loss of control (P = 0.0156 for CD38+, P = 0.0781 for HLA-DR, and P = 0.1563 for HLA-DR+ CD38+). No alterations in the expression of CD25 or CD69 were observed over time in either CD8+ (Fig. 6A) or CD4+ T-cell populations among LoC individuals and did not differ in expression levels observed in long-term controllers (data not shown). In line with previous reports (26), CD8+ T cells also expressed higher levels of PD-1 post-LoC (P = 0.0156, Fig. 6A), especially in central memory cells (CD8+ CCR7-CD45RA+), while those levels were maintained in persistent controllers (Fig. 6B). Of note, there was a significantly higher level of PD-1+ CD8+ T cells with a naive phenotype (CCR7+ CD45RA+) in the individuals with LoC than in persistent controllers. Importantly, this difference was already manifest before LoC, potentially identifying an additional risk marker for subsequent LoC.
FIG 6.

Levels of activation and exhaustion markers. (A) Percentage of expression of CD38, HLA-DR, PD-1, CD25, and CD69 on total CD8+ T cells in individuals that experience a loss of control (n = 8) tested before [pre (C)] or after [post (LoC)] loss of viral control. Values are compared to staining in samples from persistent HIV controllers taken at a median of 102 months apart. Markers were assessed following the gating strategy shown in Fig. S1. P values are shown for Wilcoxon signed rank test. (B) Levels of expression of PD-1 in subsets of CD8+ T cells populations based on CCR7 and CD45RA expression. Median and interquartile range values are shown for a group of 8 individuals that loss of control compared to 8 individuals maintained control over time. The gating strategy used is shown in Fig. S1. P values are shown for Wilcoxon signed rank test.
DISCUSSION
The search for reliable predictors of HIV control has been largely focused on cross-sectional comparisons between individuals with either controlled or uncontrolled HIV infection in the absence of antiretroviral treatment. These studies have yielded a number of correlates of control, many pointing toward an important role of the virus-specific cellular immunity in the natural control of HIV (27–30). Interpretation of such studies, however, can be limited by its cross-sectional nature and, consequently, by significant potential differences in host genetics between comparison groups, in particular, HLA class I allele frequencies (8, 31). In the present study, we have attempted to overcome some of these limitations by analyzing longitudinal samples from subjects who spontaneously lose their previously excellent and longstanding control of viral replication. The identification of markers of control, or loss thereof, could be a helpful tool for the clinical management of individuals off cART with controlled HIV infection and could have important implications for future prevention and treatment strategies in general.
One possible explanation for the abrupt HIV disease progression observed in the present cohort could be HIV superinfection. Superinfection with HIV has been associated with disease progression (17, 32) and has been previously documented, even in the setting of HIV controller cohorts (33–35). Although superinfection can be facilitated by exposure to a virus that can escape preexisting virus-specific T-cell immunity (33, 36), the first well-documented case of an uncontrolled superinfection in an HIV controller occurred in the presence of a majority of T-cell epitopes being conserved between the first and second infecting virus (17). Interestingly, in the one case of suspected superinfection in the present cohort, we observed a complete change in the pattern of CD8+ T-cell responses, where only 2 responses were maintained (in Pol and Nef), while 15 new responses toward the whole proteome of HIV were mounted; however, they were evidently not able to control the new virus. Although limited by the cohort size, the present data are in line with earlier reports of highly immunogenic but uncontrolled superinfection and suggest that superinfection may not be a major factor in the sudden loss of virus control in former HIV controllers.
An alternative explanation for LoC could be a switch in the viral tropism from CCR5 to CXCR4, which has been described to precede a decrease in CD4 T-cell counts and progression to AIDS (37–40). We found that 30% of the individuals (4 out of 13 tested) presented X4-tropic virus after progression, while none was detected during the controlled stages of infection. Interestingly, one of the individuals that showed an X4-tropic virus was the subject that acquired superinfection, suggesting that the second viral infection may have been with an X4-tropic virus. For the remaining 3 subjects who harbored X4 virus post-LoC, it remains unclear whether the change of tropism was either the cause or the consequence of the loss of control. Actually, despite significant advances in the study of relevant parameters involved in HIV control, it is still difficult to define the causality dilemma of certain factors, such as the HIV tropism evolution (28). Although a switch in coreceptor usage has been attributed to an increase in viral fitness (41, 42), we were unable to obtain any autologous virus from samples drawn at time points when the individuals were controlling the infection and thus could not compare viral replicative fitness pre- and post-LoC. Cell tropism of HIV has also been associated with cell type-specific differences in antigen processing and, hence, differences in epitope presentation on infected T cells versus infected macrophages or dendritic cells (43). An R5-to-X4 tropism change could therefore result in an effective escape from T-cell immunity that is mainly focused on epitopes presented by infected T cells but not by macrophages. In addition, it could also explain the marked increase in responses post-LoC in the subject with superinfection and that env responses were only seen after superinfection, although the epitopes were present in wild-type sequence in the controlled virus pre-LoC. As only three individuals showed evidence of a dominant X4-tropic virus post-LoC, we were not able to identify any specific response pattern that could correlate with tropism change during HIV infection progression.
As referred to above, HIV controller cohorts are oftentimes enriched in particular “beneficial” HLA class I alleles, including HLA-B*27, HLA-B*57, and others (6). Our cohort of LoC is no exception to this, as the frequency of those protective HLA class I alleles (B57/58 and B27) was the same as observed in a locally recruited elite and viremic controller cohort (44). Furthermore, we also detected broad and strong CTL responses, especially toward Gag, consistent with previous reports in comparable cohorts (2, 45–48). However, it has also been suggested that HIV controllers with beneficial or nonbeneficial HLA class I alleles may control HIV through different mechanisms, with subjects expressing nonbeneficial alleles, such as HLA-B*35, depending critically on their T-cell responses to durable control (44). As our cohort only included one subject with a strong nonbeneficial HLA class I alleles, the study of larger cohorts of LoC with unfavorable HLA class I genetics will offer a unique opportunity to determine whether the failures of these responses are indeed critical determinants in LoC. In addition, more in-depth characterization of these responses, including avidity testing and effector function profiling (49, 50), rather than its mere presence/absence pre- and post-LoC, may reveal factors that define the “failure” of these responses post-LoC. We addressed the measurement of functional activity here by conducting in vitro inhibition assays (VIA) on samples pre- and post-LoC. Our results indeed showed decreased antiviral in vitro capacity of CD8+ T cells before LoC compared to persistently controlling EC. Still, the data suggest that declined VIA activity may precede a loss of control and could serve as a predictor of failing immune control. This is in line with cross-sectional analyses that have associated antiviral capacity of CD8 T-cell in VIA with the rate of HIV disease progression and CD4+ T-cell decline (13, 24). Thus, monitoring VIA activity in untreated individuals may be a useful tool to initiate cART in a timely manner and may be especially useful in treatment interruption studies that assess outcomes of therapeutic vaccination and other HIV cure approaches.
Another well-accepted explanation for loss of effective immune control of viral replication in vivo is the occurrence of CTL escape mutations in the targeted epitope. Although most tested individuals presented an increased number of HLA-associated escape mutation in their Gag sequence between pre- and post-LoC samples, the differences in HLA footprints over time were not significant. As a single escaped CTL epitope may allow for a loss of control, it would be important to test epitope variants for their true ability to escape CTL recognition. However, we did not have sufficient samples at hand to map all responses and assess the effects of epitope mutations on CTL recognition. In addition, extended deep sequencing covering the entire viral genome and additional sampling time points close to LoC would provide more insights into the role that HLA-associated mutations outside Gag may play in the abrupt the loss of control. Deep sequencing would also reveal low-frequency mutations present that were missed by our analyses and, with additional sampling points, allow the evolution and frequencies of these mutations to be closely followed. As the frequency of individuals experiencing a marked rapid LoC is already low, having closer sampling intervals in larger cohorts will be truly challenging. Due to technical hurdles and sample availability needed to reliably amplify gag sequences in individuals with low or undetectable viremia, we cannot compare the occurrence and frequency of Gag HLA-associated escape mutations seen in our subjects with those in individuals with persistent control. However, existing data show that even in individuals with low levels of viral replication, Gag viral evolution can be detected (45, 51), suggesting that their presence does not necessarily need to lead to LoC.
In line with the loss of antiviral capacity and an increased viral replication in vivo, we observed a significant increase in the expression of markers of T-cell activation and exhaustion (in particular, CD38+ and PD-1), both of which have been linked to HIV disease progression (9, 46–48, 52–54). Of note, LoC individuals showed a significantly higher level of PD-1-expressing CD8+ T cells even before loss of control compared to individuals that maintained their viral control over time. This was especially marked for CD8+ T cells with a naive phenotype and may represent an early sign of LoC during which terminally differentiated T cells may reacquire a naive phenotype (55). These cells may not be able to cope with ongoing viral replication and immune activation pre-LoC, ultimately leading to full-blown loss of control and elevated viral loads. At the same time, the increased levels of exhaustion markers could also explain the reduced VIA activity seen pre-LoC. Moreover, the enhanced activation and effector phenotype of CD8+ T cells could lead to less proliferative capacity of cells in vitro and result in reduced VIA activity seen post-LoC. If validated in further cohorts, our analyses may thus have identified reduced VIA activity and elevated levels of PD-1 expression as predictive markers of loss of control in individuals showing natural control of HIV and may help improve their clinical management. The data may also help guide immune-based therapeutic intervention to achieve a functional HIV cure by defining minimal VIA activities and preferred T-cell phenotypes that may warrant treatment interruptions.
In light of reports showing a predictive value of reservoir size and the rebound kinetics of virus after stopping treatment (56–58) or, in on our case, after LoC, it would have been interesting to determine the size of the viral reservoir in the LoC individuals tested here. However, cell availability did not allow for such analyses. Despite some of these limitations, the present report suggests that individuals who presented an abrupt loss of HIV control had an impaired capacity to inhibit in vitro viral replication, maybe due to the increased exhaustion of their CD8+ T cells before LoC. Measuring these parameters may help identify untreated HIV controllers that are at risk of losing control and may offer a useful tool for monitoring individuals during treatment interruption phases included in therapeutic vaccine trials or other cure strategies.
Earlier studies addressing loss of control in previous HIV controllers have also been hampered by the small size of the cohorts of such particular individuals or have attempted to address this question in simian immunodeficiency virus (SIV)-infected macaques (59, 60) while assessing only one or additional aspects of host immunity and their role in persistent virus control (28, 52, 61, 62). Among these, NK function (54), polyfunctionality (18), and the occurrence of new mutations in relatively conserved viral genes have been proposed to precede the abrupt loss of control. As in our study, these analyses are limited by the difficulty of discerning cause from effect of these events or mechanisms, and further work, possibly in SIV models, may be required to validate the importance of the individual factors identified in predicting LoC.
MATERIALS AND METHODS
Study participants and samples.
Among two existing cohorts of HIV-1-infected elite and viremic controllers (defined by sustained undetectable or low-level viremia, respectively [plasma viral load {pVL}, <50 copies/ml or <2,000 copies/ml] for more than 1 year in the absence of ART), we identified 14 subjects who experienced an abrupt transition from a nonprogressive to a progressive state of HIV infection (Table 1). All individuals had stable viremia at <3 log copies/ml before LoC for a median time of 14 years since HIV diagnosis (interquartile range [IQR], 4 to 19 years). The transition to LoC was defined as (i) >1-log pVL increase and/or (ii) loss of >30% CD4+ T-cell counts or drop below 350 CD4+ T cells/ml within 1 year. Samples were obtained from the Spanish HIVHGM BioBank supported by the Spanish Instituto de Salud Carlos III and from the existing long-term sample repository at IrsiCaixa, in Badalona, Spain. All subjects were HLA-typed for HLA-A, HLA-B, and HLA-C loci at high resolution, as described previously (31). Data from a full-protein ELISPOT CTL screen of a group of 65 individuals from the IrsiCaixa Controllers Cohort with persistent HIV control for a median time of 14 years since HIV diagnosis (IQR, 5 to 21 years) were included for comparisons and included 38 EC (median viral load, 50 copies/ml; median CD4 count, 815 cells/mm3) and 27 viremic controllers (median VL, 545 copies/ml; median CD4 count, 616 cells/mm3). A subset of eight HIV-1 controllers with longer clinical follow-up and with prospective biological samples available at separated time points was selected for the viral inhibition assay comparison as long-term controllers (Table S1). All patients provided informed consent before providing samples. The study was approved by the institutional ethical review board of the Hospital Universitari Germans Trias i Pujol (reference no. EO-09-041).
Gag sequence evolution.
HIV-1 RNA was extracted from plasma using the QIAamp viral RNA kit (Qiagen) and was reverse transcribed using reverse transcription-PCR (RT-PCR) SuperScript III enzyme mix (Invitrogen). The gag gene region was then amplified by nested PCR. Sequencing was performed by the Genomics Core Facility at Germans Trias i Pujol Research Institute. Gag nucleotide alignment was performed with MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) to generate the corresponding multiple-sequence alignments (MSA). Nucleotide evolution models that best explained sequence evolution within each MSA were identified using the MEGA5 Find Best DNA/Protein Models (ML) function. The model with the lower score according to Bayesian information criteria implemented in MEGA5 was selected to construct phylogenetic trees. Gag sequences obtained were further analyzed to determine the number of HLA class I-associated immune escape mutations and were identified based on the individual’s HLA class I genotype and the HLA-associated escape mutations described by Brumme et al. (23).
Determination of viral tropism.
HIV tropism was predicted by Gp160 V3-loop region Sanger sequencing, as previously described (63). Briefly, HIV-1 RNA was extracted from plasma using the QIAamp viral RNA kit (Qiagen), and cDNA was synthetized using RT-PCR SuperScript III enzyme mix (Invitrogen). The env V3-loop-encoding region was amplified by nested PCR. Sequences were analyzed by using the Geno2Pheno [coreceptor] using a false-positive rate (FPR) cutoff of 10% to define the presence of an X4 HIV-1.
IFN-γ ELISPOT.
All individuals were screened longitudinally for IFN-γ-secreting T-cell responses to the entire HIV proteome. Briefly, cryopreserved PBMC were thawed and rested for 5 h at 37°C before plating 100,000 live cells per well in IFN-γ ELISPOT 96-well polyvinylidene plates (Millipore). PBMC were stimulated with a clade B consensus sequence set of 410 peptides (18mers overlapping by 11 amino acids [aa]) at a final concentration of 14 μg/ml. The IFN-γ Mabtech kit was used, according to the manufacturer’s instructions. Spots were counted using an automated ELISPOT reader system (ImmunoSpot S6 Versa; CTL, Germany), and the magnitude of responses was expressed as spot-forming cells (SFC) per million input cells. The threshold for positive responses was defined as at least 5 spots per well, responses exceeding the “mean number of spots in negative-control wells plus 3 standard deviations of the negative-control wells” and “three times the mean of negative-control wells,” whichever was higher.
Viral inhibition assay.
The capacity of CD8+ T cells to suppress HIV-1 replication in autologous CD4+ T cells ex vivo was assessed as described in detail elsewhere (69, 70). Briefly, CD8-depleted cells were isolated from cryopreserved PBMC by magnetic bead separation (MACS Milteny Biotec) and stimulated with phytohemagglutinin (PHA; 5 μg/ml) for 3 days in R10-RPMI medium supplemented with 10% fetal calf serum (FCS), l-glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were then washed and infected by spinoculation with an HIV-1 IIIB strain at a multiplicity of infection (MOI) of 0.01. To assess the antiviral capacity of CD8+ T cells, autologous infected CD4+ T cells were cultured alone or with unstimulated CD8+ T cells in R10 supplemented with interleukin 2 (20 IU/ml). The experiments were carried out in triplicate at different effector-to-target ratios (1:1, 1:10, and 1:2, depending on cell availability). After 6 days of coculture, intracellular p24 production was measured by flow cytometry. Antiviral capacity was expressed as percentage of inhibition, determined as [(fraction of p24+ cells in CD4+ T cells cultured alone) − (fraction of p24+ in CD4+ T cells cultured with CD8+ cells)]/(fraction of p24+ cells in CD4+ T cells cultured alone) × 100.
Flow cytometry.
CD8+-depleted cells (CD4+ T-cell enriched fraction) and CD8+-isolated T cells were stained for activation and exhaustion markers. Cells were first stained with a viability staining (Aqua LIVE/DEAD fixable dead cell stain kit; Invitrogen), followed by exclusion staining of B lymphocytes and myeloid cells using combined markers in a dump channel (using the cells/antibodies CD19/BV510 and CD14/BV510; BioLegend). For T-cell lineage and activation markers, the following cells/antibodies were used: CD3/antigen-presenting cells (APC) Cy7, CD4/peridinin chlorophyll protein (PerCP) (BD Biosciences); and CD8/PerCP, CCR7/BV421, CD45RAPECF594/HLA-DR fluorescein isothiocyanate (FITC), PD-1/phycoerythyrin (PE,) CD69/APC, CD38/BV785, and CD25/BV605 (BioLegend). After staining, cells were collected on an LSRFortessa instrument (BD), and analysis was performed using the FlowJo 10 software.
To determine the number of p24+ cells in the viral inhibition assay, cells were stained with Aqua LIVE/DEAD (Invitrogen), followed by extracellular staining with cells/antibodies CD3/APC-H7, CD4/PerCP, and CD8/APC (BD Biosciences). Cells were fixed and permeabilized (FIX & PERM cell fixation & cell permeabilization kit; Thermo Fisher Scientific) and finally stained with p24 antibody KC.57 fluorescein isothiocyanate (FITC; Beckman Coulter). Samples were acquired on an LSR-II cytometer, and data analysis was done using the FlowJo 10 software.
Statistical analysis.
GraphPad Prism version 7.0 for Windows (San Diego, CA) was used to compare response rates in both groups and subgroup analyses. A Mann-Whitney test and Wilcoxon matched paired test were used for unpaired and paired comparisons, respectively.
Data availability.
Gag sequences are available at GenBank with the accession numbers MK086511 (LP1), MK086512 (LP2 pre), MK086513 (LP2 post), MK086514 (LP3 pre), MK086515 (LP3 post), MK086516 (LP4 pre), MK086517 (LP4 post), MK086518 (LP5 pre), MK086519 (LP5 post), MK086520 (LP6 post), MK086521 (LP7 post), MK086522 (LP8 pre), MK086523 (LP8 post), MK086524 (LP9 pre), MK086525 (LP9 post), MK086526 (LP10 post), MK086527 (LP11 pre), MK086528 (LP11 post), MK086529 (LP12 pre), MK086530 (LP12 post), MK086531 (LP13 post), MK086532 (LP14 pre), and MK086533 (LP14 post).
Supplementary Material
ACKNOWLEDGMENTS
We particularly thank the patients in this study for their participation and the HIV BioBank, an integrated part of the Spanish AIDS Research Network, and collaborating centers for the generous provision of clinical samples.
The HIV BioBank is supported by Instituto de Salud Carlos III and Spanish Health Ministry (grant RD06/0006/0035). This work was supported in part by the Spanish FIPSE gant 360737-09, the Barcelona-based HIVACAT program, funding from the European Union’s Horizon 2020 research and innovation program under grant agreement 681137-EAVI2020, and a research agreement with AELIX Therapeutics, Barcelona, Spain.
Christian Brander is the CSO at AELIX Therapeutics.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01436-18.
REFERENCES
- 1.Buchbinder SP, Katz MH, Hessol NA, O’Malley PM, Holmberg SD. 1994. Long-term HIV-1 infection without immunologic progression. AIDS 8:1123–1128. doi: 10.1097/00002030-199408000-00014. [DOI] [PubMed] [Google Scholar]
- 2.Deeks SG, Walker BD, Cellerai C, Vallelian F, Bart PA, Pantaleo G, Lifson JD, O’Connor DH, Carrington M, Watkins DI. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, Baker B, Trocha A, Rosenberg R, Mackey E, Ueda P, Lu Z, Cohen D, Wrin T, Petropoulos CJ, Rosenberg ES, Walker BD. 2008. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis 197:563–571. doi: 10.1086/526786. [DOI] [PubMed] [Google Scholar]
- 4.Sáez-Cirión A, Sinet M, Shin SY, Urrutia A, Versmisse P, Lacabaratz C, Boufassa F, Avettand-Fènoël V, Rouzioux C, Delfraissy J-F, Barré-Sinoussi F, Lambotte O, Venet A, Pancino G, ANRS EP36 HIV Controllers Study Group. 2009. Heterogeneity in HIV suppression by CD8 T cells from HIV controllers: association with Gag-specific CD8 T cell responses. J Immunol 182:7828–7837. doi: 10.4049/jimmunol.0803928. [DOI] [PubMed] [Google Scholar]
- 5.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, Cozzi-Lepri A, De Luca A, Easterbrook P, Francioli P, Mallal S, Martinez-Picado J, Miro JM, Obel N, Smith JP, Wyniger J, Descombes P, Antonarakis SE, Letvin NL, McMichael AJ, Haynes BF, Telenti A, Goldstein DB. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrington M, O’Brien SJ. 2003. The influence of HLA genotype on AIDS. Annu Rev Med 54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 7.Fellay J, Ge D, Shianna KV, Colombo S, Ledergerber B, Cirulli ET, Urban TJ, Zhang K, Gumbs CE, Smith JP, Castagna A, Cozzi-Lepri A, De Luca A, Easterbrook P, Günthard HF, Mallal S, Mussini C, Dalmau J, Martinez-Picado J, Miro JM, Obel N, Wolinsky SM, Martinson JJ, Detels R, Margolick JB, Jacobson LP, Descombes P, Antonarakis SE, Beckmann JS, O’Brien SJ, Letvin NL, McMichael AJ, Haynes BF, Carrington M, Feng S, Telenti A, Goldstein DB, NIAID Center for HIV/AIDS Vaccine Immunology (CHAVI). 2009. Common genetic variation and the control of HIV-1 in humans. PLoS Genet 5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goulder PJR, Walker BD. 2012. HIV and HLA class I: an evolving relationship. Immunity 37:426–440. doi: 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sáez-Cirión A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, Barré-Sinoussi F, Delfraissy J-F, Sinet M, Pancino G, Venet A, Agence Nationale de Recherches sur le Sida EP36 HIV Controllers Study Group. 2007. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci U S A 104:6776–6781. doi: 10.1073/pnas.0611244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuñiga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, Sanchez H, Frahm N, Linde CH, Hewitt HS, Hildebrand W, Altfeld M, Allen TM, Walker BD, Korber BT, Leitner T, Sanchez J, Brander C. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J Virol 80:3122–3125. doi: 10.1128/JVI.80.6.3122-3125.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Payne R, Muenchhoff M, Mann J, Roberts HE, Matthews P, Adland E, Hempenstall A, Huang K-H, Brockman M, Brumme Z, Sinclair M, Miura T, Frater J, Essex M, Shapiro R, Walker BD, Ndung’u T, McLean AR, Carlson JM, Goulder PJR. 2014. Impact of HLA-driven HIV adaptation on virulence in populations of high HIV seroprevalence. Proc Natl Acad Sci 111:E5393–E5400. doi: 10.1073/pnas.1413339111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casadellà M, Cozzi-Lepri A, Phillips A, Noguera-Julian M, Bickel M, Sedlacek D, Zilmer K, Clotet B, Lundgren JD, Paredes R, EuroSIDA in EuroCOORD. 2017. Plasma HIV-1 tropism and the risk of short-term clinical progression to AIDS or death. PLoS One 12:e0166613. doi: 10.1371/journal.pone.0166613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okulicz JF, Marconi VC, Landrum ML, Wegner S, Weintrob A, Ganesan A, Hale B, Crum-Cianflone N, Delmar J, Barthel V, Quinnan G, Agan BK, Dolan MJ, Infectious Disease Clinical Research Program (IDCRP) HIV Working Group. 2009. Clinical outcomes of elite controllers, viremic controllers, and long-term nonprogressors in the US Department of Defense HIV natural history study. J Infect Dis 200:1714–1723. doi: 10.1086/646609. [DOI] [PubMed] [Google Scholar]
- 14.Van der Helm JJ, Geskus R, Lodi S, Meyer L, Schuitemaker H, Gunsenheimer-Bartmeyer B, Monforte A. dArminio, Olson A, Touloumi G, Sabin C, Porter K, Prins M, CASCADE Collaboration in EuroCoord. 2014. Characterisation of long-term non-progression of HIV-1 infection after seroconversion: a cohort study. Lancet HIV 1:e41–e48. doi: 10.1016/S2352-3018(14)70016-5. [DOI] [PubMed] [Google Scholar]
- 15.Noel N, Lerolle N, Lécuroux C, Goujard C, Venet A, Saez-Cirion A, Avettand-Fenoël V, Meyer L, Boufassa F, Lambotte O, Agut H, Autran B, Barin F, Costagliola D, Pancino G, Rouzioux C, Samri-Hassimi A, Taulera O, Theodorou I, Tubiana R, Viard JP, Yazdanpanah Y, Boufassa F, Lambotte O, Meyer L. 2015. Immunologic and virologic progression in HIV controllers: the role of viral “blips” and immune activation in the ANRS CO21 CODEX study. PLoS One 10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leon A, Perez I, Ruiz-Mateos E, Benito JM, Leal M, Lopez-Galindez C, Rallon N, Alcami J, Lopez-Aldeguer J, Viciana P, Rodriguez C, Grau E, Iribarren J, Gatell JM, Garcia F, EC and Immune Pathogenesis Working group of the Spanish AIDS Research Network. 2016. Rate and predictors of progression in elite and viremic HIV-1 controllers. AIDS 30:1209–1220. doi: 10.1097/QAD.0000000000001050. [DOI] [PubMed] [Google Scholar]
- 17.Altfeld M, Allen TM, Yu XG, Johnston MN, Agrawal D, Korber BT, Montefiori DC, O’Connor DH, Davis BT, Lee PK, Maier EL, Harlow J, Goulder PJR, Brander C, Rosenberg ES, Walker BD. 2002. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature 420:434–439. doi: 10.1038/nature01200. [DOI] [PubMed] [Google Scholar]
- 18.Pernas M, Tarancón-Diez L, Rodríguez-Gallego E, Gómez J, Prado JG, Casado C, Dominguez-Molina B, Olivares I, Coiras M, León A, Rodriguez C, Benito JM, Rallón N, Plana M, Martinez-Madrid O, Dapena M, Iribarren JA, del Romero J, García F, Alcamí J, Muñoz-Fernández MÁ, Vidal F, Leal M, Lopez-Galindez C, Ruiz-Mateos E, on behalf of ECRIS integrated in the Spanish AIDS Research Network . 2018. Factors leading to the loss of natural elite control of HIV-1 infection. J Virol 92:e01805-17. doi: 10.1128/JVI.01805-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, Connors M. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci U S A 97:2709–2714. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emu B, Sinclair E, Hatano H, Ferre A, Shacklett B, Martin JN, McCune JM, Deeks SG. 2008. HLA class I-restricted T-cell responses may contribute to the control of human immunodeficiency virus infection, but such responses are not always necessary for long-term virus control. J Virol 82:5398–5407. doi: 10.1128/JVI.02176-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trkola A, Kuster H, Rusert P, Joos B, Fischer M, Leemann C, Manrique A, Huber M, Rehr M, Oxenius A, Weber R, Stiegler G, Vcelar B, Katinger H, Aceto L, Günthard HF. 2005. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med 11:615–622. doi: 10.1038/nm1244. [DOI] [PubMed] [Google Scholar]
- 22.Goulder PJR, Walker BD. 1999. The great escape – AIDS viruses and immune control. Nat Med 5:1233–1235. doi: 10.1038/15184. [DOI] [PubMed] [Google Scholar]
- 23.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P, Sela J, Kadie CM, Frahm N, Brander C, Haas DW, Riddler SA, Haubrich R, Walker BD, Harrigan PR, Heckerman D, Mallal S. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4:e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger CT, Frahm N, Price DA, Mothe B, Ghebremichael M, Hartman KL, Henry LM, Brenchley JM, Ruff LE, Venturi V, Pereyra F, Sidney J, Sette A, Douek DC, Walker BD, Kaufmann DE, Brander C. 2011. High-functional-avidity cytotoxic T lymphocyte responses to HLA-B-restricted Gag-derived epitopes associated with relative HIV control. J Virol 85:9334–9345. doi: 10.1128/JVI.00460-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frahm N, Korber BT, Adams CM, Szinger JJ, Draenert R, Addo MM, Feeney ME, Yusim K, Sango K, Brown NV, SenGupta D, Piechocka-Trocha A, Simonis T, Marincola FM, Wurcel AG, Stone DR, Russell CJ, Adolf P, Cohen D, Roach T, StJohn A, Khatri A, Davis K, Mullins J, Goulder PJR, Walker BD, Brander C. 2004. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J Virol 78:2187–2200. doi: 10.1128/JVI.78.5.2187-2200.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJR, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 27.Mothe B, Ibarrondo J, Llano A, Brander C. 2009. Virological, immune and host genetics markers in the control of HIV infection. Dis Markers 27:106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prado JG, Carrillo J, Blanco-Heredia J, Brander C. 2011. Immune correlates of HIV control. Curr Med Chem 18:3963–3970. doi: 10.2174/092986711796957202. [DOI] [PubMed] [Google Scholar]
- 29.Walker-Sperling VEK, Buckheit RW, Blankson JN. 2014. Comparative analysis of the capacity of elite suppressor CD4+ and CD8+ T cells to inhibit HIV-1 replication in monocyte-derived macrophages. J Virol 88:9789–9798. doi: 10.1128/JVI.00860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Migueles SA, Mendoza D, Zimmerman MG, Martins KM, Toulmin SA, Kelly EP, Peterson BA, Johnson SA, Galson E, Poropatich KO, Patamawenu A, Imamichi H, Ober A, Rehm CA, Jones S, Hallahan CW, Follmann DA, Connors M. 2015. CD8+ T-cell cytotoxic capacity associated with human immunodeficiency virus-1 control can be mediated through various epitopes and human leukocyte antigen types. EBioMedicine 2:46–58. doi: 10.1016/j.ebiom.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olvera A, Ganoza C, Pérez-Álvarez S, Hildebrand W, Sanchez J, Brander C. 2014. HLA-B*35-PX and HLA-B*35-PY subtype differentiation does not predict observed differences in level of HIV control in a Peruvian MSM cohort. AIDS 28:2323–2325. doi: 10.1097/QAD.0000000000000403. [DOI] [PubMed] [Google Scholar]
- 32.Smith DM, Wong JK, Hightower GK, Ignacio CC, Koelsch KK, Daar ES, Richman DD, Little SJ. 2004. Incidence of HIV superinfection following primary infection. JAMA 292:1177–1178. doi: 10.1001/jama.292.10.1177. [DOI] [PubMed] [Google Scholar]
- 33.Clerc O, Colombo S, Yerly S, Telenti A, Cavassini M. 2010. HIV-1 elite controllers: beware of super-infections. J Clin Virol 47:376–378. doi: 10.1016/j.jcv.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 34.Rachinger A, Navis M, van Assen S, Groeneveld PHP, Schuitemaker H. 2008. Recovery of viremic control after superinfection with pathogenic HIV type 1 in a long‐term elite controller of HIV type 1 infection. Clin Infect Dis 47:e86–e89. doi: 10.1086/592978. [DOI] [PubMed] [Google Scholar]
- 35.Brener J, Gall A, Hurst J, Batorsky R, Lavandier N, Chen F, Edwards A, Bolton C, Dsouza R, Allen T, Pybus OG, Kellam P, Matthews PC, Goulder PJR. 2018. Rapid HIV disease progression following superinfection in an HLA-B*27:05/B*57:01-positive transmission recipient. Retrovirology 15:7. doi: 10.1186/s12977-018-0390-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Streeck H, Li B, Poon AFY, Schneidewind A, Gladden AD, Power KA, Daskalakis D, Bazner S, Zuniga R, Brander C, Rosenberg ES, Frost SDW, Altfeld M, Allen TM. 2008. Immune-driven recombination and loss of control after HIV superinfection. J Exp Med 205:1789–1796. doi: 10.1084/jem.20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaleebu P, Nankya IL, Yirrell DL, Shafer LA, Kyosiimire-Lugemwa J, Lule DB, Morgan D, Beddows S, Weber J, Whitworth JAG. 2007. Relation between chemokine receptor use, disease stage, and HIV-1 subtypes A and D. J Acquir Immune Defic Syndr 45:28–33. doi: 10.1097/QAI.0b013e3180385aa0. [DOI] [PubMed] [Google Scholar]
- 38.Solomon A, Lane N, Wightman F, Gorry PR, Lewin SR. 2005. Enhanced replicative capacity and pathogenicity of HIV-1 isolated from individuals infected with drug-resistant virus and declining CD4+ T-cell counts. J Acquir Immune Defic Syndr 40:140–148. doi: 10.1097/01.qai.0000173460.75322.93. [DOI] [PubMed] [Google Scholar]
- 39.Connor RI, Mohri H, Cao Y, Ho DD. 1993. Increased viral burden and cytopathicity correlate temporally with CD4+ T-lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected individuals. J Virol 67:1772–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swanstrom R, Coffin J. 2012. HIV-1 pathogenesis: the virus. Cold Spring Harb Perspect Med 2:a007443. doi: 10.1101/cshperspect.a007443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Troyer RM, Collins KR, Abraha A, Fraundorf E, Moore DM, Krizan RW, Toossi Z, Colebunders RL, Jensen MA, Mullins JI, Vanham G, Arts EJ. 2005. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. J Virol 79:9006–9018. doi: 10.1128/JVI.79.14.9006-9018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber J, Gibson RM, Sácká L, Strunin D, Hodek J, Weberová J, Pávová M, Alouani DJ, Asaad R, Rodriguez B, Lederman MM, Quiñones-Mateu ME. 2017. Impaired human immunodeficiency virus type 1 replicative fitness in atypical viremic non-progressor individuals. AIDS Res Ther 14:15. doi: 10.1186/s12981-017-0144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dinter J, Duong E, Lai NY, Berberich MJ, Kourjian G, Bracho-Sanchez E, Chu D, Su H, Zhang SC, Le Gall S. 2015. Variable processing and cross-presentation of HIV by dendritic cells and macrophages shapes CTL immunodominance and immune escape. PLoS Pathog 11:e1004725. doi: 10.1371/journal.ppat.1004725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koofhethile CK, Ndhlovu ZM, Thobakgale-Tshabalala C, Prado JG, Ismail N, Mncube Z, Mkhize L, van der Stok M, Yende N, Walker BD, Goulder PJR, Ndung’u T. 2016. CD8 T cell breadth and ex vivo virus inhibition capacity distinguish between viremic controllers with and without protective. J Virol 90:6818–6831. doi: 10.1128/JVI.00276-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salgado M, Garcia-Minambres A, Dalmau J, Jiménez-Moyano E, Viciana P, Alejos B, Clotet B, Prado JG, Martinez-Picado J. 2018. Control of HIV-1 pathogenesis in viremic nonprogressors is independent of Gag-specific cytotoxic T lymphocyte responses. J Virol 92:e00346-18. doi: 10.1128/JVI.00346-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choudhary SK, Vrisekoop N, Jansen CA, Otto SA, Schuitemaker H, Miedema F, Camerini D. 2007. Low immune activation despite high levels of pathogenic human immunodeficiency virus type 1 results in long-term asymptomatic disease. J Virol 81:8838–8842. doi: 10.1128/JVI.02663-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deeks SG, Kitchen CMR, Liu L, Guo H, Gascon R, Narváez AB, Hunt P, Martin JN, Kahn JO, Levy J, McGrath MS, Hecht FM. 2004. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 104:942–947. doi: 10.1182/blood-2003-09-3333. [DOI] [PubMed] [Google Scholar]
- 48.Bansal A, Sterrett S, Erdmann N, Westfall AO, Dionne-Odom J, Overton ET, Goepfert PA. 2015. Normal T-cell activation in elite controllers with preserved CD4+ T-cell counts. AIDS 29:2245–2254. doi: 10.1097/QAD.0000000000000860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mothe B, Llano A, Ibarrondo J, Zamarreño J, Schiaulini M, Miranda C, Ruiz-Riol M, Berger CT, Herrero MJ, Palou E, Plana M, Rolland M, Khatri A, Heckerman D, Pereyra F, Walker BD, Weiner D, Paredes R, Clotet B, Felber BK, Pavlakis GN, Mullins JI, Brander C. 2012. CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS One 7:e29717. doi: 10.1371/journal.pone.0029717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Betts MR, Price DA, Brenchley JM, Lore K, Guenaga FJ, Smed-Sorensen a, Ambrozak DR, Migueles SA, Connors M, Roederer M, Douek DC, Koup RA. 2004. The functional profile of primary human antiviral CD8+ T cell effector activity is dictated by cognate peptide concentration. J Immunol 172:6407–6417. doi: 10.4049/jimmunol.172.10.6407. [DOI] [PubMed] [Google Scholar]
- 51.Mens H, Kearney M, Wiegand A, Shao W, Schønning K, Gerstoft J, Obel N, Maldarelli F, Mellors JW, Benfield T, Coffin JM. 2010. HIV-1 continues to replicate and evolve in patients with natural control of HIV infection. J Virol 84:12971–12981. doi: 10.1128/JVI.00387-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benito JM, Ortiz MC, León A, Sarabia LA, Ligos JM, Montoya M, Garcia M, Ruiz-Mateos E, Palacios R, Cabello A, Restrepo C, Rodriguez C, Del Romero J, Leal M, Muñoz-Fernández MA, Alcamí J, García F, Górgolas M, Rallón N, ECRIS integrated in the Spanish AIDS Research Network. 2018. Class-modeling analysis reveals T-cell homeostasis disturbances involved in loss of immune control in elite controllers. BMC Med 16:30. doi: 10.1186/s12916-018-1026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hunt PW, Brenchley J, Sinclair E, McCune JM, Roland M, Page Shafer K, Hsue P, Emu B, Krone M, Lampiris H, Douek D, Martin JN, Deeks SG. 2008. Relationship between T cell activation and CD4+ T cell count in HIV‐seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis 197:126–133. doi: 10.1086/524143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walker-Sperling VE, Pohlmeyer CW, Veenhuis RT, May M, Luna KA, Kirkpatrick AR, Laeyendecker O, Cox AL, Carrington M, Bailey JR, Arduino RC, Blankson JN. 2017. Factors associated with the control of viral replication and virologic breakthrough in a recently infected HIV-1 controller. EBioMedicine 16:141–149. doi: 10.1016/j.ebiom.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pulko V, Davies JS, Martinez C, Lanteri MC, Busch MP, Diamond MS, Knox K, Bush EC, Sims PA, Sinari S, Billheimer D, Haddad EK, Murray KO, Wertheimer AM, Nikolich-Žugich J. 2016. Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat Immunol 17:966–975. doi: 10.1038/ni.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li JZ, Etemad B, Ahmed H, Aga E, Bosch RJ, Mellors JW, Kuritzkes DR, Lederman MM, Para M, Gandhi RT. 2015. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 30:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Namazi G, Fajnzylber JM, Aga E, Bosch R, Acosta EP, Sharaf R, Hartogensis W, Jacobson JM, Connick E, Volberding P, Skiest D, Margolis D, Sneller MC, Little SJ, Gianella S, Smith D, Kuritzkes DR, Gulick RM, Mellors JW, Mehraj V, Gandhi RT, Mitsuyasu R, Schooley RT, Henry K, Tebas P, Deeks S, Chun T-W, Collier AC, Routy J-P, Hecht FM, Walker BD, Li JZ. 2018. The Control of HIV after Antiretroviral Medication Pause (CHAMP) study: post-treatment controllers identified from 14 clinical studies. J Infect Dis 218:1954–1963. doi: 10.1093/infdis/jiy479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharaf R, Lee GQ, Sun X, Etemad B, Aboukhater LM, Hu Z, Brumme ZL, Aga E, Bosch RJ, Wen Y, Namazi G, Gao C, Acosta EP, Gandhi RT, Jacobson JM, Skiest D, Margolis DM, Mitsuyasu R, Volberding P, Connick E, Kuritzkes DR, Lederman MM, Yu XG, Lichterfeld M, Li JZ. 2018. HIV-1 proviral landscapes distinguish posttreatment controllers from noncontrollers. J Clin Invest 128:4074–4085. doi: 10.1172/JCI120549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shytaj IL, Nickel G, Arts E, Farrell N, Biffoni M, Pal R, Chung HK, LaBranche C, Montefiori D, Vargas-Inchaustegui D, Robert-Guroff M, Lewis MG, Sacha JB, Palamara AT, Savarino A. 2015. Two-year follow-up of macaques developing intermittent control of the human immunodeficiency virus homolog simian immunodeficiency virus SIVmac251 in the chronic phase of infection. J Virol 89:7521–7535. doi: 10.1128/JVI.00396-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burwitz BJ, Giraldo-Vela JP, Reed J, Newman LP, Bean AT, Nimityongskul FA, Castrovinci PA, Maness NJ, Leon EJ, Rudersdorf R, Sacha JB. 2012. CD8+ and CD4+ cytotoxic T cell escape mutations precede breakthrough SIVmac239 viremia in an elite controller. Retrovirology 9:91. doi: 10.1186/1742-4690-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thèze J, Chakrabarti LA, Vingert B, Porichis F, Kaufmann DE. 2011. HIV controllers: a multifactorial phenotype of spontaneous viral suppression. Clin Immunol 141:15–30. doi: 10.1016/j.clim.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elahi S, Dinges WL, Lejarcegui N, Laing KJ, Collier AC, Koelle DM, McElrath MJ, Horton H. 2011. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat Med 17:989–995. doi: 10.1038/nm.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bonjoch A, Pou C, Perez-Alvarez N, Bellido R, Casadella M, Puig J, Noguera-Julian M, Clotet B, Negredo E, Paredes R. 2013. Switching the third drug of antiretroviral therapy to maraviroc in aviraemic subjects: a pilot, prospective, randomized clinical trial. J Antimicrob Chemother 68:1382–1387. doi: 10.1093/jac/dks539. [DOI] [PubMed] [Google Scholar]
- 64.González-Galarza FF, Takeshita LYC, Santos EJM, Kempson F, Maia MHT, da Silva ALS, Silva ALT. e, Ghattaoraya GS, Alfirevic A, Jones AR, Middleton D. 2015. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res 43:D784–D788. doi: 10.1093/nar/gku1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 66.Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 67.Tamura K, Nei M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 68.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang H, Wu H, Hancock G, Clutton G, Sande N, Xu X, Yan H, Huang X, Angus B, Kuldanek K, Fidler S, Denny TN, Birks J, McMichael AJ, Dorrell L. 2012. Antiviral inhibitory capacity of CD8+ T cells predicts the rate of CD4+ T-cell decline in HIV-1 infection. J Infect Dis 206:552–561. doi: 10.1093/infdis/jis379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang H, Yorke E, Hancock G, Clutton G, Sande N, Angus B, Smyth R, Mak J, Dorrell L. 2013. Improved quantification of HIV-1-infected CD4 + T cells using an optimised method of intracellular HIV-1 gag p24 antigen detection. J Immunol Methods 391:174–178. doi: 10.1016/j.jim.2013.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Gag sequences are available at GenBank with the accession numbers MK086511 (LP1), MK086512 (LP2 pre), MK086513 (LP2 post), MK086514 (LP3 pre), MK086515 (LP3 post), MK086516 (LP4 pre), MK086517 (LP4 post), MK086518 (LP5 pre), MK086519 (LP5 post), MK086520 (LP6 post), MK086521 (LP7 post), MK086522 (LP8 pre), MK086523 (LP8 post), MK086524 (LP9 pre), MK086525 (LP9 post), MK086526 (LP10 post), MK086527 (LP11 pre), MK086528 (LP11 post), MK086529 (LP12 pre), MK086530 (LP12 post), MK086531 (LP13 post), MK086532 (LP14 pre), and MK086533 (LP14 post).